")
Back to Journals » Clinical and Experimental Gastroenterology » Volume 14
Nonalcoholic Fatty Liver Disease and Chronic Kidney Disease: A Review of Links and Risks
Received 1 April 2021
Accepted for publication 21 October 2021
Published 17 November 2021 Volume 2021:14 Pages 457—465
DOI https://doi.org/10.2147/CEG.S226130
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Anastasios Koulaouzidis
Amanda Cheung, Aijaz Ahmed
Division of Gastroenterology and Hepatology, Stanford University School of Medicine, Stanford, CA, USA
Correspondence: Amanda Cheung
Division of Gastroenterology and Hepatology, Stanford University School of Medicine, 750 Welch Road, Suite 210, Stanford, CA, 94304, USA
Tel +1 650-498-6080
Fax +1 650-498-5692
Email [email protected]
Abstract: Nonalcoholic fatty liver disease and chronic kidney disease are both chronic conditions with rapidly increasing prevalence and incidence worldwide that have led to a significant burden on health-care systems. The association between these two disease entities is partly attributed to shared cardiometabolic comorbidities including diabetes, hypertension, obesity, and metabolic syndrome. However, independent of these overlapping risks, there are increased rates and more severe CKD in NAFLD patients. Conversely, more progressive NAFLD is seen with advanced stages of kidney injury. In addition to overlapping risk factors, shared pathogenic mechanisms suggest these two disease entities may resemble different manifestations of a single underlying disease process.
Keywords: nonalcoholic fatty liver disease, chronic kidney disease, mortality, metabolic syndrome, gut-liver axis, gut-kidney axis, liver-kidney axis
Introduction
Nonalcoholic fatty liver disease (NAFLD) is defined by the presence of hepatic steatosis with a spectrum of severity including nonalcoholic fatty liver (NAFL), nonalcoholic steatohepatitis (NASH) with and without fibrosis, and cirrhosis. NAFL is also referred to as simple steatosis; that is, there is increased fat accumulation in hepatocytes but no significant inflammation, injury, or fibrosis. NASH is defined histologically by the presence of steatosis, lobular inflammation, and liver cell injury with hepatocyte ballooning.1
Chronic kidney disease (CKD) is defined by the Kidney Disease Improving Global Outcomes (KDIGO) work group as abnormalities of kidney structure or function for greater than three months with implications for health.2 Glomerular filtration rate (GFR) is a marker of kidney function with five categories (G1: ≥90; G2: 60–89; G3a: 45–59; G3b: 30–44; G4: 15–29; G5 <15 mL/min/1.73 m2) where CKD is defined by GFR <60 mL/min/1.73 m2. Albumin-to-creatinine ratio (ACR) is one of the markers of kidney damage with three categories (A1: <30; A2: 30–300; A3: >300 mg/g) with the threshold >30 mg/g defining CKD. Staging of CKD includes categorization based on decreased GFR and increased ACR, and these both play an important role in risk categorization and prognostication.2
Increasing rates of both NAFLD and CKD is expected with the rising trends in diabetes (DM) and obesity alongside the aging population.3 NAFLD is quickly rising to be the leading cause of liver transplantation and most rapidly increasing indication for simultaneous liver-kidney transplants.4 Understanding the interplay between these two disease entities is important for screening, early recognition, and therapeutics development.
Epidemiology
NAFLD is a growing public health concern globally with current estimated prevalence of 25%.5 These rates are predicted to increase further within the next decade to include one in three adults with NAFLD and over one in four NAFLD patients with NASH.6 The economic burden of NAFLD is significant with annual costs of $130 billion in the United States and €35 billion cumulatively in four European countries.7 Among NAFLD patients, cirrhosis and its associated complications including hepatocellular carcinoma are the most common liver-related causes of morbidity, but cardiovascular disease (CVD) events are the leading cause of overall morbidity and mortality.8
The prevalence of CKD is estimated to be 13.4% in the global population.9 Economic burden from CKD is weighted heavily on the later stages, particularly once renal replacement therapy has been initiated with an estimated annual cost of $100,593 per patient in the United States.10 Increased mortality is observed when GFR falls below 60 mL/min/1.73 m2 with an adjusted hazard ratio of 1.2, 1.8, 3.2, and 5.9 for stage 3a, 3b, 4, and 5, respectively.11 The impact of CKD on mortality is increasing over time. While it is currently ranked as the sixteenth leading cause of death in 2016, modeling data suggests a rise to the fifth leading cause of death globally by 2040.12 Risk factors that contribute to total age-standardized rate of CKD disability-adjusted life years include impaired fasting glucose, high blood pressure, and high body mass index.13
The cardiometabolic risk factors present in both NAFLD and CKD lead to atherogenic dyslipidemia and CVD events (hospitalization for coronary heart disease, heart failure, ischemic stroke, or peripheral arterial disease) which occur at greater rates in CKD.11 Increasing ACR leads to increased all-cause and cardiovascular mortality independent of GFR, including at normal GFR levels.14 In a meta-analysis of 16 observational cohort studies, NAFLD was found to be an independent risk factor for incident CVD events (OR: 1.63, CI: 1.06–2.48) with further increased rates in severe NAFLD (OR: 1.94, CI: 1.17–3.21).15 In an analysis of the Third National Health and Nutrition Survey database, presence of hepatic steatosis detected by ultrasound with concomitant CKD was associated with progressively increased rates of overall (HR: 2.31 CI: 1.8–3.16 for stage 2–3a, 4.83 CI: 2.4–9.71 for stage 3b-5) and CVD-related (2.06 CI: 1.16–3.65 for stage 2–3a, 6.04 CI: 2.39–15.26 for stage 3b–5) mortality.16 Due to the impact of coexistent NAFLD and CKD on morbidity and mortality rates, public health initiatives have included earlier identification for both disease entities and modification of known risk factors.
Association Between NAFLD and CKD
In cross-sectional studies, the prevalence of CKD was 20–55% in patients with NAFLD compared to 5–30% in patients without NAFLD with a persistent association even after adjustment for DM and other common risk factors.17 In a meta-analysis of 13 longitudinal studies, risk of incident CKD was 80% greater in NAFLD (HR: 1.79, CI: 1.65–1.95) independent of overlapping cardiometabolic risk factors.18 Similarly, in a meta-analysis of nine observational cohort studies, presence of NAFLD was associated with a 40% increased risk of incident CKD stage ≥3 (HR: 1.37, CI: 1.20–1.53).19 Importantly, more advanced NAFLD has even greater impact on incident CKD findings.
In comparison to NAFL, NASH is associated with an increased prevalence (OR: 2.53, CI: 1.58–4.05) and incidence (HR: 2.12, CI: 1.42–3.17) of CKD compared to NAFL.18 Of the histological components that define NASH, portal inflammation score ≥3 is associated with significantly increased risk of renal outcomes (HR: 6.58, p=0.001) when adjusted for age, sex, insulin resistance (IR), and hypertension (HTN).20 In comparing biopsy-proven NASH with control non-NAFLD individuals matched for age, sex, and body mass index, there is a significantly increased prevalence of CKD and albuminuria.21 Among patients with NASH, incident risk of CKD increases further with greater stages of CKD (OR: 2.49 CI: 1.21–5.13 in stage 3b, 3.45 CI: 1.15–10.39 in stage 4, 3.87 CI: 1.1–13.58 in stage 5).18 In a post hoc analysis from a trial studying impact of lifestyle changes on biopsy-proven NASH, histological resolution of NASH led to an improvement in kidney function (GFR 2.32 vs −1.04 mL/min/1.73 m2, p=0.04) irrespective of weight loss.22
In comparison to lower stage of fibrosis, NASH with advanced (stage 3–4) fibrosis is associated with greater risk of incident CKD (HR: 3.29, CI: 2.3–4.71) which is progressively increased with higher stages of CKD (OR: 7.48 CI: 2.95–18.97 in stage 3b, 7.66 CI: 2.72–21.56 in stage 4, 12.67 CI: 4.49–35.76 in stage 5).18 Fibrosis staging in NASH has a graded association with decreasing GFR independent of age, body mass index, IR, and components of the metabolic syndrome.21 Improvement of a single fibrosis stage in NASH leads to improvement in kidney function (GFR +7.6 vs −1.98 mL/min/1.73 m2, p<0.01) irrespective of weight loss.22
The Link Between NAFLD and CKD: Common Risk Factors and Shared Mechanisms
NAFLD is a multiorgan disease and has been strongly associated with type 2 DM, CVD, and CKD.23 A causal relationship between NAFLD and CKD is difficult to prove given their many shared risk factors including IR, DM, HTN, dyslipidemia, and obesity. Numerous shared risks and pathogenetic mechanisms carry out similar processes of injury with interplay between the organs (Figure 1).
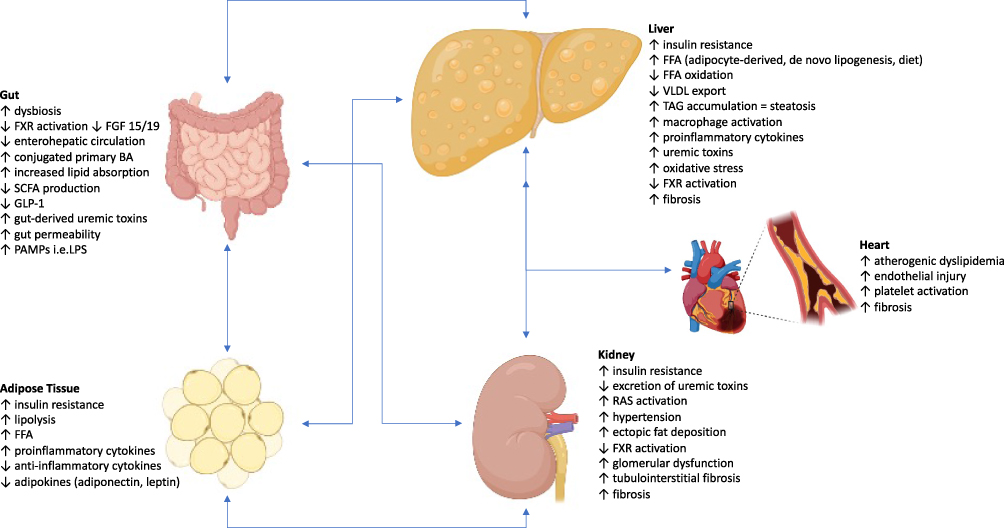 |
Figure 1 Role of a Gut-AT-Liver-Kidney Axis Leading to NAFLD and CKD. The pathogenesis of NAFLD and CKD have many shared causes including dysregulation and dysfunction of the gut and adipose tissue, chronic inflammation, atherogenic dyslipidemia, and proinflammatory cytokines and hepatokines. The primary feature of hepatic steatosis occurs when there is an imbalance of FFA leading to excess lipid droplets containing TAGs which are the storage form for FFAs in the hepatocytes. In the steady state, the concentration of TAGs in the liver is kept low by balancing the trafficking of FFAs into and out of the liver, production of FFAs by hepatocytes, consumption of FFAs by mitochondrial beta-oxidation, and TAG export from the liver as VLDL particles. The glucose uptake receptors in adipose tissue are dependent on insulin. Thus, insulin resistance leads to decreased glucose uptake and increased serum glucose concentrations, which then promotes insulin production by the pancreatic beta cells. Dysregulation of adipose tissue leads to increased lipolysis and secretion of FFAs, and this is the primary source of FFAs to the liver contributing to steatosis. Activation of macrophages in adipose tissue leads to secretion of cytokines and chemokines and further drives insulin resistance. FXR expression is downregulated by the systemic inflammation, and the altered microbiome increases conversion of primary BAs to secondary BAs, which lowers the concentration of the more potent activator of FXR and subsequently suppresses the pathways that increase GLP-1 expression, inhibit lipolysis in the adipose tissue, decrease de novo lipogenesis in the liver, and increase FFA oxidation in the liver, muscle, and adipose tissue. Activation of the renin-angiotensin-aldosterone system leads to increased production of angiotensin II and uric acid. Gut-derived uremic toxins (indole, p-cresol, trimethylamine, ammonia) increase oxidative stress and proinflammatory cytokines which further propagates gut permeability and translocation of PAMPs. Once CKD has developed, there is decreased excretion of the uremic toxins. The gut-derived toxins are shuttled to the liver where they are oxidized or conjugated into nephrotoxic compounds. Kupffer cell activation with cytokine release results in macrophage differentiation with proinflammatory and fibrogenic functions. Thus, all these aforementioned processes cumulatively activate inflammatory, oxidative, and fibrotic pathways in the kidney, liver, and heart. |
Metabolic Syndrome
Metabolic syndrome (MetS) is defined by the presence of three or more of the following risk factors:24
- Increased waist circumference (cutoffs vary by population and country of origin)
- Triglycerides ≥150 mg/dL or use of medications to lower triglycerides
- HDL-C <40 mg/dL in males <50 mg/dL in females
- Fasting glucose ≥100 mg/dL or use of medications to lower glucose
- Systolic blood pressure ≥130 mmHg, diastolic blood pressure ≥85 mmHg, or use of medications to lower blood pressure
In NAFLD patients, presence of all components of the MetS increases overall, cardiac, and liver-related mortality.25 MetS, particularly DM and HTN, have a bidirectional relationship with NAFLD.26 NAFLD is often considered the hepatic manifestation of the MetS with a greater risk of NASH (OR: 3.2, CI: 1.2–8.9, p=0.026) and advanced fibrosis (OR: 3.5, CI: 1.1–11.2, p=0.032).27 Conversely, NAFLD may also be a precursor to the future development of MetS.26
The association between MetS and CKD is not surprising given that the two leading causes for CKD, DM and HTN, are also components MetS. The presence of MetS is significantly associated with decreased eGFR <60 mL/min/1.73 m2 (OR: 1.55, CI: 1.34–1.8) which includes a contribution from each individual component of MetS and the greatest risk if all five components of MetS are present (OR: 1.96, CI: 1.71–2.24, p<0.01).28 Furthermore, reversal or resolution of an individual MetS component leads to a reduced risk of CKD.29
Insulin Resistance and Hyperinsulinemia
The underlying pathogenic mechanisms that tie NAFLD and CKD together remains under investigation but there appears to be a clear contribution from IR and the MetS. IR affects multiple organs that culminate into shared risks for development of both NAFLD and CKD. Insulin plays an important role in the body including, (1) glycolysis and glycogenesis in the liver, (2) glucose uptake, glycolysis, glycogenesis, amino acid uptake, and protein synthesis in the skeletal muscle, (3) glucose uptake and lipogenesis in adipose tissue, and (4) insulin production and secretion in the pancreas.
In the setting of IR, there is impaired glucose uptake which leads to elevated glucose levels in the blood and further production of insulin by the pancreas. Relatively higher levels of circulating glucose and insulin ultimately promote de novo lipogenesis in the liver by activating transcription factors that promote expression of lipogenic genes and suppress beta-oxidation of free fatty acids (FFA).30 In the adipose tissue, IR leads to disruption of the downstream signaling resulting in excess lipolysis with subsequent delivery of these FFAs to the liver. In NAFLD, the three main sources for triacylglycerols (TAGs) in the liver are adipocyte-derived FFAs from lipolysis, hepatic de novo lipogenesis from carbohydrates, and dietary intake, which account for approximately 59%, 26%, and 15%, respectively.31
Both hyperinsulinemia and IR contribute to kidney injury. With the increased insulin levels, there is further upregulation of the renin-angiotensin system and sympathetic nervous system activation which drives HTN and renal injury.32 High levels of glucose that occurs with IR leads to nonenzymatic glycation of proteins and lipids forming proinflammatory molecules leading to atherosclerosis and hyaline arteriosclerosis. This process contributes to the CVD commonly seen and the latter mechanism occurs in the kidneys resulting in glomerular injury and CKD even prior to the development of DM.32
Intestinal Dysbiosis
The farnesoid X receptor (FXR) is a nuclear transcription factor that plays a key role in bile acid, glucose, and lipid metabolism.33 Primary bile acids (BAs) are made from cholesterol in the liver while secondary BAs are produced in the colon from bacterial metabolism of the primary BAs. Bile acids, mainly the primary BA chenodeoxycholic acid, activate FXR in the enterocytes which promotes insulin sensitivity, decreases intestinal lipid absorption and induces fibroblast growth factor (FGF) 19 that inhibits BA production in the liver. FXR in the hepatocytes decreases de novo lipogenesis, VLDL secretion, gluconeogenesis, and glycolysis and increases FFA oxidation and glycogenesis.34
In NAFLD, numerous mechanisms are at play to ultimately decrease FXR activation.33 As a result, there is decreased FGF19 signaling, resulting in increased hepatic BA production and altered regulation of lipogenesis in the liver that culminates into hepatic TAG accumulation.34 In the kidney, FXR deficiency increases renal lipid accumulation, profibrotic growth factors, proinflammatory cytokines, and oxidative stress leading to apoptosis, fibrosis, and progression to CKD.35
High saturated fat diets promote conjugation of cholic acid with taurine (TCA) which alters the microbiome to favor sulfite-reducing bacteria since the taurine contains a sulfite group as opposed to a carboxyl group. This imbalance of microbiota, that is dysbiosis, leads to local inflammation and gut permeability, and TCA functions as an FXR antagonist. In addition, downregulation of the apical-sodium BA transporters (ASBT) on the enterocytes that are responsible for reabsorption of bile acids for enterohepatic circulation leads to decreased expression of FXR and FGF19. Thus, delivery of primary conjugated BAs to the colon is increased. The altered microbiome in the colon decreases the transformation of primary BAs to secondary BAs which typically would cause an increase in glucagon-like peptide (GLP)-1. The imbalance of primary to secondary BAs favors inflammation and increases gut permeability. While there is an excess of primary BAs, there is also increased secondary BA production, decreased lipid excretion in stool, and increased intestinal lipid absorption. The proinflammatory state further downregulates FXR expression.33
When nondigestible dietary fiber reaches the colon, it undergoes anaerobic fermentation into short-chain fatty acids (SCFA) which can affect glucose regulation by increasing incretin (i.e. GLP-1) secretion.36 A low fiber diet alongside alteration in the gut microbiota, including a decrease in SCFA-producing bacteria, result in a decreased supply of SCFAs which promotes hepatic lipogenesis and a chain of systemic inflammation.37
Byproducts from the microbiota metabolism have been implicated in the development of numerous cardiometabolic diseases.38 Increased urea concentrations in CKD is metabolized by microbiota to ammonia and contributes further to increased gut permeability with entry of pathogen-associated molecular patterns (PAMPs), increased BA absorption, and production of nephrotoxic and hepatotoxic molecules and inflammatory cytokines.39
Bacterial metabolism of ingested nutrients include trimethylamine (TMA) from carnitine and choline, indole from tryptophan, p-cresyl from tyrosine and phenylalanine, and phenylacetic acid from phenylalanine.40 Conversion of indole and p-cresyl in the liver produces uremic toxins, indoxyl sulfate and p-cresyl sulfate, that cause renal and vascular injury by producing reactive oxygen species and proinflammatory cytokines.41 Similarly, oxidation of TMA to trimethylamine-N-oxide (TMAO) occurs in the liver by an enzyme that is induced by FXR, and higher levels of TMAO have been found in CKD patients due to impaired excretion of these uremic toxins and dysbiosis in the gut.42 Higher TMAO levels suppresses reverse cholesterol transport which promotes atherosclerosis and increases the rates of cardiovascular disease in this population.43
Proinflammatory State
Insulin resistance leads to dysregulation of adipose tissue with increased release of FFAs and proinflammatory adipokines.44 Adipose tissue-derived signals of inflammation include adipocytokines (adiponectin, leptin) and proinflammatory cytokines.45 The degree of adipose tissue dysfunction, rather than the quantity of adipose tissue or obesity, has the greater effect on hepatic steatosis and necroinflammation.46,47
This mechanism of injury has been attributed to PAMPs activating the signaling cascades to macrophages that release cytokines and recruit proinflammatory cells and hepatic stellate cells which play a role in apoptosis and fibrosis.48 Reactive oxygen species (ROS) produced by oxidation of FFAs cause additional inflammation and injury to the liver. Increased transcription of proinflammatory genes in NASH contributes to systemic inflammation and production of ROS with extrahepatic effects including CKD.39
Systemic inflammation compromises the integrity of the intestinal barrier allowing endotoxins and other PAMPs from the gut to reach the hepatocytes and promotes metabolic disturbances including IR and proinflammatory cytokine production that further contributes to NAFLD, CKD, and CVD.49 Gut-derived uremic toxins increase oxidative stress and cytokines, further propagating gut permeability and translocation of PAMPs. All these aforementioned processes cumulatively activate inflammatory, oxidative, and fibrotic pathways in the kidney, liver, and heart.38
Treatment Strategies in Coexistent NAFLD and CKD
Development of therapeutics to improve both NAFLD and CKD has been forthcoming given the link between the two conditions including overlapping risks and poor outcomes. The targets for therapies are based on the proposed pathogenic mechanisms shared between the two disease entities, particularly modification of cardiometabolic risk factors and reducing IR. Additional strategies for therapeutics under investigation for NAFLD include decreasing FFA delivery to the liver and increased disposal of FFA from the liver.
Sodium‐glucose co‐transporter‐2 (SGLT2) inhibitors and glucagon-like peptide‐1 receptor agonists (GLP-1RA) are two groups of medications approved for use in DM that have additional benefits for NASH and protective effects on cardiovascular and renal outcomes.50 SGLT2 inhibitors are recommended to prevent progression of CKD in diabetic patients, particularly for those with ACR >200 mg/g or GFR 25–60 mL/min/1.73 m2.50 Meta-analysis of three RCTs comparing the use of SGLT2 inhibitors to standard of care diabetic treatment showed significant decrease in hepatic steatosis determined by magnetic resonance imaging (MRI) (−2.2, CI: −3.67 to −0.74, p=0.003) and degree of fibrosis determined by FIB-4 scores (−0.06, CI: −0.1 to −0.02, p=0.01) despite no significant difference in degree of glucose control between groups.51
GLP-1 is an incretin hormone secreted by the enterocytes that promotes peripheral insulin sensitivity and glucose uptake by adipose tissue in addition to decreasing hepatic steatosis via numerous signaling pathways that increase FFA oxidation, decrease de novo lipogenesis, and promote VLDL secretion. NASH resolution without worsening of fibrosis has been shown in two placebo-controlled RCTs with liraglutide (39% vs 9%, RR: 4.3, CI: 1–17.7, p=0.019) and semaglutide (59% vs 17%, OR: 6.87, CI: 2.6–17.62, p<0.001).52,53 A meta-analysis of data from eight cardiovascular outcomes RCTs showed that GLP-1RA treatment in diabetic patients decreased renal outcomes (HR: 0.79, CI: 0.73–0.87 p<0.0001), defined by greater than 40% decline in GFR, doubling in creatinine, new-onset macroalbuminuria, need for renal replacement therapy, or death attributed to a renal cause.54
A number of therapies currently under investigation for NASH with and without fibrosis also have potential benefit for CKD. Nuclear transcription factors, including peroxisome proliferator-activator receptor (PPAR)-α, PPAR-δ, PPAR-γ and FXR, regulate lipid trafficking and metabolism, inflammatory signaling, and fibrogenesis in the liver and kidney. As a result, these factors are targets for potential therapeutics. Thiazolidinediones are PPAR-γ agonists that improve steatosis, ballooning, and inflammation in diabetic and nondiabetic patients with NASH and also offer renal protection.55–57 However, its widespread use has been limited by side effects of treatment including a potential risk for bladder cancer and decreased bone density.58,59 In the phase II trial, PPAR-α/δ dual agonist elafibranor showed resolution of NASH without worsening of fibrosis in the post-hoc analysis (19% vs 9%, p=0.013) though interim analysis of the phase 3 trial data did not meet its endpoints and further enrollment was terminated.60 Another PPAR α/γ dual agonist decreased liver fat content measured by MRI (−19.7% vs 5.6%, p=0.004) with mild, reversible worsening of renal function on higher doses and will require further investigation.61
FXR is a targeted receptor for therapies given its role in lipid and glucose metabolism. Obeticholic acid (OCA) is synthetic analogue to the primary BA chenodeoxycholic acid that binds to and activates FXR. In the Phase 2 multicenter FLINT (FXR ligand Obeticholic Acid in NASH Treatment) trial, the OCA group had a significant improvement in NAFLD activity score (NAS) by two or more points (RR: 1.9, CI: 1.3–2.8, p=0.0002) and fibrosis (RR: 1.8, CI: 1.1–2.7, p=0.004) but also an increase in adverse events including pruritus and dyslipidemic profiles.62 At the 18 month interim analysis of the REGENERATE (Randomised Global Phase 3 Study to Evaluate the Impact on NASH with Fibrosis of Obeticholic Acid Treatment) study, there was no significant histologic resolution of NASH but there was significant improvement in fibrosis (RR: 1.9, CI: 1.4–2.8, p=0.0002) and similar reporting of adverse events.63 OCA remains under investigation as the FDA did not support accelerated approval given that it did not meet its primary endpoint for NASH resolution and longer-term safety data was requested.
FXR stimulates FGF19 transcription which is the protein that carries out most of its downstream effects, notably a decrease in hepatic bile acid synthesis and inhibition of hepatic de novo lipogenesis. Aldafermin (previously referred to as NGM282) is a synthetic FGF19 analog studied in a phase 2 trial with an absolute decrease in steatosis determined by MRI (−2.6% vs −7.8%, p=0.02) and improvement in NAS by two or more points (9% vs 62%, p<0.01) but no significant difference in fibrosis (18% vs 38%, p=0.1).64
Oxidative stress and inflammation are key contributors to both NAFLD and CKD. Vitamin E led to histologic improvement in the PIVENS (Pioglitazone vs Vitamin E vs Placebo for Treatment of Non-Diabetic Patients With Nonalcoholic Steatohepatitis) trial but is only recommended in noncirrhotic, nondiabetic patients with biopsy-proven NASH due to limited safety data.65 Numerous additional agents targeting these pathways include apoptosis signaling kinase 1 inhibitor, nuclear factor-erythroid-2-related factor 2 activator, and C-C chemokine receptor types 2/5 antagonist.66
Conclusion
Both NAFLD and CKD are rising rapidly as important public health concerns alongside the increasing rates of obesity, MetS, and DM. Causal association between NAFLD and CKD may not be possible given the observational nature of studies to date. Nonetheless, there is strong supporting evidence of the increased prevalence and incidence of CKD in NAFLD, including greater severity in NASH and advanced fibrosis. NAFLD and CKD have similar impact on outcomes, most notably cardiovascular morbidity and mortality. Thus, early recognition and screening for CKD in NAFLD patients is important to allow for earlier implementation of relevant strategies. The desire for improvement of renal function alongside histologic resolution of NASH may suggest a population who would benefit from clinical trial enrollment for NASH and fibrosis reversal as there is currently no medication approved by the regulatory agencies for this purpose.
Funding
There is no funding to report.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Kleiner DE, Brunt EM, Van Natta M, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41(6):1313–1321. doi:10.1002/hep.20701
2. Stevens PE, Levin A. Evaluation and management of chronic kidney disease: synopsis of the kidney disease: improving global outcomes 2012 clinical practice guideline. Ann Intern Med. 2013;158(11):825–830. doi:10.7326/0003-4819-158-11-201306040-00007
3. Gregg EW. Secular trends in cardiovascular disease risk factors according to body mass index in US adults. JAMA. 2005;293(15):1868. doi:10.1001/jama.293.15.1868
4. Singal AK, Hasanin M, Kaif M, Wiesner R, Kuo YF. Nonalcoholic steatohepatitis is the most rapidly growing indication for simultaneous liver kidney transplantation in the United States. Transplantation. 2016;100(3):607–612. doi:10.1097/TP.0000000000000945
5. Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease-meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology. 2016;64(1):73–84. doi:10.1002/hep.28431
6. Estes C, Razavi H, Loomba R, Younossi Z, Sanyal AJ. Modeling the epidemic of nonalcoholic fatty liver disease demonstrates an exponential increase in burden of disease. Hepatology. 2018;67(1):123–133. doi:10.1002/hep.29466
7. Younossi ZM, Blissett D, Blissett R, et al. The economic and clinical burden of nonalcoholic fatty liver disease in the United States and Europe. Hepatology. 2016;64(5):1577–1586. doi:10.1002/hep.28785
8. Ekstedt M, Hagstrom H, Nasr P, et al. Fibrosis stage is the strongest predictor for disease-specific mortality in NAFLD after up to 33 years of follow-up. Hepatology. 2015;61(5):1547–1554. doi:10.1002/hep.27368
9. Hill NR, Fatoba ST, Oke JL, et al. Global prevalence of chronic kidney disease – a systematic review and meta-analysis. PLoS One. 2016;11(7):e0158765. doi:10.1371/journal.pone.0158765
10. Elshahat S, Cockwell P, Maxwell AP, Griffin M, O’Brien T, O’Neill C. The impact of chronic kidney disease on developed countries from a health economics perspective: a systematic scoping review. PLoS One. 2020;15(3):e0230512. doi:10.1371/journal.pone.0230512
11. Go AS, Chertow GM, Fan D, Mcculloch CE, Hsu C-Y. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N Engl J Med. 2004;351(13):1296–1305. doi:10.1056/NEJMoa041031
12. Foreman KJ, Marquez N, Dolgert A, et al. Forecasting life expectancy, years of life lost, and all-cause and cause-specific mortality for 250 causes of death: reference and alternative scenarios for 2016–40 for 195 countries and territories. Lancet. 2018;392(10159):2052–2090. doi:10.1016/S0140-6736(18)31694-5
13. Bikbov B, Purcell CA, Levey AS, et al. Global, regional, and national burden of chronic kidney disease, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet. 2020;395(10225):709–733. doi:10.1016/S0140-6736(20)30045-3
14. Levey AS, De Jong PE, Coresh J, et al. The definition, classification, and prognosis of chronic kidney disease: a KDIGO Controversies Conference report. Kidney Int. 2011;80(1):17–28. doi:10.1038/ki.2010.483
15. Targher G, Byrne CD, Lonardo A, Zoppini G, Barbui C. Non-alcoholic fatty liver disease and risk of incident cardiovascular disease: a meta-analysis. J Hepatol. 2016;65(3):589–600. doi:10.1016/j.jhep.2016.05.013
16. Paik J, Golabi P, Younoszai Z, Mishra A, Trimble G, Younossi ZM. Chronic kidney disease is independently associated with increased mortality in patients with nonalcoholic fatty liver disease. Liver Int. 2019;39(2):342–352. doi:10.1111/liv.13992
17. Targher G, Chonchol MB, Byrne CD. CKD and Nonalcoholic Fatty Liver Disease. Am J Kidney Dis. 2014;64(4):638–652. doi:10.1053/j.ajkd.2014.05.019
18. Musso G, Gambino R, Tabibian JH, et al. Association of non-alcoholic fatty liver disease with chronic kidney disease: a systematic review and meta-analysis. PLoS Med. 2014;11(7):e1001680. doi:10.1371/journal.pmed.1001680
19. Mantovani A, Petracca G, Beatrice G, et al. Non-alcoholic fatty liver disease and risk of incident chronic kidney disease: an updated meta-analysis. Gut. 2020;
20. An JN, Joo SK, Koo BK, Kim JH, Oh S, Kim W. Portal inflammation predicts renal dysfunction in patients with nonalcoholic fatty liver disease. Hepatol Int. 2020;14(5):798–807. doi:10.1007/s12072-020-10063-9
21. Targher G, Bertolini L, Rodella S, Lippi G, Zoppini G, Chonchol M. Relationship between kidney function and liver histology in subjects with nonalcoholic steatohepatitis. Clin J Am Soc Nephrol. 2010;5(12):2166–2171. doi:10.2215/CJN.05050610
22. Vilar-Gomez E, Calzadilla-Bertot L, Friedman SL, et al. Improvement in liver histology due to lifestyle modification is independently associated with improved kidney function in patients with non-alcoholic steatohepatitis. Aliment Pharmacol Ther. 2017;45(2):332–344. doi:10.1111/apt.13860
23. Byrne CD, Targher G. NAFLD: a multisystem disease. J Hepatol. 2015;62(1 Suppl):S47–S64. doi:10.1016/j.jhep.2014.12.012
24. Alberti KG, Eckel RH, Grundy SM, et al. Harmonizing the metabolic syndrome: a joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation. 2009;120(16):1640–1645.
25. Golabi P, Otgonsuren M, de Avila L, Sayiner M, Rafiq N, Younossi ZM. Components of metabolic syndrome increase the risk of mortality in nonalcoholic fatty liver disease (NAFLD). Medicine (Baltimore). 2018;97(13):e0214. doi:10.1097/MD.0000000000010214
26. Lonardo A, Nascimbeni F, Mantovani A, Targher G. Hypertension, diabetes, atherosclerosis and NASH: cause or consequence? J Hepatol. 2018;68(2):335–352. doi:10.1016/j.jhep.2017.09.021
27. Marchesini G, Bugianesi E, Forlani G, et al. Nonalcoholic fatty liver, steatohepatitis, and the metabolic syndrome. Hepatology. 2003;37(4):917–923. doi:10.1053/jhep.2003.50161
28. Thomas G, Sehgal AR, Kashyap SR, Srinivas TR, Kirwan JP, Navaneethan SD. Metabolic syndrome and kidney disease: a systematic review and meta-analysis. Clin J Am Soc Nephrol. 2011;6(10):2364–2373. doi:10.2215/CJN.02180311
29. Park S, Lee S, Kim Y, et al. Reduced risk for chronic kidney disease after recovery from metabolic syndrome: a nationwide population-based study. Kidney Res Clin Pract. 2020;39(2):180–191. doi:10.23876/j.krcp.20.016
30. Luukkonen PK, Zhou Y, Sädevirta S, et al. Hepatic ceramides dissociate steatosis and insulin resistance in patients with non-alcoholic fatty liver disease. J Hepatol. 2016;64(5):1167–1175. doi:10.1016/j.jhep.2016.01.002
31. Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest. 2005;115(5):1343–1351. doi:10.1172/JCI23621
32. Sarafidis PA, Ruilope LM. Insulin resistance, hyperinsulinemia, and renal injury: mechanisms and implications. Am J Nephrol. 2006;26(3):232–244. doi:10.1159/000093632
33. Stofan M, Guo GL. Bile acids and FXR: novel targets for liver diseases. Front Med (Lausanne). 2020;7:544. doi:10.3389/fmed.2020.00544
34. Clifford BL, Sedgeman LR, Williams KJ, et al. FXR activation protects against NAFLD via bile-acid-dependent reductions in lipid absorption. Cell Metab. 2021;33(8):1671–1684.e1674. doi:10.1016/j.cmet.2021.06.012
35. Kim DH, Park JS, Choi HI, et al. The critical role of FXR is associated with the regulation of autophagy and apoptosis in the progression of AKI to CKD. Cell Death Dis. 2021;12(4):320. doi:10.1038/s41419-021-03620-z
36. Den Besten G, Van Eunen K, Groen AK, Venema K, Reijngoud D-J, Bakker BM. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J Lipid Res. 2013;54(9):2325–2340. doi:10.1194/jlr.R036012
37. Gérard C, Vidal H. Impact of gut microbiota on host glycemic control. Front Endocrinol (Lausanne). 2019;10:29. doi:10.3389/fendo.2019.00029
38. Agus A, Clément K, Sokol H. Gut microbiota-derived metabolites as central regulators in metabolic disorders. Gut. 2021;70(6):1174–1182. doi:10.1136/gutjnl-2020-323071
39. Targher G, Byrne CD. Non-alcoholic fatty liver disease: an emerging driving force in chronic kidney disease. Nat Rev Nephrol. 2017;13(5):297–310. doi:10.1038/nrneph.2017.16
40. Jovanovich A, Isakova T, Stubbs J. Microbiome and cardiovascular disease in CKD. Clin J Am Soc Nephrol. 2018;13(10):1598–1604. doi:10.2215/CJN.12691117
41. Sirich TL, Meyer TW, Gondouin B, Brunet P, Niwa T. Protein-bound molecules: a large family with a bad character. Semin Nephrol. 2014;34(2):106–117. doi:10.1016/j.semnephrol.2014.02.004
42. Xu K-Y, Xia G-H, Lu J-Q, et al. Impaired renal function and dysbiosis of gut microbiota contribute to increased trimethylamine-N-oxide in chronic kidney disease patients. Sci Rep. 2017;7(1):1–2.
43. Koeth RA, Wang Z, Levison BS, et al. Intestinal microbiota metabolism of l-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat Med. 2013;19(5):576–585. doi:10.1038/nm.3145
44. Lee BC, Lee J. Cellular and molecular players in adipose tissue inflammation in the development of obesity-induced insulin resistance. Biochim Biophys Acta. 2014;1842(3):446–462. doi:10.1016/j.bbadis.2013.05.017
45. Tilg H, Moschen AR. Evolution of inflammation in nonalcoholic fatty liver disease: the multiple parallel hits hypothesis. Hepatology. 2010;52(5):1836–1846. doi:10.1002/hep.24001
46. Musso G, Cassader M, De Michieli F, Rosina F, Orlandi F, Gambino R. Nonalcoholic steatohepatitis versus steatosis: adipose tissue insulin resistance and dysfunctional response to fat ingestion predict liver injury and altered glucose and lipoprotein metabolism. Hepatology. 2012;56(3):933–942. doi:10.1002/hep.25739
47. Lomonaco R, Ortiz-Lopez C, Orsak B, et al. Effect of adipose tissue insulin resistance on metabolic parameters and liver histology in obese patients with nonalcoholic fatty liver disease. Hepatology. 2012;55(5):1389–1397. doi:10.1002/hep.25539
48. Kazankov K, Jørgensen SMD, Thomsen KL, et al. The role of macrophages in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Nat Rev Gastroenterol Hepatol. 2019;16(3):145–159. doi:10.1038/s41575-018-0082-x
49. Schoeler M, Caesar R. Dietary lipids, gut microbiota and lipid metabolism. Rev Endocr Metab Disord. 2019;20(4):461–472. doi:10.1007/s11154-019-09512-0
50. Marx N, Davies MJ, Grant PJ, et al. Guideline recommendations and the positioning of newer drugs in type 2 diabetes care. Lancet Diabetes Endocrinol. 2021;9(1):46–52. doi:10.1016/S2213-8587(20)30343-0
51. Wei Q, Xu X, Guo L, Li J, Li L. Effect of SGLT2 inhibitors on type 2 diabetes mellitus with non-alcoholic fatty liver disease: a meta-analysis of randomized controlled trials. Front Endocrinol (Lausanne). 2021;12:635556. doi:10.3389/fendo.2021.635556
52. Armstrong MJ, Gaunt P, Aithal GP, et al. Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN): a multicentre, double-blind, randomised, placebo-controlled phase 2 study. Lancet. 2016;387(10019):679–690. doi:10.1016/S0140-6736(15)00803-X
53. Newsome PN, Buchholtz K, Cusi K, et al. A placebo-controlled trial of subcutaneous semaglutide in nonalcoholic steatohepatitis. N Engl J Med. 2020;384:1113–1124.
54. Sattar N, Lee MMY, Kristensen SL, et al. Cardiovascular, mortality, and kidney outcomes with GLP-1 receptor agonists in patients with type 2 diabetes: a systematic review and meta-analysis of randomised trials. Lancet Diabetes Endocrinol. 2021;9:653–662. doi:10.1016/S2213-8587(21)00203-5
55. Boettcher E, Csako G, Pucino F, Wesley R, Loomba R. Meta-analysis: pioglitazone improves liver histology and fibrosis in patients with non-alcoholic steatohepatitis. Aliment Pharmacol Ther. 2012;35(1):66–75. doi:10.1111/j.1365-2036.2011.04912.x
56. Cusi K, Orsak B, Bril F, et al. Long-term pioglitazone treatment for patients with nonalcoholic steatohepatitis and prediabetes or type 2 diabetes mellitus: a randomized trial. Ann Intern Med. 2016;165(5):305–315. doi:10.7326/M15-1774
57. Chung BH, Lim SW, Ahn KO, et al. Protective effect of peroxisome proliferator activated receptor gamma agonists on diabetic and non-diabetic renal diseases. Nephrology. 2005;10(s2):S40–S43. doi:10.1111/j.1440-1797.2005.00456.x
58. Mehtälä J, Khanfir H, Bennett D, Ye Y, Korhonen P, Hoti F. Pioglitazone use and risk of bladder cancer: a systematic literature review and meta-analysis of observational studies. Diabetol Int. 2019;10(1):24–36. doi:10.1007/s13340-018-0360-4
59. Portillo-Sanchez P, Bril F, Lomonaco R, et al. Effect of pioglitazone on bone mineral density in patients with nonalcoholic steatohepatitis: a 36-month clinical trial. J Diabetes. 2019;11(3):223–231. doi:10.1111/1753-0407.12833
60. Ratziu V, Harrison SA, Francque S, et al. Elafibranor, an agonist of the peroxisome proliferator-activated receptor-alpha and -delta, induces resolution of nonalcoholic steatohepatitis without fibrosis worsening. Gastroenterology. 2016;150(5):1147–1159.e1145. doi:10.1053/j.gastro.2016.01.038
61. Ruilope L, Hanefeld M, Lincoff AM, et al. Effects of the dual peroxisome proliferator-activated receptor-α/γ agonist aleglitazar on renal function in patients with stage 3 chronic kidney disease and type 2 diabetes: a phase IIb, randomized study. BMC Nephrol. 2014;15(1):180. doi:10.1186/1471-2369-15-180
62. Neuschwander-Tetri BA, Loomba R, Sanyal AJ, et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo-controlled trial. Lancet. 2015;385(9972):956–965. doi:10.1016/S0140-6736(14)61933-4
63. Younossi ZM, Ratziu V, Loomba R, et al. Obeticholic acid for the treatment of non-alcoholic steatohepatitis: interim analysis from a multicentre, randomised, placebo-controlled phase 3 trial. Lancet. 2019;394(10215):2184–2196. doi:10.1016/S0140-6736(19)33041-7
64. Harrison SA, Neff G, Guy CD, et al. Efficacy and safety of aldafermin, an engineered FGF19 analog, in a randomized, double-blind, placebo-controlled trial of patients with nonalcoholic steatohepatitis. Gastroenterology. 2021;160(1):219–231.e211. doi:10.1053/j.gastro.2020.08.004
65. Sanyal AJ, Chalasani N, Kowdley KV, et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N Engl J Med. 2010;362(18):1675–1685. doi:10.1056/NEJMoa0907929
66. Sumida Y, Yoneda M, Toyoda H, et al. Common drug pipelines for the treatment of diabetic nephropathy and hepatopathy: can we kill two birds with one stone? Int J Mol Sci. 2020;21(14):4939. doi:10.3390/ijms21144939
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.