")
Back to Journals » Journal of Pain Research » Volume 11
Nonsteroidal anti-inflammatory drugs in chronic pain: implications of new data for clinical practice
Authors Ho KY , Gwee KA , Cheng YK, Yoon KH , Hee HT, Omar AR
Received 14 March 2018
Accepted for publication 27 July 2018
Published 20 September 2018 Volume 2018:11 Pages 1937—1948
DOI https://doi.org/10.2147/JPR.S168188
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Erica Wegrzyn
Kok Yuen Ho,1 Kok Ann Gwee,2 Yew Kuang Cheng,3,4 Kam Hon Yoon,5 Hwan Tak Hee,6 Abdul Razakjr Omar7
1The Pain Clinic, Mt Alvernia Hospital, 2Stomach, Liver, and Bowel Centre, Gleneagles Hospital, 3Farrer Park Hospital, 4Yong Loo Lin School of Medicine, National University of Singapore, 5El Shaddai Arthritis and Rheumatism Specialist Medical Centre, 6Pinnacle Spine and Scoliosis Centre, Mt Elizabeth Medical Centre, 7Lee Kong Chian School of Medicine, Nanyang Technological University, Singapore
Abstract: COX2-selective and nonselective (ns) nonsteroidal anti-inflammatory drugs (NSAIDs) are widely used for chronic pain management. There are marked differences in the risk of adverse gastrointestinal (GI) and cardiovascular (CV) events among different NSAIDs. In 2017, publication of two randomized controlled trials and an individual patient-data meta-analysis provided robust data on the relative GI and CV tolerability profiles of currently available NSAIDs. The PRECISION study showed similar CV-event rates with celecoxib vs naproxen and ibuprofen, but GI tolerability was better for celecoxib. In the CONCERN study of high-GI-risk patients, celecoxib was associated with fewer adverse GI-tract events than naproxen. The meta-analysis showed no significant difference between celecoxib and ns-NSAIDs in the rate of acute myocardial infarction, and celecoxib was the only COX2-selective NSAID with a lower risk of adverse CV and GI events vs ns-NSAIDs. These data add to the body of knowledge about the relative tolerability of different NSAIDs and were used to propose an updated treatment algorithm. The decision about whether to use an NSAID and which one should be based on a patient’s risk of developing adverse GI and CV events. Lower- and upper-GI-tract events need to be considered. Celecoxib has a better lower-GI-tract tolerability profile than ns-NSAIDs plus a proton-pump inhibitor. In addition, the latest data suggest that long-term use of celecoxib 200 mg/day may be appropriate for patients at increased CV risk.
Keywords: nonsteroidal anti-inflammatory drugs, chronic pain, COX2 inhibitors, cardiovascular risk, gastrointestinal risk
Introduction
Chronic pain is defined by the International Association for the Study of Pain as “pain that persists beyond normal tissue healing time, which is assumed to be 3 months”.1 Chronic pain is one of the most prominent causes of disability worldwide and represents a major challenge for public health.2 Patients with chronic pain report that this has a significant impact on their work and daily function.3 The presence of inflammation is a common underlying mechanism of chronic pain.4 Inflammation can occur in the affected organ and/or nerves.5 Low-grade inflammation, both local and systemic, is also the hallmark of many chronic, progressing, and painful conditions, such as osteoarthritis (OA) and rheumatoid arthritis (RA).6,7
Existing guidelines recommend paracetamol (acetaminophen) as the first-line analgesic agent.8–10 However, this is not an ideal choice for chronic inflammatory pain, because paracetamol lacks anti-inflammatory activity and has been shown to be less effective than alternative nonopioid options in chronic conditions involving connective tissue.11–14 In addition, although traditionally seen as a better-tolerated analgesic option, there are safety concerns with the use of paracetamol, particularly at dosages >3 g/day, for extended periods (eg, several years).15–20
COX2-selective (c2s) and nonselective (ns) nonsteroidal anti-inflammatory drugs (NSAIDs), which display both analgesic and anti-inflammatory properties, are useful options for the management of chronic pain. However, use of these agents is limited by side effects, specifically adverse gastrointestinal (GI) and cardiovascular (CV) events.6 Nevertheless, it is increasingly being recognized that adverse NSAID-related events, including those associated with c2s agents, are not a class effect and vary substantially among different drugs. One example of this is etodolac, which has been shown in vitro to be more c2s than celecoxib,21 but like indomethacin, ketorolac, sulindac, and tolmetin is classified as a carbo- or heterocyclic acid based on chemical structure.22
All NSAIDs are inherently acidic, but there is wide interagent variation. The acid-dissociation constant (pKa) for celecoxib is 9.7 compared with 5.2, 4.2, and 4.0 for ibuprofen, naproxen and diclofenac, respectively23,24 (lower pKa values indicate higher acidity). As weak acids, ns-NSAIDs are invariably lipophilic, providing them with detergent properties. Detergent properties allow these agents to interact with phospholipids, which are essential constituents of the brush border. This causes direct damage to the surface epithelium.25 Another way that the acidity of different NSAIDs could contribute to between-agent differences in activity and toxicity is the intracellular accumulation of an NSAID being proportional to its acidity. At micromolar concentrations, NSAIDs are able to uncouple oxidative phosphorylation, which reduces intracellular ATP production. This results in the loss of intercellular integrity, because intercellular junctions are under the control of ATP-dependent actin–myosin complexes, leading to increased intestinal permeability and inflammatory enteropathy.25 On this basis, less acidic drugs would be expected to have fewer adverse GI effects.
Different NSAIDs also have different molecular structures and chemical properties. Diclofenac is a benzeneacetic derivative, ibuprofen and naproxen are propionic acid derivatives, celecoxib and valdecoxib have a sulfonamide group, and etoricoxib and rofecoxib have a sulfonyl group.26 c2s- NSAIDs with a sulfonyl group increase the susceptibility of biological lipids (eg, low-density-lipoprotein cholesterol) to oxidative modification via a nonenzymatic process independently of COX2 inhibition, an effect not seen for c2s-NSAIDs with a sulfonamide group or ns-NSAIDs.27 Oxidation of LDL is an important factor contributing to the development of atherosclerotic CV disease.
This consensus document provides an overview of the latest published data on the long-term use of NSAIDs commonly used for the management of chronic pain of musculoskeletal origin, with a focus on GI and CV risk and safety. The aim was to discuss the latest clinical trial data and use these to provide an evidence-based consensus on the most appropriate NSAID options for different groups of patients with chronic pain and propose an updated treatment algorithm.
Latest clinical trial data
Relevant studies that have been published since the last consensus document in this field are the PRECISION28 and CONCERN29 clinical trials and a meta-analysis of individual patient data authored by Bally et al.30 The PRECISION trial was the first study of NSAIDs in high-CV-risk patients and the first to compare a c2s agent with naproxen and ibuprofen.28 All three NSAIDs provided effective pain relief in patients with OA or RA (with no clinically significant differences between groups). Celecoxib was noninferior to naproxen and ibuprofen for the primary CV-event end point and was associated with significantly fewer GI and serious renal events compared with the ns-NSAIDs.28
The focus of the CONCERN study was adverse GI events.29 The trial included patients with arthritis who also had cardiothrombotic diseases requiring low-dose aspirin and a history of upper-GI-tract bleeding. Subjects were randomized to celecoxib 100 mg twice daily or naproxen 500 mg twice daily, both in combination with a prophylactic proton-pump inhibitor (PPI). The primary end point was recurrent upper-GI-tract bleeding within 18 months. The cumulative incidence of primary end-point events with celecoxib was less than half that with naproxen. The rate of serious CV events (a secondary end point of the trial) did not differ significantly between the two treatment groups.31 The key features and results of the PRECISION and CONCERN studies, along with the earlier MEDAL program that looked at long-term use of etoricoxib, are summarized in Table 1.
 | Table 1 Overview of recent key clinical trials of COX2 inhibitors vs nonselective NSAIDs Abbreviations: APTC, Antiplatelet Trialists’ Collaboration; BID, bis in die (twice daily); CSGIE, clinically significant GI event; CV, cardiovascular; GI, gastrointestinal; NSAIDs, nonsteroidal anti-inflammatory drugs; OA, osteoarthritis; PPI, proton-pump inhibitor; RA, rheumatoid arthritis; TID, ter in die (thrice daily). |
The individual patient-data meta-analysis published by Bally et al included clinical trials of ns- and c2s-NSAIDs that had acute myocardial infarction (MI) as a specific outcome, and compared the rate of this adverse event in NSAID users vs nonusers.30 All the NSAIDs studied (celecoxib, diclofenac, naproxen, ibuprofen, and rofecoxib) increased the risk of acute MI compared with no NSAID use, but there were no significant differences in the risk of acute MI between celecoxib (adjusted OR 1.24, 95% CI 0.91–1.82) and ns-NSAIDs (OR 1.50 [95% CI 1.06–2.04] for diclofenac, OR 1.48 [95% CI 1.00–2.26] for ibuprofen, and OR 1.53 [95% CI 1.07–2.33] for naproxen; the rate was highest with rofecoxib [OR 1.58 [95% CI 1.07–2.17]). The acute MI risk appeared to be dependent on the NSAID dosage, but not the duration of therapy, with usage for longer than 1 month associated with risk similar to shorter therapy durations.30
Cardiovascular side effects of NSAIDs
Concerns about the CV safety of c2s-NSAIDs first appeared when the VIGOR trial demonstrated a significantly increased risk of thromboembolic CV events with rofecoxib vs naproxen (relative risk 2.38, 95% CI 1.39–4.00; P<0.05).32,33 Rofecoxib was also shown to increase the number of Antiplatelet Trialists’ Collaboration events compared with placebo in the APPROVE trial (HR 4.61, 95% CI 1.50–18.83; P=0.008).34 Results from these studies led to the worldwide withdrawal of rofecoxib in 2004.
The Fitzgerald hypothesis suggested that increased platelet activation and aggregation occurring via selective blockade of prostacyclin formation with little inhibition of prothrombotic platelet-derived thromboxane A2 (TXA2) was the primary mechanism for increased CV risk with c2s-NSAIDs.35,36 However, COX2 selectivity varies among c2s-NSAIDs, being greater for lumiracoxib, rofecoxib, etoricoxib, and valdecoxib and comparatively lower for celecoxib.37 In addition, other potential mechanisms might influence the CV risk of an NSAID. For example, rofecoxib has prooxidant activity (potentially proatherosclerotic) and celecoxib reduces endothelial tissue-factor expression, a key initiator of the coagulation cascade (potentially protective against thrombosis).38,39
The CLASS40 trial showed a similar risk of adverse thromboembolic CV events with celecoxib 400 mg twice daily (a dose twice that recommended in RA and four times that recommended in OA) compared with diclofenac.41 Of note, 22% of the CLASS population were taking low-dose aspirin and 40% had a history of CV disease, and thus, a reasonable proportion were at risk of having a CV event during the 1-year trial.41 Around the same time, the results of the VIGOR study reported lower rates of MI with naproxen vs rofecoxib (RR 0.2, 95% CI 0.1–0.7), although the CV death rate was similar in the two groups.32 However, there were concerns that some MI events in rofecoxib recipients were not included in the published paper, leading to an underestimation of the between-group difference in the rate of this adverse event.42 Note that patients included in the CLASS study had OA, whereas the VIGOR study included patients with RA, a population that is at higher theoretical risk of thromboembolic disease, due to the association between chronic inflammatory mediators and prothrombotic factors and endothelial dysfunction.43 Furthermore, the comparator agent in VIGOR was naproxen, which may protect against thromboembolic disease.44 In contrast, it has been suggested that diclofenac (the comparator in the CLASS study) may have some intrinsic prothrombotic effects, limiting the ability to detect a difference between celecoxib and diclofenac with respect to the rate of thrombotic events.33 Data from the CLASS trial have been presented to show the incidence of MI in patients who were vs were not receiving low-dose aspirin because of the thromboembolic protection provided by aspirin and the fact that those not taking aspirin were presumably at lower risk of thromboembolic events when enrolled in the trial.33 In patients not receiving concomitant aspirin, rates of fatal and nonfatal MI were 0.2% and 0.1%, respectively, in the celecoxib and diclofenac groups (compared with 0.5% and 0.2%, respectively, in the overall population). It is also important to note that the latest analysis of PRECISION trial data showed that celecoxib had a more favorable overall safety profile than naproxen or ibuprofen when not taken with aspirin.45 The safety advantage of celecoxib over the ns-NSAIDs was attenuated by the addition of aspirin, although celecoxib was still associated with fewer GI events than ibuprofen or naproxen and fewer renal events than ibuprofen.45
In contrast, a review of all potentially serious CV events among 2,035 patients with a history of colorectal neoplasia who were enrolled in a trial comparing two doses of celecoxib (200 mg or 400 mg twice daily) with placebo for the prevention of colorectal adenomas showed a dose-dependent increase in the composite end point of CV death, MI, stroke, or heart failure in celecoxib recipients, resulting in early trial termination.46 The celecoxib doses used in this study were high compared to those used for chronic pain management (eg, in RA or OA). Another study with a similar indication (prevention of colorectal adenomatous polyps) failed to find any significant difference in the serious CV-event rate with celecoxib 400 mg once daily vs placebo (RR 1.3, 95% CI 0.65–2.62). In the ADAPT study, use of naproxen 220 mg twice daily was associated with a 60% increase in the risk of MI, congestive heart failure, cerebrovascular accident or transient ischemic attack compared with placebo (HR 1.40, 95% CI 1.12–1.75; P<0.05), but the higher rate of CV events in the celecoxib group (200 mg twice daily) did not reach statistical significance (HR 1.10, 95% CI 0.67–1.79).47 However, it should be noted that safety was not a prespecified end point in this trial.
On April 7, 2005, the US Food and Drug Administration (FDA) added a black-box warning for all NSAIDs (both c2ns and c2s) highlighting the risk of adverse CV events. In the CNT collaboration study, the only NSAID that did not increase vascular risk was naproxen,48 but the FDA maintained the black-box warning for all NSAIDs. There are several differences between rofecoxib and celecoxib that might account for the different trial outcomes26,39 and suggest that increased risk of CV events might not be a class effect of c2s-NSAIDs (Table 2).
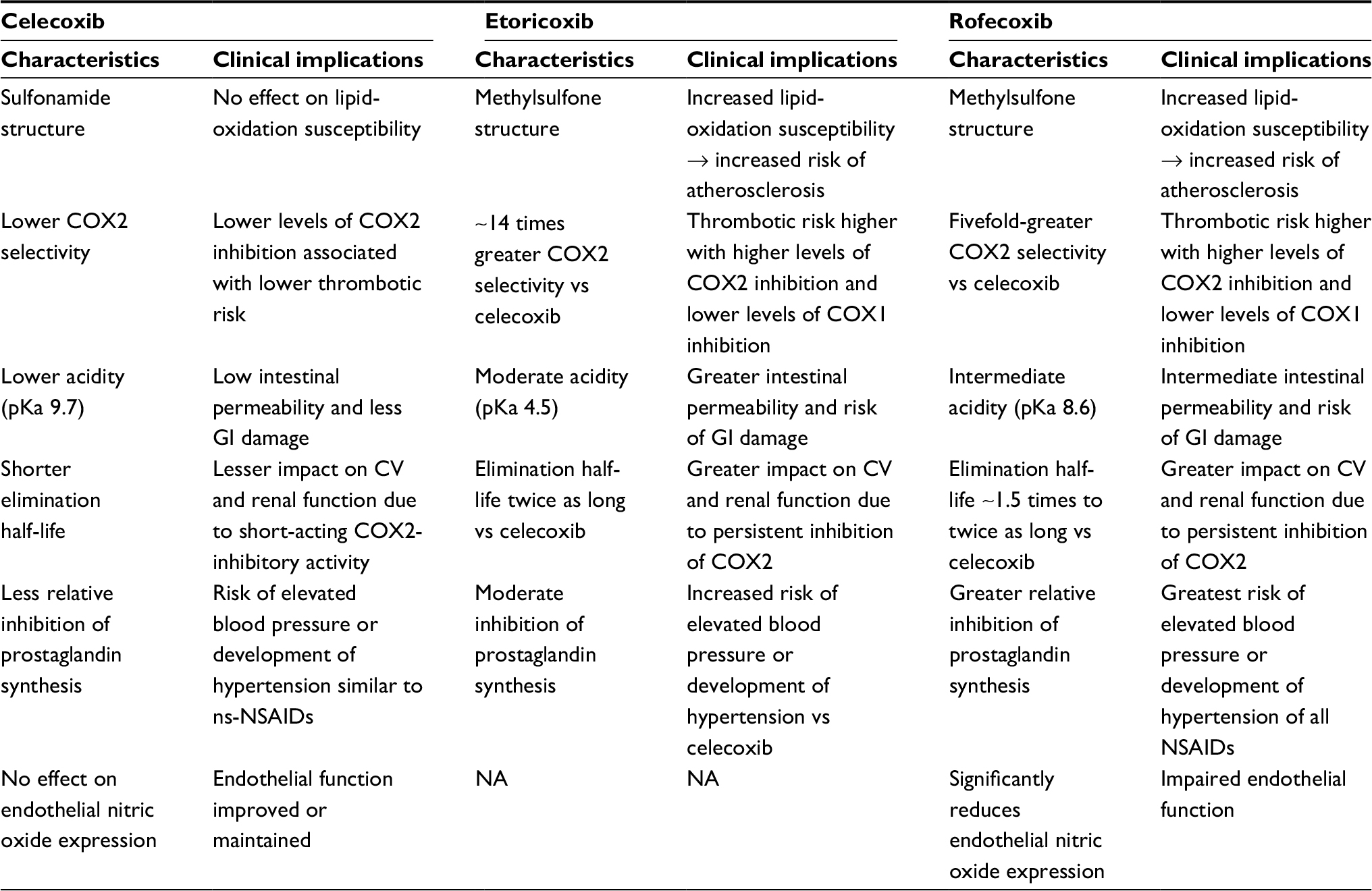 | Table 2 Differential features of celecoxib, etoricoxib, and rofecoxib Note: Data from these studies.16,23,26,48,61,87,92–101 Abbreviations: CV, cardiovascular; GI, gastrointestinal; NA, not available; ns, nonselective; NSAIDs, nonsteroidal anti-inflammatory drugs. |
Data from an FDA nested case–control study using a US managed-care organization database showed that the risk of acute MI and sudden death was by far the highest with rofecoxib at doses >25 mg.49 The only other NSAIDs associated with a significant increase in CV risk compared with control were indomethacin and diclofenac.49 In a meta-analysis, celecoxib and valdecoxib were the only two c2s-NSAIDs that were associated with lower CV- and GI-event rates than ns-NSAIDs.50 Based on the data analyzed, treatment of 1,000 patients for a year with celecoxib rather than with an ns-NSAID would be associated with 12 fewer upper-GI-tract complications and two fewer fatal or nonfatal MIs or strokes.50 The MEDAL program was the first CV-outcome analysis prospectively designed to evaluate the relative thrombotic CV risk of a c2s inhibitor and a traditional NSAID.51 The results showed no difference in risk of thrombotic CV events in arthritis patients on long-term therapy with etoricoxib or diclofenac. However, both drugs increased blood pressure, and the rate of discontinuation due to hypertension was higher in the etoricoxib group.51 As already described, the PRECISION trial did not show any increased CV risk with celecoxib compared with naproxen or ibuprofen.28
There were a number of differences between the MEDAL and PRECISION studies (Table 1), including lower overall CV risk, less aspirin use, and less use of gastroprotective therapy in patients in the MEDAL trial. In addition, and perhaps most importantly, the ns-NSAID comparator in MEDAL was diclofenac, whereas naproxen and ibuprofen were used in PRECISION. This choice of comparator for PRECISION was made because naproxen had been shown to be the ns-NSAID associated with the lowest vascular risk (followed by ibuprofen).13,48,52 In addition, the Arthritis and Drug Safety and Risk Management Advisory Committee of the US FDA recommended in 2005 that naproxen be the preferred comparator for large trials of c2s-NSAIDs. The FDA voted against approval of etoricoxib, based on the fact that CV-safety studies were conducted using diclofenac as the comparator.53 In a recently published trial, SCOT,54 there was no difference in the incidence of adverse CV events between celecoxib and ns-NSAIDs; similar results were seen in an observational Japanese study of patients with RA or OA.55
Assessment of a patient’s CV risk is mandatory before initiation of NSAID therapy. Patients with New York Heart Association class I or II heart failure were included in the PRECISION study,28 and the results demonstrated acceptable long-term CV tolerability with the use of NSAIDs in this patient group. The results of a meta-analysis based on pooled individual patient data (n=446,763) supported the main conclusions of the PRECISION study in terms of noninferiority of CV risk with celecoxib compared with ibuprofen or naproxen.30 Other important findings of the meta-analysis were that NSAID-related CV risk was greater when daily doses were higher, CV risk did not appear to increase as duration of NSAID therapy increased, and the risk of MI during the use of rofecoxib (especially at >25 mg/day) was greater than that associated with use of any other NSAID, including celecoxib.30 It is suggested that physicians should use adequate, but not high, doses of c2s-NSAIDs, to obtain analgesic and anti-inflammatory benefit without markedly increasing CV risk.
Gastrointestinal side effects of NSAIDs
Potential GI side effects of NSAID therapy are a concern for physicians. Traditionally, adverse upper-GI-tract effects have been the focus, but there is increasing recognition that lower-GI complications are also an important issue.56–58 Infection with Helicobacter pylori is also relevant and may be more common in some populations.59–61 H. pylori infection increases the risk of GI complications during NSAID therapy,62 and eradication of H. pylori prior to commencing long-term antiplatelet therapy is recommended to reduce GI risk.63
Other risk factors for upper-GI-tract complications during NSAID therapy include older age (≥60 years), history of dyspepsia, concomitant use of corticosteroids and/or oral anticoagulants, and a history of peptic ulcer bleeding.64 In the last few decades, mortality rates in patients with NSAID-associated upper-GI bleed or perforation have increased.65 Serious GI complications necessitating emergency surgery or hospitalization can occur without prior symptoms. Approximately 60%–80% of arthritis patients hospitalized or requiring surgery for serious GI complications during NSAID therapy report no previous GI symptoms.66,67 This highlights the need for a proactive approach to risk management, given that lack of symptoms makes it difficult to intervene before GI events become life-threatening.
NSAIDs are associated with different GI risks, and it is important that this is taken into account in clinical practice. Data from a nested case–control study showed that celecoxib was the only NSAID that did not have a significantly higher rate of upper-GI-tract events compared with placebo.68 In contrast, the risk of adverse upper-GI-tract events with rofecoxib, ibuprofen, diclofenac, naproxen, and aspirin was significantly higher than in controls.68 Celecoxib has also been shown to be associated with a significantly lower rate of GI intolerability than naproxen, ibuprofen, or diclofenac in elderly patients (age ≥65 years).69
The addition of a PPI for ulcer prophylaxis significantly decreases the proportion of NSAID recipients who will develop a gastric or duodenal ulcer during chronic NSAID therapy.6,58,70,71 In very high-risk subjects (defined as those who had previous NSAID-induced ulcer bleeding and continued need for anti-inflammatory analgesics), combining celecoxib with PPI provides even greater gastroprotection (cumulative incidence of recurrent ulcer bleeding over 13 months’ follow-up: 0% for celecoxib+ esomeprazole vs 8.9% for celecoxib alone; P=0.0004).31 However, PPIs do not have any protective effects in the lower GI tract.58,72,73 It is also important to note that PPIs are not a risk-free therapy, and thus, care needs to be taken in the setting of long-term PPI use.58 Adverse events associated with a PPI include increased risk of Clostridium difficile infection and hospital-acquired pneumonia, hypersensitivity with resulting organ damage (eg, interstitial nephritis), increased fractures in postmenopausal women, altered absorption of vitamins and minerals, increased risk of CV complications, and potential for drug interactions.74–78 In addition, PPI treatment has been shown to increase the incidence of small-intestine bacterial overgrowth.79
Epidemiological data from Spain showed that although the number of NSAID-related GI events decreased over the period 1996–2005 (probably due to the addition of PPI therapy), there was a corresponding increase in the number of lower-GI-tract events associated with NSAID use.80 NSAID-induced enteropathy and bleeding may occur more often than NSAID-induced gastropathy.81,82 One study estimated that approximately 70% of chronic NSAID users have significant small-intestine damage and bleeding.56,57 In addition, lower-GI-tract events are more severe than upper-GI-tract events in terms of higher mortality rate, prolonged length of hospital stay, and more diagnostic tests required.80
The most likely mechanism for lower-GI-tract damage with ns-NSAIDs is increased lower-GI-tract mucosal permeability and inflammation.83 ns-NSAIDs cause direct damage to the surface epithelium, increasing intestinal permeability, which results in a tissue reaction known as inflammatory enteropathy.25 Anemia, occult blood loss, malabsorption, and protein loss are common.83 Clinically significant GI bleeding, perforation, diarrhea, mucosal ulceration, diverticular disease, and strictures may also occur.83 Capsule-endoscopy studies have increased understanding of the adverse lower-GI-tract effects of NSAIDs, with one showing biochemical and direct evidence of macroscopic injury to the small intestine in 68%–75% of volunteers after 2 weeks’ treatment with slow-release diclofenac.84 Such changes appear to develop even after the addition of a PPI.72,73,85
Lower-GI-tract injury has been shown to be significantly lower with celecoxib 200 mg twice daily compared with either naproxen 500 mg twice daily or ibuprofen 800 mg three times daily, both given in combination with omeprazole.72,73 The CONDOR study was a randomized, double-blind trial comparing H. pylori-negative OA or RA patients with high GI and low CV risk treated with celecoxib 200 mg twice daily or diclofenac SR 75 mg twice daily with omeprazole 20 mg once daily for 6 months.86 A composite of clinically significant adverse upper- and lower-GI-tract events was the primary end point. The cumulative proportion of primary end-point events was significantly lower in patients receiving celecoxib compared with the diclofenac and omeprazole group (0.9% vs 3.8%, P<0.0001). The lower rate of adverse GI events with celecoxib was evident from 1 month of therapy onward.86 The same clinically significant upper- and lower-GI-tract event end point was used in the GI-REASONS study, in which approximately 8,000 patients in the US were randomized to celecoxib 200 mg once daily or ns-NSAIDs for 6 months (dosages could be adjusted based on US prescribing information).87 The rate of primary end-point events was significantly lower in the celecoxib group (1.3%) than in patients treated with ns-NSAIDs (2.4%, P=0.0003).87 In the MEDAL study, upper-GI-tract tolerability of etoricoxib was significantly better than that of diclofenac, but there was no significant difference between the two groups with respect to the cumulative incidence of lower-GI-tract clinical events (bleeding, perforation, or obstruction).88
GI safety was investigated in the PRECISION trial, which had clinically significant GI events as a secondary outcome and iron-deficiency anemia of GI origin as a tertiary outcome.28 GI outcomes for the on-treatment population showed that among the three agents studied (celecoxib, naproxen, and ibuprofen), significantly fewer clinically significant GI events occurred in the celecoxib group. Similarly, there was a lower incidence of anemia in celecoxib recipients compared with those receiving ns-NSAIDs.28
In the CONCERN study on arthritis patients with both high GI and high CV risk, the cumulative incidence of recurrent upper-GI-tract bleeding (the primary end point) was 5.6% in the celecoxib group compared with 12.3% in the naproxen group (HR 0.44, 95% CI 0.23–0.82; P=0.01), and no treatment-related deaths occurred. On the basis of their findings, the authors suggested that naproxen be avoided in high-GI/high-CV-risk patients, “despite its perceived cardiovascular safety”.29
Treatment recommendations – an update
Prior to publication of the PRECISION trial, the most recent consensus document on NSAID use and the balance between GI and CV risks was by Scarpignato et al.6 Key treatment-related recommendations and levels of evidence from that paper are summarized in Table 3, and quality grading for levels of evidence is defined in Table 4. The risk of adverse events throughout the GI tract not being prevented by PPIs, better GI-tolerability profile of celecoxib, and the similarity of celecoxib to ns-NSAIDs in terms of CV risk were statements made in that document. This is consistent with the results of a systematic review of data from and meta-analyses of randomized controlled trials that showed that celecoxib and valdecoxib were the only NSAIDs associated with lower rates of both GI and CV adverse events compared with ns-NSAIDs.50 In contrast, although the rate of GI events was lower with other c2s-NSAIDs (including etoricoxib and rofecoxib), the rate of CV events was higher than with ns-NSAIDs.50
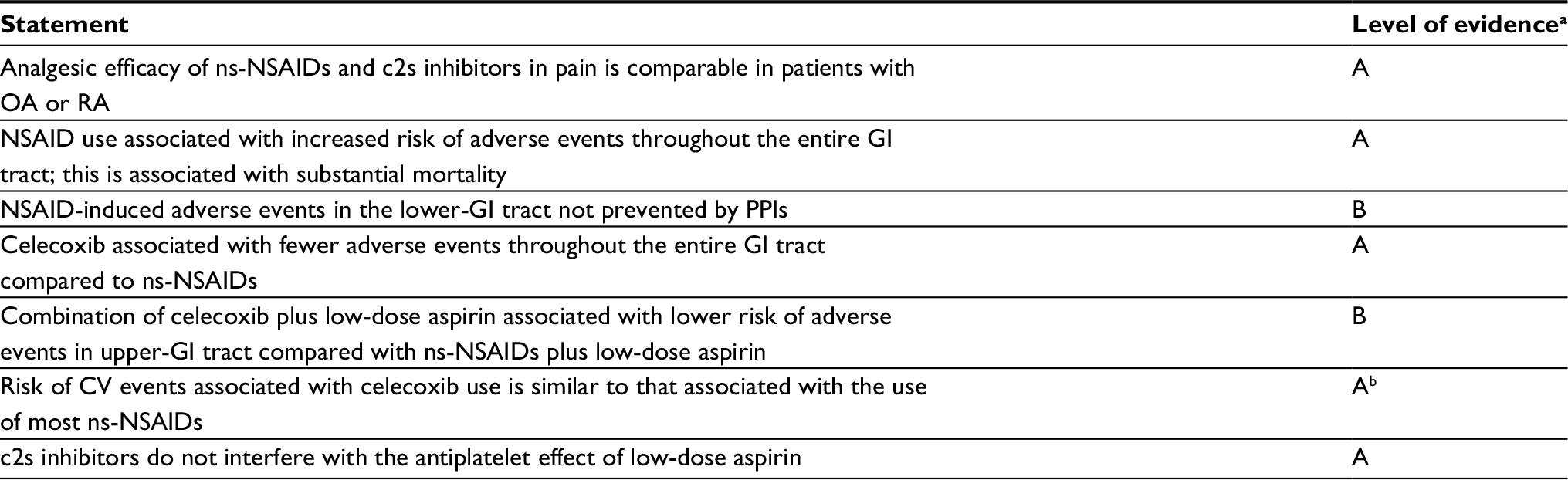 | Table 3 Summary of NSAID treatment-related statements from consensus guidelines by Scarpignato et al6 Notes: aLevels of evidence defined in Table 4; bPRECISION results consistent with this statement, and thus the study contributes to the high level of evidence for this statement. Abbreviations: CV, cardiovascular; GI, gastrointestinal; ns, nonselective; NSAIDs, nonsteroidal anti-inflammatory drugs; OA, osteoarthritis; PPIs, proton-pump inhibitors; RA, rheumatoid arthritis; c2s, COX2-selective. |
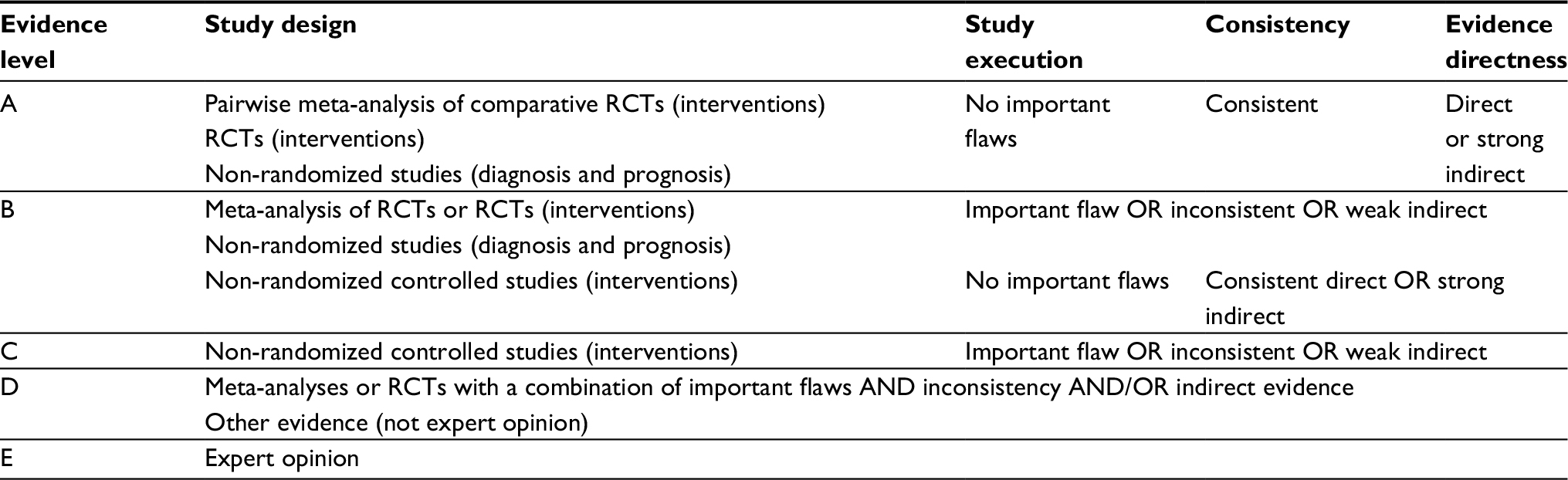 | Table 4 Evidence-quality grading based on the GRADE system Abbreviation: RCTs, randomized controlled trials. |
Given the new findings from PRECISION,28 CONCERN,29 and the most recent meta-analysis by Bally et al,30 an updated and simplified treatment algorithm has been proposed (Figure 1). The decision on whether to use an NSAID and the choice of NSAID should be based on a patient’s risk of developing adverse GI and CV events. However, the new algorithm takes into consideration data showing that adverse lower-GI-tract events need to be considered during NSAID therapy, that the risk of adverse CV events is similar with celecoxib and naproxen/ibuprofen at the dosages used in the PRECISION trial,28 the lack of any significant difference in the rate of acute MI between celecoxib and ns-NSAIDs,30 and the suggestion that naproxen should be avoided in high-GI/high-CV-risk patients.29 Based on these data, celecoxib appears to be the c2s-NSAID with the best tolerability profile. However, direct comparisons between celecoxib and etoricoxib are lacking. Nevertheless, the facts that etoricoxib contains a sulfonyl group (like rofecoxib) and the NSAID comparator in the MEDAL program was diclofenac (rather than the better-tolerated agents naproxen and ibuprofen) need to be taken into account when assessing the available evidence and applying it in clinical practice. Based on the latest data, long-term use of celecoxib 200 mg/day may be appropriate for patients at increased CV risk. However, the 2007 scientific statement from the American Heart Association stating that COX2 inhibitors should be used at the lowest possible dose and for the shortest possible time to minimize the risk of CV events remains valid.89
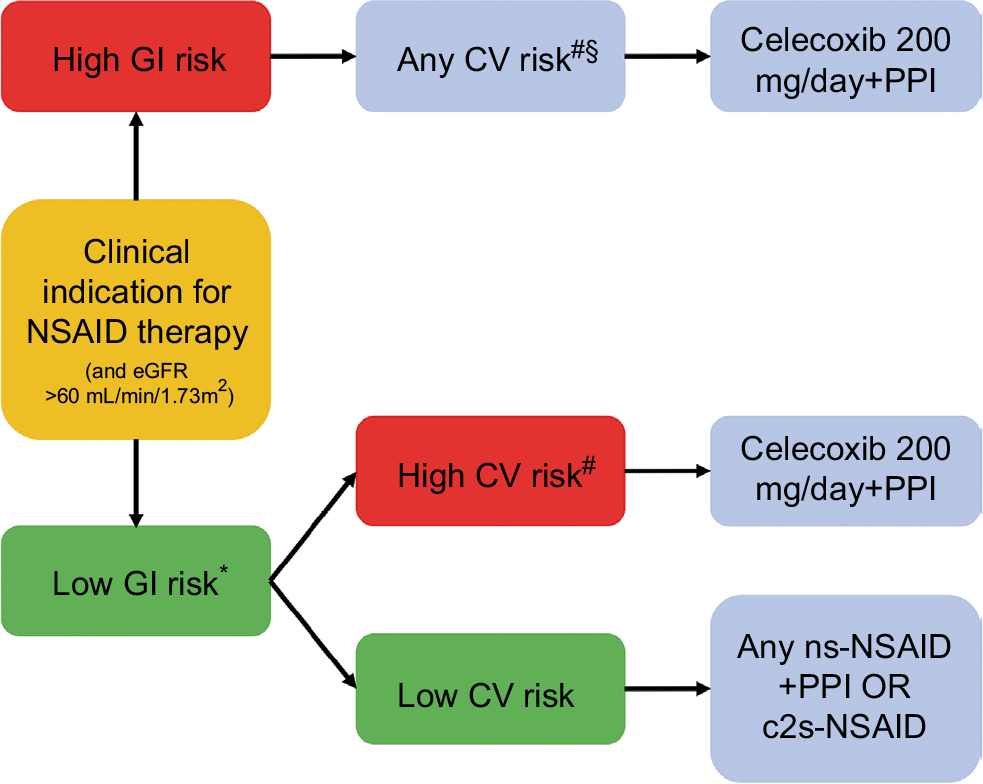 | Figure 1 Updated simplified treatment algorithm for the use of NSAIDs in different patient groups. Notes: *Assess upper- and lower-GI risk and note that PPIs do not prevent lower GI-tract NSAID-related side effects; #celecoxib currently contraindicated in patients with existing CVD, and all NSAIDs should be avoided in these patients; §recommendations the same, irrespective of CV-risk status. Abbreviations: CV, cardiovascular; CVD, CV disease; eGFR, estimated glomerular filtration rate; GI, gastrointestinal; ns, nonselective; NSAID, nonsteroidal anti-inflammatory drug; PPI, proton-pump inhibitor. |
The consideration of both upper- and lower-GI risk is also very important. Given the increased risk of lower-GI-tract side effects with all ns-NSAIDs and the inability of PPIs to protect against these adverse effects, the algorithm considered ns-NSAIDs inappropriate for use in a patient with high GI risk (Figure 1). Of the c2s agents, celecoxib has consistently been shown to have a better lower-GI-tract tolerability profile than ns-NSAIDs ± PPI,86,90,91 but etoricoxib is not superior to ns-NSAIDs with respect to lower-GI-tract tolerability.88
Conclusion
The choice of NSAID for an individual patient should be based on their risk of developing both GI and CV adverse events. Based on the latest published evidence, both lower- and upper-GI adverse events need to be considered when evaluating the tolerability profile of NSAIDs. The latest published evidence suggests that celecoxib is associated with a lower risk of both upper- and lower-GI-tract side effects than ns-NSAIDs. In addition to the choice of NSAID, use of prophylactic PPIs and eradication of H. pylori can help improve GI tolerability, particularly for preventing adverse upper-GI-tract events. Data from the most recent studies on long-term use of moderate doses of celecoxib showed that it also has a favorable CV-risk profile that is not worse than ns-NSAIDs.
Acknowledgments
Editorial assistance for manuscript preparation was provided by Nicola Ryan, independent medical writer, and See Mee Yen of Medica Comms Pte Ltd. Manuscript development is funded by Pfizer Pte Ltd, Singapore.
Author contributions
All authors were involved in study conception and design and analysis and interpretation of data. All authors were also involved in preparation of the manuscript, revising it for important intellectual content, and final approval before submission for publication.
Disclosure
This paper was compiled based on discussions during an expert advisory meeting convened in Singapore on April 1, 2017 attended by the coauthors and sponsored by Pfizer Pte Ltd, Singapore. The content reflects the opinion of the authors, and Pfizer had no role in drafting or editing the manuscript. The authors received an honorarium for their attendance at the meeting, but no honorarium for the preparation of the manuscript. Dr KYH declares receipt of speaker fees from Pfizer, MSD Pharma, and Mundipharma. The other authors report no conflicts of interest in this work.
References
Classification of chronic pain. Descriptions of chronic pain syndromes and definitions of pain terms. Prepared by the International Association for the Study of Pain, Subcommittee on Taxonomy. Pain Suppl. 1986;3:S1-226. | ||
Vos T, Flaxman AD, Naghavi M, et al. Years lived with disability (YLDs) for 1160 sequelae of 289 diseases and injuries 1990–2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380(9859):2163–2196. | ||
Yeo SN, Tay KH. Pain prevalence in Singapore. Ann Acad Med Singapore. 2009;38(11):937–942. | ||
Lipnik-Stangelj M. Mediators of inflammation as targets for chronic pain treatment. Mediators Inflamm. 2013;2013:783235. | ||
Tal M. A role for inflammation in chronic pain. Curr Rev Pain. 1999;3(6):440–446. | ||
Scarpignato C, Lanas A, Blandizzi C, Lems WF, Hermann M, Hunt RH. Safe prescribing of non-steroidal anti-inflammatory drugs in patients with osteoarthritis: an expert consensus addressing benefits as well as gastrointestinal and cardiovascular risks. BMC Med. 2015;13:55. | ||
Berenbaum F. Osteoarthritis as an inflammatory disease (osteoarthritis is not osteoarthrosis!). Osteoarthritis Cartilage. 2013;21(1):16–21. | ||
Hochberg MC, Altman RD, April KT, et al. American College of Rheumatology 2012 recommendations for the use of nonpharmacologic and pharmacologic therapies in osteoarthritis of the hand, hip, and knee. Arthritis Care Res (Hoboken). 2012;64(4):465–474. | ||
Jordan KM, Arden NK, Doherty M, et al. EULAR Recommendations 2003: an evidence based approach to the management of knee osteoarthritis: report of a task force of the Standing Committee for International Clinical Studies Including Therapeutic Trials (ESCISIT). Ann Rheum Dis. 2003;62(12):1145–1155. | ||
Simon LS, Jacox AK, Caudill-Slosberg M, Gilll LH, Keefe FJ, Kerr KL. Pain in osteoarthritis, rheumatoid arthritis, and juvenile chronic arthritis. In: Clinic Practice Guidelines. 2nd ed. Glenview, IL: American Pain Society; 2002:1–179. | ||
Lee C, Straus WL, Balshaw R, Barlas S, Vogel S, Schnitzer TJ. A comparison of the efficacy and safety of nonsteroidal antiinflammatory agents versus acetaminophen in the treatment of osteoarthritis: a meta-analysis. Arthritis Rheum. 2004;51(5):746–754. | ||
Towheed TE, Maxwell L, Judd MG, Catton M, Hochberg MC, Wells G. Acetaminophen for osteoarthritis. Cochrane Database Syst Rev. 2006;(1):CD004257. | ||
Trelle S, Reichenbach S, Wandel S, et al. Cardiovascular safety of non-steroidal anti-inflammatory drugs: network meta-analysis. BMJ. 2011;342:c7086. | ||
Zhang W, Jones A, Doherty M. Does paracetamol (acetaminophen) reduce the pain of osteoarthritis? A meta-analysis of randomised controlled trials. Ann Rheum Dis. 2004;63(8):901–907. | ||
Chan AT, Manson JE, Albert CM. Nonsteroidal antiinflammatory drugs, acetaminophen, and the risk of cardiovascular events. Circulation. 2006;113(12):1578–1587. | ||
Curhan GC, Willett WC, Rosner B, Stampfer MJ. Frequency of analgesic use and risk of hypertension in younger women. Arch Intern Med. 2002;162(19):2204–2208. | ||
Forman JP, Rimm EB, Curhan GC. Frequency of analgesic use and risk of hypertension among men. Arch Intern Med. 2007;167(4):394–399. | ||
González-Pérez A, Rodríguez LA. Upper gastrointestinal complications among users of paracetamol. Basic Clin Pharmacol Toxicol. 2006;98(3):297–303. | ||
Graham GG, Day RO, Graudins A, Mohamudally A. FDA proposals to limit the hepatotoxicity of paracetamol (acetaminophen): are they reasonable? Inflammopharmacology. 2010;18(2):47–55. | ||
Sudano I, Flammer AJ, Periat D, et al. Acetaminophen increases blood pressure in patients with coronary artery disease. Circulation. 2010;122(18):1789–1796. | ||
Warner TD, Giuliano F, Vojnovic I, Bukasa A, Mitchell JA, Vane JR. Nonsteroid drug selectivities for cyclo-oxygenase-1 rather than cyclo-oxygenase-2 are associated with human gastrointestinal toxicity: a full in vitro analysis. Proc Nat Acad Sci U S A. 1999;96(13):7563–7568. | ||
Henderson TA. Chemical classes of non-steroidal anti-inflammatories (NSAIDs) in US; 2014. Available from: http://paindr.com/wp-content/uploads/2014/07/NSAIDS-Chemical-Classes_2014_Shahzad-Henderson-Fudin.pdf. Accessed July 6, 2018. | ||
Bjarnason I, Scarpignato C, Takeuchi K, Rainsford KD. Determinants of the short-term gastric damage caused by NSAIDs in man. Aliment Pharmacol Ther. 2007;26(1):95–106. | ||
Khazaeinia T, Jamali F. A comparison of gastrointestinal permeability induced by diclofenac-phospholipid complex with diclofenac acid and its sodium salt. J Pharm Pharm Sci. 2003;6(3):352–359. | ||
Bjarnason I, Takeuchi K. Intestinal permeability in the pathogenesis of NSAID-induced enteropathy. J Gastroenterol. 2009;44(Suppl 19):23–29. | ||
Chang IJ, Harris RC. Are all COX-2 inhibitors created equal? Hypertension. 2005;45(2):178–180. | ||
Walter MF, Jacob RF, Day CA, Dahlborg R, Weng Y, Mason RP. Sulfone COX-2 inhibitors increase susceptibility of human LDL and plasma to oxidative modification: comparison to sulfonamide COX-2 inhibitors and NSAIDs. Atherosclerosis. 2004;177(2):235–243. | ||
Nissen SE, Yeomans ND, Solomon DH, et al. Cardiovascular safety of celecoxib, naproxen, or ibuprofen for arthritis. N Engl J Med. 2016;375(26):2519–2529. | ||
Chan FK, Ching JY, Tse YK, et al. Gastrointestinal safety of celecoxib versus naproxen in patients with cardiothrombotic diseases and arthritis after upper gastrointestinal bleeding (CONCERN): an industry-independent, double-blind, double-dummy, randomised trial. Lancet. 2017;389(10087):2375–2382. | ||
Bally M, Dendukuri N, Rich B, et al. Risk of acute myocardial infarction with NSAIDs in real world use: Bayesian meta-analysis of individual patient data. BMJ. 2017;357:j1909. | ||
Chan FK, Wong VW, Suen BY, et al. Combination of a cyclo-oxygenase-2 inhibitor and a proton-pump inhibitor for prevention of recurrent ulcer bleeding in patients at very high risk: a double-blind, randomised trial. Lancet. 2007;369(9573):1621–1626. | ||
Bombardier C, Laine L, Reicin A, et al. Comparison of upper gastrointestinal toxicity of rofecoxib and naproxen in patients with rheumatoid arthritis. N Engl J Med. 2000;343(21):1520–1528. | ||
Mukherjee D, Nissen SE, Topol EJ. Risk of cardiovascular events associated with selective COX-2 inhibitors. JAMA. 2001;286(8):954–959. | ||
Bresalier RS, Sandler RS, Quan H, et al. Cardiovascular events associated with rofecoxib in a colorectal adenoma chemoprevention trial. N Engl J Med. 2005;352(11):1092–1102. | ||
Fitzgerald GA, Patrono C. The coxibs, selective inhibitors of cyclooxygenase-2. N Engl J Med. 2001;345(6):433–442. | ||
Fitzgerald GA. Coxibs and cardiovascular disease. N Engl J Med. 2004;351(17):1709–1711. | ||
Warner TD, Mitchell JA. COX-2 selectivity alone does not define the cardiovascular risks associated with non-steroidal anti-inflammatory drugs. Lancet. 2008;371(9608):270–273. | ||
Howes LG. Selective COX-2 inhibitors, NSAIDs and cardiovascular events: is celecoxib the safest choice? Ther Clin Risk Manag. 2007;3(5):831–845. | ||
Steffel J, Luscher TF, Ruschitzka F, Tanner FC. Cyclooxygenase-2 inhibition and coagulation. J Cardiovasc Pharmacol. 2006;47(Suppl 1):S15–S20. | ||
Silverstein FE, Faich G, Goldstein JL, et al. Gastrointestinal toxicity with celecoxib vs nonsteroidal anti-inflammatory drugs for osteoarthritis and rheumatoid arthritis – the CLASS study: a randomized controlled trial. JAMA. 2000;284(10):1247–1255. | ||
White WB, Faich G, Whelton A, et al. Comparison of thromboembolic events in patients treated with celecoxib, a cyclooxygenase-2 specific inhibitor, versus ibuprofen or diclofenac. Am J Cardiol. 2002;89(4):425–430. | ||
Curfman GD, Morrissey S, Drazen JM. Expression of concern: Bombardier et al, “Comparison of upper gastrointestinal toxicity of rofecoxib and naproxen in patients with rheumatoid arthritis”. N Engl J Med. 2005;353(26):2813–2814. | ||
Choi HK, Rho YH, Zhu Y, Cea-Soriano L, Aviña-Zubieta JA, Zhang Y. The risk of pulmonary embolism and deep vein thrombosis in rheumatoid arthritis: a UK population-based outpatient cohort study. Ann Rheum Dis. 2013;72(7):1182–1187. | ||
Rahme E, Pilote L, Lelorier J. Association between naproxen use and protection against acute myocardial infarction. Arch Intern Med. 2002;162(10):1111–1115. | ||
Reed GW, Abdallah MS, Shao M, et al. Effect of aspirin coadministration on the safety of celecoxib, naproxen, or ibuprofen. J Am Coll Cardiol. 2018;71(16):1741–1751. | ||
Solomon SD, McMurray JJ, Pfeffer MA, et al. Cardiovascular risk associated with celecoxib in a clinical trial for colorectal adenoma prevention. N Engl J Med. 2005;352(11):1071–1080. | ||
ADAPT Research Group. Cardiovascular and cerebrovascular events in the randomized, controlled Alzheimer’s Disease Anti-Inflammatory Prevention Trial (ADAPT). PLoS Clin Trials. 2006;1(7):e33. | ||
Bhala N, Emberson J. Vascular and upper gastrointestinal effects of non-steroidal anti-inflammatory drugs: meta-analyses of individual participant data from randomised trials. Lancet. 2013;382(9894):769–779. | ||
Graham DJ, Campen D, Hui R, et al. Risk of acute myocardial infarction and sudden cardiac death in patients treated with cyclo-oxygenase 2 selective and non-selective non-steroidal anti-inflammatory drugs: nested case-control study. Lancet. 2005;365(9458):475–481. | ||
Moore RA, Derry S, McQuay HJ. Cyclo-oxygenase-2 selective inhibitors and nonsteroidal anti-inflammatory drugs: balancing gastrointestinal and cardiovascular risk. BMC Musculoskelet Disord. 2007;8:73. | ||
Combe B, Swergold G, McLay J, et al. Cardiovascular safety and gastrointestinal tolerability of etoricoxib vs diclofenac in a randomized controlled clinical trial (the MEDAL study). Rheumatology (Oxford). 2009;48(4):425–432. | ||
McGettigan P, Henry D. Cardiovascular risk with non-steroidal anti-inflammatory drugs: systematic review of population-based controlled observational studies. PLoS Med. 2011;8(9):e1001098. | ||
Avorn J. Keeping science on top in drug evaluation. N Engl J Med. 2007;357(7):633–635. | ||
MacDonald TM, Hawkey CJ, Ford I, et al. Randomized trial of switching from prescribed non-selective non-steroidal anti-inflammatory drugs to prescribed celecoxib: the Standard care vs. Celecoxib Outcome Trial (SCOT). Eur Heart J. 2017;38(23):1843–1850. | ||
Hirayama A, Tanahashi N, Daida H, et al. Assessing the cardiovascular risk between celecoxib and nonselective nonsteroidal antiinflammatory drugs in patients with rheumatoid arthritis and osteoarthritis. Circ J. 2014;78(1):194–205. | ||
Bjarnason I, Hayllar J, MacPherson AJ, Russell AS. Side effects of nonsteroidal anti-inflammatory drugs on the small and large intestine in humans. Gastroenterology. 1993;104(6):1832–1847. | ||
Graham DY, Opekun AR, Willingham FF, Qureshi WA. Visible small-intestinal mucosal injury in chronic NSAID users. Clin Gastroenterol Hepatol. 2005;3(1):55–59. | ||
Gwee KA, Goh V, Lima G, Setia S. Coprescribing proton-pump inhibitors with nonsteroidal anti-inflammatory drugs: risks versus benefits. J Pain Res. 2018;11:361–374. | ||
Fock KM, Ang TL. Epidemiology of Helicobacter pylori infection and gastric cancer in Asia. J Gastroenterol Hepatol. 2010;25(3):479–486. | ||
Goh KL, Parasakthi N. The racial cohort phenomenon: seroepidemiology of Helicobacter pylori infection in a multiracial South-East Asian country. Eur J Gastroenterol Hepatol. 2001;13(2):177–183. | ||
Eusebi LH, Zagari RM, Bazzoli F. Epidemiology of Helicobacter pylori infection. Helicobacter. 2014;19(9):1–5. | ||
Lanza FL, Chan FK, Quigley EM. Guidelines for prevention of NSAID-related ulcer complications. Am J Gastroenterol. 2009;104(3):728–738. | ||
Bhatt DL, Scheiman J, Abraham NS, et al. ACCF/ACG/AHA 2008 expert consensus document on reducing the gastrointestinal risks of antiplatelet therapy and NSAID use: a report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents. J Am Coll Cardiol. 2008;52(18):1502–1517. | ||
Lanas A, Tornero J, Zamorano JL. Assessment of gastrointestinal and cardiovascular risk in patients with osteoarthritis who require NSAIDs: the LOGICA study. Ann Rheum Dis. 2010;69(8):1453–1458. | ||
Straube S, Tramèr MR, Moore RA, Derry S, McQuay HJ. Mortality with upper gastrointestinal bleeding and perforation: effects of time and NSAID use. BMC Gastroenterol. 2009;9:41. | ||
Armstrong CP, Blower AL. Non-steroidal anti-inflammatory drugs and life threatening complications of peptic ulceration. Gut. 1987;28(5):527–532. | ||
Singh G, Ramey DR, Morfeld D, Shi H, Hatoum HT, Fries JF. Gastrointestinal tract complications of nonsteroidal anti-inflammatory drug treatment in rheumatoid arthritis: a prospective observational cohort study. Arch Intern Med. 1996;156(14):1530–1536. | ||
Hippisley-Cox J, Coupland C, Logan R. Risk of adverse gastrointestinal outcomes in patients taking cyclo-oxygenase-2 inhibitors or conventional non-steroidal anti-inflammatory drugs: population based nested case-control analysis. BMJ. 2005;331(7528):1310–1316. | ||
Mallen SR, Essex MN, Zhang R. Gastrointestinal tolerability of NSAIDs in elderly patients: a pooled analysis of 21 randomized clinical trials with celecoxib and nonselective NSAIDs. Curr Med Res Opin. 2011;27(7):1359–1366. | ||
Scheiman JM, Yeomans ND, Talley NJ, et al. Prevention of ulcers by esomeprazole in at-risk patients using non-selective NSAIDs and COX-2 inhibitors. Am J Gastroenterol. 2006;101(4):701–710. | ||
Scarpignato C, Hunt RH. Nonsteroidal antiinflammatory drug-related injury to the gastrointestinal tract: clinical picture, pathogenesis, and prevention. Gastroenterol Clin North Am. 2010;39(3):433–464. | ||
Goldstein JL, Eisen GM, Lewis B, et al. Small bowel mucosal injury is reduced in healthy subjects treated with celecoxib compared with ibuprofen plus omeprazole, as assessed by video capsule endoscopy. Aliment Pharmacol Ther. 2007;25(10):1211–1222. | ||
Goldstein JL, Eisen GM, Lewis B, Gralnek IM, Zlotnick S, Fort JG. Video capsule endoscopy to prospectively assess small bowel injury with celecoxib, naproxen plus omeprazole, and placebo. Clin Gastroenterol Hepatol. 2005;3(2):133–141. | ||
Johnson DA, Oldfield EC. Reported side effects and complications of long-term proton pump inhibitor use: dissecting the evidence. Clin Gastroenterol Hepatol. 2013;11(5):458–464. | ||
Katz MH. Failing the acid test: benefits of proton pump inhibitors may not justify the risks for many users. Arch Intern Med. 2010;170(9):747–748. | ||
Lin HJ, Hung YP, Liu HC, et al. Risk factors for Clostridium difficile-associated diarrhea among hospitalized adults with fecal toxigenic C. difficile colonization. J Microbiol Immunol Infect. 2015;48(2):183–189. | ||
Pello Lázaro AM, Cristóbal C, Franco-Peláez JA, et al. Use of proton-pump inhibitors predicts heart failure and death in patients with coronary artery disease. PLoS One. 2017;12(1):e0169826. | ||
Sukhovershin RA, Cooke JP. How may proton pump inhibitors impair cardiovascular health? Am J Cardiovasc Drug. 2016;16(3):153–161. | ||
Lombardo L, Foti M, Ruggia O, Chiecchio A. Increased incidence of small intestinal bacterial overgrowth during proton pump inhibitor therapy. Clin Gastroenterol Hepatol. 2010;8(6):504–508. | ||
Lanas A, García-Rodríguez LA, Polo-Tomás M, et al. Time trends and impact of upper and lower gastrointestinal bleeding and perforation in clinical practice. Am J Gastroenterol. 2009;104(7):1633–1641. | ||
Adebayo D, Bjarnason I. Is non-steroidal anti-inflammatory drug (NSAID) enteropathy clinically more important than NSAID gastropathy? Postgrad Med J. 2006;82(965):186–191. | ||
McCarthy DM. GI bleeding: problems that persist. Gastrointest Endosc. 2009;70(2):225–228. | ||
Lanas A, Hunt R. Prevention of anti-inflammatory drug-induced gastrointestinal damage: benefits and risks of therapeutic strategies. Ann Med. 2006;38(6):415–428. | ||
Maiden L, Thjodleifsson B, Theodors A, Gonzalez J, Bjarnason I. A quantitative analysis of NSAID-induced small bowel pathology by capsule enteroscopy. Gastroenterology. 2005;128(5):1172–1178. | ||
Fujimori S, Gudis K, Takahashi Y, et al. Distribution of small intestinal mucosal injuries as a result of NSAID administration. Eur J Clin Invest. 2010;40(6):504–510. | ||
Chan FK, Lanas A, Scheiman J, Berger MF, Nguyen H, Goldstein JL. Celecoxib versus omeprazole and diclofenac in patients with osteoarthritis and rheumatoid arthritis (CONDOR): a randomised trial. Lancet. 2010;376(9736):173–179. | ||
Cryer B, Li C, Simon LS, Singh G, Stillman MJ, Berger MF. GI-REASONS: a novel 6-month, prospective, randomized, open-label, blinded endpoint (PROBE) trial. Am J Gastroenterol. 2013;108(3):392–400. | ||
Laine L, Curtis SP, Langman M, et al. Lower gastrointestinal events in a double-blind trial of the cyclo-oxygenase-2 selective inhibitor etoricoxib and the traditional nonsteroidal anti-inflammatory drug diclofenac. Gastroenterology. 2008;135(5):1517–1525. | ||
Antman EM, Bennett JS, Daugherty A, Furberg C, Roberts H, Taubert KA. Use of nonsteroidal antiinflammatory drugs – an update for clinicians: a scientific statement from the American Heart Association. Circulation. 2007;115(12):1634–1642. | ||
Chan FK, Cryer B, Goldstein JL, et al. A novel composite endpoint to evaluate the gastrointestinal (GI) effects of nonsteroidal antiinflammatory drugs through the entire GI tract. J Rheumatol. 2010;37(1):167–174. | ||
Moore A, Makinson G, Li C. Patient-level pooled analysis of adjudicated gastrointestinal outcomes in celecoxib clinical trials: meta-analysis of 51,000 patients enrolled in 52 randomized trials. Arthritis Res Ther. 2013;15(1):R6. | ||
Chan CC, Reid CM, Aw TJ, Liew D, Haas SJ, Krum H. Do COX-2 inhibitors raise blood pressure more than nonselective NSAIDs and placebo? An updated meta-analysis. J Hypertens. 2009;27(12):2332–2341. | ||
Fanelli A, Ghisi D, Aprile PL, Lapi F. Cardiovascular and cerebrovascular risk with nonsteroidal anti-inflammatory drugs and cyclooxygenase 2 inhibitors: latest evidence and clinical implications. Ther Adv Drug Saf. 2017;8(6):173–182. | ||
Rodríguez LA, Tacconelli S, Patrignani P. Role of dose potency in the prediction of risk of myocardial infarction associated with nonsteroidal anti-inflammatory drugs in the general population. J Am Coll Cardiol. 2008;52(20):1628–1636. | ||
Huntjens DR, Danhof M, Della Pasqua OE. Pharmacokinetic–pharmacodynamic correlations and biomarkers in the development of COX-2 inhibitors. Rheumatology (Oxford). 2005;44(7):846–859. | ||
Sowers JR, White WB, Pitt B, et al. The effects of cyclooxygenase-2 inhibitors and nonsteroidal anti-inflammatory therapy on 24-hour blood pressure in patients with hypertension, osteoarthritis, and type 2 diabetes mellitus. Arch Intern Med. 2005;165(2):161–168. | ||
Okumu A, Dimaso M, Löbenberg R. Computer simulations using GastroPlus to justify a biowaiver for etoricoxib solid oral drug products. Eur J Pharm Biopharm. 2009;72(1):91–98. | ||
Riendeau D, Percival MD, Brideau C, et al. Etoricoxib (MK-0663): preclinical profile and comparison with other agents that selectively inhibit cyclooxygenase-2. J Pharmacol Exp Ther. 2001;296(2):558–566. | ||
Aw TJ, Haas SJ, Liew D, Krum H. Meta-analysis of cyclooxygenase-2 inhibitors and their effects on blood pressure. Arch Intern Med. 2005;165(5):490–496. | ||
Widlansky ME, Price DT, Gokce N, et al. Short- and long-term COX-2 inhibition reverses endothelial dysfunction in patients with hypertension. Hypertension. 2003;42(3):310–315. | ||
Craig DG, Bates CM, Davidson JS, Martin KG, Hayes PC, Simpson KJ. Staggered overdose pattern and delay to hospital presentation are associated with adverse outcomes following paracetamol-induced hepatotoxicity. Br J Clin Pharmacol. 2012;73(2):285–294. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.