")
Back to Journals » Drug Design, Development and Therapy » Volume 16
Novel 2-(5-Aryl-4,5-Dihydropyrazol-1-yl)thiazol-4-One as EGFR Inhibitors: Synthesis, Biological Assessment and Molecular Docking Insights
Authors Al-Warhi T, El Kerdawy AM , Said MA , Albohy A , Elsayed ZM, Aljaeed N, Elkaeed EB , Eldehna WM , Abdel-Aziz HA , Abdelmoaz MA
Received 4 January 2022
Accepted for publication 12 April 2022
Published 16 May 2022 Volume 2022:16 Pages 1457—1471
DOI https://doi.org/10.2147/DDDT.S356988
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Tin Wui Wong
Tarfah Al-Warhi,1 Ahmed M El Kerdawy,2,3 Mohamed A Said,4 Amgad Albohy,5 Zainab M Elsayed,6 Nada Aljaeed,1 Eslam B Elkaeed,7 Wagdy M Eldehna,8,9 Hatem A Abdel-Aziz,10 Miral A Abdelmoaz11
1Department of Chemistry, College of Science, Princess Nourah Bint Abdulrahman University, Riyadh, Saudi Arabia; 2Department of Pharmaceutical Chemistry, Faculty of Pharmacy, Cairo University, Cairo, Egypt; 3Department of Pharmaceutical Chemistry, School of Pharmacy, Newgiza University (NGU), Newgiza, Cairo, Egypt; 4Department of Pharmaceutical Chemistry, Faculty of Pharmacy, Egyptian Russian University, Badr City, Cairo, 11829, Egypt; 5Department of Pharmaceutical Chemistry, Faculty of Pharmacy, The British University in Egypt (BUE), El-Sherouk City, Cairo, 11837, Egypt; 6Scientific Research and Innovation Support Unit, Faculty of Pharmacy, Kafrelsheikh University, Kafrelsheikh, Egypt; 7Department of Pharmaceutical Sciences, College of Pharmacy, Almaarefa University, Riyadh, 13713, Saudi Arabia; 8School of Biotechnology, Badr University in Cairo, Badr City, Cairo, 11829, Egypt; 9Department of Pharmaceutical Chemistry, Faculty of Pharmacy, Kafrelsheikh University, Kafrelsheikh, 33516, Egypt; 10Department of Applied Organic Chemistry, National Research Center, Dokki, Giza, 12622, Egypt; 11Department of Pharmaceutical Chemistry, Faculty of Pharmacy, Sinai University, Kantra, Egypt
Correspondence: Wagdy M Eldehna, Department of Pharmaceutical Chemistry, Faculty of Pharmacy, Kafrelsheikh University, Kafrelsheikh, 33516, Egypt, Tel +201068837640, Email [email protected]
Introduction: Epidermal growth factor receptor (EGFR) regulates several cell functions which include cell growth, survival, multiplication, differentiation, and apoptosis. Currently, EGFR kinase inhibitors are of increasing interest as promising targeted antitumor therapeutic agents.
Methods: Different thiazolyl-pyrazoline derivatives ( 7a-o) were synthesized and were first tested for anti-proliferative effect towards the A549 lung cancer cell line and the T-47D breast cancer cell line in MTT assay. Thereafter, thiazolyl-pyrazolines ( 7b, 7g, 7l, and 7m) were subsequently evaluated for their PK inhibition for EGFR. Moreover, representative promising derivatives ( 7g and 7m) in cytotoxic and PK inhibition assays were tested to investigate their impact on the apoptosis and cell cycle phases in T-47D cells in order to explore more insights into the antitumor actions of the target thiazolyl-pyrazolines. Furthermore, docking studies were accomplished to evaluate the patterns of binding of thiazolyl-pyrazolines 7b, 7g, 7l, and 7m in the EGFR active pocket (PDB ID: 1M17).
Results: Testing the thiazolyl pyrazoline compounds 7a-o on A549 and T-47D cell lines showed IC50 arrays between 3.92 and 89.03 μM, and between 0.75 and 77.10 μM, respectively. Also, the tested thiazolyl-pyrazolines ( 7b, 7g, 7l, and 7m) demonstrated significant sub-micromolar EGFR inhibitory actions with IC50 values 83, 262, 171 and 305 nM, respectively, in comparison to erlotinib (IC50 =57 nM).
Discussion: Generally, it was observed that the tested thiazolyl pyrazolines showed more potent antiproliferative activity toward breast cancer cells T-47D than toward lung cancer cell lines A549. In particular, thiazolyl pyrazolines 7g and 7m showed the best activity against A549 cells (IC50 = 3.92 and 6.53 μM) and T-47D cells (IC50 = 0.88 and 0.75 μM). Compounds 7g and 7m provoked a sub-G1 phase arrest and cell apoptosis which are in agreement with the expected outcome of EGFR inhibition. Finally, the molecular docking of 7g and 7m in the active site of EGFR revealed a common binding pattern similar to that of erlotinib which involves the accommodation of the 1,3 thiazol-4-one ring and pyrazoline ring of target compounds in the binding region of erlotinib’s quinazoline ring and anilino moiety.
Keywords: antitumor, EGFR inhibitors, pyrazolo, molecular docking, breast cancer, lung cancer
Introduction
Cancer stands as a baleful fatal challenge, in the current medical era, with regard to morbidity and mortality ranks second only to cardiovascular diseases and is expected to overtake the leading death cause globally in the near future.1 Accordingly, World Health Organization (WHO) reported that the number of cancer deaths could reach 13 million by the end of 2030.1,2 It is estimated that one out of every five people under the age of 75 will develop cancer during their lifetime.1–3 Most cancers are characterized by uncontrolled cell division, growth, and impaired proliferation because of the deregulation of the necessary proteins and enzymes that control cell division.4 Despite the numerous efforts to treat cancer diseases and the significant progress from cancer diagnosis to cancer treatment, drug developers worldwide face a slew of issues stemming from the several undesirable side effects of conventional non-selective chemotherapeutic agents such as systemic toxicity, decreased bioavailability, and drug resistance.5
The emergence of targeted therapies specifically the signaling-pathways-targeting chemotherapeutic agents construct a new era of selective chemotherapeutic hits that could attack cancer cells and/or the tumor microenvironment responsible for tumor proliferation, resulting in greater efficacy and minor side effects on normal cells.6,7 One of the most principal signaling pathways was the tyrosine kinase (TK) Epidermal Growth Factor Receptor (EGFR) that regulates several cell functions which include cell growth, survival, multiplication, differentiation, and apoptosis. The EGFR is activated as a result of conformational changes caused by binding interactions of the endogenous EGF ligand within EGFR extracellular binding domain.8,9 As a result, several tyrosine residues are autophosphorylated resulting in a downstream activation of Mitogen Activated Protein Kinase (MAPK) pathway through the phosphorylation of following proteins in the pathway. Remarkable mutations in multiple proteins of the MAPK pathway, including EGFR, have been linked to various cancer types which include the colorectal carcinoma, hepatocellular carcinoma, pancreatic cancer, breast cancer, as well as the non-small cell lung cancer.10 This explains why EGFR kinase inhibitors are of increasing interest as promising targeted antitumor therapeutic agents.
Both pyrazoline- and thiazole-bearing small molecules have variable forms of biological activities, including anti-inflammatory, anti-cholinesterase, antimalarial, anti-carbonic anhydrase, and antimicrobial.11–17 In particular, pyrazolo- and thiazolo-based small molecules are significant organic compounds that have been extensively studied and reported for their antitumor activity.18–21 Surveying literature showed that there has been a remarkable rise in the amount of published studies investigating the antitumor and kinase inhibitory activities of both pyrazoles and thiazoles.9,21–27 For example, Akhtar et al28 reported new pyrazoline-bearing EGFR inhibitors (Compound I, Figure 1) with potent cytotoxic effect against several human cancer cell lines which include breast cells MDA-MB231, and lung cells A549. Moreover, in 2021 Kamonpan et al29 investigated the antitumor effect of a series of thiazole-based chalcone derivatives against lung carcinoma cells A549 overexpressing EGFR. Interestingly, compound II (Figure 1) showed promising inhibitory activities against both lung carcinoma cells (A549) growth and EGFR TK (IC50 of 16.30 µM and 33 nM, respectively) upon comparison to the reference drug erlotinib.
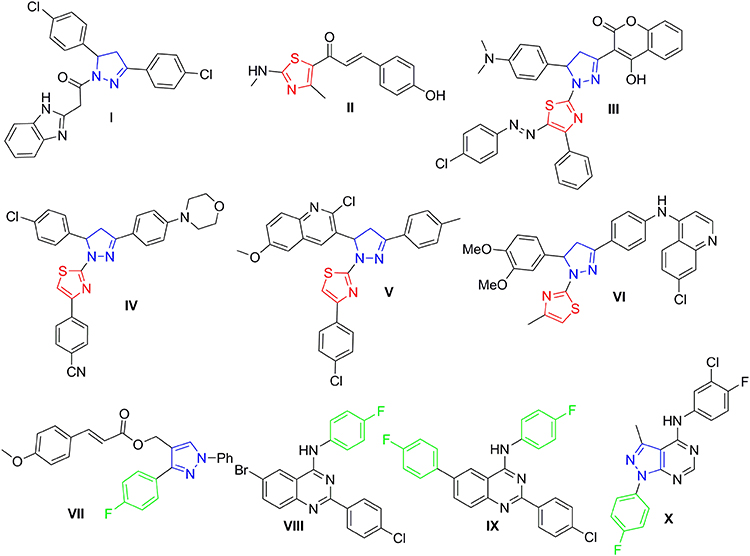 |
Figure 1 Structures of certain pyrazoline- and/or thiazole-bearing small molecules (I–X) endowed with anticancer and kinases inhibitory activities. |
In the last few years, many research groups were interested in designing and synthesizing diverse thiazolyl-pyrazoline hybrids in order to gain the most beneficial antitumor activity of both rings.30–33 In 2019, Mohamed et al34 synthesized novel series of thiazolylpyrazolyl coumarin derivatives as new effective vascular endothelial growth factor receptor-2 (VEGFR-2) inhibitors. In particular, the polycyclic lead compound III (Figure 1) exhibited a remarkably potent VEGFR-2 inhibitory effect (IC50= 34 nM), in addition to a significant growth inhibitory effect against different human cancer cell lines especially breast MCF-7 cells (IC50= 5.41 μM). Moreover, another new series of dual EGFR/HER2 inhibitors based on a hybrid thiazolyl/pyrazoline scaffold has been reported recently which exhibited promising anti-proliferative activity towards lung A549 and breast MCF-7 cancer cell lines in MTT cytotoxicity assay.35 Compound IV (Figure 1) inhibited HER2 and EGFR kinases possessing IC50 of 2.28 μM and 4.34 μM, respectively. Moreover, it exerted moderate anti-proliferative effect against the two examined cell lines with IC50 of 10.76 μM and 8.05 μM, respectively.35 By the end of 2019, another study reported two series of dihydropyrazole thiazole hybrids, one of the promising derivatives was compound V (Figure 1) that exerted strong anti-proliferative impact against breast cancer cell line (MCF-7) (IC50 of 0.227 μM) through inhibition of EGFR with an IC50 of 31.8 nM with a positive control (Gefitinib) IC50 of 29.16 nM.36 Recently in 2021, novel thiazolylpyrazoline-based small molecules were reported by Batran et al;37 from which, compound VI (Figure 1) showed dual kinase inhibitory effect towards EGFR and HER2 with IC50 of 60 nM and 80 nM, respectively, versus 40 nM for erlotinib.
Several studies reported 4-fluorophenyl moiety as a key part of several potent EGFR inhibitors with pronounced cytotoxic activity on several cancer cell lines.38–40 The 4-fluorophenyl pyrazole derivative VII (Figure 1) showed a potent cytotoxic activity on MCF-7 breast cancer cell line with IC50 of 3.87 μM and a promising EGFR and HER-2 inhibitory activity with IC50 of 4.87 and 6.23 μM, respectively.38 The 4-fluorophenyl quinazolines VIII and IX (Figure 1) exhibited promising cytotoxic activity on MCF-7 and HeLa cell lines with IC50 of 1.44 μM and 2.70 μM, respectively, on MCF-7 and IC50 of 4.97 μM and 1.39 μM, respectively, on HeLa cell lines. Moreover, they showed effective EGFR inhibitory activity with IC50 of 37.66 nM and 59.21 nM, respectively.39 Finally, the 4-fluorophenyl pyrazolo[3,4-d]pyrimidine derivative X (Figure 1) proved prominent anticancer activity against OVCAR-4, NCI-H460, and ACHN cell lines with IC50 of 1.74, 4.44, and 5.53 μM, respectively, more potent than the used reference standard (erlotinib); it inhibited EGFR and HER2 kinases at sub-micromolar level (IC50 of 0.18 and 0.25 μM, respectively).40
The pronounced cytotoxic activity of the pyrazoline/thiazole hybrid compounds III–VI and the 4-fluorophenyl derivatives VIII–X which attributed to their remarkable kinase inhibitory activity on EGFR, HER2, and/or VEGFR2 encouraged us to design and synthesize a novel series of 4-fluorophenyl substituted thiazolyl-pyrazoline derivatives (7a-o) (Figure 2). They were further evaluated for their cytotoxic activity against the lung cancer cell line A549 and the breast cancer cell line T-47D using MTT assay. Derivatives exhibited potent anti-proliferative action were subsequently evaluated for their PK inhibition for EGFR. Representative promising derivatives in cytotoxic and PK inhibition were further tested for their effect on cell apoptosis and cell cycle progression in breast cancer cell line (T-47D). In addition, molecular docking simulations were performed to predict the binding pattern of the target thiazolyl-pyrazoline hybrids in EGFR kinase domain (PDB ID: 1M17).
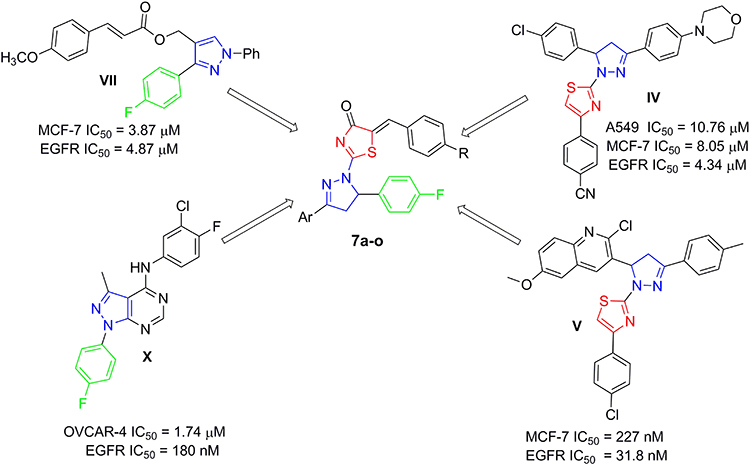 |
Figure 2 Design of the target thiazolyl pyrazolines 7a-o, based on the structure of compounds IV, V, VII and X. |
 |
Figure 3 Pro-apoptotic effect of thiazolyl pyrazolines 7g and 7m toward breast cancer cells T-47D. |
Materials and Methods
Chemical Synthesis
General
The proton NMR spectra were collected on a Bruker 400 MHz spectrometer for all herein prepared novel intermediates and final target thiazolyl pyrazolines. 13C NMR spectra were done in deuterated dimethylsulfoxide (DMSO-d6) using 100 MHz frequency. Chemical shifts (δH) have been reported relative to the solvent (DMSO-d6) peaks. IR spectra were collected on Shimadzu FT-IR 8400S spectrophotometer. Elemental analyses (EI) were measured at the Regional Center for Microbiology and Biotechnology, Al-Azhar University, Egypt. Compounds 3a-c, 4a-c and 5a-b were reported previously.41,42
Synthesis of Thiazolidenone Derivatives 5a-c
The appropriate pyrazoline thioamide derivative 4a-c (6 mmol) was heated under reflux with equimolar amount of ethyl bromoacetate (1 g, 6 mmol) in absolute EtOH (30 mL), with TLC monitoring. After 4 hours, the starting materials were consumed, and the whole mixture was cooled to lab temperature. The resulting precipitate was collected with filtration, washed with petroleum ether and recrystallized from dioxane to produce the key thiazolidenone intermediates 5a-c in a good yield (87–91%).
Synthesis of the Target Thiazolyl-Pyrazolines 7a-o
To a hot stirring solution of thiazolidenone intermediates 5a-c (1 mmol) in glacial acetic acid (10 mL), the appropriate aldehydes 6a-e (1.1 mmol) and sodium acetate (0.1 g, 1.2 mmol) were mixed. The whole solution was heated for 5 hours then the resulted precipitate was collected by filtration on hot, washed using hexane and crystallized from DMF/ethanol mixture to provide the thiazolyl-pyrazolines 7a-o in a good yield (78–90%).
Full characterisation (physical and spectral data) for the key intermediate (5c) and the target thiazolyl pyrazolines (7a-o) were presented in the Supporting Information.
Biological Evaluation
All experimental procedures utilized in the biological cytotoxicity,43,44 Annexin V-FITC Apoptosis,45 cell cycle,46 and EGFR TK47 assays were carried out as reported earlier and were shown in the Supporting Information.
Molecular Docking Studies
Molecular docking was carried out using Molecular Operating Environment software (MOE, 2020.0901), using X-ray crystallographic structure of EGFR co-crystallized with erlotinib (PDB code: 1M17). The details of the utilized protocol as well as its validation were provided in the Supporting Information.
Results and Discussion
Synthesis of Compounds
The preparation of pyrazoline derivatives 7a-o was done through the synthetic scheme depicted in Scheme 1. The chalcones intermediate 3a-c were prepared through a condensation reaction of different acetophenones 1a-c with 4-flourobenzaldehyde 2 via the reported procedures.48 The previously prepared chalcones 3a-c subjected to heterocyclization with thiosemicarbazide in basic condition (using NaOH) to furnish the pyrazoline derivatives 4a-c. The pyrazoline derivatives 4a-c were further cyclized with ethyl bromoacetate in absolute ethyl alcohol to produce the key thiazolidenone intermediates 5a-c. Then, a knoevenagel condensation reaction between thiazolidenones 5a-c and different aldehydes 6a-e, in refluxing acetic acid and sodium acetate, led to the production of thiazolyl-pyrazoline derivatives 7a-o. Scheme 1 illustrates these steps.
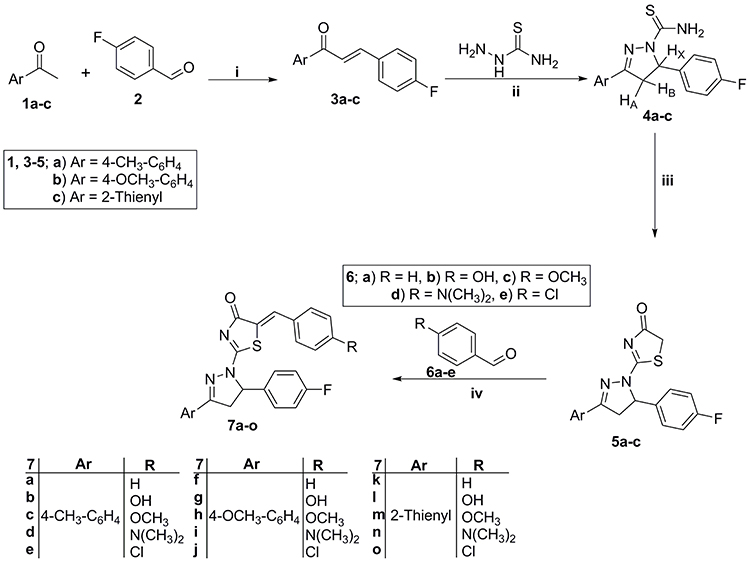 |
Scheme 1 Preparation of key thiazolyl-pyrazolines 7a-o products; Reagents and conditions: (i) 40% NaOH, 95% ethanol, stirring R.T. 8 hr; (ii) NaOH, ethanol, reflux 2 hr; (iii) Ethyl bromoacetate, absolute ethyl alcohol, reflux 4 hr; (iv) Glacial acetic acid, sodium acetate, reflux 5 hr. |
All of the prepared thiazolyl-pyrazolines 7a-o showed satisfactory spectroscopic and analytical data that have complete agreement with respective structures. The proton NMR spectra of the targeted thiazolyl-pyrazolines clearly show a couple of doublets of doublet signals attributable to the diastereotopic protons (HA and HB) of -CH2- group at C-4 of the pyrazoline ring around δ 3.50 and 4.0 ppm. Moreover, a third doublets of doublet signal corresponding to the C-5 proton of the pyrazoline motif around δ 5.50 ppm because of the vicinal coupling between the non-equivalent methylene hydrogens at C-4 of the pyrazoline moiety.
In addition, the 1H NMR spectra of thiazolyl-pyrazolines 7c, 7f–j and 7m confirmed the presence of extra aliphatic signals attributable to the protons of the methoxy group around δ 3.85 ppm, while the spectra of thiazolyl-pyrazolines 7a–e revealed the presence of another aliphatic signals for CH3 group protons around δ 2.30 ppm. Moreover, the structure of thiazolyl-pyrazolines 7d, 7i and 7n was confirmed with the existence of a singlet signal of (N(CH3)2) protons at δ 3.0 ppm, whereas, the structure of compounds 7b, 7g and 7l was proven with the presence of an exchangeable proton of OH functionality at δ 10.10–10.21 ppm. Moreover, the 13C NMR spectra of the targeted thiazolyl pyrazoline derivatives 7a–o confirmed the presence of the two characteristic aliphatic signals of pyrazoline ring (C-4 and C-5) around δ 44.50 and 67.50 ppm, respectively.
Biological Evaluation
Using MTT assay, the novel new prepared thiazolyl-pyrazolines 7a–o were first evaluated for their anti-proliferative effect on the lung cancer cell line A549 and the breast cancer cell line T-47D. Compounds exhibited potent anti-proliferative action, were subsequently evaluated for their PK inhibition for EGFR. Representative promising derivatives in cytotoxic and PK inhibition were tested on T-47D cell line apoptosis and cell cycle progression.
In vitro Cytotoxic Activity
It is well-reported that EGFR is frequently overexpressed in diverse human malignancies such as non‐small cell lung cancer and breast cancer, as well as, it stands out as a key molecule in the initiation and progression for these tumors.49–52 In this study, A549 and T-47D cell lines were selected as representative ones for the NSCLC and breast cancer respectively. Using the MTT assay, all the prepared thiazolyl-pyrazolines 7a–o were tested for their inhibitory effect on two different cell lines including A549 lung cancer and the T-47D breast cancer cell lines, and they were compared to Staurosporine and erlotinib as a standard reference anticancer drugs.53 IC50 values of target compounds against both cell lines are shown in Table 1.
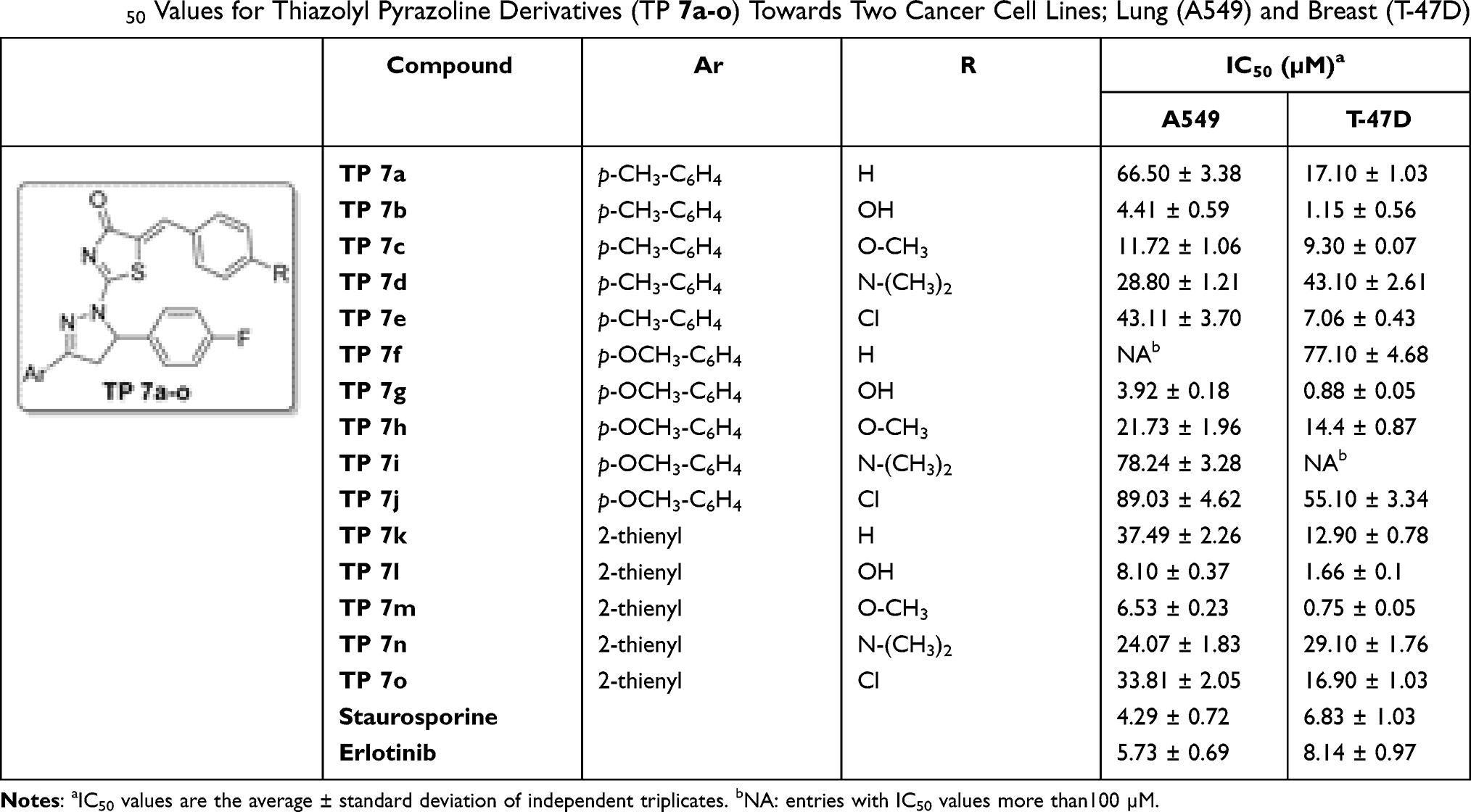 |
Table 1 IC50 Values for Thiazolyl Pyrazoline Derivatives (TP 7a-o) Towards Two Cancer Cell Lines; Lung (A549) and Breast (T-47D) |
Testing the thiazolyl pyrazoline compounds 7a–o on A549 cell line showed an IC50 array between 3.92 and 89.03 µM, with compounds 7b and 7g showing the most potent in vitro anti-proliferative impact with (IC50 of 4.41 and 3.92 µM, respectively) comparable to staurosporine (IC50 = 4.29 µM). Moreover, compounds 7l and 7m showed a moderate antiproliferative activity (IC50 = 8.10 and 6.53 µM, respectively), whereas the remainder of compounds showed modest antiproliferative activities (IC50 are between 10 and 100 µM) (Table 1).
Testing the thiazolyl pyrazoline compounds on T-47D cell line exhibited an IC50 array between 0.75 and 77.10 µM. Compounds 7b, 7g, 7l and 7m showed more potent antiproliferative activity (IC50 of 0.75, 0.88, 1.15, and 1.66 µM, respectively) than the used reference standard (staurosporine) (IC50 = 6.83 µM). Moreover, compounds 7e and 7c showed moderate cytotoxic activity with IC50 of 7.06 and 9.30 µM, respectively, while the remainder compounds showed modest antiproliferative activities with IC50 ranged between 10 and 100 µM. Generally, the tested pyrazoline derivatives showed more potent antiproliferative activity on the breast cancer cell line (T-47D) than on the lung cancer cell line (A549) (Table 1).
As can be seen in Table 1, the hydrophilic p-hydroxy substituted phenyl ring improves the antiproliferative activity of the tested derivatives against both used cell lines as indicated by the significant potency of thiazolyl pyrazoline 7b, 7g and 7l with IC50 of 4.41, 3.92 and 8.10 µM, respectively, towards A549 cell line and IC50 of 1.15, 0.88 and 1.66 µM, respectively, against T-47D cell line compared to staurosporine (IC50 = 4.29 and 6.83 µM, respectively). Moreover, the p-methoxyphenyl derivatives 7c, 7h and 7m showed potent to moderate antiproliferative activity against both used cell lines with IC50 of 11.72, 21.73 and 6.53 µM, respectively, against A549 cell line and IC50 of 9.30, 14.40 and 0.75 µM, respectively, against T-47D cell line. Thiazolyl pyrazoline 7g and 7m showed the most potent antiproliferative activity toward the tested cell lines with IC50 equal 3.92 and 6.53 µM, respectively, against A549 cell line and IC50 of 0.88 and 0.75 µM, respectively, toward T-47D cell line.
From the presented IC50 values in Table 1, we can draw valuable structure activity relationships (SARs). First, the obtained cell growth inhibition data ascribed to the target thiazolyl pyrazoline derivatives 7a–o more enhanced activity toward breast T-47D cell line than lung A549 cell line, with an exception for N,N-dimethylamino bearing compounds 7d, 7i and 7n which displayed slight better activity against A549 cell line (IC50 = 28.80 ± 1.21, 78.24 ± 3.28 and 24.07 ± 1.83 µM, respectively) than T-47D cell line (IC50 = 43.10 ± 2.61, > 100 and 29.10 ± 1.76 µM, respectively), Table 1.
Upon investigation of impact of the substitution of the benzylidine motif, appended on C-5 of the thiazol-4-one ring, we can see that grafting of different substituents within the para positon such as hydroxyl, methoxy, N,N-dimethylamino and chloro substituents, resulted in an enhanced anti-proliferative activity against lung A549 cell line. This could be evidenced by the lower IC50 values of the substituted thiazolyl pyrazoline derivatives 7b–d (IC50 range: 4.41 ± 0.59–43.11 ± 3.70 µM), 7g–j (IC50 range: 3.92 ± 0.18–89.03 ± 4.62 µM), and 7l–o (IC50 range: 6.53 ± 0.23–33.81 ± 2.05 µM), comparing to their unsubstituted analogues 7a, 7f and 7k (IC50 = 66.50 ± 3.38, > 100 and 37.49 ± 2.26 µM), respectively. In like manner, the para substitution of the benzylidine motif in the target thiazolyl pyrazolines with hydroxyl, methoxy, and chloro substituents boosted the anti-proliferative activity against breast T-47D cell line. Compounds 7b, 7c and 7e displayed better anti-proliferative activity (IC50 = 1.15 ± 0.56, 9.30 ± 0.07 and 7.06 ± 0.43 µM, respectively) than compound 7a (IC50 = 17.10 ± 1.03 µM), as well as thiazolyl pyrazolines 7g, 7h and 7j were more potent than their unsubstituted counterpart 7f (IC50 = 77.10 ± 4.68 µM). On the other hand, para substitution of the benzylidine moiety with N,N-dimethylamino functionality resulted in target thiazolyl pyrazolines 7d, 7i and 7n with decreased anti-proliferative activity against T-47D cell line (IC50 = 43.10 ± 2.61, > 100 and 29.10 ± 1.76 µM, respectively) than their unsubstituted counterparts 7a, 7f and 7k (IC50 = 17.10 ± 1.03, 77.10 ± 4.68 and 12.90 ± 0.78 µM, respectively).
Concerning the SAR about the aryl moiety appended to C-3 of the pyrazoline ring, it is worth mentioning that the bioisosteric replacement of the 4-methyl/methoxy phenyl ring with the 2-thienyl heterocycle led to an enhancement of the anti-proliferative activity toward both examined cell lines (A549 and T-47D), except the 4-hydroxyl derivatives 7b and 7g.
Furthermore, the cytotoxic activity of thiazolyl-pyrazolines 7b, 7g, 7l and 7m was assessed against non-tumorigenic breast MCF-10A cell line to explore their selectivity toward the cancer cells. Interestingly, the examined thiazolyl-pyrazolines exerted non-significant toxicity towards the normal breast MCF-10A cells with IC50s equal 48.19 ± 3.71, 55.03 ± 4.53, 72.84 ± 4.02 and 39.70 ± 2.68, respectively (Table 2).
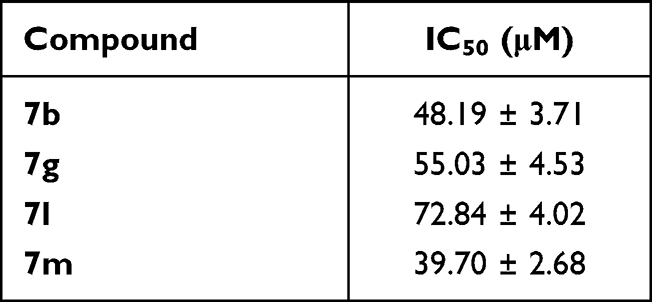 |
Table 2 Cytotoxic Activity of Thiazolyl-Pyrazolines 7b, 7g, 7l and 7m Against Non-Tumorigenic Breast MCF-10A Cell Line |
EGFR Inhibitory Activity
Compounds with significant antiproliferative impact against the investigated cell lines, viz, 7b, 7g, 7l, and 7m, were biochemically assessed for their EGFR inhibitory activity and erlotinib was used as a reference standard, Table 3 summarizes these results. When compared to the employed reference standard, erlotinib (IC50 = 57 nM), the tried compounds demonstrated significant nanomolar inhibitory activities with IC50 values of 83, 262, 171 and 305 nM, respectively.
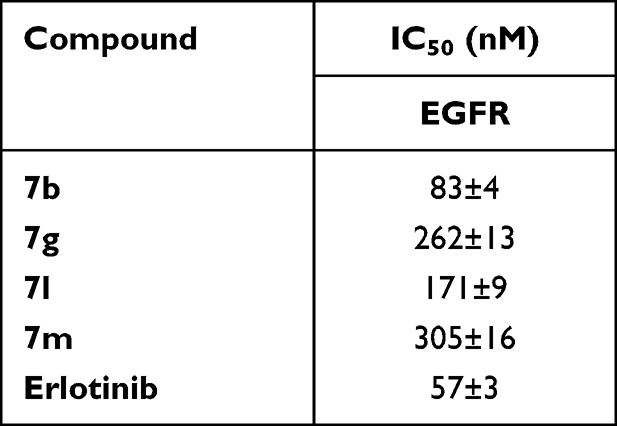 |
Table 3 Inhibitory Activity of 7b, 7g, 7l and 7m Against EGFR |
Regarding the correlation of cytotoxicity with EGFR inhibition, it is worth mentioning that the cell growth inhibition profile of thiazolyl-pyrazolines 7b, 7g, 7l and 7m was found to be rather flat, where the measured IC50 toward lung (A549) and breast (T-47D) cell lines ranged from 3.92 ± 0.18–8.10 ± 0.37 and 0.75 ± 0.05–1.66 ± 0.1 μM, respectively. The same could be observed for the resulting inhibitory activity against EGFR since the obtained IC50 values span from 83 ± 4 to 305 ± 16 nM, with about 3-fold difference in activity.
Cell Cycle Analysis
Compounds with significant antiproliferative and EGFR inhibitory activities 7g and 7m were next evaluated for the impact on cell cycle using flow cytometry technique utilizing propidium iodide (PI) staining. Cell cycle parameters for T-47D cells were compared after incubation of test compounds for 24 hours and after using DMSO as a negative control. Table 4 and Figure S3 show the results of this experiment.
 |
Table 4 Effect of Thiazolyl-Pyrazolines 7g and 7m on the Cell Cycle Phases of T-47D Cells |
According to Table 4, a significant increase of cells in the sub-G1 phase is seen, from 2.14% in control to 31.41 and 26.78% in cells incubated with 7g and 7m, respectively, which suggest a sub-G1 phase arrest and cell apoptosis which is the expected outcome of EGFR inhibition.54,55
The higher efficiency of compound 7g in cell arrest in the sub-G1 phase and in cell apoptosis which is seen in Table 4 compared to its 7m analogue agrees with its more potent antiproliferative effect on T-47D cell line as indicated by its IC50 of 3.92 μM in comparison to that of 7m (IC50 of 6.53 μM).
Apoptosis Assay
The potent thiazolyl-pyrazolines 7g and 7m were investigated for action on cells apoptosis using Annexin V-FITC/propidium iodide dual-staining.56 The apoptotic markers of the tested cell line, T-47D, were analyzed in the presence and absence of compounds 7g and 7m. Programmed cell death is characterized by the phosphatidylserine (PS) phospholipid translocation to the apoptotic cell surface. PS is stained with annexin V fluorescent conjugate then detected with flow cytometry. Additionally, T-47D cancer cells have been stained by the propidium iodide (PI) which is able to penetrate only the cells with broken plasma membranes; facilitating the differentiation among the early apoptotic cells (positive for PS, but negative for PI), late apoptotic and necrotic cells (positive for both PS and PI).
As can be seen in Table 5 and Figure 3, the total percentage of apoptotic cells in T-47D cell line increases because of compounds 7g and 7m treatment (30.38 and 25.25%, respectively) compared to non-treated cells (0.70%) which is a clear evidence of the apoptotic action of these two target compounds. Moreover, compounds 7g and 7m showed an increase in both early and late apoptotic phases where early phase was increased from 0.52 to 3.22 and 6.28 for 7g and 7m, respectively, while late phase was also increased from 0.18 to 27.16 and 18.97, respectively.
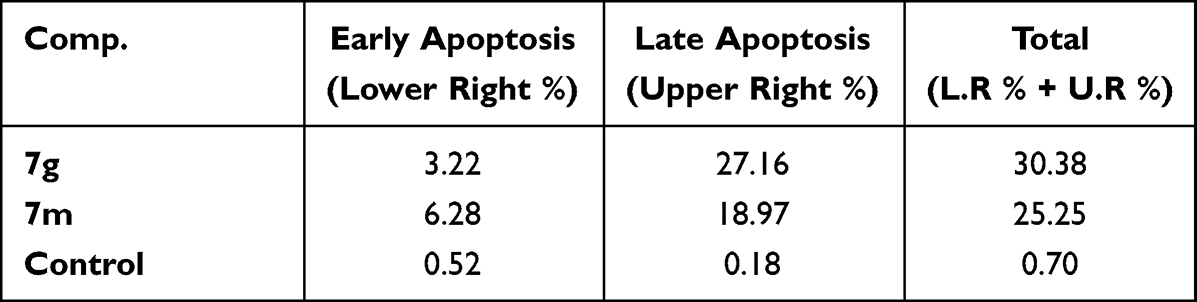 |
Table 5 Distribution of the Apoptotic Cells in the AnnexinV Assay in T-47D Cells Upon Treatment with Thiazolyl Pyrazolines 7g and 7m |
Likewise, The higher apoptotic effect of compound 7g (Table 5) relative to its 7m analogue agrees with its more potent antiproliferative effect on T-47D cell line, as indicated by its IC50 of 3.92 μM in comparison to that of 7m (IC50 of 6.53 μM).
Docking Study
The molecular docking was performed to study the binding interactions between thiazolyl pyrazolines 7b, 7g, 7l, and 7m and the EGFR active pocket (PDB ID: 1M17)57. The molecular docking procedure was first validated by self-docking of the known active co-crystallized ligand (erlotinib) in the proximity of the active site showing docking score (S) of −10.86 kcal/mol and RMSD of 1.46 Å. The ability of the docking protocol to predict the correct crystal structure pose was demonstrated by the presence of all key interactions between the residues in the active site and docked erlotinib indicating the suitability of it for the intended docking study. The key interactions are represented by the hydrogen bonding with Met 769, a water Hydrogen bond bridge with Thr 766 and cation-pi bond with Lys 721 (for further details, see Supporting Information Figures S1 and S2).
The investigated compounds showed a common binding pattern which involves the accommodation of the 1,3-thiazol-4-one ring in the binding region of erlotinib’s quinazoline ring involved in an essential hydrogen bond with the important residue Met 769 directing the 5-(p-hydroxy/p-methoxy phenyl) substitution towards the vicinity of the hydrophobic side chains of the amino acids Leu 694 and Leu 820.
The pyrazoline ring of the target compounds is accommodated in the binding region of erlotinib’s anilino moiety directing the substitutions in position 3 and 5 of the pyrazoline ring towards both sides of the binding site interacting through several interactions with Glu738, Met742, and Asp831 on one side and Arg817 on the other side. The good activity of the tested thiazolyl pyrazoline derivatives was rationalized by their ability to interact with the key amino acids in the binding site as indicated by their docking binding pattern and docking score which ranged between −11.14 and −10.64 kcal/mol that was comparable to that of erlotinib (S = −10.86 kcal/mol) (Figures 4–7, and Table 6).
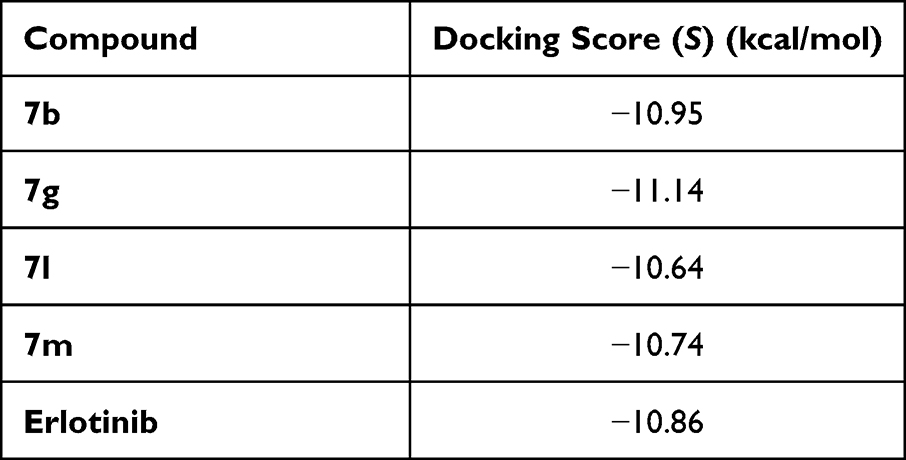 |
Table 6 Docking Energy Scores (S) of Reference and Tested Thiazolyl Pyrazoline |
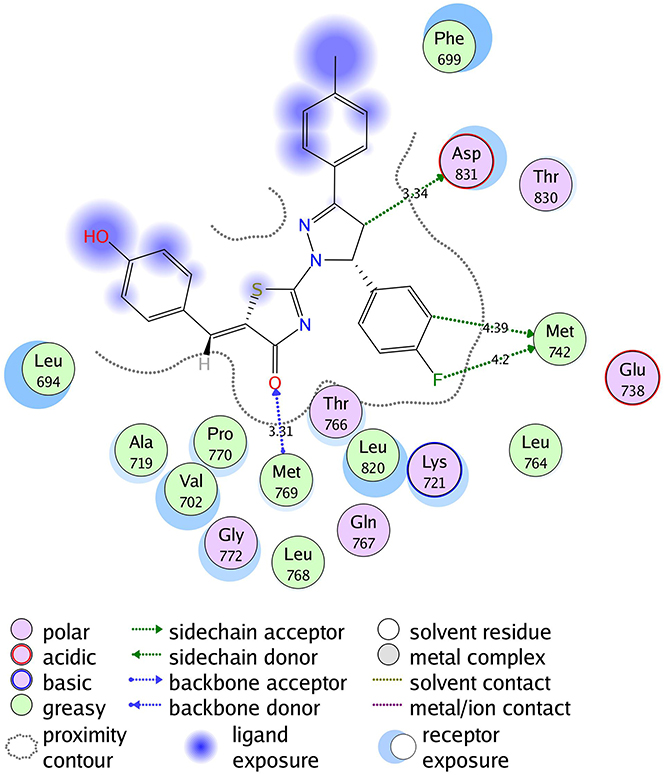 |
Figure 4 2D diagram of thiazolyl pyrazoline 7b in the binding site of EGFR. |
 |
Figure 5 2D diagram for thiazolyl pyrazoline 7g in the binding site of EGFR. |
 |
Figure 6 2D diagram of thiazolyl pyrazoline 7l in the binding site of EGFR. |
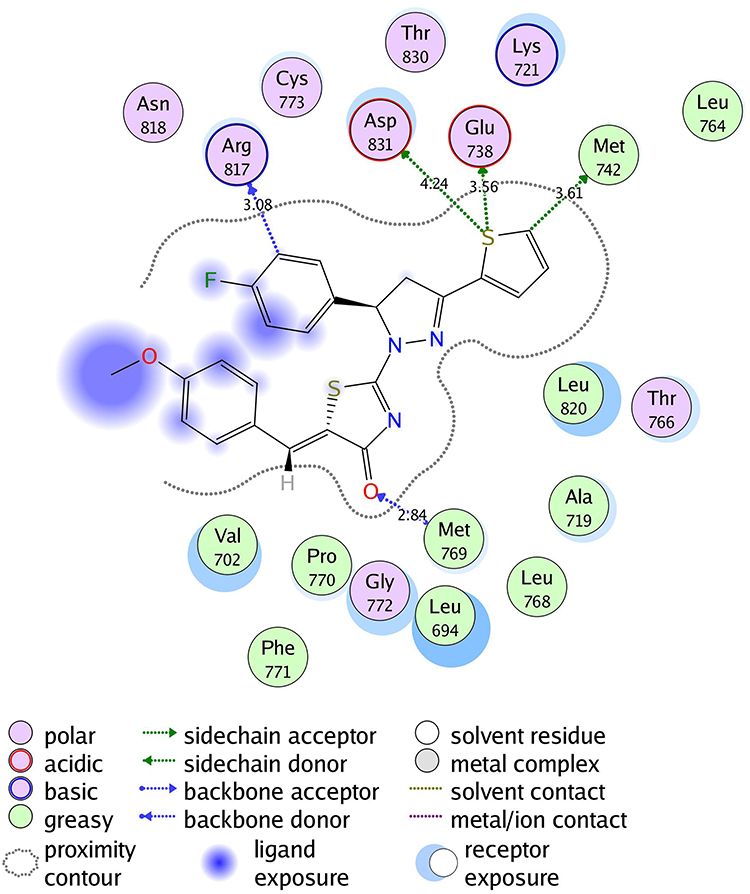 |
Figure 7 2D diagram of thiazolyl pyrazoline 7m in the EGFR binding site. |
Conclusions
Novel synthesized thiazolyl-pyrazoline derivatives (7a–o) were investigated in vitro for their potential anticancer effect against the lung cancer cell line A549 and the breast cancer cell line T-47D in MTT assay. Generally, it was observed that the tested thiazolyl pyrazolines showed more potent antiproliferative activity toward breast cancer cells T-47D than toward lung cancer cell lines A549. Four thiazolyl pyrazolines (7b, 7g, 7l, 7m), out of fifteen, appeared to be potent cell growth inhibitors for both the examined cell lines. In particular, thiazolyl pyrazolines 7g and 7m showed the best activity against A549 cells (IC50 = 3.92 and 6.53 µM, respectively) and T-47D cells (IC50 = 0.88 and 0.75 µM, respectively). Further in vitro biochemical evaluation was performed for the potent hits (7b, 7g, 7l, 7m) so as to assay their inhibitory effect for the EGFR kinase with respect to the approved EGFR inhibitor erlotinib. Target compounds demonstrated potent nanomolar inhibition activity with IC50 of 83, 262, 171 and 305 nM, respectively, in comparison to erlotinib (IC50 =57 nM). Furthermore, a flow-cytometric cell cycle assay was carried out on the breast T-47D cells for the promising thiazolyl pyrazolines 7g and 7m so as to explore their cellular mechanism of action. Compounds 7g and 7m provoked a sub-G1 phase arrest and cell apoptosis which are in agreement with the expected outcome of EGFR inhibition. Finally, the molecular docking of 7g and 7m in the active site of EGFR revealed a common binding pattern similar to that of erlotinib which involves the accommodation of the 1,3 thiazol-4-one ring and pyrazoline ring of target compounds in the binding region of erlotinib’s quinazoline ring and anilino moiety. Moreover, the ability of the tested thiazolyl pyrazolines to interact with the key amino acids in the binding site rationalizes their good activity as indicated by their docking binding pattern and docking score which ranged between −11.14 and −10.64 kcal/mol which is comparable to that of erlotinib (S = −10.86 kcal/mol).
Acknowledgments
This work is funded by the Deanship of Scientific Research at Princess Nourah bint Abdulrahman University, through the Research Groups Program Grant no (RGP-1440 -0025)(2).
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Kim SK, Kalimuthu S. Introduction to anticancer drugs from marine origin. In: Handbook of Anticancer Drugs from Marine Origin. Cham: Springer; 2015:1–13.
2. Emami S, Dadashpour S. Current developments of coumarin-based anti-cancer agents in medicinal chemistry. Eur J Med Chem. 2015;102:611–630. doi:10.1016/j.ejmech.2015.08.033
3. Ferlay J. GLOBOCAN 2008 v1. 2, cancer incidence and mortality world-wide: IARC Cancer Base No. 10; 2010. Available from: http://globocan.iarc.
4. Kang J, Brajanovski N, Chan KT, Xuan J, Pearson RB, Sanij E. Ribosomal proteins and human diseases: molecular mechanisms and targeted therapy. Sign Transduct Target Ther. 2021;6(1):1–22. doi:10.1038/s41392-020-00451-w
5. Sancar A, Van Gelder RN. Clocks, cancer, and chronochemotherapy. Science. 2021;371:6524. doi:10.1126/science.abb0738
6. Montoya S, Soong D, Nguyen N, Affer M, Munamarty SP, Taylor J. Targeted therapies in cancer: to be or not to be, selective. Biomedicines. 2021;9(11):1591. doi:10.3390/biomedicines9111591
7. Allam HA, Aly EE, Farouk AK, El Kerdawy AM, Rashwan E, Abbass SE. Design and synthesis of some new 2, 4, 6-trisubstituted quinazoline EGFR inhibitors as targeted anticancer agents. Bioorg Chem. 2020;98:103726. doi:10.1016/j.bioorg.2020.103726
8. Okamoto I. Epidermal growth factor receptor in relation to tumor development: EGFR‐targeted anticancer therapy. FEBS J. 2010;277(2):309–315. doi:10.1111/j.1742-4658.2009.07449.x
9. George RF, Kandeel M, El-Ansary DY, El Kerdawy AM. Some 1, 3, 5-trisubstituted pyrazoline derivatives targeting breast cancer: design, synthesis, cytotoxic activity, EGFR inhibition and molecular docking. Bioorg Chem. 2020;99:103780. doi:10.1016/j.bioorg.2020.103780
10. Ayati A, Moghimi S, Salarinejad S, Safavi M, Pouramiri B, Foroumadi A. A review on progression of epidermal growth factor receptor (EGFR) inhibitors as an efficient approach in cancer targeted therapy. Bioorg Chem. 2020;99:103811. doi:10.1016/j.bioorg.2020.103811
11. Nehra B, Rulhania S, Jaiswal S, Kumar B, Singh G, Monga V. Recent advancements in the development of bioactive pyrazoline derivatives. Eur J Med Chem. 2020;205:112666. doi:10.1016/j.ejmech.2020.112666
12. Varghese B, Al-Busafi SN, Suliman FO, Al-Kindy SM. Unveiling a versatile heterocycle: pyrazoline–a review. RSC Adv. 2017;7(74):46999–47016. doi:10.1039/C7RA08939B
13. Salian VV, Narayana B, Sarojini BK, Byrappa K. A comprehensive review on recent developments in the field of biological applications of potent pyrazolines derived from chalcone precursors. Lett Drug Des Discov. 2018;15(5):516–574. doi:10.2174/1570180814666170703164221
14. Shaaban MR, Mayhoub AS, Farag AM. Recent advances in the therapeutic applications of pyrazolines. Expert Opin Ther Pat. 2012;22(3):253–291. doi:10.1517/13543776.2012.667403
15. Nayak S, Gaonkar SL. A review on recent synthetic strategies and pharmacological importance of 1, 3-thiazole derivatives. Mini Rev Med Chem. 2019;19(3):215–238. doi:10.2174/1389557518666180816112151
16. Singh IP, Gupta S, Kumar S. Thiazole compounds as antiviral agents: an update. Med Chem. 2019;10:1573406415666190614101253.
17. Kashyap A, Adhikari N, Das A, et al. Review on synthetic chemistry and antibacterial importance of thiazole derivatives. Curr Drug Discov Technol. 2018;15(3):214–228. doi:10.2174/1570163814666170911144036
18. Jain S, Pattnaik S, Pathak K, et al. Anticancer potential of thiazole derivatives: a retrospective review. Mini Rev Med Chem. 2018;18(8):640–655. doi:10.2174/1389557517666171123211321
19. Sharma PC, Bansal KK, Sharma A, Sharma D, Deep A. Thiazole-containing compounds as therapeutic targets for cancer therapy. Eur J Med Chem. 2020;188:112016. doi:10.1016/j.ejmech.2019.112016
20. Guerrero-Pepinosa NY, Cardona-Trujillo MC, Garzón-Castaño SC, Veloza LA, Sepúlveda-Arias JC. Antiproliferative activity of thiazole and oxazole derivatives: a systematic review of in vitro and in vivo studies. Biomed Pharmacother. 2021;138:111495. doi:10.1016/j.biopha.2021.111495
21. Singh N, Gupta M. Therapeutic journey of pyrazolines as EGFR tyrosine kinase inhibitors: an insight into structure-activity relationship. Curr Bioact Compd. 2020;16(9):1260–1272. doi:10.2174/1573407216666200128155640
22. Nawaz F, Alam O, Perwez A, et al. Design, synthesis, molecular docking, and anticancer evaluation of pyrazole linked pyrazoline derivatives with carbothioamide tail as EGFR kinase inhibitors. Anticancer Agents Med Chem. 2021;21(1):42–60. doi:10.2174/1871520620666200727093613
23. Halim PA, Hassan RA, Mohamed KO, et al. Synthesis and biological evaluation of halogenated phenoxychalcones and their corresponding pyrazolines as cytotoxic agents in human breast cancer. J Enzyme Inhib Med Chem. 2022;37(1):189–201. doi:10.1080/14756366.2021.1998023
24. Ayati A, Emami S, Moghimi S, Foroumadi A. Thiazole in the targeted anticancer drug discovery. Future Med Chem. 2019;11(16):1929–1952. doi:10.4155/fmc-2018-0416
25. Yuan JW, Wang SF, Luo ZL, et al. Synthesis and biological evaluation of compounds which contain pyrazole, thiazole and naphthalene ring as antitumor agents. Bioorg Med Chem Lett. 2014;24(10):2324–2328. doi:10.1016/j.bmcl.2014.03.072
26. Bilodeau MT, Rodman LD, McGaughey GB, et al. The discovery of N-(1, 3-thiazol-2-yl) pyridin-2-amines as potent inhibitors of KDR kinase. Bioorg Med Chem Lett. 2004;14(11):2941–2945. doi:10.1016/j.bmcl.2004.03.052
27. El-Miligy MM, Abd El Razik HA, Abu-Serie MM. Synthesis of piperazine-based thiazolidinones as VEGFR2 tyrosine kinase inhibitors inducing apoptosis. Future Med Chem. 2017;9(15):1709–1729. doi:10.4155/fmc-2017-0072
28. Lu Y, Li CM, Wang Z, et al. Discovery of 4-substituted methoxybenzoyl-aryl-thiazole as novel anticancer agents: synthesis, biological evaluation, and structure− activity relationships. J Med Chem. 2009;52(6):1701–1711. doi:10.1021/jm801449a
29. Sanachai K, Aiebchun T, Mahalapbutr P, et al. Discovery of novel JAK2 and EGFR inhibitors from a series of thiazole-based chalcone derivatives. RSC Med Chem. 2021;12(3):430–438. doi:10.1039/D0MD00436G
30. Havrylyuk D, Roman O, Lesyk R. Synthetic approaches, structure activity relationship and biological applications for pharmacologically attractive pyrazole/pyrazoline–thiazolidine-based hybrids. Eur J Med Chem. 2016;113:145–166. doi:10.1016/j.ejmech.2016.02.030
31. Matiadis D, Sagnou M. Pyrazoline hybrids as promising anticancer agents: an up-to-date overview. Int J Mol Sci. 2020;21(15):5507. doi:10.3390/ijms21155507
32. El Azab IH, Bakr RB, Elkanzi NA. Facile one-pot multicomponent synthesis of pyrazolo-thiazole substituted pyridines with potential anti-proliferative activity: synthesis, in vitro and in silico studies. Molecules. 2021;26(11):3103. doi:10.3390/molecules26113103
33. Gümüş M, Yakan M, Koca İ. Recent advances of thiazole hybrids in biological applications. Future Med Chem. 2019;11(16):1979–1998. doi:10.4155/fmc-2018-0196
34. Mohamed TK, Batran RZ, Elseginy SA, Ali MM, Mahmoud AE. Synthesis, anticancer effect and molecular modeling of new thiazolylpyrazolyl coumarin derivatives targeting VEGFR-2 kinase and inducing cell cycle arrest and apoptosis. Bioorg Chem. 2019;85:253–273. doi:10.1016/j.bioorg.2018.12.040
35. Sever B, Altıntop MD, Radwan MO, et al. Design, synthesis and biological evaluation of a new series of thiazolyl-pyrazolines as dual EGFR and HER2 inhibitors. Eur J Med Chem. 2019;182:111648. doi:10.1016/j.ejmech.2019.111648
36. George RF, Samir EM, Abdelhamed MN, Abdel-Aziz HA, Abbas SE. Synthesis and anti-proliferative activity of some new quinoline based 4, 5-dihydropyrazoles and their thiazole hybrids as EGFR inhibitors. Bioorg Chem. 2019;83:186–197. doi:10.1016/j.bioorg.2018.10.038
37. Batran RZ, El‐Kashak WA, El‐Daly SM, Ahmed EY. Dual kinase inhibition of EGFR/HER2: design, synthesis and molecular docking of thiazolylpyrazolyl‐based aminoquinoline derivatives as anticancer agents. ChemistrySelect. 2021;6(40):11012–11021. doi:10.1002/slct.202102917
38. Zhang WM, Xing M, Zhao TT, et al. Synthesis, molecular modeling and biological evaluation of cinnamic acid derivatives with pyrazole moieties as novel anticancer agents. RSC Adv. 2014;4(70):37197–37207. doi:10.1039/C4RA05257A
39. Mphahlele MJ, Paumo HK, Choong YS. Synthesis and in vitro cytotoxicity of the 4-(halogenoanilino)-6-bromoquinazolines and their 6-(4-fluorophenyl) substituted derivatives as potential inhibitors of epidermal growth factor receptor tyrosine kinase. Pharmaceuticals. 2017;10(4):87. doi:10.3390/ph10040087
40. Maher M, Kassab AE, Zaher AF, Mahmoud Z. Novel pyrazolo [3, 4-d] pyrimidines: design, synthesis, anticancer activity, dual EGFR/ErbB2 receptor tyrosine kinases inhibitory activity, effects on cell cycle profile and caspase-3-mediated apoptosis. J Enzyme Inhib Med Chem. 2019;34(1):532–546. doi:10.1080/14756366.2018.1564046
41. Qiu KM, Wang HH, Wang LM, et al. Design, synthesis and biological evaluation of pyrazolyl-thiazolinone derivatives as potential EGFR and HER-2 kinase inhibitors. Bioorg Med Chem. 2012;20:2010–2018. doi:10.1016/j.bmc.2012.01.051
42. Kaplancıklı ZA, Turan-Zitouni G, Özdemir A, Revial G, Güven K. Synthesis and antimicrobial activity of some thiazolyl-pyrazoline derivatives. Phosphorus Sulfur Silicon Relat Elem. 2007;182:749–764. doi:10.1080/10426500601047529
43. Eldehna WM, Rashood ST, Al-Warhi T, Eskandrani RO, Alharbi A, El Kerdawy AM. Novel oxindole/benzofuran hybrids as potential dual CDK2/GSK-3β inhibitors targeting breast cancer: design, synthesis, biological evaluation, and in silico studies. J Enzyme Inhib Med Chem. 2021;36:270–285. doi:10.1080/14756366.2020.1862101
44. Eldehna WM, El Hassab MA, Abo-Ashour MF, et al. Development of isatin-thiazolo[3,2-a]benzimidazole hybrids as novel CDK2 inhibitors with potent in vitro apoptotic antiproliferative activity: synthesis, biological and molecular dynamics investigations. Bioorg Chem. 2021;110:104748. doi:10.1016/j.bioorg.2021.104748
45. Sabt A, Eldehna WM, Al-Warhi T, et al. Discovery of 3,6-disubstituted pyridazines as a novel class of anticancer agents targeting cyclin-dependent kinase 2: synthesis, biological evaluation and in silico insights. J Enzyme Inhib Med Chem. 2020;35:1616–1630. doi:10.1080/14756366.2020.1806259
46. Al-Rashood ST, Hamed AR, Hassan GS, et al. Antitumor properties of certain spirooxindoles towards hepatocellular carcinoma endowed with antioxidant activity. J Enzym Inhib Med Chem. 2020;35:831–839. doi:10.1080/14756366.2020.1743281
47. Abdelsalam EA, Zaghary WA, Amin KM, et al. Synthesis and in vitro anticancer evaluation of some fused indazoles, quinazolines and quinolines as potential EGFR inhibitors. Bioorg Chem. 2019;89:102985. doi:10.1016/j.bioorg.2019.102985
48. Masoud DM, Azzam RA, Hamdy F, Mekawey AA, Abdel‐Aziz HA. Synthesis of some novel pyrazoline‐thiazole hybrids and their antimicrobial activities. J Heterocycl Chem. 2019;56(11):3030–3041. doi:10.1002/jhet.3698
49. Zhou P, Hu J, Wang X, Wang J, Zhang Y, Wang C. Epidermal growth factor receptor expression affects proliferation and apoptosis in non-small cell lung cancer cells via the extracellular signal-regulated kinase/microRNA 200a signaling pathway. Oncol Lett. 2018;15(4):5201–5207. doi:10.3892/ol.2018.7961
50. Bethune G, Bethune D, Ridgway N, Xu Z. Epidermal growth factor receptor (EGFR) in lung cancer: an overview and update. J Thorac Dis. 2010;2(1):48.
51. Masuda H, Zhang D, Bartholomeusz C, Doihara H, Hortobagyi GN, Ueno NT. Role of epidermal growth factor receptor in breast cancer. Breast Cancer Res Treat. 2012;136(2):331–345. doi:10.1007/s10549-012-2289-9
52. Davidson NE, Gelmann EP, Lippman ME, Dickson RB. Epidermal growth factor receptor gene expression in estrogen receptor-positive and negative human breast cancer cell lines. Mol Endocrinol. 1987;1(3):216–223. doi:10.1210/mend-1-3-216
53. Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65(1):55–63. doi:10.1016/0022-1759(83)90303-4
54. Cunningham MP, Thomas H, Fan Z, Modjtahedi H. Responses of human colorectal tumor cells to treatment with the anti–epidermal growth factor receptor monoclonal antibody ICR62 used alone and in combination with the EGFR tyrosine kinase inhibitor gefitinib. Cancer Res. 2006;66(15):7708–7715. doi:10.1158/0008-5472.CAN-06-1000
55. Zhou X, Zheng M, Chen F, et al. Gefitinib inhibits the proliferation of pancreatic cancer cells via cell cycle arrest. Anat Rec. 2009;292(8):1122–1127. doi:10.1002/ar.20938
56. Gorczyca W. Cytometric analyses to distinguish death processes. Endocr Relat Cancer. 1999;6(1):17–19. doi:10.1677/erc.0.0060017
57. Stamos J, Sliwkowski MX, Eigenbrot C. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J Biol Chem. 2002;277:46265–46272. doi:10.1074/jbc.M207135200
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.