")
Back to Journals » OncoTargets and Therapy » Volume 9
Non-secretory multiple myeloma: from biology to clinical management
Authors Dupuis MM, Tuchman SA
Received 13 September 2016
Accepted for publication 17 October 2016
Published 15 December 2016 Volume 2016:9 Pages 7583—7590
DOI https://doi.org/10.2147/OTT.S122241
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Samir Farghaly
Megan Murray Dupuis,1 Sascha A Tuchman2
1Department of Medicine, Duke University, Durham, 2Division of Hematology/Oncology, Lineberger Comprehensive Cancer Center, University of North Carolina at Chapel Hill, Chapel Hill, NC, USA
Abstract: Multiple myeloma (MM) is the second most common hematologic malignancy in the US. It is typically characterized by production of large amounts of defective immunoglobulin (Ig). Diagnosing MM and monitoring treatment response, including eventual relapse, are largely based on sequential measurements of Ig. However, a small subset of MM called non-secretory multiple myeloma (NSMM) produces no detectable Ig. This subset of true NSMM has become even smaller over time, as the advent of the serum free light chain assay has resulted in the majority of NSMM patients being recategorized as light-chain MM – that is, MM cells that produce only the light-chain component of Ig. True forms of NSMM, meaning MM that secretes no monoclonal proteins whatsoever, constitute a distinct entity that is reviewed; definition of NSMM using current detection methods, discuss the biology underpinning NSMM development, and share recommendations for how NSMM should be managed clinically with respect to detection, treatment, and monitoring.
Keywords: multiple myeloma, non-secretory, immunoglobulin
Introduction
Multiple myeloma (MM) is the second most common hematologic neoplasm in the US, with ~30,000 new cases annually.1 It is a malignancy of terminally differentiated, bone marrow-resident plasma cells (PCs), which normally function to support long-term humoral immunity. Normal PCs are uniquely programmed to generate significant amounts of antibody/immunoglobulin (Ig) while surviving indefinitely in the bone marrow microenvironment.2 As MM cells are the transformed version of PCs, they often produce large amounts of Ig, albeit completely non-functional. It is hence unsurprising that the complications from MM arise not only from invasive MM cell growth primarily in the bone and bone marrow but also from the production of aberrant Ig. Symptoms from the former include bone pain, osteolytic lesions, hypercalcemia, and cytopenias.3 The latter can result in a panoply of problems, including neuropathy and renal injury, which can occur through myriad mechanisms.4 In the extreme, MM cells can produce enough Ig to increase the viscosity of serum two- to fourfold, resulting in the rare but potentially fatal hyperviscosity syndrome.5
The exact nature of measurable malignant Ig can vary; MM Ig typically can be detected in serum and/or urine as 1) high concentrations of a full Ig molecule consisting of heavy and light chains bound together; 2) high concentrations of the full Ig molecule plus high concentrations of light chains unbound to heavy chain (free light chains [FLCs]); or 3) primarily FLC in the presence of very small amounts or even no complete Ig whatsoever. A fourth entity exists, which is production of free heavy chain in the absence of bound light chain, but this is very rare.
MM cells generally remain in the bone marrow with usually very low numbers of MM cells circulating in blood, but Ig circulates and its concentration in serum and urine generally correlates with total PC burden. Drawing blood and collecting urine are far simpler than repeat bone marrow biopsies, and so longitudinal monitoring of the concentration of monoclonal Ig as a surrogate for direct measurements of tumor burden has evolved as critical to the assessment of treatment responses and disease progression in MM. The tests most useful for following Ig are serum protein electrophoresis (SPEP) and urine protein electrophoresis (UPEP), serum and urine immunofixation electrophoresis (IFE), and the serum free light chain (SFLC) assay.6,7 Most patients’ MM can be accurately monitored using some combination of these tests. Consensus response criteria for determining effectiveness of MM therapy in clinical trials and off protocol are largely based on this panel.8
Interestingly, it has been observed since the 1950s that a very small subset of the myeloma population is functionally non-secreting, that is, there is no detectable monoclonal Ig by electrophoresis of the serum or urine.9–11 Initial reports estimated that these non-secretory multiple myelomas (NSMMs) represented anywhere from 3% to 5% of the total MM population.9 However, advances in the detection of SFLCs by high-sensitivity enzyme-linked immunosorbent assay (ELISA) have demonstrated that most of these NSMMs were probably oligosecretors – that is, their MM produced primarily or solely SFLC in the absence of heavy chain. FLCs are difficult to detect by standard SPEP and serum IFE.12 The routine use of the ELISA-based SFLC assay has revealed in most recent studies that the proportion of true NSMM, meaning MM that secretes no measurable monoclonal heavy or light chain at all, is closer to <1%–2% of all MMs.13
This review discusses the epidemiology of NSMM, the known physiologic underpinnings of non-secretion, and the clinical implications of non-secretion for diagnosis, treatment, and prognosis.
Ig synthesis and secretion by normal long-lived PCs
To understand the mechanisms underpinning NSMM, it is critical to first understand the biology of Ig synthesis and secretion by PCs, which is detailed in a 2005 review by Shapiro-Shelef and Calame.14 In brief, it has been shown that a specific cascade of genetic signals mediated by BLIMP1 and IRF4 are necessary to begin derepression and enhancement, respectively, of the Ig genes. Fully formed Ig is composed of two heavy chains (IgH) and two light chains (IgL). The heavy chains define the Ig isotype (IgM, IgG, IgA, and IgE), and the light chains are one of two isotypes, either kappa or lambda. By the time a B cell is terminally differentiated into a PC, the appropriate gene rearrangements have already occurred such that the Ig produced will be restricted to one IgH isotype, one IgL isotype, and one idiotype (defined as the antigen-specific sequence of the variable region of Ig), resulting in an antigen-specific, symmetrical Ig molecule. BLIMP1 is also required to derepress the secretable (mu) form of the IgH mRNA, so that the full Ig molecule may be secreted into the extracellular environment. The PC also depends upon a physiologic unfolded protein response for appropriate endoplasmic reticulum expansion required to produce significant amounts of secretable Ig. This process is further enhanced by a number of chaperone and folding proteins, which help to post-translationally modify and shuttle the Ig molecule to the cell membrane for eventual secretion. Unsurprisingly, errors at any step in the pathway – be it heavy-chain synthesis, light-chain synthesis, Ig assembly, transport, or secretion – could lead to altered production and secretion pathways seen in both secretory MM and NSMM. Figure 1 visually depicts this process.
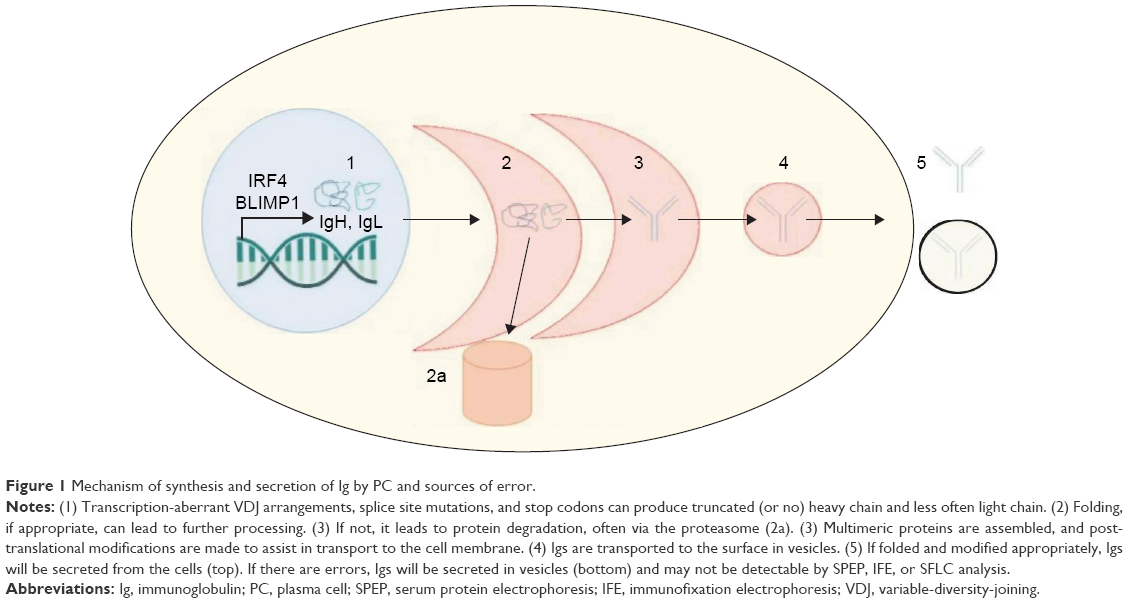 | Figure 1 Mechanism of synthesis and secretion of Ig by PC and sources of error. |
The initial reports hinting at the existence of a non-secretory variant of MM came decades ago from patients who had MM by bone marrow biopsy and yet did not produce detectable Ig on either SPEP or UPEP.15 At the time, it was not clear whether the MM cells produced no Ig whatsoever or if Ig was synthesized but not secreted from the cell. In 1970, Hurez et al16 demonstrated that some NSMM cells do indeed produce Ig that becomes trapped within the cytoplasm and is rapidly degraded. In a follow-up study by the same group,17 they observed that the surface or intracellular Ig of NSMM is restricted to either the light or heavy chain alone. This is in direct contrast to secretory myeloma, which is observed to secrete both heavy and light chains in 75% of cases.18 These data suggest that the “evolution” of NSMM cells may be stepwise from fully secretory MM to MM that loses production of heavy chain and then in a subsequent step loses production of light chain. Consistent with this hypothesis is the fact that in this study and as it has been observed elsewhere as discussed earlier in this manuscript, most of the NSMMs initially identified were actually FLC secretors and the “non-secretory” finding was largely attributable to undetected FLCs. The International Myeloma Working Group still defines NSMM as MM lacking monoclonal protein by serum or urine immunofixation, which can include light-chain MM with quite high levels of monoclonal FLCs detected solely by the SFLC assay.12,19 However, one can argue that this definition is perhaps a misnomer in these cases, since the MM indeed is actively secreting a component of Ig.
One can more accurately subclassify cases of NSMM into at least four distinct categories with separate molecular mechanisms (Figure 2):
- Oligosecretors/FLC-restricted MMs: Oligosecretors/FLC-restricted MM has been discussed, and most cases can be followed by SFLC assay.
- Non-producers: At least 12 patients have been described whose MM is non-secretory due to a complete, true absence of any Ig production whatsoever.20 Such rare patients would not be able to be monitored by either traditional methods or intracellular immunofluorescence, which can be used to detect monoclonal Ig in the cytoplasm of many cases of NSMM. It is hypothesized that the mechanism of non-production is the loss of SFLC secretion by MM clones, which were initially FLC secretors, although this has not been definitively proven.
- True non-secretors: These MM cells produce Ig molecules but are unable to secrete them (the variety of mechanisms by which this occurs is discussed in detail in the following).10,21
- False non-secretors: Initially Decourt et al21 and since then others have referred to certain cases as “false non-secretors.” These are MM variants or related plasma cell diseases that had measurable intracellular Ig by immunofluorescence but no measurable extracellular component by typical testing, despite clear pathological evidence that they were being secreted (such as Ig deposits found in renal biopsies, as can be found as part of the recently described entity monoclonal gammopathy of renal significance).21–23 There are some data that suggest that these Igs are being secreted in vesicles via budding off of the cell membrane, rendering them undetectable in the serum. This, too, would represent a challenge for detection and treatment.
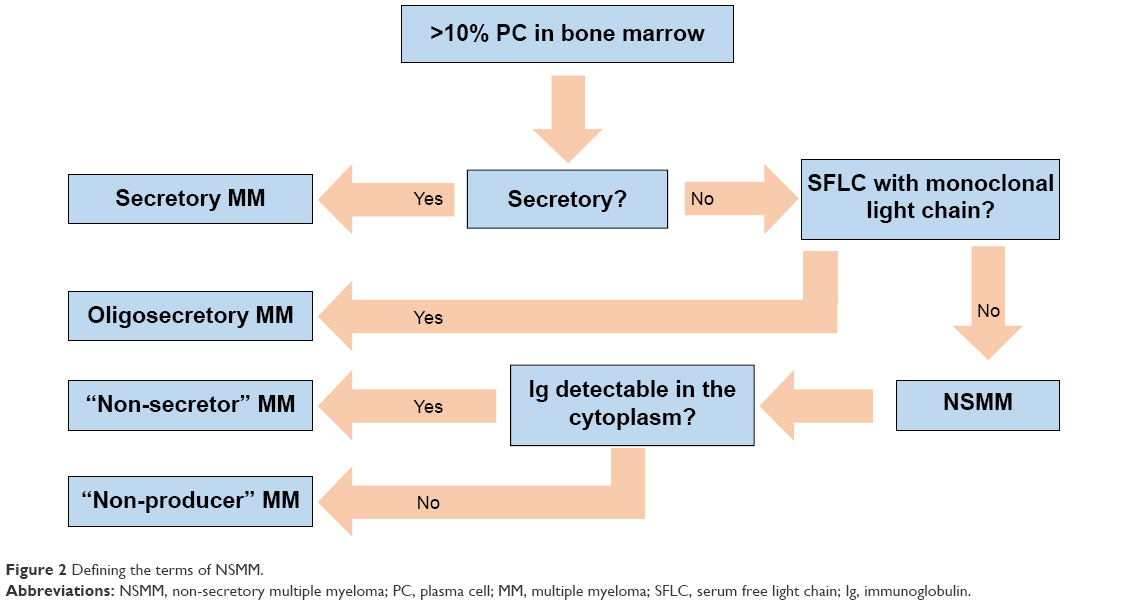 | Figure 2 Defining the terms of NSMM. |
In this review, further use of the term NSMM refers to non-producers and true non-secretors, ie, subtypes of MM that truly have no serum or urine monoclonal Ig, partial (meaning light chain), or otherwise (Figure 2).
Mechanism of NSMM
The exact mechanisms that prevent either production or secretion of monoclonal Ig by NSMM remain poorly understood. As mentioned earlier, one competing hypothesis argues that true NSMMs arise due to a sequential loss of secretion of first heavy chains and then light chains.17 In individual case reports, it has been shown that true non-secretors have lost the polyadenylation site that is necessary for extracellular Ig secretion.24 Other reports have shown that loss of the V domain of the heavy chain also prevents secretion and stimulates intracellular degradation.25 Interestingly, in the same patient, the MM cells had also entirely lost light-chain production due to kappa-chain mutations.25 A case report in 2004 also detailed a frameshift mutation in a patient with kappa-restricted MM. This mutation altered the location of cysteine residues that are critical for appropriate light-chain folding, resulting in an inability to interact with chaperone proteins and ultimately proteasome-mediated degradation.26
Interestingly, loss of ability to secrete the heavy chain (ie, light-chain restriction) appears to be a pro-survival characteristic during clonal evolution in MM. The current leading hypothesis is that production of heavy chain may be toxic to PCs.21 This toxicity may be related to the intracellular inclusions themselves (seen clinically in the so-called Mott cells), which can activate downstream apoptotic signaling pathways. Mechanistically, many cases of light-chain MM have been shown to have interruption of the IgH domain, typically via translocation, and the other locus may be defective due to non-functional VDJ (variable-diversity-joining) arrangements.27 One study in 2002 found that 11 out of 14 NSMM patients had a t(11;14)(q13;q32) rearrangement, which the authors postulated gave the cells a more “lymphoplasmacytic morphology” with a lower secreting capacity than MM cells without the translocation.20,28 Interestingly, the same translocation was detected in the MM case report detailed earlier that also demonstrated the frameshift mutation in the gene coding the light-chain constant region, functionally preventing secretion of the kappa light chain.26 Taken together, these examples bolster the hypothesis for a stepwise loss in secretion of the heavy and light chains as a possible mechanism for NSMM development.
Implications for prognosis
Given the rarity of NSMM in the overall MM population, its clinical course and prognosis are not thoroughly characterized. Moreover, since monitoring of the Ig is an essential metric for tracking response to therapy and detecting relapse, NSMM patients are almost universally excluded from clinical trials.29 The result is a substantive dearth of data for this population.
In 1986, Smith et al30 released a case series that included 13 NSMM patients, in which NSMM patients had a median survival of 46 months compared to 22 months for secretors. At that time, ELISA-based SFLC testing was not commercially available and therefore it is unclear how many of the NSMM patients actually had light-chain MMs. In a retrospective study by Chawla et al13 in 2015, the survival and prognosis of 124 NSMM patients were examined, and the study included SFLC testing where available. They demonstrated that time to progression, progression-free survival (PFS), and overall survival (OS) prior to 2001 were similar between NSMM patients and secretory MM patients. When they examined the years 2001–2012 – the time frame during which new therapeutics were broadly introduced – there was a marked increase in OS in both groups. However, it appeared that the improvement in OS was greater in the NSMM group. Interestingly, the survival gain was not seen in patients treated with high-dose chemotherapy and autologous stem cell transplantion.13,31
Diagnosis, treatment, and monitoring of NSMM in the clinic
The standardized workup put forth by the International Myeloma Working Group for any patient suspected of having MM includes measurement of monoclonal Ig by SPEP, UPEP, serum and urine IFE, and SFLC analysis.32 All patients with suspected MM, including NSMM, should undergo bone marrow aspiration and biopsy with standard studies, including flow cytometry and CD138-enriched fluorescent in situ hybridization testing. Samples should also be stained for intracellular Ig if NSMM is suspected. As in all other forms of MM, NSMM requires myeloma-defining events and/or evidence of MM-mediated end organ damage such as hypercalcemia, anemia, or bone lesions to differentiate an asymptomatic MM precursor from true MM.7
Therapeutically, no data exist to suggest that NSMM responds differently to standard MM treatments. In fact, retrospective studies examining standard approaches13 and autologous stem cell transplantation (ASCT)13,33 are underpowered for specifically examining NSMM, but as mentioned already, it appears that NSMM may do equally well if not better than secretory MM. Consequently, until data emerge to suggest other pathways, treatment of NSMM should follow the same guidelines as those provided for secretory MM.
Monitoring response, however, is where practice clearly deviates from secretory MM, in that clinicians’ inability to use serum and urine Ig studies as reliable surrogates for tumor burden presents a unique dilemma for clinical decision making in NSMM. Serial bone marrow studies facilitate direct examination of tumor burden and are the gold standard, but the cost, time, and patient discomfort associated with frequent bone marrow aspirations and biopsies make them suboptimal from the standpoint of practicality; hence, the focus turns to imaging.
Radiological findings by conventional X-rays frequently lag behind tumor response, and hence, X-rays are inadequate and more advanced imaging modalities are necessary. Specific data are lacking for imaging in NSMM, and as a result, they are extrapolated from secretory MM, where magnetic resonance imaging (MRI) and positron emission tomography (PET) are most well established.
The first relevant question is whether specific imaging techniques are abnormal in MM at baseline, prior to initiation of therapy, since response can only be monitored if abnormalities exist pre-intervention. Multiple case series examining the relative utility of MRI vs PET vs X-rays have been published but are difficult to interpret comparatively due to small numbers and heterogeneity. Walker et al34 showed that 74% of patients in the Arkansas Total Therapy series had focal lesions in bone marrow that were detectable by MRI. Similarly, in one other small study (n=46) in which PET/CT, MRI, and X-rays were compared, MRI of spine–pelvis detected diagnostic abnormalities in 92% of patients, whereas 72% had lesions on whole-body PET/CT.35 A relatively recent systematic review by Regelink et al36 observed that both MRI and PET had a sensitivity of >90% using X-rays as the gold standard (ie, MRI and PET individually detected abnormalities in >90% of patients who had abnormal findings on X-ray) and both modalities detected a higher total number of lesions than X-rays, suggesting that both techniques are more sensitive. These findings have been confirmed in one more recent study.37 Taken together, one concludes that PET/CT and MRI are more sensitive than X-rays for diagnosing MM, and MRI may be the most sensitive technique of all.
Equally important is determining whether abnormalities uncovered by these modalities at baseline change with therapy, ie, whether they can be reliably used to follow response. One Italian study found that 76% of 192 patients going for ASCT for MM had detectable lesions by PET/CT at diagnosis and 35% still had detectable lesions after stem cell transplant. Lack of complete normalization of standardized uptake value activity post transplant strongly predicted both PFS and OS (hazard ratios [HRs] of 1.89 and 3.9, respectively, P=0.03 for both).38 Similarly, the Arkansas group found that post induction, pre-transplant suppression of focal lesions detected on PET was predictive of improved OS (HR 0.33, P=0.001) and PFS (HR 0.47, P=0.13).34
Conversely, MRI has been the subject of fewer and smaller studies, and data are conflicting. For patients with diffuse marrow abnormalities by MRI at diagnosis, normalization of the findings on whole-body MRI correlated well with response measured by conventional Ig markers in one study of 30 subjects. On the other hand, for patients presenting with focal marrow lesions on MRI (small myelomatous lesions that are measurable in size), only 33.5% of patients who achieved very good partial response or better response by standard response criteria8 had shrinkage of these lesions, suggesting inadequate sensitivity for detecting response.39 This finding has been reproduced in one other similar study.40 Hence, MRI may be inadequately sensitive for monitoring response in MM presenting with certain radiological findings in secretory MM, which in turn detracts from its appeal in NSMM. Add to that, the cost and time required for whole-body MRI make it clinically impractical, and one realizes that MRI is likely to be suboptimal.
It is therefore concluded that whole-body PET/CT is imperfect but currently arguably the optimal non-invasive modality for following response in NSMM, when lesions are detectable at diagnosis. It is abnormal in the majority of patients, changes with overall response to chemotherapy, and beyond that has been shown to be prognostic for both PFS and OS. MRI is likely more sensitive for detecting lesions at diagnosis, but the practical limitations and its relatively static nature despite tumor kill (ie, MRI does not reliably change with treatment response) make it inadequate for monitoring.
Finally, the gold standard for monitoring response in any form of MM, including NSMM, is directly measuring the tumor burden via bone marrow aspiration and biopsy. “Going to the source” is of course desirable from the perspective of avoiding surrogate response markers, including imaging. That said, the prospect of bone marrow biopsies every 6–12 weeks indefinitely is unpalatable for patients. One other consideration is the fact that MM can be patchy; one thinks of MM as a diffuse process that uniformly invades the bone marrow throughout the body, but the fact that one can see focal marrow lesions on MRI and heterogeneous marrow 18F-fluorodeoxyglucose-avidity on PET both suggest that the extent of marrow involvement at different sites can be heterogeneous within a single patient, and one can further infer that response determination via blindly obtained marrow samples may be subject to sampling error. That said, one small study suggested that bilaterally obtained marrow samples in MM showed similar results on both sides for both immunohistochemistry and flow cytometry,41 providing some reassurance that standard unilateral posterior iliac crest bone marrow collections are indeed accurate for following NSMM.
Translating the data to actual clinical practice, for NSMM patients with detectable lesions on PET/CT at diagnosis, whole-body PET/CT was performed every 6–12 weeks with the interval decided based on the duration of treatment cycles and the clinical circumstances (more often for patients with aggressive disease and/or lack of other reliable clinical indicators of response versus less often for patients with indolent disease and/or other clinical indicators that the MM is clearly responding, such as improvement in symptoms or cell counts). For patients in remission and undergoing long-term monitoring, perhaps on maintenance chemotherapy, PET/CT every is undertaken 3–6 months. In these patients, bone marrow aspirations and biopsies were pursued approximately every 3–6 months during induction, depending on the level of confidence in other response assessments and then space those out as well once patients move into the remission/maintenance phase. Specifically, for a patient with PET/CT that clearly and believably reflects the MM disease burden and who is clinically doing well, bone marrow testing were pursued approximately yearly. For patients who need to be monitored more closely, bone marrow testing is done every 3–6 months.
In patients with NSMM that cannot be followed by PET/CT, one must follow their disease by serial bone marrow aspirations and biopsies approximately every 4–8 weeks during induction, again depending on clinical factors described earlier, more often, especially early in therapy, for patients requiring closer monitoring versus less often for patients who are felt to warrant a longer interval between assessments. Once stably in remission, we perform bone marrow testing roughly once every 3–6 months.
Discussion
NSMM is a rare variant of MM that is becoming even more uncommon as methods of Ig detection become more sensitive. Because of its rarity and its systematic and purposeful exclusion from clinical trials, there is a paucity of data regarding prognosis. Based on the current studies available, however, NSMM appears to perhaps be less aggressive than secretory MM.
From a biological perspective, this seems counterintuitive. Ig production is metabolically disadvantageous to the cell (since a cell is required to expend energy in its production). More efficient energy management should lead to a proliferative and survival advantage for NSMM, which conceivably should confer a poorer prognosis clinically. One would also perhaps expect that eventually all MM would become NSMM and thereby appropriate that metabolic advantage, although that rarely occurs clinically. We hypothesize that that incongruence could be explained by: 1) the fact that renal insufficiency is uncommon in NSMM,11,30 and renal dysfunction is associated with worse mortality4,42,43; 2) preclinical models that show that NSMM may express light chains on class I major histocompatibility complex receptors, thereby stimulating anti-NSMM cytotoxic T-cell responses44,45; 3) differential response to therapy. Because many NSMM cells are likely dependent on the proteasome for degradation of retained protein products, they may be more sensitive to proteasome-targeting agents such as bortezomib. Unfortunately, due to systematic exclusion of NSMM from clinical trials, there are no good data to definitively suggest that NSMM patients respond more rapidly or durably to proteasome inhibitors.
In thinking about why MM does not more frequently become NSMM, it may be that in NSMM subclones may develop frequently but may undergo preferential elimination by drugs, the immune system, or both, for reasons listed above.
A limitation in hypotheses 2 and 3 is that both are grounded in the inaccurate assumption that all NSMM cells retain misfolded Ig proteins. MM in a vanishingly small number of patients produces neither heavy nor light chain of any kind. These non-producers would appear to lack any of the hypothesized disadvantages of Ig synthesis, such as increased susceptibility to immune-mediated cytotoxicity or proteasome inhibition, which should increase the prevalence of this phenomenon but does not. Ultimately, it may simply be the number of gene mutations necessary to render all heavy-chain and light-chain genes inactive that limits the conversion of secretory MM to NSMM, but clearly further studies in PC biology and MM pathogenesis are needed.
Overall, NSMM is rare and its biology is incompletely understood. Therapy based on contemporary data should match that for secretory MM. In contrast, monitoring NSMM relies far more heavily on PET/CT scans and frequent bone marrow sampling. Ultimately, clinical trials dedicated to NSMM would be very useful, although the challenges of NSMM’s rarity and the fact that an NSMM trial would require a complete revision of the well-established response criteria used to follow secretory forms of MM make such a prospective trial unlikely.
Disclosure
Dr Tuchman reports receiving research support, honoraria and speakers’ bureau from Takeda Pharmaceuticals Inc. The authors report no further conflicts of interest in this work.
References
NIH. Myeloma – SEER Stat Fact Sheets. Available from: http://seer.cancer.gov/statfacts/html/mulmy.html. Accessed July 31, 2016. | ||
Slifka MK, Ahmed R. Long-lived plasma cells: a mechanism for maintaining persistent antibody production. Curr Opin Immunol. 1998;10(3):252–258. | ||
Fonseca R, Bergsagel PL, Drach J, et al. International Myeloma Working Group molecular classification of multiple myeloma: spotlight review. Leukemia. 2009;23(12):2210–2221. | ||
Hutchison CA, Vecihi B, Judith B, et al. The pathogenesis and diagnosis of acute kidney injury in multiple myeloma. Nat Rev Nephrol. 2011;8(1):43–51. | ||
Mehta J, Singhal S. Hyperviscosity syndrome in plasma cell dyscrasias. Semin Thromb Hemost. 2003;29(5):467–471. | ||
Dispenzieri A, Kyle R, Merlini G, et al. International Myeloma Working Group guidelines for serum-free light chain analysis in multiple myeloma and related disorders. Leukemia. 2009;23(2):215–224. | ||
Rajkumar SV, Vincent Rajkumar S, Dimopoulos MA, et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014;15(12):e538–e548. | ||
Durie BGM, Harousseau JL, Miguel JS, et al; International Myeloma Working Group. International uniform response criteria for multiple myeloma. Leukemia. 2007;21(5):1134. | ||
Bladé J, Kyle RA. Nonsecretory myeloma, immunoglobulin D myeloma, and plasma cell leukemia. Hematol Oncol Clin North Am. 1999;13(6):1259–1272. | ||
Middela S, Kanse P. Nonsecretory multiple myeloma. Indian J Orthop. 2009;43(4):408–411. | ||
Cavo M, Galieni P, Gobbi M, et al. Nonsecretory multiple myeloma. Presenting findings, clinical course and prognosis. Acta Haematol. 1985;74(1):27–30. | ||
Drayson M, Tang LX, Drew R, Mead GP, Carr-Smith H, Bradwell AR. Serum free light-chain measurements for identifying and monitoring patients with nonsecretory multiple myeloma. Blood. 2001;97(9):2900–2902. | ||
Chawla SS, Kumar SK, Dispenzieri A, et al. Clinical course and prognosis of non-secretory multiple myeloma. Eur J Haematol. 2015;95(1):57–64. | ||
Shapiro-Shelef M, Calame K. Regulation of plasma-cell development. Nat Rev Immunol. 2005;5(3):230–242. | ||
Serre H, Jaffiol C. [Protein abnormalities in multiple myeloma: atypic biological forms and origin of the myelomatous proteins]. Presse Med. 1958;66(91):2044–2047. French. | ||
Hurez D, Preud’Homme JL, Seligmann M. Intracellular “monoclonal” immunoglobulin in non-secretory human myeloma. J Immunol. 1970;104(1):263–264. | ||
Preud’Homme JL, Hurez D, Danon F, Brouet JC, Seligmann M. Intracytoplasmic and surface-bound immunoglobulins in “nonsecretory” and Bence-Jones myeloma. Clin Exp Immunol. 1976;25(3):428–436. | ||
Kyle RA, Gertz MA, Witzig TE, et al. Review of 1027 patients with newly diagnosed multiple myeloma. Mayo Clin Proc. 2003;78(1):21–33. | ||
The International Myeloma Working Group. Criteria for the classification of monoclonal gammopathies, multiple myeloma and related disorders: a report of the International Myeloma Working Group. Br J Haematol. 2003;121(5):749–757. | ||
Mancilla R, Raul M, Davis GL. Nonsecretory multiple myeloma. Am J Med. 1977;63(6):1015–1022. | ||
Decourt C, Galea HR, Sirac C, Cogné M. Immunologic basis for the rare occurrence of true nonsecretory plasma cell dyscrasias. J Leukoc Biol. 2004;76(3):528–536. | ||
Turesson I, Grubb A. Non-secretory or low-secretory myeloma with intracellular kappa chains. Report of six cases and review of the literature. Acta Med Scand. 1978;204(6):445–451. | ||
Leung N, Bridoux F, Hutchison CA, et al. Monoclonal gammopathy of renal significance: when MGUS is no longer undetermined or insignificant. Blood. 2012;120(22):4292–4295. | ||
Cogné M, Preud’homme JL. Gene deletions force nonsecretory alpha-chain disease plasma cells to produce membrane-form alpha-chain only. J Immunol. 1990;145(8):2455–2458. | ||
Cogné M, Guglielmi P. Exon skipping without splice site mutation accounting for abnormal immunoglobulin chains in nonsecretory human myeloma. Eur J Immunol. 1993;23(6):1289–1293. | ||
Coriu D, Weaver K, Schell M, et al. A molecular basis for nonsecretory myeloma. Blood. 2004;104(3):829–831. | ||
Magrangeas F, Cormier M-L, Descamps G, et al. Light-chain only multiple myeloma is due to the absence of functional (productive) rearrangement of the IgH gene at the DNA level. Blood. 2004;103(10):3869–3875. | ||
Avet-Loiseau H, Garand R, Lodé L, Harousseau JL, Bataille R; Intergroupe Francophone du Myélome. Translocation t(11;14)(q13;q32) is the hallmark of IgM, IgE, and nonsecretory multiple myeloma variants. Blood. 2002;101(4):1570–1571. | ||
Bross PF, Kane R, Farrell AT, et al. Approval summary for bortezomib for injection in the treatment of multiple myeloma. Clin Cancer Res. 2004;10(12 pt 1):3954–3964. | ||
Smith DB, Harris M, Gowland E, Chang J, Scarffe JH. Non-secretory multiple myeloma: a report of 13 cases with a review of the literature. Hematol Oncol. 1986;4(4):307–313. | ||
Jacobs RW, Saliba RM, Sasaki K, et al. Outcome of patients with nonsecretory multiple myeloma after autologous hematopoietic stem cell transplantation. Clin Lymphoma Myeloma Leuk. 2016;16(1):36–42. | ||
Dimopoulos M, Kyle R, Fermand JP, et al; Myeloma Workshop Consensus Panel 3. Consensus recommendations for standard investigative workup: report of the International Myeloma Workshop Consensus Panel 3. Blood. 2011;117(18):4701–4705. | ||
Terpos E, Apperley JF, Samson D, et al. Autologous stem cell transplantation in multiple myeloma: improved survival in nonsecretory multiple myeloma but lack of influence of age, status at transplant, previous treatment and conditioning regimen. A single-centre experience in 127 patients. Bone Marrow Transplant. 2003;31(3):163–170. | ||
Walker R, Barlogie B, Haessler J, et al. Magnetic resonance imaging in multiple myeloma: diagnostic and clinical implications. J Clin Oncol. 2007;25(9):1121–1128. | ||
Zamagni E, Nanni C, Patriarca F, et al. A prospective comparison of 18F-fluorodeoxyglucose positron emission tomography-computed tomography, magnetic resonance imaging and whole-body planar radiographs in the assessment of bone disease in newly diagnosed multiple myeloma. Haematologica. 2007;92(1):50–55. | ||
Regelink JC, Minnema MC, Terpos E, et al. Comparison of modern and conventional imaging techniques in establishing multiple myeloma-related bone disease: a systematic review. Br J Haematol. 2013;162(1):50–61. | ||
Caers J, Withofs N, Hillengass J, et al. The role of positron emission tomography-computed tomography and magnetic resonance imaging in diagnosis and follow up of multiple myeloma. Haematologica. 2014;99(4):629–637. | ||
Zamagni E, Patriarca F, Nanni C, et al. Prognostic relevance of 18-F FDG PET/CT in newly diagnosed multiple myeloma patients treated with up-front autologous transplantation. Blood. 2011;118(23):5989–5995. | ||
Lin C, Luciani A, Belhadj K, et al. Multiple myeloma treatment response assessment with whole-body dynamic contrast-enhanced MR imaging. Radiology. 2010;254(2):521–531. | ||
Bannas P, Hentschel HB, Bley TA, et al. Diagnostic performance of whole-body MRI for the detection of persistent or relapsing disease in multiple myeloma after stem cell transplantation. Eur Radiol. 2012;22(9):2007–2012. | ||
Manasanch EE, Korde N, Zingone A, et al. The proteasome: mechanisms of biology and markers of activity and response to treatment in multiple myeloma. Leuk Lymphoma. 2014;55(8):1707–1714. | ||
Knudsen LM, Martin H, Erik H; Nordic Myeloma Study Group. Renal failure in multiple myeloma: reversibility and impact on the prognosis. Eur J Haematol. 2000;65(3):175–181. | ||
Haynes RJ, Read S, Collins GP, Darby SC, Winearls CG. Presentation and survival of patients with severe acute kidney injury and multiple myeloma: a 20-year experience from a single centre. Nephrol Dial Transplant. 2010;25(2):419–426. | ||
Galea HR, Cogné M. GM-CSF and IL-12 production by malignant plasma cells promotes cell-mediated immune responses against monoclonal Ig determinants in a light chain myeloma model. Clin Exp Immunol. 2002;129(2):247–253. | ||
Galea HR, Denizot Y, Cogné M. Light chain myeloma plasma cells induce a strong cell-mediated immune response mainly directed against the monoclonal light chain determinants in a murine experimental model. Cancer Immunol Immunother. 2002;51(4):229–234. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.