")
Back to Journals » Drug Design, Development and Therapy » Volume 16
Non-Inferior Efficacy of Tenofovir Disoproxil to Tenofovir Disoproxil Fumarate in Virologically Suppressed Chronic Hepatitis B Patients
Authors Yim HJ , Kim JH , Cho YK, Kweon YO, Cho HC, Hwang JS, Lee C , Koh MS, Baek YH, Park YM, Lee JH , Kim SU , Kang MK , Park NH, Lee JS , Chon YE, Cheon GJ, Chae HB, Sohn JH, Lim YS
Received 16 June 2022
Accepted for publication 16 September 2022
Published 23 September 2022 Volume 2022:16 Pages 3263—3274
DOI https://doi.org/10.2147/DDDT.S376821
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Anastasios Lymperopoulos
Hyung Joon Yim,1,* Ji Hoon Kim,2,* Yong Kyun Cho,3 Young Oh Kweon,4 Hyun Chin Cho,5 Jae Seok Hwang,6 Changhyeong Lee,7 Moon Soo Koh,8 Yang-Hyun Baek,9 Young-Min Park,10 Jeong-Hoon Lee,11 Seung Up Kim,12 Min-Kyu Kang,13 Neung Hwa Park,14 June Sung Lee,15 Young Eun Chon,16 Gab Jin Cheon,17 Hee Bok Chae,18 Joo Hyun Sohn,19 Young-Suk Lim20
1Department of Internal Medicine, Korea University Ansan Hospital, Korea University College of Medicine, Seoul, Korea; 2Department of Internal Medicine, Korea University Guro Hospital, Korea University College of Medicine, Seoul, Korea; 3Division of Gastroenterology and Hepatology, Department of Internal Medicine, Kangbuk Samsung Hospital, Sungkyunkwan University School of Medicine, Seoul, Korea; 4Department of Internal Medicine, Kyungpook National University, School of Medicine, Daegu, Korea; 5Department of Internal Medicine, Gyeongsang National University Hospital, Gyeongsang National University School of Medicine, Jinju, Korea; 6Department of Internal Medicine, Keimyung University School of Medicine, Daegu, Korea; 7Department of Internal Medicine, Daegu Catholic University School of Medicine, Daegu, Korea; 8Department of Internal Medicine, Dongguk University Ilsan Hospital, College of Medicine, Dongguk University, Goyang, Korea; 9Department of Gastroenterology, DongA University College of Medicine, Busan, Korea; 10Department of Internal Medicine, Bundang Jesaeng General Hospital, Seongnam, Korea and Hepatology Center, Bundang Jesaeng General Hospital, Seongnam, Korea; 11Department of Internal Medicine and Liver Research Institute, Seoul National University College of Medicine, Seoul, Korea; 12Department of Internal Medicine, Yonsei University College of Medicine, Seoul, Korea, and Yonsei Liver Center, Severance Hospital, Seoul, Korea; 13Department of Internal Medicine, Yeungnam University College of Medicine, Daegu, Korea; 14Department of Internal Medicine, Ulsan University Hospital, University of Ulsan College of Medicine, Ulsan, Korea; 15Department of Internal Medicine, Ilsan Paik Hospital, Inje University College of Medicine, Goyang, Korea; 16Department of Internal Medicine, CHA Bundang Medical Center, CHA University, Seongnam, Korea; 17Department of Medicine, GangNeung Asan Hospital, Ulsan University College of Medicine, Gangwon-do, Korea; 18Department of Internal Medicine, Chungbuk National University Hospital, Cheongju, Korea; 19Department of Internal Medicine, Hanyang University College of Medicine, Seoul, Korea, and Hanyang University Guri Hospital, Guri, Korea; 20Department of Gastroenterology, Liver Center, Asan Medical Center, University of Ulsan College of Medicine, Seoul, Korea
*These authors contributed equally to this work
Correspondence: Young-Suk Lim, Department of Gastroenterology, Liver Center, Asan Medical Center, University of Ulsan College of Medicine, 43-Gil 88, Olympic-Ro, Songpa-Gu, Seoul, Korea, Tel +82-2-3010-3190, Fax +82-2-485-5782, Email [email protected]
Purpose: Tenofovir disoproxil (TD), modified from tenofovir disoproxil fumarate (TDF), was developed as a salt-free formulation, removing fumarate to improve the ease of oral intake by reducing the tablet’s size. We evaluated the maintenance of antiviral effects and overall safety profile of TD 245 mg after switching from TDF 300 mg in patients with chronic hepatitis B (CHB).
Patients and Methods: CHB patients with HBV-DNA < 69 IU/mL after ≥ 24 weeks of TDF therapy were enrolled. The primary efficacy endpoint was the HBV-DNA suppression rate (HBV-DNA < 69 IU/mL) at week 48; We evaluated the non-inferiority (10% margin) of TD to TDF in terms of efficacy. Safety was assessed based on adverse events (AEs), laboratory tests, bone mineral density, and renal function abnormalities.
Results: Overall, 189 subjects were randomized in a 2:1 ratio, and 117 and 66 subjects in the TD and TDF groups, respectively, completed the study. In the per-protocol set, the HBV-DNA suppression rate at week 48 was 99.1% and 100% in the TD and TDF groups, respectively. The lower limit of the 97.5% one-sided confidence interval for the intergroup difference in HBV-DNA suppression rate was − 2.8%, which was greater than the prespecified margin of non-inferiority. The changes in creatinine clearance from baseline to week 48 was significantly less in the TD group and in the TDF group; − 0.8 ± 9.8 versus − 2.4 ± 12.8 mL/min, respectively (P=0.017).
Conclusion: TD was non-inferior to TDF for maintaining viral suppression in CHB patients, showing the less decline of renal function.
Keywords: viral DNA, bone density, antiviral agents, viral suppression
Introduction
Chronic hepatitis B (CHB) is associated with a wide range of hepatic complications. In patients with CHB, the annual incidence of liver cirrhosis is 5.1%, with a 5-year cumulative incidence of 23%, while the annual incidence of hepatocellular carcinoma is reported to be 0.8%, with a 5-year cumulative incidence of 3%.1,2 Globally, more than 780,000 deaths are reported, annually, in patients with CHB due to liver cirrhosis and hepatocellular carcinoma.3
The goal of CHB treatment is to inhibit hepatitis B virus (HBV) replication, thereby alleviating necroinflammation and preventing fibrosis, which, in turn, prevents liver cirrhosis, hepatic failure, and progression to hepatocellular carcinoma, and ultimately reduces death rates due to liver disease.1,5
One of the most important and challenging aspects of decreasing and controlling the progression of liver diseases is early diagnosis and treatment of CHB.4,5 Early detection of the chronic HBV infection through various types of molecular biology such as DNA microarray or nucleic acid techniques and proper treatment has the potential to prevent or inhibit the sequelae.6,7
The ideal treatment endpoint would be the loss of serum hepatitis B surface antigen (HBsAg), but this is a clinically rare event because covalently closed circular DNA persists despite long-term antiviral therapy.8 Consequently, therapeutic aims in general clinical practice are normalization of alanine transaminase (ALT), undetectable serum HBV-DNA, loss or seroconversion of hepatitis B e antigen (HBeAg), as surrogate markers of treatment response.9 Nevertheless, HBsAg loss is considered the hallmark of a functional cure, so antiviral treatment should be continued for an extended period, even for lifetime. Therefore, compliance to taking medication is very important.
Among the many antivirals currently available, tenofovir disoproxil fumarate (TDF) shows strong antiviral effects and low resistance rates, even after 8 years of treatment.10 Hence, the drug is widely used globally, including in South Korea.11,12 However, the relatively larger size of the TDF tablet is one of the drawbacks, causing inconvenience, particularly in older individuals and children, given that CHB patients require long-term daily medication. As poor medication compliance is an important factor in virological breakthrough, with antiviral therapy for HBV,13,14 there was a need to develop a new formulation with improved medication convenience to enhance treatment efficacy.
Tenofovir disoproxil (TD) (Virehepa Tab, Daewoong Pharmaceutical Co., Ltd., Seoul, South Korea), a drug incrementally modified from TDF, was developed as a salt-free formulation with the removal of fumarate. By reducing the active ingredient from 300 mg to 245 mg, the tablets are smaller, which makes them easier to swallow. The new formulation was proven to be pharmacokinetically equivalent to TDF and was approved in Korea in 2017.15
To date, no studies have evaluated the antiviral effects and safety of TD in CHB patients. This clinical trial evaluated the maintenance of antiviral effects after switching to TD in CHB patients who had confirmed viral suppression with TDF. We also investigated the overall safety profile of TD, including treatment-emergent adverse events as well as long-term side effects of tenofovir, such as renal tubular damage and decreased bone mineral density.16–18
Materials and Methods
Study Design
This Phase 4 clinical trial was conducted at 20 study sites from January 2018 to November 2019. The study was performed in compliance with the International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use guidelines. This study was approved by the institutional review board committee of Asan Medical Center, Korea University Ansan Hospital, Korea University Guro Hospital, Kangbuk Samsung Hospital, Kyungpook National University Hospital, Gyeongsang National University Hospital, Keimyung University Hospital, Daegu Catholic University Hospital, Dongguk University Ilsan Hospital, DongA University Hospital, Bundang Jesaeng General Hospital, Seoul National University Hospital, Severance Hospital, Yeungnam University Hospital, Ulsan University Hospital, Ilsan Paik Hospital, CHA Bundang Medical Center, GangNeung Asan Hospital, Chungbuk National University Hospital and Hanyang University Hospital. The ClinicalTrials.gov registration number of this study is NCT03485534. Written informed consent was obtained from all subjects before study enrollment or procedures. In compliance with the Declaration of Helsinki, Good Clinical Practice, and all applicable regulations, the study was conducted ethically and scientifically.
The study was conducted on patients with CHB who had confirmed viral suppression (HBV-DNA <69 IU/mL) after ≥24 weeks of TDF treatment. Eligible subjects, based on the inclusion and exclusion criteria described below, were randomized in a 2:1 ratio to the test group (TD 245 mg once a day) or the comparator group (TDF 300 mg once a day) and stratified by HBeAg status (positive or negative). The 2:1 ratio was determined to obtain sufficient safety data from the TD group.19 The subjects received investigational product (IP; TD or TDF) orally for 48 weeks in an open-label manner, according to the planned dosage and administration for the assigned group. Those who had decreased renal function (creatinine clearance rate [Ccr]: 30–49 mL/min) during the study period received the IP once every other day, according to the label. At weeks 24 and 48, the safety and efficacy of the drug in the subjects were assessed based on the maintenance of antiviral effects. Medication compliance was checked by telephone at weeks 12 and 36. For the TD group, an optional on-site visit was performed at week 12 for a safety assessment, at the investigator’s discretion.
Patients
This study enrolled Korean CHB patients aged 19–69 years who had documented HBsAg-positivity for ≥24 weeks or a history of hepatitis B for ≥24 weeks and were anti-HBc IgM-negative. Other inclusion criteria included patients who were on TDF monotherapy for CHB, had received prior treatment with TDF for ≥24 weeks, demonstrated good medication compliance (≥80%) based on interviews, confirmed viral suppression, and were expected to require tenofovir monotherapy for ≥48 weeks.
Patients with a history of organ or bone marrow transplants or confirmed malignant tumors within the past 5 years and those with decompensated liver disease were excluded. Other exclusion criteria included co-infection with hepatitis C virus, human immunodeficiency virus, or hepatitis delta virus, decreased renal function (Ccr <50 mL/min), and ALT >5 × upper limit of normal (ULN).
Endpoints
The primary efficacy endpoint was the proportion of subjects with HBV-DNA suppression (HBV-DNA <69 IU/mL) by quantitative polymerase chain reaction at week 48; We evaluated the non-inferiority (10% margin) of TD to TDF in terms of efficacy.
The secondary efficacy endpoints were as follows. First, virological endpoints were the proportion of subjects with HBV-DNA suppression at week 24; rate of undetectable HBV-DNA (HBV-DNA <20 IU/mL) at weeks 24 and 48, and HBV-DNA level and change in HBV-DNA from baseline at weeks 24 and 48. Second, serological endpoints were, among those with positive HBeAg at randomization, the proportion of patients with loss of HBeAg and the seroconversion rate at weeks 24 and 48, and the proportion of patients with loss of HBsAg and the seroconversion rate at weeks 24 and 48. Third, biochemical endpoints were the proportion of patients with normal ALT levels (male: ≤41 U/L, female: ≤33 U/L) at weeks 24 and 48; the proportion of patients with ALT levels ≤ ULN (male: ≤30 U/L, female: ≤19 U/L) at weeks 24 and 48, according to the American Association for the Study of Liver Disease criteria;20 and the change from baseline ALT levels at weeks 24 and 48. Fourth, failure of antiviral effect was determined based on the rate of virological breakthrough (an increase of 1 log10 from the individual nadir in two consecutive HBV-DNA levels, measured ≥2 weeks apart) and cumulative genetic mutation rate at weeks 24 and 48.
Additionally, subgroup analyses were performed on the primary and secondary endpoints in patients positive for HBeAg at the time of enrollment and those with a history of antiviral therapy before TDF.
Safety was assessed based on treatment-emergent adverse events and laboratory test abnormalities. Additionally, the proportions of subjects with ALT flare (ALT >10 × ULN), an increase of ≥0.5 mg/dL from baseline in serum creatinine, and Ccr <50 mL/min were assessed. One of the safety endpoints was the percentage change from baseline in bone mineral density of the lumbar spine and hip as measured by dual-energy X-ray absorptiometry. Furthermore, to determine the stage of liver fibrosis in a non-invasive manner, changes in the aspartate aminotransferase‒platelet ratio index and Fibrosis-4 score were analyzed.21
Statistical Analysis
The sample size (TD group, n = 126; TDF group; n = 63) was sufficient to demonstrate the non-inferiority of the TD group to the TDF group, with 80% power at a one-sided significance level of 0.025, assuming a 95% treatment success rate in the TDF group.22 A 10% dropout rate was considered.
For demographic information and baseline disease information, the number of subjects, mean ± standard deviation (SD), and median (range) were presented for continuous data, and frequencies and percentages were presented for categorical data. Unless otherwise specified, two-sided tests were performed at a significance level of 5%.
Efficacy was assessed primarily in the per-protocol set (PPS) and additionally in the full analysis set. The PPS included subjects who completed the study without any major protocol deviations. The full analysis set included subjects who were randomized and treated with IP and who underwent HBV-DNA quantification tests at least once.
If the primary efficacy endpoint (HBV-DNA suppression rate at week 48) was missing, HBV-DNA was considered ≥69 IU/mL. Subjects who had a study visit that deviated from the scheduled visit window (±14 days) were handled as missing and classified as a failure for the maintenance of antiviral effect for analysis.
In terms of the primary efficacy endpoint, the non-inferiority of the TD group to the TDF group should be declared if the lower limit of the 97.5% one-sided confidence interval (CI) for the inter-group difference in the HBV-DNA suppression rate at week 48 was smaller than the non-inferiority margin of 10%.
Safety results were analyzed in the safety set, which included all subjects who were randomized and treated with at least one dose of IP. Adverse events (AEs), number of subjects, incidence, and number of events were presented by the treatment group and analyzed using Fisher’s exact test for treatment-emergent AEs, adverse drug reactions, serious AEs (SAEs), serious adverse drug reactions, and AEs leading to treatment withdrawal. Additionally, the number of subjects, incidence, and number of events were presented by treatment group for AEs classified by system organ class and the preferred term of the Medical Dictionary for Regulatory Activities v21.0.23
Efficacy and safety variables, presented as proportion, frequency, and percentage, were presented by the treatment group, and analyzed using Pearson’s chi-square test or Fisher’s exact test. For variables presented as change, the number of subjects, mean, and standard deviation were presented by the treatment group and analyzed using the two-sample t-test or Wilcoxon’s rank-sum test.
Medication compliance was calculated as the percentage of the quantity administered relative to the quantity planned to be administered based on the unused IPs retrieved.
Results
Patient Disposition and Baseline Characteristics
A total of 244 subjects were screened, and 189 were randomized. Among 122 subjects assigned to the TD group, 117 subjects completed the study, while 5 subjects dropped out due to withdrawal of informed consent, violation of the inclusion/exclusion criteria, treatment with prohibited concomitant drugs, loss to follow-up, and other reasons based on the investigator’s judgment (one subject each). Among the 67 subjects assigned to the TDF group, 66 subjects completed the study, with the remaining subject dropping out due to violation of the inclusion/exclusion criteria. None withdrew from the study due to virological breakthrough, drug resistance, or AEs (Figure 1).
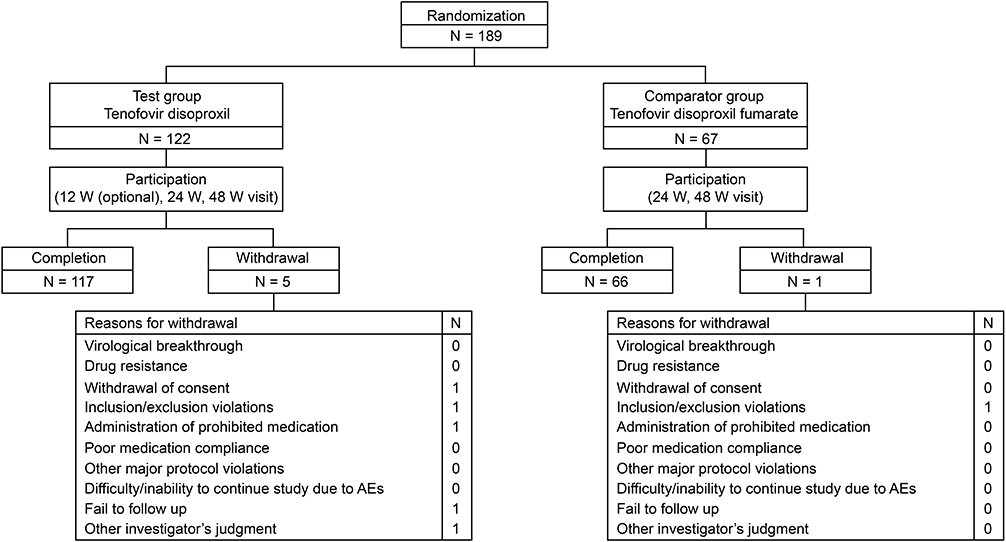 |
Figure 1 Disposition of study subjects. |
Demographic and baseline information were generally comparable between the two groups (Table 1).
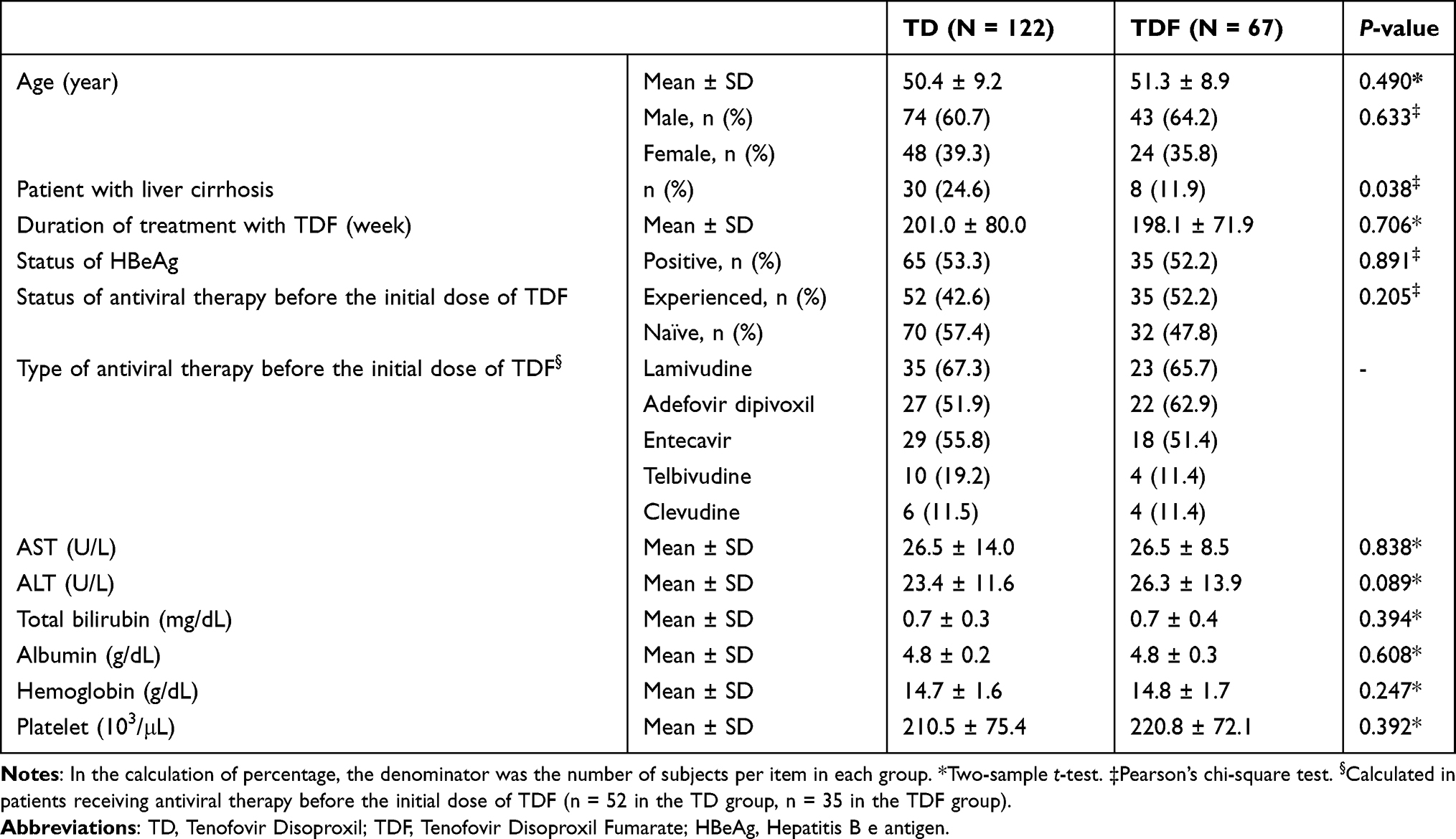 |
Table 1 Assessment of Demographics and Baseline Information |
Efficacy Outcome
Mean medication compliance was comparable at 97.3% and 97.9% in the TD and TDF groups, respectively. The results of the primary and secondary efficacy endpoints are presented in Tables 2 and 3, respectively. The results were validated using different analytical equipment (Supplementary File 1).
 |
Table 2 Primary Efficacy Endpoint |
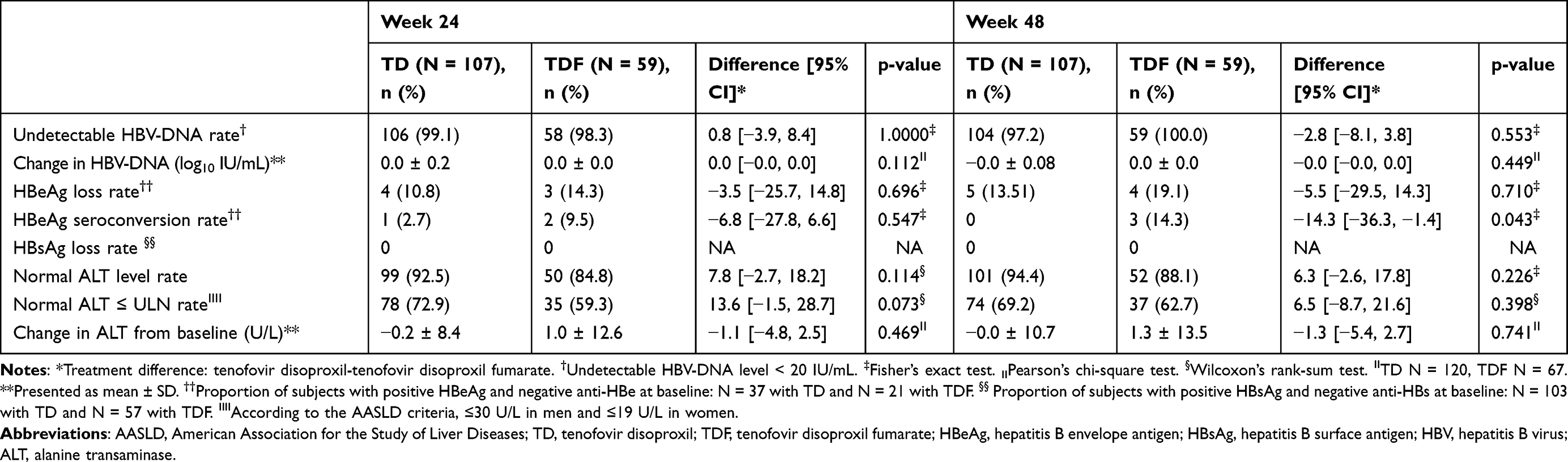 |
Table 3 Secondary Efficacy Endpoints (per-Protocol Set) |
In the PPS main analysis set, the HBV-DNA suppression rate (<69 IU/mL) at week 48 was 99.1% (106/107 subjects) in the TD group versus 100.0% (59/59 subjects) in the TDF group (Figure 2). The adjusted difference was −0.9%, and the lower limit of the 97.5% one-sided CI for the intergroup difference was −2.8%, which was greater than the prespecified −10% margin used to demonstrate non-inferiority for the primary efficacy endpoint at week 48.
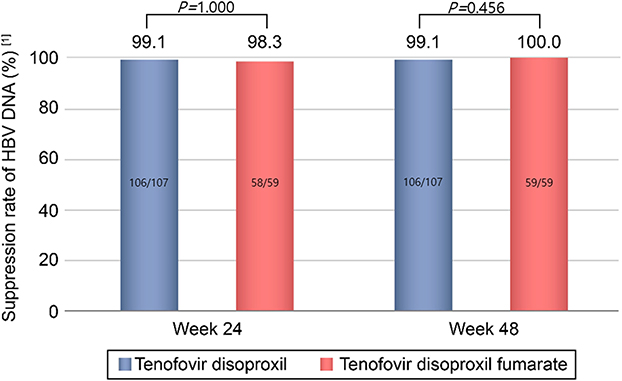 |
Figure 2 HBV-DNA Suppression Rate at Weeks 24 and 48 after Randomization (<69 IU/mL). The adjusted difference was 0.8% (95% CI −3.9% to 8.4, P = 1.0000) and −0.9% (97.5% CI −2.8% to ∞, P = 0.456) at week 24 and 48, respectively. [1]Subjects with HBV DNA measurements less than 69 IU/mL out of the subjects on follow-up at 24 and 48 weeks after randomization. |
Failure of HBV-DNA suppression was reported in one subject in the TD group who had a rapid increase in HBV-DNA levels at week 24 (1120 IU/mL). After approximately 1 month, an unscheduled visit was performed to assess a virological breakthrough. At this visit, the HBV-DNA level had declined to 293 IU/mL and then continued to decline to 96.1 IU/mL by week 48.
The proportion of patients with undetectable HBV DNA (<20 IU/mL) at weeks 24 and 48 was 99.1% (106/107) and 97.2% (104/107) of those receiving TD, respectively, compared to 98.3% (58/59) and 100.0% (59/59) of those receiving TDF. The adjusted difference was 0.8% (95% CI −3.9% to 8.4%, P=1.000) and −2.8% (95% CI −8.1% to 3.8%, P=0.553) at week 24 and 48, respectively.
The rate of HBeAg loss among HBeAg-positive subjects receiving TD was 10.8% (4/37) and 13.5% (5/37) at week 24 and 48, respectively, which was not statistically different from the rate of 14.3% (3/21) and 19.1% (4/21) among subjects receiving TDF (P=0.696 and P=0.710, respectively). The rate of HBeAg seroconversion was numerically higher among patients receiving TDF at week 24, but the difference did not achieve statistically significant (2.7% (1/37) in TD group vs 9.5% (2/21) in TDF group, P=0.547). However, the rate of HBeAg seroconversion at week 48 was statistically significant between groups (0.0% (0/37) in TD group vs 14.29% (3/21) in TDF group, P=0.043). During the study, no patients in either group had HBsAg loss or seroconversion.
The proportion of subjects with normal ALT at weeks 24 and 48 of treatment was 92.5% (99/107) and 94.4% (101/107) in those receiving TD, respectively, compared to 84.8% (50/59) and 88.1% 52/59) among subjects receiving TDF (P=0.114 and P=0.226, respectively). The proportion of subjects with ALT levels ≤ULN, according to the American Association for the Study of Liver Disease (AASLD) criteria (male: ≤30 U/L, female: ≤19 U/L), at weeks 24 and 48 of treatment was 72.9% (78/107) and 69.2% (74/107) in TD group, compared to 59.3% (35/59) and 62.7% (37/59) in TDF group (P=0.073 and P=0.398, respectively). The differences were not significantly different.
The subgroup analysis in HBeAg-positive subjects at enrollment showed that in the PPS main analysis set, the lower limit of the 97.5% one-sided CI for the intergroup difference in the rate of HBV-DNA suppression at week 48 was −5.2%, which was above the non-inferiority margin of −10% (Supplementary Table S1). Results from the subgroup analysis in subjects with a history of other antiviral therapy before TDF showed that the HBV-DNA suppression rate at week 48 was 100.0% in both groups (Supplementary Table S2). These results are similar to the results of the main analysis.
Neither group exhibited virological breakthrough or genetic mutations at weeks 24 and 48.
Safety Outcomes
Safety results from the study are presented in Table 4.
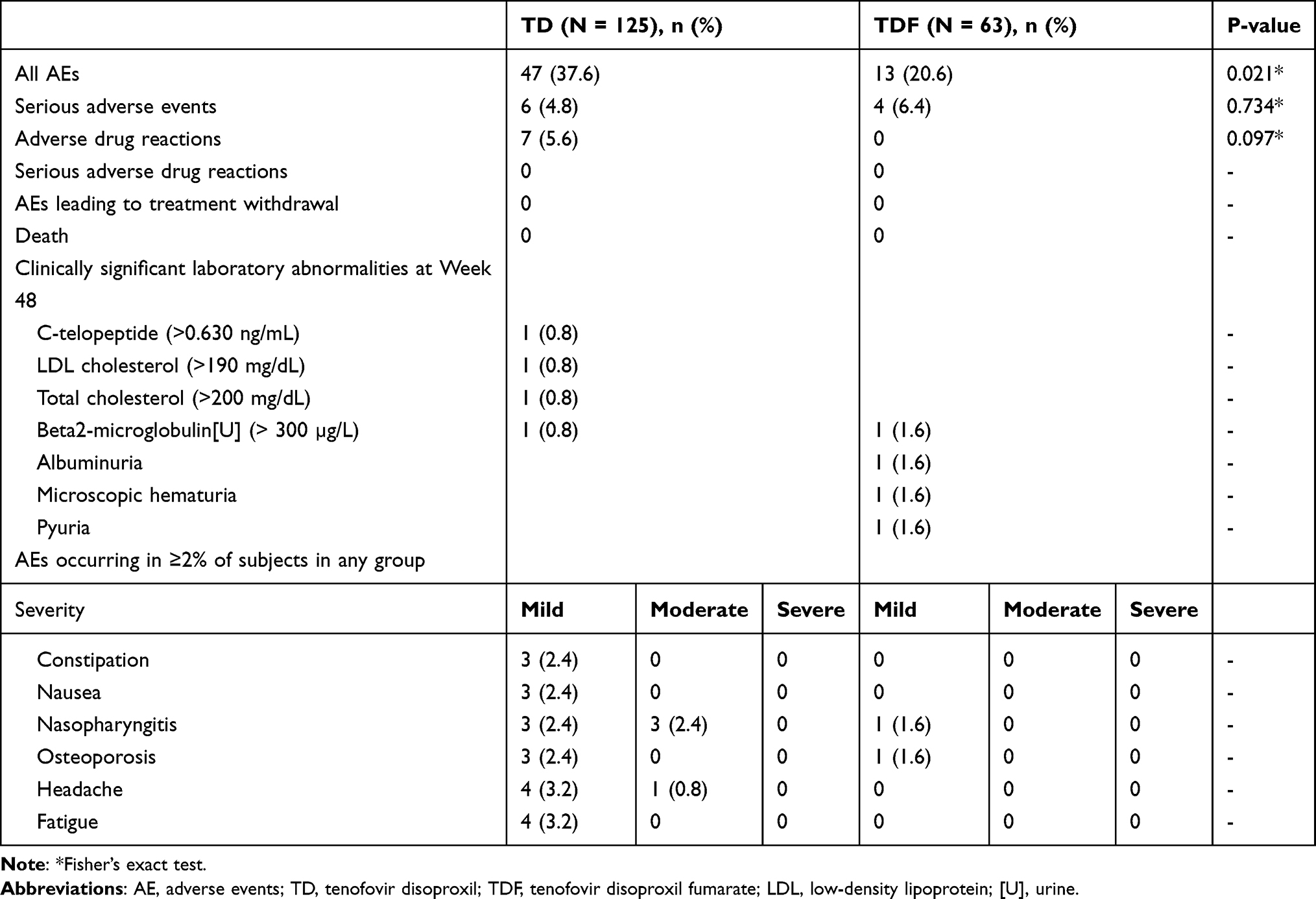 |
Table 4 Summary of Treatment-Emergent Adverse Events Reported Up to Week 48 |
In this study, the AE incidence was higher in the TD group (37.6% in the TD group versus 20.6% in the TDF group; P=0.021). The most common AE was nasopharyngitis. As the greater number of AEs in the TD group was thought to be affected by the more frequent visits in that group, the two groups were compared based on data collected after week 12. There were no intergroup differences (Table 5).
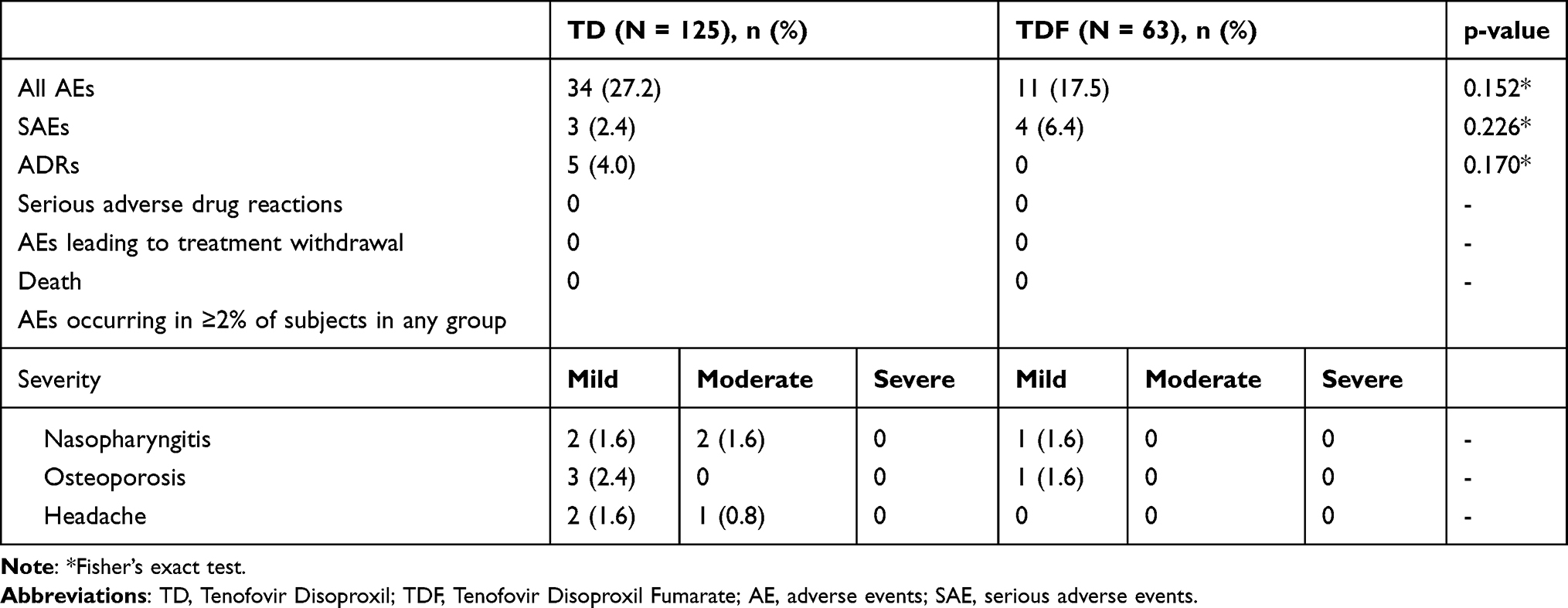 |
Table 5 Summary of Treatment-Emergent Adverse Events Reported from Week 12 to Week 48 |
The incidence of adverse drug reactions was 5.6% in the TD group and none in the TDF group. The incidence of SAEs was not significant: 4.8% in the TD group versus 6.4% in the TDF group. None of the subjects experienced serious adverse drug reactions, AEs leading to treatment withdrawal, or death. One subject in the TD group with a history of liver cirrhosis was diagnosed with hepatocellular carcinoma 36 days after TD initiation. However, this subject continued to participate in the study and maintained HBV-DNA below the lower limit of detection up to week 48.
The majority of AEs were mild to moderate, had resolved or were resolving at the end of the study, and were assessed as “possibly” related in 5.6% (7/125 subjects, 7 events) in the TD group and “unlikely” in all other subjects, in terms of the causal relationship to the IP. None of the subjects required an IP dose decrease due to AEs.
The laboratory test results that evaluated clinically significant abnormalities at 48 weeks after administration are presented in Table 4. None in either group had ALT flares or an increase of ≥0.5 mg/dL in serum creatinine. The proportion of subjects with Ccr <50 mL/min at week 48 was not significant: 1.7% (2/125 subjects) in the TD group versus 3.3% (2/63 subjects) in the TDF group.
The change in Ccr from baseline at week 48 was significant, −0.8 ± 9.8 mL/min in the TD group versus −2.4 ± 12.8 mL/min in the TDF group (P=0.017) (Figure 3). At week 48, there were no significant differences in the change from baseline in bone mineral density of the total lumbar spine, total hip, or femur neck, and the change from baseline of the aspartate aminotransferase‒platelet ratio index or Fibrosis-4 (P=0.211 and P=0.253, respectively) (Supplementary Figure S1).
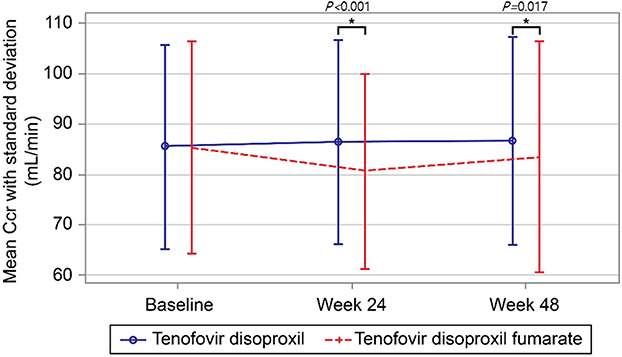 |
Figure 3 Changes in Renal Function Estimated by serial Creatinine Clearance Rate (Ccr). The mean Ccr significantly declined less in the TD group than in the TDF group at week 24 (P < 0.001) and at week 48 (P = 0.017). *Inter-group difference in change from baseline is statistically significant at 5% two-sided significance level. |
One subject in the TD group was pregnant at week 48. At this point, the subject completed the planned TD treatment. Pregnancy follow-up after the end of the study found that the subject gave birth to a 3.6-kg female baby and that the baby was generally healthy.
Discussion
This clinical trial demonstrated that TD could be safely used to maintain antiviral effects in patients with CHB who had previously achieved viral suppression with ≥24 weeks of TDF treatment.
In the PPS main analysis set, the proportion of subjects with HBV-DNA suppression at week 48 of treatment was 99.1% in the TD group versus 100.0% in the TDF group. These findings (>95%) indicated good maintenance of antiviral effects in both groups and demonstrated the non-inferiority of the TD group to the TDF group. Furthermore, less decline of renal function assessed by Ccr was observed in patients who received TD than in those receiving TDF. These findings suggest switching to TD could be recommended in CHB patients receiving TDF if there is a risk of renal dysfunction.
Failure of HBV-DNA suppression was noted in one subject in the TD group at week 24, but the HBV-DNA level spontaneously decreased without further change in medication. Concomitant medications, drug compliance, and other possible factors were scrutinized; however, the cause of the transient HBV-DNA elevation could not be identified. The subject was confirmed to have maintained undetectable HBV-DNA until after the clinical trial was completed (September 2021).
Results from the analysis of the primary efficacy endpoint in the full analysis set indicated that the 97.5% upper CI for the intergroup difference in HBV-DNA suppression rate was −12.8%, which was slightly different from the results in the main analysis set (PPS analysis set). The difference appears to be due to subjects who had a week 48 visit that deviated from the visit window, as their HBV-DNA measurements (5.8% [7/120 subjects] in the TD group and 4.5% [3/67 subjects] in the TDF group) were handled as missing and were classified as the failure of HBV-DNA suppression. Additional analysis (modified full analysis set) that included subjects whose HBV-DNA levels were measured at visits outside the stipulated visit window (±14 days) indicated that the 97.5% upper CI for the intergroup difference was −7.28%, which was within the non-inferiority margin (Table 2).
An additional subgroup analysis was performed to investigate differences in therapeutic effects in patients who were HBeAg-positive at screening or had a history of other antiviral therapy before TDF. Results from this subgroup analysis indicated that antiviral suppression in these subgroups was similar to that in the overall subjects. Based on these data, the status of HBeAg-positivity and prior treatment history were not factors affecting viral suppression with TD and TDF (Table 2).
Outstanding antiviral efficacy was also demonstrated in terms of the rate of undetectable HBV-DNA (<20 IU/mL) at week 48 of treatment: 97.2% in the TD group and 100.0% in the TDF group. These results were similar to those from a similar clinical trial that evaluated the efficacy of tenofovir alafenamide (TAF) after switching from TDF.24
In this study, none of the subjects experienced HBsAg loss or seroconversion, but both groups had 13.5–19.1% of subjects who experienced HBeAg loss at week 48 of treatment. Nevertheless, no subject in the TD group and only three subjects in the TDF group formed antibodies during the study (resulting in a significant intergroup difference). Likewise, no HBeAg seroconversion was reported in a previous study on TAF after switching from TDF.24 TAF was previously compared with TDF, and it was shown that TAF was non-inferior to TDF in antiviral efficacy and improved renal as well as bone safety up to 96 weeks.25 With this result, TAF was recommended switching from TDF in patients with risk factors of renal dysfunction and decreased bone density.5 It would be worth comparing TD with TAF in the future.
Although the AE incidence was somewhat higher in the TD than in the TDF group, the AE incidence in the TD group (37.6%) was not considered high, particularly considering the AE incidence in TDF groups in previous studies: 74% (317/426 subjects) reported by Marcellin et al,26 48% (118/245 subjects) reported by Lampertico et al,24 and 66% (192/292 subjects) reported by Chan et al.19 The test drug, TD, was developed as a salt-free formulation with the removal of fumarate from TDF. The impact of the different ingredients of the two drugs on the incidence of AEs is considered minimal. No significant difference in single-dose safety between groups was shown in a Phase 1 clinical trial that demonstrated pharmacokinetic equivalence between the two drugs.15
As tenofovir is primarily excreted by the kidney, it can cause renal impairment by renal tubular damage, cell death, and mitochondrial toxicity.27,28 To evaluate renal function in the study subjects, the proportion of subjects with abnormal serum creatinine and Ccr was assessed as a safety variable. The change from baseline in Ccr at week 48 was −0.8 ± 9.8 mL/min in the TD group versus −2.4 ± 12.8 mL/min in the TDF group, which was significantly different (P=0.017, Figure 3). Therefore, TD demonstrated protective effects against renal function impairment, and hence better renal safety at the 48-week comparison. We think TD could have comparable benefits with TAF in terms of renal safety, which needs to be validated further.25
As the development of TD mainly aimed to improve medication convenience, the drug was expected to bring about greater clinical benefits than TDF, particularly in older individuals and children, by improving their medication compliance. However, the study only recruited patients with good medication compliance, and it did not investigate whether TD improved patient satisfaction in terms of medication convenience and quality of life. Consequently, it was difficult to evaluate improvements in medication compliance and clinical benefits in populations with high unmet needs. Additionally, considering the relatively short study period, a further long-term study will help collect long-term efficacy data. Safety and compliance information should also be investigated in further assessments.
Conclusion
Concluding, when patients with CHB who had confirmed antiviral effects after ≥24 weeks of TDF switched to TD or continued TDF for another 48 weeks of treatment, TD was non-inferior to TDF in terms of viral suppression. Additionally, while the two drugs showed similar favorable virological, serological, and biochemical efficacy outcomes, TD showed a protective effect against renal function impairment. TD is considered effective and safe for CHB patients.
Data Sharing Statement
The datasets generated and analyzed during the study are available from the corresponding author, Professor Young-Suk Lim ([email protected]), upon reasonable request.
Acknowledgments
The authors thank the investigators and the participants of the study.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This research received funding from Daewoong Pharmaceuticals Co., Ltd.
Disclosure
Y-S.L. and the authors received research funds from Daewoong Pharmaceuticals Co., Ltd. The authors have indicated that they have no other conflicts of interest regarding the content of this article.
References
1. Yim HJ, Lok AS. Natural history of chronic hepatitis B virus infection: what we knew in 1981 and what we know in 2005. Hepatology. 2006;43(Suppl 1):S173–181. doi:10.1002/hep.20956
2. Lee KJ, Han KH, Chun JY, et al. Natural history of chronic hepatitis type B throughout long-term follow-up. Korean J Gastroenterol. 1997;29(3):343–351.
3. Lozano R, Naghavi M, Foreman K, et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380(9859):2095–2128. doi:10.1016/S0140-6736(12)61728-0
4. Ebrahimi M, Asadi M, Akhavan O. Graphene-based nanomaterials in fighting the most challenging viruses and immunogenic disorders. ACS Biomater Sci Eng. 2022;8(1):54–81. doi:10.1021/acsbiomaterials.1c01184
5. Korean Association for the Study of the Liver. KASL clinical practice guidelines for management of chronic hepatitis B. Clin Mol Hepatol. 2019;25(2):93–159. doi:10.3350/cmh.2019.1002
6. Behzadi P, Behzadi E, Alavian SM. DNA microarray technology in HBV genotyping. Minerva Med. 2017;108(5):473–476. doi:10.23736/S0026-4806.17.05059-5
7. Behzadi P, Ranjbar R, Alavian SM. Nucleic acid-based approaches for detection of viral hepatitis. Jundishapur J Microbiol. 2015;8(1):e17449. doi:10.5812/jjm.17449
8. Calvaruso V, Craxì A. Fibrosis in chronic viral hepatitis. Best Pract Res Clin Gastroenterol. 2011;25(2):219–230. doi:10.1016/j.bpg.2011.02.012
9. Yim HJ, Kim JH, Park JY, et al. Comparison of clinical practice guidelines for the management of chronic hepatitis B: when to start, when to change, and when to stop. Clin Mol Hepatol. 2020;26(4):411–429. doi:10.3350/cmh.2020.0049
10. Liu Y, Corsa AC, Buti M, et al. No detectable resistance to tenofovir disoproxil fumarate in HBeAg+ and HBeAg- patients with chronic hepatitis B after 8 years of treatment. J Viral Hepat. 2017;24(1):68–74. doi:10.1111/jvh.12613
11. Korean Pharmaceutical Information Center. [In Korean] (VIREAD® Tablet Korean Product Manual). Available from: http://www.health.kr/images/insert_pdf/IN_2010062500026_00.pdf.
12. U.S. Food & Drug Administration. VIREAD® (tenofovir disoproxil fumarate) US package insert. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/label/2012/021356s042,022577s002lbl.pdf.
13. Snow-Lampart A, Chappell B, Curtis M, et al. No resistance to tenofovir disoproxil fumarate detected after up to 144 weeks of therapy in patients monoinfected with chronic hepatitis B virus. Hepatology. 2011;53(3):763–773. doi:10.1002/hep.24078
14. Kitrinos KM, Corsa A, Liu Y, et al. No detectable resistance to tenofovir disoproxil fumarate after 6 years of therapy in patients with chronic hepatitis B. Hepatology. 2014;59(2):434–442. doi:10.1002/hep.26686
15. Korean Pharmaceutical Information Centre. [In Korean] (Virehepa Tablet Korean Product Manual). Available from: http://www.health.kr/images/insert_pdf/IN_2017081400014_00.pdf.
16. Duarte-Rojo A, Heathcote EJ. Efficacy and safety of tenofovir disoproxil fumarate in patients with chronic hepatitis B. Therap Adv Gastroenterol. 2010;3(2):107–119. doi:10.1177/1756283X09354562
17. Gupta SK. Tenofovir-associated Fanconi syndrome: review of the FDA adverse event reporting system. AIDS Patient Care STDS. 2008;22(2):99–103. doi:10.1089/apc.2007.0052
18. Essig M, Duval X, Kaied FA, et al. Is phosphatemia the best tool to monitor renal tenofovir toxicity? J Acquir Immune Defic Syndr. 2007;46(2):256–258. doi:10.1097/QAI.0b013e3181142f31
19. Chan HLY, Fung S, Seto WK, et al. Tenofovir alafenamide versus tenofovir disoproxil fumarate for the treatment of patients with HBeAg-negative chronic hepatitis B virus infection: a randomised, double-blind, Phase 3, non-inferiority trial. Lancet Gastroenterol Hepatol. 2016;1(3):185–195. doi:10.1016/S2468-1253(16)30024-3
20. Terrault NA, Bzowej NH, Chang KM, Hwang JP, Jonas MM, Murad MH. American Association for the Study of Liver Diseases. AASLD guidelines for treatment of chronic hepatitis B. Hepatology. 2016;63(1):261–283. doi:10.1002/hep.28156
21. Kim WR, Berg T, Asselah T, et al. Evaluation of APRI and FIB-4 scoring systems for non-invasive assessment of hepatic fibrosis in chronic hepatitis B patients. J Hepatol. 2016;64(4):773–780. doi:10.1016/j.jhep.2015.11.012
22. Heathcote EJ, Marcellin P, Buti M, et al. Three-year efficacy and safety of tenofovir disoproxil fumarate treatment for chronic hepatitis B. Gastroenterology. 2011;140(1):132–143. doi:10.1053/j.gastro.2010.10.011
23. Medical Dictionary for Regulatory Activities. Introductory Guide MedDRA version 21.0; 2018. Available from: https://admin.new.meddra.org/sites/default/files/guidance/file/intguide_21_0_english.pdf.
24. Lampertico P, Buti M, Fung S, et al. Switching from tenofovir disoproxil fumarate to tenofovir alafenamide in virologically suppressed patients with chronic hepatitis B: a randomised, double-blind, phase 3, multicentre non-inferiority study. Lancet Gastroenterol Hepatol. 2020;5(5):441–453. doi:10.1016/S2468-1253(19)30421-2
25. Agarwal K, Brunetto M, Seto WK, et al. 96 weeks treatment of tenofovir alafenamide vs. tenofovir disoproxil fumarate for hepatitis B virus infection. J Hepatol. 2018;68(4):672–681. doi:10.1016/j.jhep.2017.11.039
26. Marcellin P, Heathcote EJ, Buti M, et al. Tenofovir disoproxil fumarate versus Adefovir dipivoxil for chronic hepatitis B. N Engl J Med. 2008;359(23):2442–2455. doi:10.1056/NEJMoa0802878
27. Fernandez-Fernandez B, Montoya-Ferrer A, Sanz AB, et al. Tenofovir Nephrotoxicity: 2011 update. AIDS Res Treat. 2011;2011:354908. doi:10.1155/2011/354908
28. Yang YM, Choi EJ. Renal safety of tenofovir and/or entecavir in patients with chronic HBV monoinfection. Ther Clin Risk Manag. 2017;13:1273–1285. doi:10.2147/TCRM.S143286
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.