")
Back to Journals » International Journal of Nanomedicine » Volume 18
Non-Viral Gene Delivery to Hepatocellular Carcinoma via Intra-Arterial Injection
Authors Vaughan HJ, Zamboni CG, Luly KM , Li L , Gabrielson KL, Hassan LF , Radant NP, Bhardwaj P , Selaru FM, Pomper MG , Green JJ
Received 5 October 2022
Accepted for publication 29 March 2023
Published 11 May 2023 Volume 2023:18 Pages 2525—2537
DOI https://doi.org/10.2147/IJN.S390384
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Phong A Tran
Hannah J Vaughan,1,2 Camila G Zamboni,1,2 Kathryn M Luly,1,2 Ling Li,3 Kathleen L Gabrielson,4 Laboni F Hassan,1,2 Nicholas P Radant,1,2 Pranshu Bhardwaj,1,2 Florin M Selaru,3 Martin G Pomper,1,5,6 Jordan J Green1,2,6,7
1Department of Biomedical Engineering and the Institute for NanoBioTechnology, Johns Hopkins University School of Medicine, Baltimore, MD, USA; 2Translational Tissue Engineering Center, Johns Hopkins University School of Medicine, Baltimore, MD, USA; 3Division of Gastroenterology and Hepatology, Department of Medicine and Sidney Kimmel Comprehensive Cancer Center, Johns Hopkins Medical Institutions, Baltimore, MD, USA; 4Department of Molecular and Comparative Pathobiology, Johns Hopkins University School of Medicine, Baltimore, MD, USA; 5Russell H. Morgan Department of Radiology and Radiological Science, Johns Hopkins Medical Institutions, Baltimore, MD, USA; 6Department of Materials Science and Engineering and the Department of Chemical and Biomolecular Engineering, Johns Hopkins University, Baltimore, MD, USA; 7Departments of Neurosurgery, Oncology, Ophthalmology, and Bloomberg~Kimmel Institute for Cancer Immunotherapy, Johns Hopkins University School of Medicine, Baltimore, MD, USA
Correspondence: Jordan J Green, Department of Biomedical Engineering, Johns Hopkins University School of Medicine, 400 N Broadway, Smith 5017, Baltimore, MD, 21231, USA, Tel +1 410 614-9113, Email [email protected]
Purpose: Hepatocellular carcinoma (HCC) has limited treatment options, and modest survival after systemic chemotherapy or procedures such as transarterial chemoembolization (TACE). There is therefore a need to develop targeted therapies to address HCC. Gene therapies hold immense promise in treating a variety of diseases, including HCC, though delivery remains a critical hurdle. This study investigated a new approach of local delivery of polymeric nanoparticles (NPs) via intra-arterial injection for targeted local gene delivery to HCC tumors in an orthotopic rat liver tumor model.
Methods: Poly(beta-amino ester) (PBAE) nanoparticles were formulated and assessed for GFP transfection in N1-S1 rat HCC cells in vitro. Optimized PBAE NPs were next administered to rats via intra-arterial injection with and without orthotopic HCC tumors, and both biodistribution and transfection were assessed.
Results: In vitro transfection of PBAE NPs led to > 50% transfected cells in adherent and suspension culture at a variety of doses and weight ratios. Administration of NPs via intra-arterial or intravenous injection demonstrated no transfection of healthy liver, while intra-arterial NP injection led to transfection of tumors in an orthotopic rat HCC model.
Conclusion: Hepatic artery injection is a promising delivery approach for PBAE NPs and demonstrates increased targeted transfection of HCC tumors compared to intravenous administration, and offers a potential alternative to standard chemotherapies and TACE. This work demonstrates proof of concept for administration of polymeric PBAE nanoparticles via intra-arterial injection for gene delivery in rats.
Keywords: nanoparticle, gene therapy, liver cancer, poly(beta-amino ester), targeted
Introduction
Liver cancer is the 5th most common type of cancer globally and has a high mortality.1 Hepatocellular carcinoma (HCC) is the most common type of primary liver tumor, accounting for >80% of cases.2 HCC is most often caused by chronic liver disease or cirrhosis, and screening for HCC in patients with risk factors such as hepatitis B infection, alcoholic cirrhosis, or a family history of HCC, may be beneficial.3 Patients with early-stage disease are eligible for tumor resection or liver transplantation, which can be curative, though transplantation requires lifelong immunosuppressive drugs.4 However, many HCC patients are diagnosed at a later stage when treatment options are generally limited to palliative care.5 Treatment regimens for these patients are complicated due to the high prevalence of underlying liver disease. Globally, approximately 80% of HCC cases can be attributed to chronic hepatitis infection, and in the United States, non-alcoholic fatty liver disease underlies 10–20% of HCC cases.6 These comorbidities cause inflammation and cirrhosis, significantly damaging the normal function of the liver. Therefore, systemic or non-targeted treatments such as general chemotherapy have a high risk of liver failure for these patients.7
Locoregional approaches are preferable to target tumor tissue while avoiding off-target toxicity to the rest of the liver or other organs.8 One locoregional approach for intermediate stage HCC is trans arterial chemoembolization (TACE).9 This is an interventional radiology procedure which involves accessing the hepatic artery non-invasively, then delivering chemotherapy followed by an embolic agent. Because the hepatic artery preferentially supplies blood to HCC tumor while the rest of the liver is fed by the portal vein, embolizing the hepatic artery cuts off blood supply to the tumor while maintaining blood supply to the healthy liver. Local chemotherapy is a standard component of TACE, but clinical trials have shown only modest benefit to adding chemotherapeutic agents over the embolic agent alone.10 This may be due to the poor pharmacokinetics of small molecule drugs which have short half lives. A potential solution is drug eluting bead TACE (DEB-TACE), in which microspheres are used to embolize the artery and release drug over an extended period, resulting in sustained chemotherapy in the tumor. In terms of effectiveness, DEB-TACE has not shown proven benefit over conventional TACE.11
There is great interest in improving the therapeutic benefit of TACE by replacing chemotherapeutic and/or embolic agents with newer generations of therapeutics for enhanced efficacy and safety. Nanoparticles (NPs) have different pharmacokinetics from small molecule drugs and are observed in animal models to have an enhanced permeation and retention (EPR) effect that enables passive tumor targeting.12 Further, NPs may be modified for enhanced targeting by optimizing the material of the delivery vehicle or adding targeting ligands to the surface.13 Additionally, NP delivery vehicles have been developed for targeted delivery of nucleic acids, which can specifically target genetic abnormalities of HCC tumors. Other groups have investigated intra-arterial gene delivery using cationic lipid nanocomplexes in rat livers14 or Bletilla striata-derived polysaccharide microspheres in a VX2 rabbit model of HCC,15 but hepatic artery administration of polymeric nanoparticles has not previously been described in the literature.
Poly(beta-amino ester)s (PBAE) are a class of biodegradable polymers that have been optimized for targeted non-viral nucleic acid delivery to multiple cancer types in vitro and in vivo.16–18 We recently identified NPs comprised of 2-((3-aminopropyl)amino) ethanol end-modified poly(1,5-pentanediol diacrylate-co-3-amino-1-propanol) (PBAE “536”) using high throughput screening for preferential transfection in nine HCC cell lines over healthy hepatocytes.19 PBAE 536 NPs effectively delivered therapeutic DNA and significantly slowed the growth of subcutaneous HCC tumors.17 We hypothesized that the biomaterial-mediated specificity of PBAE NPs when combined with a regioselective delivery method would lead to improved delivery to orthotopic HCC tumors with decreased off-target effects. The PBAE class of nanoparticles has not been previously investigated for treating tumors in rats, including in orthotopic tumors, which are difficult to treat and can serve as a good translational model. PBAE 536, a relatively new biodegradable material for gene delivery, has never before been evaluated in rat cells in vitro or in vivo or any PBAE evaluated previously for intra-arterial delivery. Although some groups report benefit from the proposed EPR effect, this is a passive targeting strategy and results have not shown uniform effects across cancer models and development of methods for local delivery are therefore necessary. Such approaches have the potential to be beneficial for nanoparticles of various sizes, including larger sized particles with reduced EPR penetration. Therefore, we sought to develop a protocol for trans-arterial administration of PBAE NPs in a rat orthotopic liver tumor model and investigate this route for targeted non-viral DNA delivery to HCC.
Methods
PBAE Synthesis and Characterization
PBAEs were synthesized as described previously.19 Briefly, backbone monomers, 1,4-Butanediol diacrylate (B4, Alfa Aesar, Haverhill, MA) or 1,5-Pentanediol diacrylate (B5, Santa Cruz Biotechnology, Dallas, TX), were mixed at a 1.1:1 ratio with sidechain monomers 3-amino-1-propanol (S3, Alfa Aesar, Haverhill, MA), 4-amino-1-butanol (S4, Fisher Scientific, Hampton, NH), or 5-amino-1-pentanol (S5, Alfa Aesar, Haverhill, MA) and allowed to react overnight at 85°C. B-S polymers were then resuspended in tetrahydrofuran (THF) and endcapped with 2-(3-Aminopropylamino)ethanol (E6, Sigma, St. Louis, MO) or 1-(3-Aminopropyl)-4-methylpiperazine (E7, Alfa Aesar, Haverhill, MA) for 2 h at room temperature. Resulting polymers B4-S4-E6 (446), B4-S5-E7 (457), and B5-S3-E6 (536) were ether precipitated, dried, and stored at −20°C on desiccant. To determine the molecular weight and polydispersity index of PBAE 536, ether purified polymers were resuspended in tetrahydrofuran and gel permeation chromatography was performed using polystyrene reference standards and a refractive index detector (Agilent, Santa Clara, CA).
Tissue Culture
N1-S1 cells (ATCC, Manassas, VA) were cultured in IMDM (ThermoFisher Scientific, Waltham, MA) supplemented with 10% FBS (Sigma, St. Louis, MO) and 1% penicillin-streptomycin (ThermoFisher Scientific, Waltham, MA). Cells were grown at 37°C and 5% CO2. For in vitro transfections, cells were plated at 10k/well in 96-well plates coated with laminin (Sigma, St. Louis, MO) for adherent culture, or at 20k/well and 30k/well in round-bottom 96-well plates. Cells were seeded and then incubated overnight before transfection.
In vitro Nanoparticle Transfection
PBAE (446, 457, or 536) and eGFP-N1 plasmid DNA were each dissolved in 25 mM sodium acetate (25 mM, pH 5), then combined for final polymer:DNA weight ratios of 50 and 25 w/w. DNA and polymer were allowed to complex for 10 minutes, then incubated on cells for 2 h at 37°C and 5% CO2 followed by complete media change. 10X images were acquired using Axiovision 5 software and an Zeiss Axio Observer (Zeiss, White Plains, NY) and overlaid images were generated in ImageJ. GFP transfection was assessed by flow cytometry at 48 h post-transfection utilizing an Attune NxT Flow Cytometer (ThermoFisher Scientific, Waltham, MA). Cellular viability was measured 24 h post-transfection via MTS CellTiter 96® AQueous One Solution Cell Proliferation Assay (Promega, Madison, WI) according to the instructions of the manufacturer.
In vivo Nanoparticle Preparation
Firefly luciferase (fLuc) DNA was amplified by Aldevron (Fargo, ND) and used for in vivo transfection studies. For biodistribution studies, fLuc DNA was functionalized using Label IT Nucleic Acid Modifying Reagent (Mirus Bio Madison, WI) and labeled with IRDye 800RS NHS Ester Infrared Fluorescent Dye (LI-COR, Lincoln, NE). 10% labeled DNA was used for biodistribution studies.
PBAE 536 was diluted in sodium acetate (40.4 mM, pH 5), then added to 1 mg/mL plasmid DNA in water to obtain a polymer:DNA weight ratio of 25 w/w and a 0.25 mg/mL DNA concentration. DNA and polymer were allowed to complex for 10 minutes, then sucrose was added to obtain a concentration of 90 mg/mL. NPs were frozen at −80°C for storage and thawed immediately prior to injection.
Nanoparticle Characterization
NP solutions were prepared as described above with and without sucrose. Frozen formulations were stored at −80°C for three days, then thawed immediately prior to characterization studies. Three independently prepared replicates were used for each condition with mean values reported. NPs were diluted 1:10 in phosphate-buffered saline (PBS, pH 7.4) and size, represented by hydrodynamic diameter, was determined via dynamic light scattering. NPs were also diluted 1:10 in 10 mM NaCl and surface charge, represented by zeta potential, was determined via electrophoretic mobility. Both size and surface charge measurements were obtained using a Malvern Zetasizer Pro (Malvern Panalytical).
Statistics
Statistical analysis was performed using GraphPad Prism (GraphPad Software, San Diego, CA). One-way ANOVA was performed with Dunnett’s multiple comparisons post-tests to assess differences in NP size and surface charge. Significance is denoted: ns, P > 0.05; *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001.
Animal Experiments
Johns Hopkins Institutional Animal Care and Use Committee (IACUC) approved and oversaw all in vivo procedures. Johns Hopkins complies with US Department of Agriculture regulations, with the Public Health Service Policy on Humane Care and Use of Laboratory Animals, and with other applicable United States government and institutional guidelines and policies. RNU athymic rats were purchased from Charles River Laboratories (Wilmington, MA) or bred at Johns Hopkins from two homozygous RNU athymic rats. For all surgical procedures, animals were anesthetized with 3% isoflurane in 100% oxygen until a sufficient plane of anesthesia was reached and anesthesia was confirmed by monitoring response to stimuli (toe pinch). Ophthalmic ointment was applied to eyes while animals were under anesthesia. During surgical procedures, rats were placed onto a 37°C heated surface to ensure optimum thermoregulation and minimize post-op recovery time. The hair was removed from the surgical site (abdomen) using clippers and/or depilatory cream and prepared using a wash of povidone-iodine soap then rinsed with 70% ethanol. After surgery, the peritoneum was closed using simple continuous suturing of the muscle layer with absorbable sutures and skin layer was closed using wound clips or simple interrupted suturing with non-absorbable sutures. Finally, Vetbond tissue adhesive was applied topically to prevent dehiscence and to improve post-procedure healing. Warm, sterile isotonic fluids at 3–5% of the body weight was injected subcutaneously prior to and at the end of surgery. Meloxicam (2 mg/kg) was delivered subcutaneously prior to surgery and again at 24 and 48 h after surgery for analgesia.
Liver Tumor Implantation Surgery
Liver tumor implantation was performed as previously described by Sheu et al.20 To access the liver, a laparotomy was performed extending caudally from the xiphoid process. Under direct visualization, 1×106 N1-S1 cancer cells in a 1:1 solution of HBSS and High Concentration Matrigel (Corning, Corning, NY) were injected under the liver capsule. A white protrusion at the point of injection validated successful inoculation with liver cancer cells. Gentle compression was applied for at least 15 seconds using a sterile cotton tipped applicator to prevent cancer cell leakage or bleeding. Healthy RNU rats were used as controls.
Jugular Injection
Under careful dissection, the right or left jugular was visualized. A 27G insulin needle was inserted into the overlying pectoral muscle, then directed cranially into the jugular vein lumen.21 Prior to injection, the plunger was pulled back, and blood in the syringe confirmed proper placement. PBAE NPs were injected slowly at 100 µL per minute in a total volume of 500 µL and were visualized intravenously in the jugular vein during delivery.
Hepatic Artery Injection
The microsurgical hepatic artery injection was adapted from Sheu et al.20 The procedure was performed under a dissecting microscope to improve visualization of small delicate structures. Animals were prepared for sterile technique surgery as described above, then an incision of approximately 3 cm was made with a scalpel extending caudally from the xiphoid process. The peritoneum was cut with scissors along the linea alba. The lateral left lobe of the liver was externalized on a sterile piece of gauze soaked with saline and retracted. Similarly, the duodenum was externalized on gauze and retracted. The mesenteric connective tissues were dissected, and common bile duct was retracted using silk suture to allow visualization of the common hepatic artery, proper hepatic artery and gastroduodenal artery (GDA). The GDA was freed from surrounding tissue and ligated at the distal end. A few drops of 2% lidocaine were applied to the GDA to promote vasodilation. The common hepatic artery was clamped using a bulldog clamp, occluding blood flow. A 27-gauge needle was inserted into the gastroduodenal artery, and 500 µL of PBAE NPs was slowly hand injected at a rate of 100 µL per minute. Successful injection was confirmed by visualizing the displacement of blood in the proper hepatic artery. Following injection, the needle was slowly withdrawn from the vessel. A suture was used to ligate the GDA proximal to the needle insertion site, and the clamp was removed from the common hepatic artery to restore blood flow through the proper hepatic artery. The duodenum and liver were replaced in the abdominal cavity, and the incision was closed as described above.
In vivo Imaging
Biodistribution and transfection were monitored by IVIS (IVIS Spectrum imaging system, Perkin Elmer, Waltham, MA). 150 mg/kg D-luciferin (Gold Biotechnology, St. Louis, MO) was first intraperitoneally administered. Eight minutes later, animals were sacrificed and livers were harvested. Bioluminescence and fluorescence images were acquired. Regions of interest (ROI) of the images were evaluated using Living Image software (Perkin Elmer, Waltham, MA).
Results
In Vitro Nanoparticle Assessment
Three PBAE polymers were synthesized using various acrylate backbone (B), amine sidechain (S), and small molecule endcap (E) monomers (Figure 1A). Resulting polymers, B4-S4-E6 (446), B4-S5-E7 (457), and B5-S3-E6 (536) (Figure 1B) were then assessed for transfection efficacy in N1-S1 rat HCC cells. Transfection with 446, 457, or 536 NPs harboring GFP plasmid DNA resulted in >50% GFP-positive cells at various DNA doses and weight ratios either in adherent or suspension culture (Figure 2). The MTS assay indicated >70% cellular viability for all formulations in adherent and suspension cultures, except for the highest dose and weight ratio of 457 and 536 particles (Figure 2). Higher weight ratio (50 w/w vs 25 w/w) led to higher transfection across all three polymeric nanoparticle types for both adherent culture and suspension culture as determined by flow cytometry. The pattern for percent of cells positively transfected with GFP matched the pattern of geometric mean expression of GFP across the nanoparticle formulations. Fluorescence microscopy also demonstrated the successful in vitro transfection of N1-S1 cells by PBAE NPs 446, 457, and 536 containing GFP plasmid DNA (Figure 3).
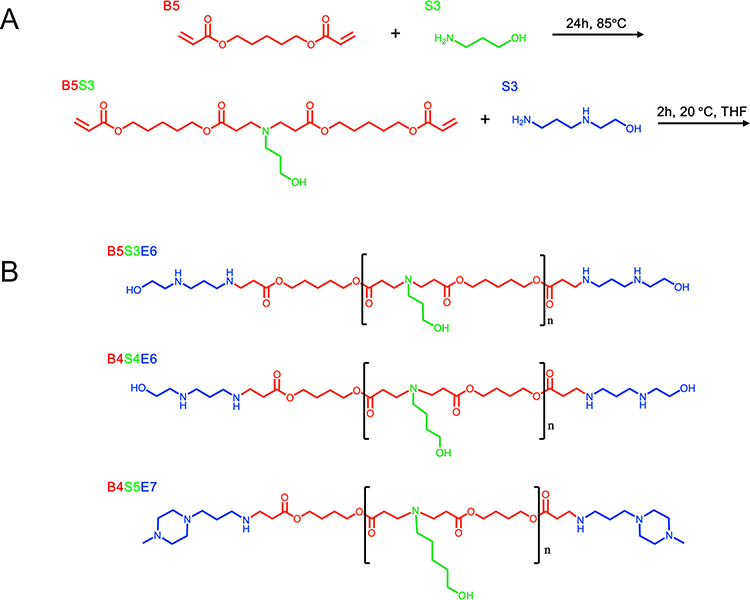 |
Figure 1 PBAE Synthesis. (A) Synthetic route of model PBAE 536. (B) Resulting structures of PBAEs 536, 446, and 457. |
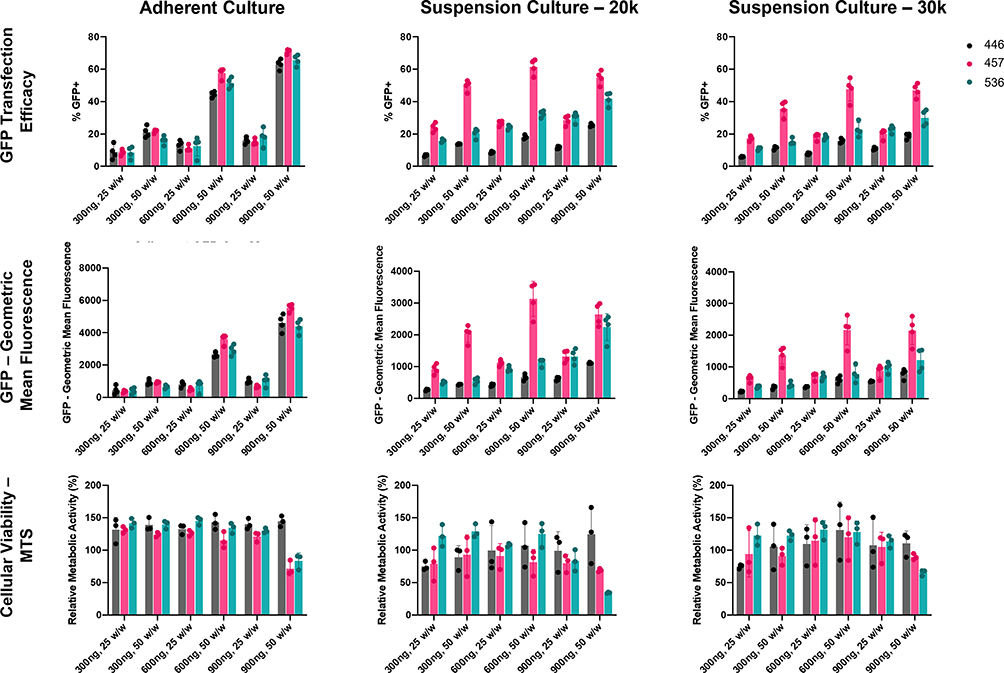 |
Figure 2 PBAE NP Screening in N1-S1 cells. Three PBAEs, 446, 457, and 536, were tested in N1-S1 cells harboring 300, 600, or 900 ng of a GFP plasmid at 25 and 50 w/w. GFP transfection efficacy and geometric mean fluorescence were assessed via flow cytometry after 48 h, and cellular viability was assessed via MTS assay after 24 h. Cells were plated on laminin coated plates for adherent culture or seeded at 20k or 30k cell per well in suspension culture. |
 |
Figure 3 Fluorescence Microscopy of PBAE NPs in N1-S1 cells. 446, 457, and 536 NPs harboring 600 ng GFP DNA at 50 w/w were administered to N1-S1 cells grown in adherent culture (10k cells/well) or in suspension culture (20k or 30k cells/well). GFP transfection was visualized 48 h following transfection. Scalebars represent 200 µm. |
Nanoparticle Characterization
With intermediate levels of transfection efficacy and good cell viability, and given demonstrated efficacy of this material in the context of HCC,22 PBAE 536 NPs were chosen for subsequent experiments. Gel permeation chromatography revealed that PBAE 536 has a molecular weight of 4370 g/mol and a polydispersity index of 2.40. Biophysical characterization of 536 NPs formulated for in vivo use indicated that fresh or frozen NPs, with and without sucrose, maintained a hydrodynamic particle diameter of approximately 300 nm with no significant differences between formulations (Figure 4). Measurements of zeta potential indicated a similar range of 20–30 mV for all nanoparticle formulations as well as a modest decrease in positive zeta potential in the formulations containing sucrose compared to fresh particles without sucrose (fresh+sucrose, p=0.0288; frozen+sucrose, p<0.0001) (Figure 4).
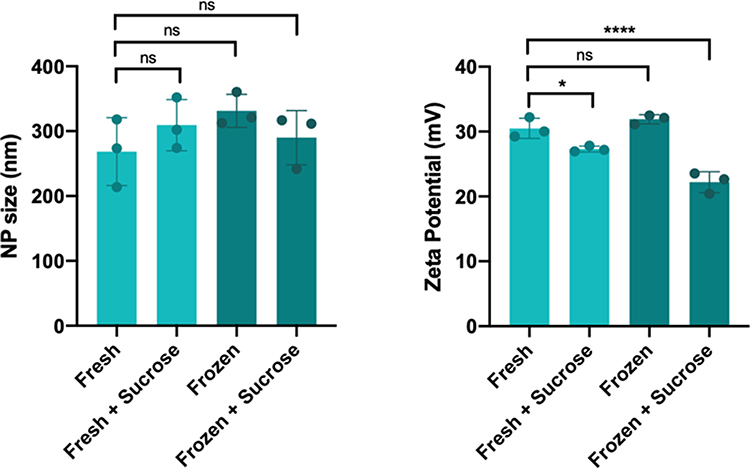 |
Figure 4 536 NP Biophysical Characterization. NP size (as hydrodynamic diameter) and surface charge (as zeta potential) were determined for 536 NPs. Formulations were either prepared fresh, with and without 90 mg/mL sucrose, or frozen at −80°C for 72 h with and without sucrose. Size measurements were conducted in physiological PBS, and zeta potential measurements were conducted in 10 mM NaCl. Significance denoted as follows: *p<0.05; ****p<0.0001. Abbreviation: ns, not significant. |
Hepatic Artery Injection
A microsurgical procedure was developed based on Sheu et al20 to administer NPs via the hepatic artery. A laparotomy was performed, and the liver and duodenum were retracted on sterile gauze. The gastroduodenal artery (GDA) was located near the caudate lobe, anterior to the portal vein and posterior to the common bile duct (Figure 5A). To prevent bleeding, the GDA was selected as the injection site, as the vessel may be permanently ligated without complications. The GDA was isolated and ligated at the distal end using silk suture. Next, a few drops of 2% lidocaine were applied directly to the GDA to induce vasodilation. The common hepatic artery was clamped temporarily using a micro bulldog clamp. A 27G insulin needle was used to inject 500 µL of NPs into the GDA, and flow was directed to the common hepatic artery. Successful injection was confirmed by visualizing the displacement of blood in the proper hepatic artery. Injections were performed at a rate of 100 µL per minute to minimize risk of embolism. After injection, the needle was carefully removed, and the proximal end of the GDA was ligated before removing the clamp on the common hepatic artery. To visualize the injection route and anatomy, a separate injection was performed with a blue latex solution using this procedure (Figure 5B). The injection was directed into the proper hepatic artery and into the left and right branches and into the liver.
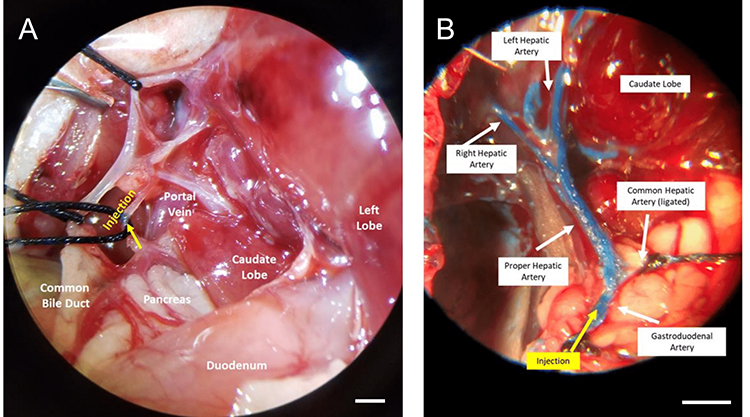 |
Figure 5 Intra-arterial Injection Procedure. (A) Portal triad anatomy. The common biliary duct is retracted to visualize the hepatic artery. Arrow (yellow) indicates the injection site in the gastroduodenal artery. (B) Intra-arterial injection to the hepatic artery visualized using a blue latex solution. Injection into the gastroduodenal artery progressed into the proper hepatic artery and into the lobar arteries. Images were acquired under microscope magnification. |
In Vivo Nanoparticle Biodistribution and Transfection
To assess biodistribution and transfection, NPs were synthesized with fLuc DNA labeled (10%) with near-IR fluorescent dye (NIR-fLuc NPs). DNA at 0.25 mg/mL was combined with PBAE 536 polymer in sodium acetate (pH 5, 25 mM) and allowed to self-assemble. The tonicity of the solution was balanced using sucrose, for a final concentration of 90 mg/mL. NIR-fLuc NPs were administered intravenously or via hepatic artery, and biodistribution (fluorescence) and transfection (luminescence) were assessed 6 h later. In control RNU rats without tumors, fluorescent NPs administered by both the intra-arterial (N=2 rats) and intravenous (N=3 rats) routes accumulated in the liver (Figure 6). Yet, no transfection was observed in the liver of these animals, validating recent in vitro results indicating poor transfection of healthy hepatocytes with optimized PBAE-NPs.19
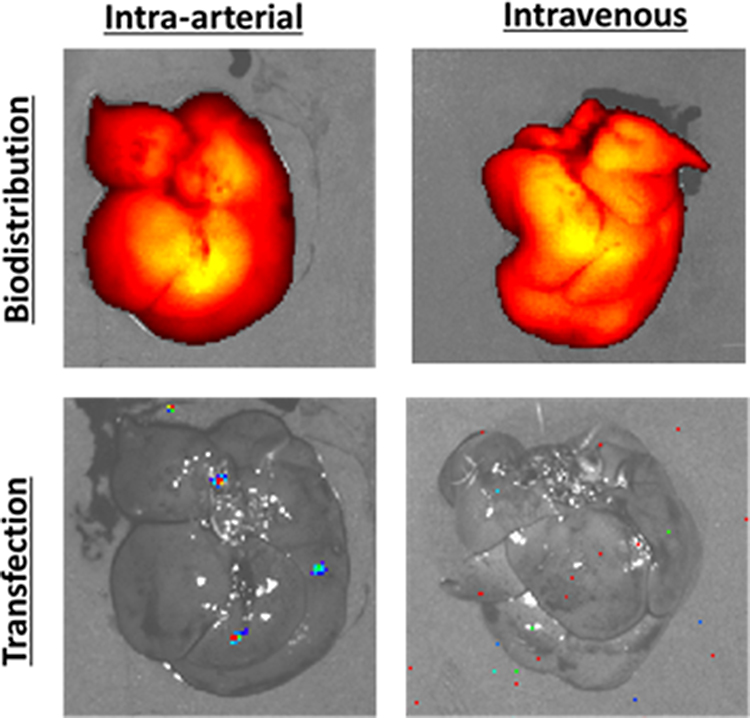 |
Figure 6 NIR-fLuc NP biodistribution and transfection following intra-arterial or intravenous injection in animals without liver tumors. Healthy rats were injected with 500 μL PBAE NPs harboring 125 μL of fLuc DNA, with 10% of DNA mass covalently labeled with near infrared fluorophore. With both intravenous and intra-arterial administration, NPs were localized to liver tissue, but no transfection was detected by bioluminescence imaging. |
Orthotopic N1-S1 HCC tumors were next established in the livers of RNU athymic rats. Athymic rats were used in this model due to spontaneous tumor regression of N1-S1 tumors in syngeneic Sprague Dawley rats.23 Tumor-bearing rats were injected with NIR-fLuc NPs by hepatic artery (N=2) or intravenous injection (N=1) two weeks after N1-S1 HCC cell implantation in the liver. Intravenously injected NPs were observed to accumulate in the healthy liver, but not in the tumor tissue (Figure 7). This biodistribution may be due to the dual vascular supply to the liver, in which two thirds of the blood supply to the liver is from the portal vein, while the hepatic artery supplies blood to HCC tumors. NPs administered via the hepatic artery were observed to accumulate in both the tumor and in the surrounding liver parenchyma. Moreover, successful transfection was detected in the tumor following intra-arterial injection (27.9-fold increase in average radiance for the ROI over background), whereas animals injected intravenously showed no meaningful transfection in the tumor or liver (1.61-fold increase in average radiance for the ROI over background) (Figure 7). This observation supports the hypothesis that loco-regional administration has the potential improve both biodistribution and transfection efficacy of non-viral nanoparticles in orthotopic liver tumors. This result also validates that PBAE NPs, and polymeric NP 536 in particular, may be promising as a delivery vehicle for liver cancer gene therapy. With PBAE 536 NPs, we also observed transfection outside the primary tumor mass. This may indicate transfection of micrometastases that have spread through the liver, or it may signal that additional targeting methods may also be needed, such as transcriptional targeting, to prevent off-target expression in the liver parenchyma.
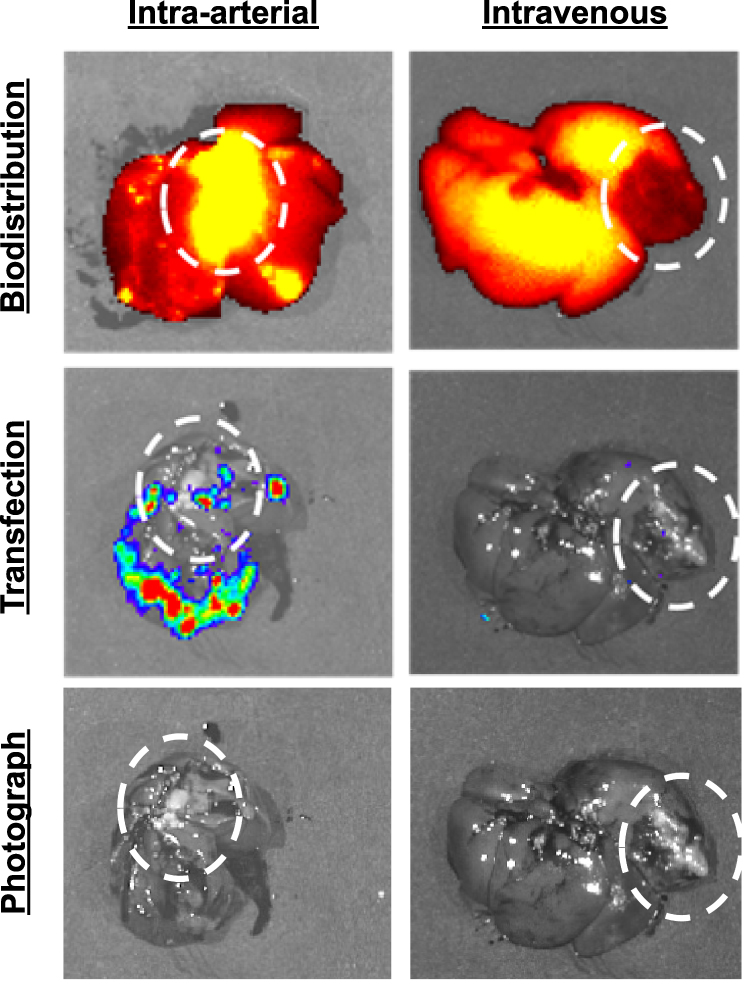 |
Figure 7 NIR-fLuc NP biodistribution and transfection following intra-arterial or intravenous injection in animals with N1-S1 tumors (circled). 2 weeks after N1-S1 tumor implantation, rats were injected with 500 μL PBAE NPs harboring 125 μL of fLuc DNA, with 10% of DNA mass covalently labeled with near infrared fluorophore. NPs administered by intravenous injection accumulate in the liver, but not into the tumor. NPs administered via the hepatic artery accumulate in the liver and tumor, with gene expression shown by bioluminescence imaging. |
Discussion
Tumor-specific delivery is an important goal to improve outcomes of treatments for HCC. A challenge is that many drug agents have poor pharmacokinetics and can cause systemic toxicity. To address these concerns, innovative advanced therapies, such as immunotherapy, cell therapy, and gene therapy, are being investigated for HCC. For example, in a Phase 3 trial, antibodies against PD-L1 and VEGFA, when dosed in combination, demonstrated increased overall survival compared to frontline small-molecule sorafenib,24 and a variety of other antibodies and immunotherapy combinations are currently being explored in clinical trials.25 Cellular therapies have also been explored to treat HCC. In a Phase 1 clinical trial, patients received autologous CAR-GPC3 T-cell therapy which showed promising effects, though toxicity was a major concern, and additional trials are ongoing.26
Gene therapy nanoparticle technologies also hold particular promise in addressing liver cancer. This is because many different types of agents, ranging from tumor suppressors to cytotoxic agents to immunogenic agents, can be genetically encoded to be locally expressed in liver cancer. This gene delivery can lead to the death of the tumor cells where the nanoparticles were taken up in addition to having a bystander effect in neighboring tumor cells. For example, Asialofetuin-modified Poly(D,L-lactic-co-glycolic) acid (PLGA) 1,2-dioleoyl-3-(trimethylammonium) propane (DOTAP) blended cationic NPs delivering IL-12 DNA led to significantly smaller BNL HCC tumors compared to control and led to long-term survival in 75% of mice.27 In another study, magnetic NPs delivering tumor suppressor RASSF1A DNA to HepG2 tumors led to smaller tumors and increased tumor Casp-3 expression.28 In addition, gene delivery nanoparticles can also be utilized to deliver other types of nucleic acids to HCC tumors. Researchers have used gold NPs to deliver a tumor suppressor miRNA, miR-375, to HepG2 tumors and this resulted in decreased tumor volume compared to control.29 Additional studies have shown the promise of delivering siRNAs with lipid NPs30,31 and selenium-polyethyleneamine-folate NPs32 to achieve significant tumor regression off HCC in vivo.
To improve outcomes further and overcome extracellular delivery barriers, NP formulations can be designed for superior retention in tumor tissue, which has been exploited in the development of many systemically administered therapeutic nanoformulations.33 While passive targeting by the EPR effect has shown promise in preclinical studies, this effect has not translated well to clinical trials in patients with solid tumors.34 Thus, new administration methods for nanomedicines are a priority to advance the field, in particular to advance cancer therapy. Cancer gene therapy, as a modality, has great promise for the delivery of both therapeutic agents and imaging agents to tumors, but both extracellular and intracellular delivery barriers remain formidable challenges.
We first evaluated and validated biodegradable polymeric nanoparticles composed of PBAEs for effective and safe delivery to N1-S1 rat HCC. By optimizing polymer structure and nanoparticle formulation w/w ratio, we were able to overcome intracellular delivery barriers, resulting in high levels of non-viral gene delivery across cell culture conditions. PBAE 536 NPs, selected here as optimal for rat HCC studies, have also recently shown promise with human HCC cell lines and tumors in work by Zamboni et al.19 Moving to in vivo studies, we found that in an orthotopic rat model of HCC, polymeric nanoparticle gene therapy intravenously administered distributed to healthy liver, but did not accumulate in tumor tissue. To meet the challenge posed by this extracellular delivery barrier and to further develop administration methods for nanomedicines, an intra-arterial injection procedure for nanomedicine gene therapies was developed. Through intra-arterial injection via the proper hepatic artery, intratumoral accumulation of the nanoparticles was significantly increased. Thus by taking advantage of the differential blood supply to tumor and liver tissue, NP biodistribution in the tumor was improved.
While trans-arterial injection improved transfection over intravenous delivery, off-target expression was observed in the surrounding liver tissue. While this preclinical model in rats enabled effective NP delivery, it does not recapitulate the full selectivity of the TACE procedure, where specific branches feeding the tumor are selectively catheterized using real-time imaging. Larger animal models may allow for more accurate simulation of this procedure to direct NPs specifically to the tumor mass. In addition, NP properties could be further optimized for improved delivery. PBAE 536 NPs are relatively large (~300 nm) and cationic, which we can limit diffusion into a tumor. Tumors can exhibit leaky vasculature which can enable transport of larger NPs, but systematic studies of NP size have shown improved intratumoral delivery with smaller (~100 nm) NPs.35
Nucleic acid therapeutics may be used to target dysregulated oncogenes or tumor suppressor genes in cancer cells. Further, plasmid DNA may be targeted for cancer cell expression by employing a cancer-specific promoter for therapeutic gene expression.36,37 Delivery efficiency is still a major barrier to DNA therapy for cancer, and local administration can lead to an increased concentration of NPs at the target site.38 Further, local administration routes lead to decreased exposure to systematic circulation, slowing clearance and reducing the risk of systemic toxicity.39 Cancer gene therapy requires the designed nanomedicine to be able to cross both the extracellular and intracellular delivery barriers with the same particle. We observed gene delivery via the expression of fLuc reporter DNA in HCC liver tumors following intra-arterial injection of PBAE NPs. On the other hand, traditional intravenous administration of the NPs was not observed to transfect tumor or liver tissue.
While local delivery may not be a feasible option for many types of solid tumors, HCC tumors are uniquely accessible by the proper hepatic artery, and this has been harnessed in a clinical setting with the TACE procedure.9 This procedure is minimally invasive, and branches of the artery can be accessed to target a patient’s tumor more specifically. By optimizing a preclinical surgical procedure to access this artery for NP delivery, this route may be explored for the delivery of alternative agents, including biologics such as DNA, siRNA, or mRNA. Additionally, active targeting agents which specifically bind to tumor cells may be developed and tested for further improvements to targeting.40 The rat model described here recapitulates many of the barriers of delivery to HCC tumors. Future model development could also look at toxin-induced or genetic HCC models which may better model the vasculature of these tumors as well as their immunological responses. For example, a s1pr1 knockout genetic model recapitulates “capillarization” of the tumor, in which fibrosis triggers decreased fenestration in the tissue.41 Capillarization is observed in patient biopsies of HCC tumors and may significantly impact NP transport and diffusion with this locoregional approach.42 With a potential gene delivery nanocarrier identified, further therapeutic development could focus on new anti-cancer nucleic acid cargos, used individually or in combination within the same nanoparticles, to be delivered to the HCC tumors.
Conclusions
In this study, we developed biodegradable nanoparticles for non-viral gene therapy to HCC in rat models in vitro and in vivo. We described an approach for locoregional targeting of nanomedicines to HCC via the proper hepatic artery in rats. We showed that this route of administration had the potential to provide an advantage over intravenous administration for the delivery of PBAE NPs, to improve both accumulation of NPs in orthotopic liver tumor and successful intracellular delivery and expression of a reporter gene. By combining advanced nanomedicine and a translational clinical method of administering the nanomedicine, non-viral gene therapy capable of transversing both extracellular and intracellular delivery barriers may be achieved. This cancer gene therapy nanobiotechnology may be useful in the treatment of hepatocellular carcinoma and other liver cancers.
Abbreviations
HCC, hepatocellular carcinoma; PBAE, poly(beta-amino ester); NP, nanoparticle; TACE, trans arterial chemoembolization; EPR, enhanced permeation and retention; B4, 1,4-Butanediol diacrylate; B5, 1,5-Pentanediol diacrylate; S3, 3-amino-1-propanol; S4, 4-amino-1-butanol; S5, 5-amino-1-pentanol; E6, 2-(3-Aminopropylamino)ethanol; E7, 1-(3-Aminopropyl)-4-methylpiperazine; THF, tetrahydrofuran; fLuc, firefly luciferase; NIR, near-IR fluorescent dye; PBS, phosphate-buffered saline; GDA, gastroduodenal artery; ROI, regions of interest, DEB-TACE, drug eluting bead TACE.
Data Sharing Statement
The data associated with this study are contained within this article or are available from the corresponding author upon request.
Consent for Publication
The authors have all approved the manuscript for publication.
Acknowledgments
A preliminary non-peer reviewed version of this research was written and published as a part of HJV’s Johns Hopkins University PhD dissertation.43 The authors thank Dr. Luke Higgins for introducing helpful literature and concepts to the study. The authors also thank Dr. Cory Brayton and James Potter for advising on surgical protocols for the study.
Funding
The authors thank the NIH for support (R01EB022148, R01CA228133, P41EB024495, P41EB028239). HV was supported by the NIH F31 Predoctoral Fellowship (F31CA250365).
Disclosure
Johns Hopkins filed patents related to the technology discussed in the manuscript with JJG as a co-inventor. JJG is also a board member, CSO, and co-founder of Cove Therapeutics, a manager, CTO, and co-founder of Dome Therapeutics, and a manager and co-founder of OncoSwitch Therapeutics. MGP is on the scientific advisory board of Dome Therapeutics. Any potential conflicts of interest are managed by the Johns Hopkins University Committee on Outside Interests. There are no competing interests from the other authors.
References
1. Shaw JJ, Shah SA. Rising incidence and demographics of hepatocellular carcinoma in the USA: what does it mean? Expert Rev Gastroenterol Hepatol. 2011;5(3):365–370. doi:10.1586/egh.11.20
2. Ayuso C, Rimola J, Vilana R, et al. Diagnosis and staging of hepatocellular carcinoma (HCC): current guidelines. Eur J Radiol. 2018;101:72–81. doi:10.1016/j.ejrad.2018.01.025
3. Yang JD, Roberts LR. Hepatocellular carcinoma: a global view. Nat Rev Gastroenterol Hepatol. 2010;7(8):448–458. doi:10.1038/nrgastro.2010.100
4. Onghena L, Develtere W, Poppe C, et al. Quality of life after liver transplantation: state of the art. World J Hepatol. 2016;8(18):749. doi:10.4254/wjh.v8.i18.749
5. Golabi P, Fazel S, Otgonsuren M, Sayiner M, Locklear CT, Younossi ZM. Mortality assessment of patients with hepatocellular carcinoma according to underlying disease and treatment modalities. Medicine. 2017;96(9):e5904. doi:10.1097/MD.0000000000005904
6. Yang JD, Hainaut P, Gores GJ, Amadou A, Plymoth A, Roberts LR. A global view of hepatocellular carcinoma: trends, risk, prevention and management. Nat Rev Gastroenterol Hepatol. 2019;16(10):589–604. doi:10.1038/s41575-019-0186-y
7. El–Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology. 2007;132(7):2557–2576. doi:10.1053/j.gastro.2007.04.061
8. Forner A, Reig M, Varela M, et al. Diagnóstico y tratamiento del carcinoma hepatocelular. Actualización del documento de consenso de la AEEH, SEOM, SERAM, SERVEI y SETH. Med Clin. 2016;146(11):511.e1–511.e22. doi:10.1016/j.medcli.2016.01.028
9. Tsurusaki M, Murakami T. Surgical and locoregional therapy of HCC: TACE. Liver Cancer. 2015;4(3):165–175. doi:10.1159/000367739
10. Brown KT, Do RK, Gonen M, et al. Randomized trial of hepatic artery embolization for hepatocellular carcinoma using doxorubicin-eluting microspheres compared with embolization with microspheres alone. J Clin Oncol. 2016;34(17):2046. doi:10.1200/JCO.2015.64.0821
11. Zou JH, Zhang L, Ren ZG, Ye SL. Efficacy and safety of cTACE versus DEB‐TACE in patients with hepatocellular carcinoma: a meta‐analysis. 2016.
12. Maeda H, Wu J, Sawa T, Matsumura Y, Hori K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. J Control Release. 2000;65(1–2):271–284. doi:10.1016/S0168-3659(99)00248-5
13. Zhong Y, Meng F, Deng C, Zhong Z. Ligand-directed active tumor-targeting polymeric nanoparticles for cancer chemotherapy. Biomacromolecules. 2014;15(6):1955–1969. doi:10.1021/bm5003009
14. Choi BY, Chung JW, Park JH, et al. Gene delivery to the rat liver using cationic lipid emulsion/DNA complex: comparison between intra-arterial, intraportal and intravenous administration. Korean J Radiol. 2002;3(3):194. doi:10.3348/kjr.2002.3.3.194
15. Xia X, Li X, Feng G, Zheng C, Liang H, Zhou G. Intra-arterial interleukin-12 gene delivery combined with chemoembolization: anti-tumor effect in a rabbit hepatocellular carcinoma (HCC) model. Acta Radiol. 2013;54(6):684–689. doi:10.1177/0284185113480072
16. Gao X, Jin Z, Tan X, et al. Hyperbranched poly(β-amino ester) based polyplex nanoparticles for delivery of CRISPR/Cas9 system and treatment of HPV infection associated cervical cancer. J Control Release. 2020;321(October2019):654–668. doi:10.1016/j.jconrel.2020.02.045
17. Vaughan HJ, Zamboni CG, Radant NP, et al. Poly(beta-amino ester) nanoparticles enable tumor-specific TRAIL secretion and a bystander effect to treat liver cancer. Mol Ther Oncolytics. 2021;21(4):377–388. doi:10.1016/j.omto.2021.04.004
18. Kim J, Mondal SK, Tzeng SY, et al. Poly(ethylene glycol)–poly(beta-amino ester)-based nanoparticles for suicide gene therapy enhance brain penetration and extend survival in a preclinical human glioblastoma orthotopic xenograft model. ACS Biomater Sci Eng. 2020;6(5):2943–2955. doi:10.1021/acsbiomaterials.0c00116
19. Zamboni CG, Kozielski KL, Vaughan HJ, et al. Polymeric nanoparticles as cancer-specific DNA delivery vectors to human hepatocellular carcinoma. J Control Release. 2017;263:18–28. doi:10.1016/j.jconrel.2017.03.384
20. Sheu AY, Zhang Z, Omary RA, Larson AC. Invasive catheterization of the hepatic artery for preclinical investigation of liver-directed therapies in rodent models of liver cancer. Am J Transl Res. 2013;5(3):269–278.
21. Ishiyama S, Kissel C, Guo X, et al. A syngeneic orthotopic osteosarcoma Sprague Dawley rat model with amputation to control metastasis rate. J Visual Exp. 2021;2021(171):1–18. doi:10.3791/62139
22. Vaughan HJ, Zamboni CG, Hassan LF, et al. Polymeric nanoparticles for dual-targeted theranostic gene delivery to hepatocellular carcinoma. Sci Adv. 2022;8(29):1–13. doi:10.1126/sciadv.abo6406
23. Buijs M, Geschwind JFH, Syed LH, et al. Spontaneous tumor regression in a syngeneic rat model of liver cancer: implications for survival studies. J Vasc Intervent Radiol. 2012;23(12):1685–1691. doi:10.1016/j.jvir.2012.08.025
24. Finn RS, Qin S, Ikeda M, et al. Atezolizumab plus bevacizumab in unresectable hepatocellular carcinoma. N Engl J Med. 2020;382(20):1894–1905. doi:10.1056/nejmoa1915745
25. Llovet JM, Castet F, Heikenwalder M, et al. Immunotherapies for hepatocellular carcinoma. Nat Rev Clin Oncol. 2022;19(3):151–172. doi:10.1038/s41571-021-00573-2
26. Rochigneux P, Chanez B, De Rauglaudre B, Mitry E, Chabannon C, Gilabert M. Adoptive cell therapy in hepatocellular carcinoma: biological rationale and first results in early phase clinical trials. Cancers. 2021;13(2):1–17. doi:10.3390/cancers13020271
27. Díez S, Navarro G, de ILarduya CT. In vivo targeted gene delivery by cationic nanoparticles for treatment of hepatocellular carcinoma. J Gene Med. 2009;11(1):38–45. doi:10.1002/jgm.1273
28. Xue WJ, Feng Y, Wang F, et al. Asialoglycoprotein receptor-magnetic dual targeting nanoparticles for delivery of RASSF1A to hepatocellular carcinoma. Sci Rep. 2016;6(1):1–13. doi:10.1038/srep22149
29. Xue HY, Liu Y, Liao JZ, et al. Gold nanoparticles delivered miR-375 for treatment of hepatocellular carcinoma. Oncotarget. 2016;7(52):86675–86686. doi:10.18632/oncotarget.13431
30. Fitamant J, Kottakis F, Benhamouche S, et al. YAP inhibition restores hepatocyte differentiation in advanced HCC, leading to tumor regression. Cell Rep. 2015;10(10):1692–1707. doi:10.1016/j.celrep.2015.02.027
31. Younis MA, Khalil IA, Elewa YHA, Kon Y, Harashima H. Ultra-small lipid nanoparticles encapsulating sorafenib and midkine-siRNA selectively-eradicate sorafenib-resistant hepatocellular carcinoma in vivo. J Control Release. 2021;331(September2020):335–349. doi:10.1016/j.jconrel.2021.01.021
32. Xia Y, Zhao M, Chen Y, et al. Folate-targeted selenium nanoparticles deliver therapeutic siRNA to improve hepatocellular carcinoma therapy. RSC Adv. 2018;8(46):25932–25940. doi:10.1039/c8ra04204g
33. Kang H, Rho S, Stiles WR, et al. Size‐dependent EPR effect of polymeric nanoparticles on tumor targeting. Adv Healthc Mater. 2020;9(1):1901223. doi:10.1002/adhm.201901223
34. Maeda H. Toward a full understanding of the EPR effect in primary and metastatic tumors as well as issues related to its heterogeneity. Adv Drug Deliv Rev. 2015;91:3–6. doi:10.1016/j.addr.2015.01.002
35. Zhang M, Gao S, Yang D, et al. Influencing factors and strategies of enhancing nanoparticles into tumors in vivo. Acta Pharm Sin B. 2021;11(8):2265–2285. doi:10.1016/j.apsb.2021.03.033
36. Qiao J, Doubrovin M, Sauter BV, et al. Tumor-specific transcriptional targeting of suicide gene therapy. Gene Ther. 2002;9(3):168–175. doi:10.1038/sj.gt.3301618
37. Robson T, Hirst DG. Transcriptional targeting in cancer gene therapy. Biomed Res Int. 2003;2003(2):110–137.
38. Wolinsky JB, Colson YL, Grinstaff MW. Local drug delivery strategies for cancer treatment: gels, nanoparticles, polymeric films, rods, and wafers. J Control Release. 2012;159(1):14–26. doi:10.1016/j.jconrel.2011.11.031
39. Hu J, Dong Y, Ding L, et al. Local delivery of arsenic trioxide nanoparticles for hepatocellular carcinoma treatment. Signal Transduct Target Ther. 2019;4(1):1–7. doi:10.1038/s41392-018-0034-5
40. Choi CHJ, Alabi CA, Webster P, Davis ME. Mechanism of active targeting in solid tumors with transferrin-containing gold nanoparticles. Proc Natl Acad Sci. 2010;107(3):1235LP-- 1240. doi:10.1073/pnas.0914140107
41. Kawai H, Osawa Y, Matsuda M, et al. Sphingosine-1-phosphate promotes tumor development and liver fibrosis in mouse model of congestive hepatopathy. Hepatology. 2022;76(1):112–125. doi:10.1002/hep.32256
42. Kin M, Torimura T, Ueno T, Inuzuka S, Tanikawa K. Sinusoidal capillarization in small hepatocellular carcinoma. Pathol Int. 1994;44(10–11):771–778. doi:10.1111/j.1440-1827.1994.tb02925.x
43. Vaughan HJ. Engineered Therapeutic Plasmids and Nanoparticles Delivery Vehicles for Targeted Treatment of Hepatocellular Carcinoma. Ph.D. Thesis. Johns Hopkins University; 2021.
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.