")
Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 14
Non-Enzymatic Glycation of Transferrin and Diabetes Mellitus
Authors Ma Y, Cai J, Wang Y, Liu F , Fu S
Received 1 February 2021
Accepted for publication 10 May 2021
Published 8 June 2021 Volume 2021:14 Pages 2539—2548
DOI https://doi.org/10.2147/DMSO.S304796
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Antonio Brunetti
Yanqi Ma,1,* Jing Cai,1,* Ying Wang,1 Jingfang Liu,1,2 Songbo Fu1,2
1The First Clinical Medical College, Lanzhou University, Lanzhou, Gansu, People’s Republic of China; 2Department of Endocrinology,The First Hospital of Lanzhou University, Lanzhou, Gansu, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Jingfang Liu
Department of Endocrinology, The FirstHospital of Lanzhou University, 1 Donggang West Road, Lanzhou, Gansu, 730000, People’s Republic of China
Tel +86-931-8356242
Email [email protected]
Abstract: Diabetes is a metabolic disease characterized by high blood sugar. Its complications may damage multiple organs, such as eyes, kidneys, heart, blood vessels, and nerves, severely threatening human health. Transferrin (Tf) is a major iron transport protein in the body. Recent studies have shown that the degree of non-enzymatic glycated modification of Tf is increased in diabetic patients, and glycated Tf is closely related to the occurrence and development of diabetes and diabetic complications. However, the molecular mechanisms underlying this glycated modification in diabetes and diabetic complications are still unclear. It is speculated that the mechanism may be that glycated modification reduces the binding ability of Tf and its receptor TfR, followed by excessive iron accumulation in the body. Iron overload in the body may further lead to the death of pancreatic beta cells and insulin resistance by increasing oxidative stress, inducing iron death, interfering with the insulin signaling pathway, and causing autophagy deficiency. In addition, non-enzymatic glycation affects the binding of Tf with chromium and reduces the ability of Tf to transport chromium into tissues, resulting in a decrease in the levels of chromium in tissues and ultimately affecting the sensitivity of tissues to insulin. In diabetic patients, the concentrations of glycated Tf in serum were significantly correlated with those of fructosamine.Tf has a shorter half-life, and not affected by anemia or hypoalbuminemia and less negative charge under physiological conditions, while glycated modification could not change the isoelectric point of Tf, which easily passes through the negatively charged basement membrane of the glomerulus. Therefore, compared to glucosamine, HbA1C, etc., glycated Tf may be a future biomarker for evaluating short-term glycemic control and early renal damage in diabetic patients.
Keywords: transferrin, glycation, diabetes, diabetic complication
Introduction
Diabetes is a metabolic disease characterized by high blood sugar. The incidence of diabetes is increasing worldwide. The International Diabetes Federation estimated that the number of adults with diabetes was about 451 million in 2017 and that it would increase to 693 million in 2045.1 Diabetic complications, such as nephropathy, retinopathy, atherosclerosis, and neuropathy, threaten human health severely.2
Non-enzymatic glycation reaction, also known as “Maillard” reaction, is a spontaneous, cascading, non-enzymatic, site-specific complex reaction.3 It is different from glycosylation (enzyme-mediated conjugation) that can site-specifically link the sugar at lower temperatures. This mild modification has been used to enhance the sugar-conjugation efficiency and improve the functional properties of proteins.4
Non-enzymatic glycation of proteins sets the stage for the formation of advanced glycation end-products and the development of chronic complications of diabetes5 Transferrin (Tf) is a critical iron transport protein in the body.6,7 In recent years, with the continuous deepening of research technologies, such as proteomics and glycometabonomics, and the rapid development of related detection technologies, such as high-performance liquid chromatography/electrospray ionization-mass spectrometry (HPLC/ESI-MS), studies have shown that non-enzymatic glycated modification of Tf is closely related to the occurrence and development of diabetes.8–12 In vitro, under high sugar state, Lys 206 and Lys 534 in the structure of apo-transferrin9 or Lys103, Lys312, and Lys382 in the structure of holo-transferrin10 are most sensitive to glycation modification. The specific glycation of these sites may affect the normal structure and function of Tf. Compared to non-diabetic patients, the degree of Tf glycation in the serum of diabetic patients was increased.13 The glycation modification damages to the iron-binding capacity of Tf,11 contributing to increased oxidative stress14 and the occurrence of non-Tf-bound iron species (NTBI). The prevalence of NTBI in diabetes has been described.12
The role of iron overload has been verified in the occurrence and development of diabetes.15 The patients with hereditary hemochromatosis (HH),16 delta-beta thalassemia,17 or excessive heme iron intake18 may have higher morbidity of type 2 diabetes mellitus. The mechanism may be that the free iron accumulated in the pancreas participates in the Fenton reaction to generate reactive oxygen species (ROS), which further induces oxidative stress and pancreatic β-cell apoptosis19 and eventually affects insulin secretion. In addition, the accumulation of iron in the liver may also lead to insulin resistance.20
Clinical studies have shown that the concentrations of glycated Tf in the serum of diabetic patients were significantly positively correlated with serum fructosamine concentrations. The degree of Tf glycation and albumin was higher than that of other serum proteins, while the half-life of Tf was shorter than that of albumin. Therefore, glycated Tf has the potential to be a biological indicator reflecting the short-term glycemic control status of diabetic patients in the future.21 This study reviewed the roles and possible mechanisms of Tf non-enzymatic glycation in the occurrence and development of diabetes.
Structure and Function of Tf
Human Tf, a ferroprotein with a molecular weight of about 79 kDa, is composed of 679 amino acids. It is synthesized primarily by the liver and then released into the blood with a stable content. The serum concentration is generally 2–3 g/L.6 If the serum Tf concentration is <0.1 g/L, the chances of infection, growth retardation, and anemia increase significantly.22 The Tf molecule can be divided into two domains: the amino-terminal (N-terminal, including 336 amino acids) and the carboxy-terminal (C-terminal, including 343 amino acids). The N-terminal and C-terminal domains are composed of two small subunits with the same size, and the gap between the small subunits is the Fe3+ binding sites, which can reversibly bind Fe3+.6,7 Tf can be divided into apo-Tf, mono-Tf, and holo-Tf, according to the binding numbers of Fe3+.23
For many years, Tf has been regarded as the core of iron metabolism.7 As a powerful iron chelator, it binds iron tightly and reversibly and allows iron to circulate in the body as a soluble and non-toxic form. Also, it transports iron to the corresponding tissues and organs in a receptor-mediated pH-dependent way. The ability of Tf to transport iron is related to its special structure. As shown in Figure 1, iron (Fe3+) in food is reduced to “Fe2+” under the action of gastric acid and reducing substances, which is absorbed by intestinal mucosa cells, then the absorbed “Fe2+ ” is oxidized as “Fe3+” by ceruloplasmin, then “Fe3+” binds to Tf, and the conformation of Tf changes immediately after binding with “Fe3+”, which helps Tf to bind to the Tf receptor (TfR) on the cell membrane surface to form a “TfR- Tf-Fe3+” complex. Subsequently, the complex enters the cell through receptor-mediated endocytosis to form an endosome coated with clathrin. In the acidic milieu (pH<5.5) in the endosome, “Fe3+” dissociates from Tf and is reduced to “Fe2+” by metalloreductases such as six-transmembrane epithelial antigen of the prostate 3 (STEAP3) followed by transportation into the cytosol by divalent metal transporter 1 (DMT1). Following, the Tf/TfR complex recirculates to the cell surface, separates, and enters into the blood circulation for the next process of iron ion transport.23–25 In addition to the main function of binding and transporting iron, Tf has antibacterial activity and participates in the growth, differentiation, and cell protection.6 For example, apo-Tf may inhibit bacterial adhesion,26 remove free iron ions from the body, and protect tissues from the toxic effects of ROS. It also improves the anemia state in the animal model of beta-thalassemia.27,28
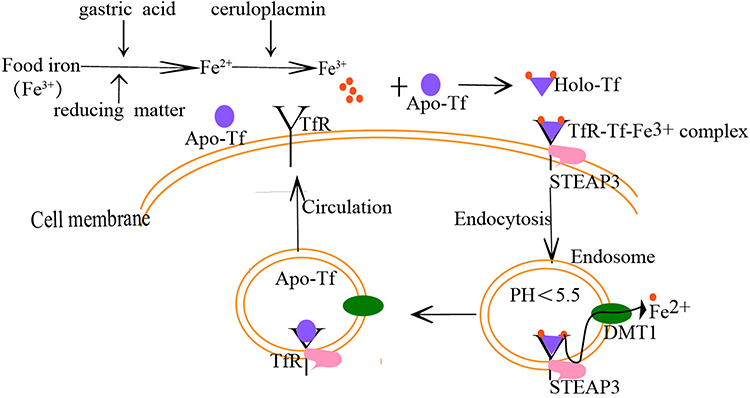 |
Figure 1 The process of Tf binding to a receptor to transport iron. The iron (Fe3+) in food is reduced to “Fe2+ ”under the action of gastric acid and reducing substances, which is absorbed by intestinal mucosa cells, the absorbed “Fe2+ ” is oxidized as “Fe3+ by ceruloplasmin, then Fe3+ binds to Tf, and the conformation of Tf changes immediately after binding with Fe3+, which helps Tf to bind to the Tf receptor (TfR) on the cell membrane surface to form a “TfR-Tf-Fe3+” complex. Subsequently, the complex enters the cell through receptor-mediated endocytosis to form an endosome coated with clathrin. In the acidic milieu (pH<5.5) in the endosome, Fe3+ dissociates from Tf and is reduced to Fe2+ by metalloreductases such as six-transmembrane epithelial antigen of the prostate 3 (STEAP3) followed by transportation into the cytosol by divalent metal transporter 1 (DMT1).Following, the Tf/TfR complex recirculates to the cell surface, separates, and enters into the blood circulation for the next process of iron ion transport. Abbreviations: Apo-Tf, non-iron bound transferrin; Holo-Tf, diferric transferrin; TfR, Tf receptor; STEAP3, six-transmembrane epithelial antigen of the prostate 3; DMT1, divalent metal transporter 1. |
Non-Enzymatic Glycated Modification of Tf and Related Diseases
Glycation is a common post-translational modification. In the early stage of the reaction, the aldehyde or ketone groups of reducing sugars (glucose, fructose, or galactose) are condensed with the ε-amino groups of lysine or hydroxylysine in the protein molecules to form an unstable Schiff base, the unstable base rearranges to form stable ketamine, known as Amadori products.
Then, Amadori products are oxidized, degraded, dehydrated, and rearranged to produce intermediate products, such as aldehydes, two-carbon compounds, and reduced ketones. Eventually, these intermediate products may be further polymerized or reacted with amino acids and nucleic acids to form a variety of compounds collectively termed as advanced glycated end-products (AGEs).3,29 The glycation process is shown in Figure 3. To date, many AGEs-modified proteins, such as hemoglobin, albumin, collagen, lens proteins,and erythrocyte membrane proteins, have been reported.30–33
The formation of AGEs is accompanied by the production of ROS, and their cross-linking properties of AGEs can impair the normal protein function3 AGEs bind to the cell surface receptors (receptor for AGE (RAGE)) to activate NF-κB or other signaling pathways that regulate transcription of a number of proteins, including E-selectin, pro-inflammatory cytokines, and tissue factors, leading to a series of pathophysiological effects,34,35 which might be the foundation for one of the diabetic pathogenesis: “metabolic memory” theory.35,36
In recent years, the liquid-mass spectrometry technology of the glycol-proteomics has shown abnormal Tf glycation in a variety of diseases. This abnormal modification may be used as a critical biological marker in the clinical diagnosis of some diseases. A controlled clinical study on patients with ovarian cancer showed differences in the glycated characteristics of serum Tf between normal individuals and patients with borderline ovarian cancer, which might be superior to CA125 in the diagnosis of borderline ovarian cancer.37 Penezić et al showed that the structure of serum Tf glycans differed between the different healthy people with varied ages and type 2 diabetes or colon cancer patients, as assessed by lectin-based protein chip technology.38 Compared to healthy controls, oral squamous cell carcinoma patients have different expression levels of N-glycotypes of several serum proteins, such as Tf, IgG1, IgG4, and haptoglobin.39 The serum profile of Tf isoforms is altered in patients with rheumatoid arthritis (RA) and RA patients treated with rituximab. Thus, we can predict that the levels of trisialylated isoforms of Tf serve as a biochemical marker of the RA activity, and the relative concentration of trisialotransferrin, tetrasialotransferrin, and pentasialotransferrin can be a useful tool for monitoring the therapy.40,41
Non-Enzymatic Glycated Tf, Diabetes, and Diabetic Complications
Diabetes, a metabolic disease characterized by hyperglycemia only when inadequately treated, is caused by various factors, such as genetics and environmental conditions. The complications may cause damage to multiple organs (eyes, kidneys, heart, blood vessels, and nerves), seriously threatening human health. Hitherto, diabetes has not been cured, and hence, early diagnosis and effective glycemic control may be necessary.
While low levels of glycation do occur at normal blood glucose concentrations, the rate is very low and so is the accumulation of glycated protein end-products. Studies showed that the degree of glycation of total plasma proteins in diabetic patients with poor blood sugar control was increased. The relative degree of glycation of different plasma proteins is a complex function of glucose concentration and the half-life chemical properties of each protein.42 Thus, effective biological markers for early diagnosis of diabetes and blood sugar monitoring through protein glycation are still under intensive research focus. Glycated hemoglobin (HbA1c) and glycated serum protein (GSP) are common indicators used clinically to evaluate the mid- and long-term glycemic control status of diabetic patients, among which the former reflects average blood glucose levels of diabetes from 4 to 8 weeks, while the latter reflects the same from 1 to 3 weeks. However, these indicators are not suitable for diabetic patients with anemia or hypoalbuminemia as they are affected by moderate-to-severe anemia and serum albumin concentration.43
A clinical cross-sectional study revealed a correlation between the ratios of AGEs/sRAGE in serum and the ratios of urine albumin/serum creatinine in type 2 diabetic patients with renovascular complications, speculating that AGEs and sRAGE may be early prediction indicators of vascular complications in uncontrolled type 2 diabetic patients.44
Tf is one of the iron transport proteins, and the correlation between its glycation modification and type 2 diabetes gained increasing attention. Soboleva et al detected the difference in glycated sites of high-content plasma proteins between type 2 diabetic patients and healthy subjects of the same age and sex using LC-MS and found six glycated sites of plasma Tf in type 2 diabetic patients.45 Golizeh et al reported that compared to normal controls,6 lysine and 1 arginine residues were modified by glycation in the serum Tf of type 2 diabetic patients, as assessed by shotgun proteomics and heavy water (2H2O)-based metabolic labeling method.8 Amadori peptides, as early glycated products, the contents in the plasma of type 2 diabetic patients were significantly higher than that of healthy individuals, and the high glucose status had varying degrees of influence on the glycated sites of human plasma proteins. It also revealed the K683 site that was modified glycation of Tf, demonstrated by MS analysis.46
The diabetic patients showed an increased degree of non-enzymatic glycation of serum Tf,13,42 and the degree of glycation increased from 1% to 2% in healthy individuals to 5% in adult diabetic patients,42 while it increased from 4% in healthy children to 11% in children with type 1 diabetes.21 Lactoferrin is a special form of Tf that exists in tears and colostrum. The levels of glycated lactoferrin in the tears of patients with diabetic retinopathy were increased,47 and the degree of N-glycated of lactoferrin in the milk of patients with gestational diabetes also increased significantly.48 The glycated modification of lactoferrin might affect the natural antibacterial activity, thereby increasing the susceptibility of people with diabetes to bacteria.49
Animal experiments have shown that not only the serum levels of glycated Tf in the diabetic rats,50 but also the levels of glycated Tf in the kidney tissues were increased significantly and were positively correlated with diabetes and blood sugar levels.51 Dextran gel electrophoresis showed that the degree of glycated albumin and glycated Tf in the serum of diabetic patients was higher than that of other proteins.52 Tf has a shorter half-life (8 days) than albumin. Therefore, glycated Tf may be a sensitive indicator for the glycemic control status of diabetic patients.21 Other studies have shown that glycated Tf is positively correlated with the concentration of fructosamine in serum and the levels of glycated albumin. Under physiological conditions, Tf is less negatively charged than albumin, and glycation fails to change the isoelectric points of Tf. Glycated Tf permeates the negatively charged glomerular basement membrane, and hence, is sensitive to the early damage of the glomerular charge barrier in patients with diabetic nephropathy.53
Molecular Mechanisms of Non-Enzymatic Glycated Tf in Diabetes and Diabetic Complications
Sustained hyperglycemia caused by diabetes contributes to the non-enzymatic glycation of Tf and the production of AGEs-modified Tf.
AGEs are a complex class of heterogeneous molecules that cause protein cross-linking. Their cross-linking properties could confer effects by directly altering the structure of proteins and disrupting their cellular functions. The glycated proteins and the gradual accumulation of AGEs reduce enzyme activity, reduce ligand binding, and change the half-life and immunogenicity of proteins.3,54
Increased AGEs in the circulation may upregulate the expression levels of RAGE. The accumulation of AGEs and the increased expression levels of their receptors play a critical role in diabetes and its complications.54 Animal models showed that AGEs and RAGEs were related to several complications of diabetes, such as nephropathy,55 retinopathy,56 and neuropathy.57 Clinical studies have also shown a correlation between serum AGEs/sRAGE ratios and urine albumin/serum creatinine ratios in type 2 diabetic patients with reno-vascular complications.44
AGEs can activate intracellular signal pathways, such as mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/ERK), phosphatidylinositol 3 kinase/protein kinase (PI3K/AKT), janus kinase/signal transducer and activator of transcription 1 (JAK2/STAT1), or NADPH oxidase/ROS after binding to RAGE, the main signal receptor on the cell membrane surface. These pathways act in synergy on the nuclear factor NF-κB to induce the production and release of pro-inflammatory cytokines (TNF-α and IL-6), oxidative stress indicators (ROS), and pro-fibrotic cytokines (TGF-β and IL-13), thereby promoting the occurrence and development of diabetes and its complications.29,54,58,59
Carboxymethyl lysine (CML) is one of the members of the AGEs family, which might interfere with the regulatory feedback mechanism of intracellular cholesterol to cause intracellular lipid accumulation and diabetic nephropathy, and AGEs inhibitor aminoguanidine hydrochloride may delay the progress of diabetic nephropathy.60
Furthermore, AEGs overload is related to the occurrence of diabetic nephropathy, probably by interfering with the autophagy-lysosomal pathway.61 AGEs also induce the nuclear accumulation of FoxO1, this in turn reduced pancreaticoduodenal homeobox-1 (Pdx-1) expression by decreasing its protein stability, ultimately contributing to a deficiency in insulin synthesis.62
Tf is a specific protein, and the effect and mechanism of its glycated modification on the occurrence and development of diabetes and its complications are yet to be clarified. The non-enzymatic glycation of Tf may hinder its function as a high-affinity iron-binding protein.9 The molecular dynamics model showed that the reduction in the iron-binding capacity of glycated apo-Tf might be related to the stereochemical effect caused by the glycation of the lysine residue in the molecule, which interfered with the conformational change required for its binding to the metal.63 As the glycated sites of holo-Tf were distal from the iron-binding site, the abnormal iron release caused by glycation could be related to the cumulative effect of the instability of the overall structure due to the destruction of the hydrogen bond in the Tf molecule and the electrostatic interaction.10
The specificity of Tf glycated modification sites under high glucose conditions might affect their normal structure and function. We observed that in vitro, after incubation of apo-Tf or holo-Tf with different concentrations of glucose, followed by HPLC/ESI-MS analysis, Lys 206 and Lys 534 sites in the structure of apo-Tf9 and Lys103, Lys312, and Lys382 sites in the structure of holo-Tf10 were most sensitive to glycation. Fujimoto et al discovered that the generation of oxygen free radicals (O2-) and hydroxyl (OH) by the glycated holo-Tf was more than that by glycated apo-Tf.64
In addition to affecting iron transport, Tf glycation may also hinder its binding to TfR. X-ray crystallography and cryoelectron microscopy mapped the Tf c-lobe region that interacts with TfR to the amino acid sequence residues 349–378 of Tf, and the residues K354, K365, and K380 located at or near these regions and glycated undergo conformational changes, destroying the binding between Tf and TfR.10
Tf glycation may weaken its affinity to TfR. In addition, TfR glycation under high sugar conditions may affect the interaction between TfR and Tf. Georgieff et al65 demonstrated that TfR isolated from the placenta of diabetic patients had a higher molecular weight than that of non-diabetics; this difference could be attributed to the occurrence of glycation of N-chain oligosaccharides in the TfR of placenta in the hyperglycemia state. This glycated modification of TfR might change its three-dimensional structure or charge, thereby reducing its binding affinity to Tf.
The above effects led to the continued existence of NTBI in serum. Iron is an indispensable element for maintaining the normal function of pancreatic β-cells and glucose metabolism. However, iron overload may also cause a series of toxic effects. Additional studies confirmed the correlations between iron metabolism and diabetes. Iron metabolism affects sugar metabolism and vice versa; these phenomena are mediated by oxidative stress and inflammatory factors.66
The mechanism of iron overload leading to diabetes and its complications could be that free iron deposits in various cells, such as the pancreas and liver, generate some O2- and OH by participating in the Fenton and Haber–Weiss reactions. These O2- and OH further damage intracellular proteins, lipids, and nucleic acids, resulting in abnormal mitochondrial function, leading to cytotoxic damage and apoptosis.67 Moreover, pancreatic β cells are extremely sensitive to oxidative stress, ultimately leading to insulin resistance and decreased secretion.68,69 In addition, iron-chelating agents reduce ferritin levels and increase insulin sensitivity in diabetic patients.70 The iron-overloaded diabetic rat model showed that iron aggravates diabetic kidney damage by promoting oxidative stress and reducing the body’s antioxidant capacity.71
Abundant iron is deposited in the liver. It interferes with the production of liver glycogen, reduces the uptake of insulin in liver, and interferes with insulin signal transduction, eventually leading to peripheral hyperinsulinemia.72
The chronic iron overload may cause autophagy defects by inhibiting the mTOR complex 1 (mTORC1)-UVRAG and could be a novel mechanism for insulin resistance; however, removing excess iron in the body eliminates abnormal autophagosomes in the body and increases insulin sensitivity. This phenomenon is consistent with the clinical use of iron chelator therapy to increase insulin sensitivity, thereby alleviating the progression of type 2 diabetes.73 In addition, studies have shown that glycation of insulin (INS) itself as well as insulin receptor (IR), insulin-like growth hormones and their receptors, etc., contribute to INS resistance, and may provide a mechanism for the development of INS resistance in diabetes.74–77 Therefore, glycation of these proteins including Tf may play important roles in diabetes pathogenesis.
Iron overload is a specific death method different from apoptosis, cell necrosis, and autophagy, called “iron death.” Recently, Li et al detected the characteristic changes of “iron death” in the kidneys of diabetic mouse models and human proximal tubular epithelial cells (HK-2) cultured with high glucose and iron overload. The weakened antioxidant capacity, large amounts of deposition of ROS and lipid peroxidation, and decreased expression of NFE2-related factor 2 (Nrf2) were observed in diabetic nephropathy models. Fenofibrate inhibits diabetes-related iron death by upregulating the expression of Nrf2 and delays the progression of nephropathy.78 Quercetin may have a beneficial effect on type 2 diabetic patients by inhibiting iron deposition in the pancreas and effectuating iron death of pancreatic β-cells.79
Clinical studies have shown that serum iron metabolism indexes (iron, ferritin, and transferrin saturation) and insulin resistance index (HOMA-IR) in patients with gestational diabetes were higher than in the control group, and regression analysis indicated that serum ferritin and Tf saturation were significantly positively correlated with HOMA-IR index.80 A longitudinal and retrospective study encompassing 111 type 2 diabetic patients with kidney diseases showed that the iron accumulation confirmed by biopsy in renal tubular epithelial cells of patients with diabetic nephropathy was higher than that in the control group.81 Therefore, the iron overload caused by the non-enzymatic glycated Tf under high-glucose state may be closely related to the occurrence and development of diabetes and its complications.
Chromium improves the sensitivity of tissues to insulin in animal models of obesity, insulin resistance, or diabetes; however, the premise of this effect is chromium transportation to insulin-sensitive tissues, wherein Tf plays a critical role. Animal experiments have shown that non-enzymatic glycated modification under high glucose conditions alters the conformation of Tf and affects its binding to chromium, thereby reducing the ability of Tf to transport chromium into tissues.82
From what has been discussed above, the degree of non-enzymatic glycated modification of Tf is increased in diabetic patients, but the roles and mechanism of this glycation in diabetes and diabetic complications are yet to be clarified. Herein, we speculated that the mechanism might be that glycated modification reduces the binding ability of Tf and its receptor TfR, resulting in excessive iron accumulation in the body. The iron overload in the body may further lead to the death of pancreatic beta cells and insulin resistance by increasing oxidative stress, inducing iron death, interfering with the insulin signaling pathway, and causing autophagy deficiency. In addition, non-enzymatic glycation affects the binding of Tf with chromium, reducing the ability of Tf to transport chromium into tissues, resulting in a decrease in the levels of chromium in tissues and ultimately affecting the sensitivity of tissues to insulin. The roles and mechanism of non-enzymatic glycated Tf in diabetes mellitus and the related complications are summarized in Figure 2.
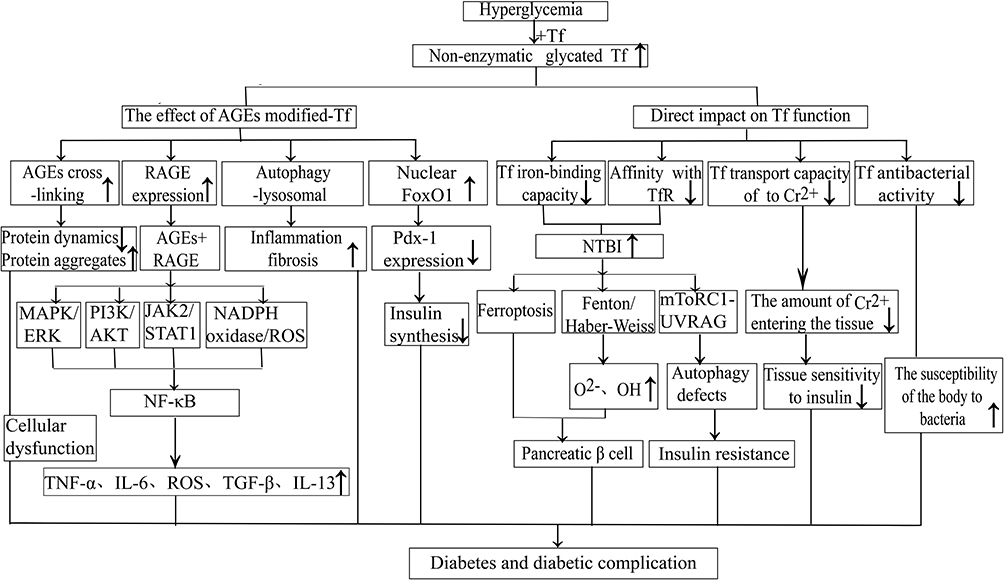 |
Figure 2 The roles and mechanism of non-enzymatic glycated Tf in diabetes and its complications. Abbreviations: Tf, transferrin; AGEs, advanced glycated end-products; RAGE, AGEs receptor; TfR, Tf receptor; Pdx-1, pancreaticoduodenal homeobox-1; NTBI, non-Tf-bound iron species; ROS, reactive oxygen species; MAPK/ERK, mitogen-activated protein kinase/extracellular signal-regulated kinase; PI3K/AKT, phosphatidylinositol 3 kinase/protein kinase; JAK2/STAT1, janus kinase/signal transducer and activator of transcription 1; mTORC1-UVRAG, mTOR complex 1-UVRAG-2−oxygen free radicals; OH, hydroxyl; TNF-α, tumor necrosis factor-α; IL-6, interleukin-6; TGF-β, transforming growth factor-β; IL-13, interleukin-13; |
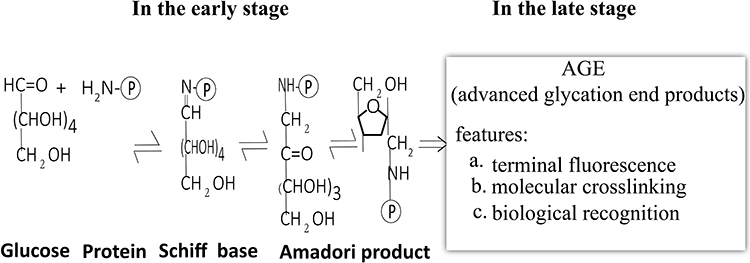 |
Figure 3 The non-enzymatic glycation process. |
In conclusion, in diabetic patients, the concentrations of glycated Tf in serum were significantly correlated with fructosamine concentrations. Tf has a shorter half-life, and is not affected by anemia or hypoalbuminemia, and less of a negative charge under physiological conditions, while glycation could not change the isoelectric point of Tf. In addition, Tf passes through the negatively charged basement membrane of the glomerulus easily, and therefore, compared to glucosamine, HbA1C, etc., glycated Tf may be a promising biomarker for evaluating short-term glycemic control and early renal damage in diabetic patients in the future. However, there are still relatively few studies on the mechanism of glycation of Tf in diabetes mellitus and its complications, and the monitoring technology is relatively complex. Therefore, its clinical application in the diagnosis and treatment of diabetes and its complications still needs more verification.
So far, there is no definite method to reduce the glycation of protein. Studies have shown that the supplementation of vitamin C can decrease the glycation degree of insulin and ameliorate aspects of the obesity-diabetes syndrome in ob/ob mice.83 In vivo and in vitro studies also showed that L-lysine could significantly improve the structure and function of glycated lysozyme in type 2 diabetes, thereby reducing the risk of infection in these patients.84 However, it still needs a lot of studies to confirm whether these substances may reduce the glycation of Tf.
Acknowledgments
The authors gratefully acknowledge the financial support from the National Natural Science Foundation of China (No.81960155) and the Health industry scientific research project of Gansu Province (No.GSWSKY-2019-07).
Disclosure
The authors declare that they have no competing interests.
References
1. Cho NH, Shaw JE, Karuranga S, et al. IDF Diabetes Atlas: global estimates of diabetes prevalence for 2017 and projections for 2045. Diabetes Res Clin Pract. 2018;138:271–281.
2. Diagnosis and classification of diabetes mellitus. Diabetes Care. 2013;36 Suppl 1(Suppl1):S67–74.
3. Younus H, Anwar S. Prevention of non-enzymatic glycosylation (glycation): implication in the treatment of diabetic complication. Int J Health Sci (Qassim). 2016;10(2):261–277.
4. Yuan F, Ahmed I, Lv L, et al. Impacts of glycation and transglutaminase-catalyzed glycosylation with glucosamine on the conformational structure and allergenicity of bovine β-lactoglobulin. Food Funct. 2018;9(7):3944–3955.
5. Zhang Q, Monroe ME, Schepmoes AA, et al. Comprehensive identification of glycated peptides and their glycation motifs in plasma and erythrocytes of control and diabetic subjects. J Proteome Res. 2011;10(7):3076–3088.
6. Gomme PT, McCann KB, Bertolini J. Transferrin: structure, function and potential therapeutic actions. Drug Discov Today. 2005;10(4):267–273.
7. Gkouvatsos K, Papanikolaou G, Pantopoulos K. Regulation of iron transport and the role of transferrin. Biochim Biophys Acta. 2012;1820(3):188–202.
8. Golizeh M, Lee K, Ilchenko S, et al. Increased serotransferrin and ceruloplasmin turnover in diet-controlled patients with type 2 diabetes. Free Radic Biol Med. 2017;113:461–469.
9. Silva AM, Sousa PR, Coimbra JT, et al. The glycation site specificity of human serum transferrin is a determinant for transferrin’s functional impairment under elevated glycaemic conditions. Biochem J. 2014;461(1):33–42.
10. Silva AMN, Coimbra JTS, Castro MM, et al. Determining the glycation site specificity of human holo-transferrin. J Inorg Biochem. 2018;186:95–102.
11. Van Campenhout A, Van Campenhout C, Lagrou AR, Manuel YKB. Effects of in vitro glycation on Fe3+ binding and Fe3+ isoforms of transferrin. Clin Chem. 2004;50(9):1640–1649.
12. Lee DH, Liu DY, Jacobs DR
13. Van Campenhout A, Van Campenhout C, Olyslager YS, Van Damme O, Lagrou AR, Manuel-y-Keenoy B. A novel method to quantify in vivo transferrin glycation: applications in diabetes mellitus. Clin Chim Acta. 2006;370(1–2):115–123.
14. Van Campenhout A, Van Campenhout C, Lagrou AR, Moorkens G, De Block C, Manuel-y-Keenoy B. Iron-binding antioxidant capacity is impaired in diabetes mellitus. Free Radic Biol Med. 2006;40(10):1749–1755.
15. Kunutsor SK, Apekey TA, Walley J, Kain K. Ferritin levels and risk of type 2 diabetes mellitus: an updated systematic review and meta-analysis of prospective evidence. Diabetes Metab Res Rev. 2013;29(4):308–318.
16. Crownover BK, Covey CJ. Hereditary hemochromatosis. Am Fam Physician. 2013;87(3):183–190.
17. Rattanaporn P, Tongsima S, Mandrup-Poulsen T, Svasti S, Tanyong D. Combination of ferric ammonium citrate with cytokines involved in apoptosis and insulin secretion of human pancreatic beta cells related to diabetes in thalassemia. PeerJ. 2020;8:e9298.
18. Bao W, Rong Y, Rong S, Liu L. Dietary iron intake, body iron stores, and the risk of type 2 diabetes: a systematic review and meta-analysis. BMC Med. 2012;10:119.
19. Hansen JB, Moen IW, Mandrup-Poulsen T. Iron: the hard player in diabetes pathophysiology. Acta Physiol (Oxf). 2014;210(4):717–732.
20. Varghese J, James J, Vaulont S, McKie A, Jacob M. Increased intracellular iron in mouse primary hepatocytes in vitro causes activation of the Akt pathway but decreases its response to insulin. Biochim Biophys Acta Gen Subj. 2018;1862(9):1870–1882.
21. Kemp SF, Creech RH, Horn TR. Glycosylated albumin and transferrin: short-term markers of blood glucose control. J Pediatr. 1984;105(3):394–398.
22. Hayashi A, Wada Y, Suzuki T, Shimizu A. Studies on familial hypotransferrinemia: unique clinical course and molecular pathology. Am J Hum Genet. 1993;53(1):201–213.
23. Kawabata H. Transferrin and transferrin receptors update. Free Radic Biol Med. 2019;133:46–54.
24. Luck AN, Mason AB. Transferrin-mediated cellular iron delivery. Curr Top Membr. 2012;69:3–35.
25. Gao G, Li J, Zhang Y, Chang YZ. Cellular iron metabolism and regulation. Adv Exp Med Biol. 2019;1173:21–32.
26. Ardehali R, Shi L, Janatova J, Mohammad SF, Burns GL. The inhibitory activity of serum to prevent bacterial adhesion is mainly due to apo-transferrin. J Biomed Mater Res A. 2003;66(1):21–28.
27. Li H, Rybicki AC, Suzuka SM, et al. Transferrin therapy ameliorates disease in beta-thalassemic mice. Nat Med. 2010;16(2):177–182.
28. Li H, Choesang T, Bao W, et al. Decreasing TfR1 expression reverses anemia and hepcidin suppression in β-thalassemic mice. Blood. 2017;129(11):1514–1526.
29. Shen CY, Lu CH, Wu CH, et al. The development of Maillard reaction, and Advanced Glycation End Product (AGE)-Receptor for AGE (RAGE) signaling inhibitors as novel therapeutic strategies for patients with age-related diseases. Molecules. 2020;25:23.
30. Vetter SW. Glycated Serum Albumin and AGE Receptors. Adv Clin Chem. 2015;72:205–275.
31. Bernstein RE. Nonenzymatically glycosylated proteins. Adv Clin Chem. 1987;26:1–78.
32. Lee JH, Shin DH, Lupovitch A, Shi DX. Glycosylation of lens proteins in senile cataract and diabetes mellitus. Biochem Biophys Res Commun. 1984;123(3):888–893.
33. Miller JA, Gravallese E, Bunn HF. Nonenzymatic glycosylation of erythrocyte membrane proteins. Relevance to diabetes. J Clin Invest. 1980;65(4):896–901.
34. Tang SC, Chan LY, Leung JC, et al. Differential effects of advanced glycation end-products on renal tubular cell inflammation. Nephrology (Carlton). 2011;16(4):417–425.
35. Ahmed N. Advanced glycation end products–role in pathology of diabetic complications. Diabetes Res Clin Pract. 2005;67(1):3–21.
36. Testa R, Bonfigli AR, Prattichizzo F, La Sala L, De Nigris V, Ceriello A. The “Metabolic memory” theory and the early treatment of hyperglycemia in prevention of diabetic complications. Nutrients. 2017;9:5.
37. O’Flaherty R, Muniyappa M, Walsh I, et al. A Robust and Versatile Automated Glycoanalytical Technology for Serum Antibodies and Acute Phase Proteins: ovarian Cancer Case Study. Mol Cell Proteomics. 2019;18(11):2191–2206.
38. Penezić A, Križakova M, Miljuš G, Katrlik J, Nedić O. Diagnostic Potential of Transferrin Glycoforms-A Lectin-Based Protein Microarray Approach. Proteomics Clin Appl. 2019;13(5):e1800185.
39. Saraswat M, Mäkitie A, Tohmola T, et al. Tongue cancer patients can be distinguished from healthy controls by specific N-glycopeptides found in serum. Proteomics Clin Appl. 2018;12(6):e1800061.
40. Gudowska M, Gruszewska E, Wrona A, et al. The profile of serum transferrin isoforms in rheumatoid arthritis. J Clin Rheumatol. 2019;25(4):159–162.
41. Cylwik B, Gruszewska E, Gindzienska-Sieskiewicz E, Kowal-Bielecka O, Chrostek L. Serum profile of transferrin isoforms in rheumatoid arthritis treated with biological drugs. Clin Biochem. 2019;74:31–35.
42. Austin GE, Mullins RH, Morin LG. Non-enzymic glycation of individual plasma proteins in normoglycemic and hyperglycemic patients. Clin Chem. 1987;33(12):2220–2224.
43. Suo M, Wen D, Wang W, Zhang T. Comparative study on hemoglobin A1c, glycated albumin and glycosylated serum protein in aplastic anemia patients with Type 2 diabetes mellitus. Biosci Rep. 2020;40:5.
44. Farhan SS, Hussain SA. Advanced glycation end products (AGEs) and their soluble receptors (sRAGE) as early predictors of reno-vascular complications in patients with uncontrolled type 2 diabetes mellitus. Diabetes Metab Syndr. 2019;13(4):2457–2461.
45. Soboleva A, Mavropulo-Stolyarenko G, Karonova T, et al. Multiple glycation sites in blood plasma proteins as an integrated biomarker of Type 2 diabetes mellitus. Int J Mol Sci. 2019;20:9.
46. Frolov A, Blüher M, Hoffmann R. Glycation sites of human plasma proteins are affected to different extents by hyperglycemic conditions in type 2 diabetes mellitus. Anal Bioanal Chem. 2014;406(24):5755–5763.
47. Zhao Z, Liu J, Shi B, He S, Yao X, Willcox MD. Advanced glycation end product (AGE) modified proteins in tears of diabetic patients. Mol Vis. 2010;16:1576–1584.
48. Smilowitz JT, Totten SM, Huang J, et al. Human milk secretory immunoglobulin a and lactoferrin N-glycans are altered in women with gestational diabetes mellitus. J Nutr. 2013;143(12):1906–1912.
49. Li YM. Glycation ligand binding motif in lactoferrin. Implications in diabetic infection. Adv Exp Med Biol. 1998;443:57–63.
50. Liu J, Cheng J, Tang X, Fu S, Ma L, Tian Y. Puerarin reduces the levels of AGE-modified proteins in serum and retinal tissues to improve the retinal damage in diabetic rats. Lat.Am.J.Pharm. 2019;38(2):396–405.
51. Liu J, Wang L, Tang X, Fu S, Tian Y, Ma L. Analysis of differentially expressed advanced glycation end product-modified proteins in diabetic rat kidney. Int J Diabetes Dev Ctries. 2018;38:417–423.
52. Kumari K, Bansal V, Jagmohan ACG, Rastogi AK, Sahib MK. Retrospective glycemic status of diabetic patients: glycosylation of blood proteins in diabetes and chronic renal failure. Acta Diabetol Lat. 1987;24(2):91–99.
53. Kunika K, Yamaoka T, Itakura M. Damage of charge-dependent renal tubular reabsorption causes diabetic microproteinuria. Diabetes Res Clin Pract. 1997;36(1):1–9.
54. Sanajou D, Ghorbani Haghjo A, Argani H, Aslani S. AGE-RAGE axis blockade in diabetic nephropathy: current status and future directions. Eur J Pharmacol. 2018;833:158–164.
55. Xia X, Mao D, Dai H, et al. Effect of Cyclocarya paliurus polysaccharides on streptozotocin-induced diabetic nephropathy in rats. J Tradit Chin Med. 2020;40(6):956–964.
56. Wu Q, Liu H, Zhou M. Fangchinoline Ameliorates Diabetic retinopathy by inhibiting Receptor for Advanced Glycation End-Products (RAGE)-Nuclear Factor Kappa-Light-Chain-Enhancer of Activated B Cells (NF-κB) pathway in Streptozotocin (STZ)-induced diabetic rats. Med Sci Monit. 2019;25:1113–1121.
57. Momeni Z, Bautista M, Neapetung J, et al. RAGE signaling is required for AMPA receptor dysfunction in the hippocampus of hyperglycemic mice. Physiol Behav. 2021;229:113255.
58. Walke PB, Bansode SB, More NP, Chaurasiya AH, Joshi RS, Kulkarni MJ. Molecular investigation of glycated insulin-induced insulin resistance via insulin signaling and AGE-RAGE axis. Biochim Biophys Acta Mol Basis Dis. 2021;1867(2):166029.
59. Yamagishi SI. Role of Advanced Glycation Endproduct (AGE)-Receptor for Advanced Glycation Endproduct (RAGE) Axis in Cardiovascular Disease and Its Therapeutic Intervention. Circ J. 2019;83(9):1822–1828.
60. Yuan Y, Sun H, Sun Z. Advanced glycation end products (AGEs) increase renal lipid accumulation: a pathogenic factor of diabetic nephropathy (DN). Lipids Health Dis. 2017;16(1):126.
61. Takahashi A, Takabatake Y, Kimura T, et al. Autophagy inhibits the accumulation of advanced glycation end products by promoting lysosomal biogenesis and function in the kidney proximal tubules. Diabetes. 2017;66(5):1359–1372.
62. Shu T, Zhu Y, Wang H, Lin Y, Ma Z, Han X. AGEs decrease insulin synthesis in pancreatic β-cell by repressing Pdx-1 protein expression at the post-translational level. PLoS One. 2011;6(4):e18782.
63. Ghanbari Z, Housaindokht MR, Bozorgmehr MR, Izadyar M. The effect of glycosylation on the transferrin structure: a molecular dynamic simulation analysis. J Theor Biol. 2016;404:73–81.
64. Fujimoto S, Kawakami N, Ohara A. Nonenzymatic glycation of transferrin: decrease of iron-binding capacity and increase of oxygen radical production. Biol Pharm Bull. 1995;18(3):396–400.
65. Georgieff MK, Petry CD, Mills MM, McKay H, Wobken JD. Increased N-glycosylation and reduced transferrin-binding capacity of transferrin receptor isolated from placentae of diabetic women. Placenta. 1997;18(7):563–568.
66. Fernández-Real JM, López-Bermejo A, Ricart W. Cross-talk between iron metabolism and diabetes. Diabetes. 2002;51(8):2348–2354.
67. Simcox JA, McClain DA. Iron and diabetes risk. Cell Metab. 2013;17(3):329–341.
68. Fernández-Real JM, McClain D, Manco M. Mechanisms linking glucose homeostasis and iron metabolism toward the onset and progression of Type 2 diabetes. Diabetes Care. 2015;38(11):2169–2176.
69. Shu T, Lv Z, Xie Y, Tang J, Mao X. Hepcidin as a key iron regulator mediates glucotoxicity-induced pancreatic β-cell dysfunction. Endocr Connect. 2019;8(3):150–161.
70. Chuansumrit A, Pengpis P, Mahachoklertwattana P, et al. Effect of iron chelation therapy on glucose metabolism in non-transfusion-dependent thalassaemia. Acta Haematol. 2017;137(1):20–26.
71. Gao W, Li X, Gao Z, Li H. Iron increases diabetes-induced kidney injury and oxidative stress in rats. Biol Trace Elem Res. 2014;160(3):368–375.
72. Niederau C, Berger M, Stremmel W, et al. Hyperinsulinaemia in non-cirrhotic haemochromatosis: impaired hepatic insulin degradation? Diabetologia. 1984;26(6):441–444.
73. Jahng JWS, Alsaadi RM, Palanivel R, et al. Iron overload inhibits late stage autophagic flux leading to insulin resistance. EMBO Rep. 2019;20(10):e47911.
74. Farah MA, Bose S, Lee JH, Jung HC, Kim Y. Analysis of glycated insulin by MALDI-TOF mass spectrometry. Biochim Biophys Acta. 2005;1725(3):269–282.
75. Abdel-Wahab YH, O’Harte FP, Boyd AC, Barnett CR, Flatt PR. Glycation of insulin results in reduced biological activity in mice. Acta Diabetol. 1997;34(4):265–270.
76. Hunter SJ, Boyd AC, O’Harte FP, et al. Demonstration of glycated insulin in human diabetic plasma and decreased biological activity assessed by euglycemic-hyperinsulinemic clamp technique in humans. Diabetes. 2003;52(2):492–498.
77. Rhinesmith T, Turkette T, Root-Bernstein R. Rapid Non-enzymatic glycation of the insulin receptor under hyperglycemic conditions inhibits insulin binding in vitro: implications for insulin resistance. Int J Mol Sci. 2017;18:12.
78. Li S, Zheng L, Zhang J, Liu X, Wu Z. Inhibition of ferroptosis by up-regulating Nrf2 delayed the progression of diabetic nephropathy. Free Radic Biol Med. 2021;162:435–449.
79. Li D, Jiang C, Mei G, et al. Quercetin Alleviates Ferroptosis of Pancreatic β Cells in Type 2 Diabetes. Nutrients. 2020;12:10.
80. Feng Y, Feng Q, Lv Y, Song X, Qu H, Chen Y. The relationship between iron metabolism, stress hormones, and insulin resistance in gestational diabetes mellitus. Nutr Diabetes. 2020;10(1):17.
81. Zhao L, Zou Y, Zhang J, et al. Serum transferrin predicts end-stage Renal Disease in Type 2 diabetes mellitus patients. Int J Med Sci. 2020;17(14):2113–2124.
82. Deng G, Dyroff SL, Lockart M, Bowman MK, Vincent JB. The effects of the glycation of transferrin on chromium binding and the transport and distribution of chromium in vivo. J Inorg Biochem. 2016;164:26–33.
83. Abdel-Wahab YH, O’Harte FP, Mooney MH, Barnett CR, Flatt PR. Vitamin C supplementation decreases insulin glycation and improves glucose homeostasis in obese hyperglycemic (ob/ob) mice. Metabolism. 2002;51(4):514–517.
84. Mirmiranpour H, Khaghani S, Bathaie SZ, et al. The preventive effect of l-lysine on lysozyme glycation in Type 2 diabetes. Acta Med Iran. 2016;54(1):24–31.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.