")
Back to Journals » Journal of Inflammation Research » Volume 13
NLRP3 Regulated CXCL12 Expression in Acute Neutrophilic Lung Injury
Authors Peng Y , Wu Q, Tang H, Chen J, Wu Q, Yuan X, Xiong S , Ye Y, Lv H
Received 30 April 2020
Accepted for publication 2 July 2020
Published 23 July 2020 Volume 2020:13 Pages 377—386
DOI https://doi.org/10.2147/JIR.S259633
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Yanwen Peng,1 Qiongli Wu,2 Hao Tang,3 Jingrou Chen,1 Qili Wu,1 Xiaofeng Yuan,4 Shiqiu Xiong,5 Yujin Ye,6 Haijin Lv7
1The Biotherapy Center, The Third Affiliated Hospital, Sun Yat-sen University, Guangzhou 510630, People’s Republic of China; 2Department of Immunology, Zhongshan School of Medicine, Sun Yat-sen University, Guangzhou 510080, People’s Republic of China; 3Department of General Practice, The First Affiliated Hospital, Sun Yat-sen University, Guangzhou 510080, People’s Republic of China; 4The General Intensive Care Unit, The Third Affiliated Hospital, Sun Yat-sen University, Guangzhou 510630, People’s Republic of China; 5Cell Biology Group, National Measurement Lab, LGC Fordham, Cambridgeshire CB7 5WW, UK; 6Department of Rheumatology, The First Affiliated Hospital, Sun Yat-sen University, Guangzhou 510080, People’s Republic of China; 7The Surgical and Transplant Intensive Care Unit, The Third Affiliated Hospital, Sun Yat-sen University, Guangzhou 510630, People’s Republic of China
Correspondence: Haijin Lv; Yanwen Peng Email [email protected]; [email protected]
Background and Purpose: Both NLRP3 inflammasome and chemokines are involved in the initiation and development of acute lung inflammation, but the underlying mechanism is still elusive. The present study investigated the role of chemokines and NLRP3 in recruiting neutrophils in the early phase of acute lung injury.
Methods: In an endotoxin (lipopolysaccharide [LPS])-induced acute lung injury model, we measured the lung injury severity, myeloperoxidase (MPO) activity and chemokine profiles in wild-type (WT) and NLRP3 knockout (NLRP3–/–) mice, and then identified the key chemokines by specific antibody blockage.
Results: The results showed that NLRP3 deficiency was associated with alleviating lung damage, by reducing alveolar epithelial cell apoptosis and decreasing neutrophil accumulation. Furthermore, compared with WT mice, IL-1β, CCL2, CXCL1, CXCL5 and CXCL12 levels from the serum of NLRP3–/– mice were much lower after exposure to LPS. However, in lung tissue, only lower CXCL12 levels were observed from the NLRP3–/– ALI mice, and higher levels of CXCR4 were expressed in NLRP3–/– neutrophils. Blockage of CXCL12 dramatically relieved the severity of ALI and reduced neutrophil accumulation in the lung.
Conclusion: NLRP3 alters CXCL12 expression in acute lung injury. CXCL12 is crucial for neutrophil recruitment in NLRP3-mediated neutrophilic lung injury.
Keywords: NLRP3, acute lung injury, ALI, neutrophils, chemokines, CXCL12
Introduction
Acute respiratory distress syndrome (ARDS) or acute lung injury (ALI) is a devastating condition and contributes to considerable mortality in critically ill patients. Bacterial endotoxins, such as lipopolysaccharide (LPS), induce a severe systemic inflammatory response, which disrupts the alveolar endothelium with a subsequent cellular and fluid influx, and epithelial destruction is the hallmark of septic ARDS.1 Owing to the elusive molecular and cellular mechanisms of ARDS, the clinical strategy is largely unresolved.
The innate immune system plays an integral role in the pathophysiology of lung homeostasis.2 Neutrophils are the key effector cells for host defense against microbial invasion. In sepsis-induced ALI, neutrophils are recruited and primed in the injury site by insults released from activated residential cells in circulation,3 which subsequently clear the pathogen by efferocytosis, NETosis and proteinases.4,5 However, the excessive accumulation of primed neutrophils contributes to uncontrolled inflammation and extreme lung damage, which are correlated with poor clinical outcome in ARDS.6–8 In animal models, the depletion of neutrophils reduces the severity of lung injury.9 A promising therapeutic target would be to inhibit neutrophil recruitment, rather than eliminating them, in ALI.4
In ARDS, chemokine levels are elevated in the lungs and correlate with outcome.10,11 Neutralization of chemokines or their receptors has been shown to attenuate injury in animal models of ALI.12 Several chemokines and receptors are involved in neutrophil recruitment or migration.3,11–13 CCL2 and CCL7 are elevated in bronchoalveolar lavage fluid (BALF) and synergize with CXCL8, promoting human neutrophil migration in ARDS.10 CXCR2 and its ligands CXCL1 and CXCL5 are critical for neutrophil trafficking in ALI.14,15 The CXCL12/CXCR4 axis has been found to be involved in neutrophilic inflammation.16,17 Inhibition of CXCR4 decreases the transendothelial and transepithelial migration of neutrophils in pulmonary inflammation.18 However, its role in ALI is still unclear.
Inflammasomes were initially described in 2002, in relation to several inflammatory disorders. Accumulating data suggest that NLRP3 inflammasome contributes to the pathogenesis of ALI.19 NLRP3 inflammasome mediates neutrophil recruitment to the sterile injured lung.20 Inhibition or depletion of NLRP3 attenuates airway neutrophilic inflammation.20–22 Chemokines are also the primers of inflammasomes. CXCL1 and CXCL2 regulate NLRP3 activation in macrophages.23 However, the role of chemokines in NLRP3-mediated neutrophil recruitment to the injury site is still unknown.
In this study, we investigated the critical role of NLRP3 in the pathogenesis of LPS-induced ALI and explored the profiles of candidate cytokines and chemokines in the lungs and serum of two genotypic mice. Among all candidates, only CXCL12 blockage could dramatically alleviate the symptoms of ALI and reduce the infiltration of neutrophils into the lung. This suggests that CXCL12 is one of the main drivers in neutrophil recruitment during lung injury.
Materials and Methods
Animals
Eight-to-twelve-week-old male C57BL/6 mice were purchased from the Nanjing Model Experimental Animal Center and maintained under specific pathogen-free conditions at Sun Yat-sen University. NLRP3 knockout mice (B6.129S6-Nlrp3tm1Bhk/J) were obtained from Jackson Laboratory (Maine, USA), and housed and bred in Guangdong Laboratory Animals Monitoring Institute. Age- and weight-matched mice were used for all mouse-related experiments.
Acute Lung Injury (ALI) Model
NLRP3 knockout (NLRP3–/–) or wild-type (WT) (C57BL/6) mice in the model group received 10 mg/kg of 200 μL LPS (E. coli O111:B4; Sigma-Aldrich) via intraperitoneal injection to induce ALI.9 The control mice were administered with 200 μL PBS. After 6 or 12 hours, mice were anesthetized. Retro-orbital blood and BALF were collected, and the left lungs were resected. Samples were stored for biochemical, protein or nucleus assays. For histological analysis, blood cells were washed out from the right ventricle by PBS perfusion before lung harvest. The Animal Care and Use Committee of the Third Affiliated Hospital of Sun Yat-sen University approved the experimental procedure in this study (no. 00129136) according to the NIH Guide for the Care and Use of Laboratory Animals.
Bronchoalveolar Lavage and Neutrophil Counts in BALF
After deep sedation and tracheostomy, a 30-gauge needle was inserted into the mouse trachea and BALF was collected by 0.5 mL 4°C PBS lavage three times. Cells were precipitated by centrifugation at 300 g for 5 minutes, then resuspended in 1 mL PBS. The total cell number in BALF was counted and the neutrophil proportion were identified by Wright–Giemsa stain. The neutrophil counts were calculated as total cell number × proportion.
Lung Injury Score
Murine lungs were immersed in paraformaldehyde for more than 24 hours. Paraffin slides were prepared and stained with hematoxylin and eosin (HE) for histological and morphometric analysis. Lung injury scores were acquired as previously described.24 In brief, five random high-power fieldsfor each slide were chosen for the calculation. Each typical pathological change was assigned a score of 0, 1or 2: neutrophil counts in the interstitial (A) or alveolar spaces (B), 0, 1–5, >5; presenting with hyaline membranes (C) or proteinaceous debris filling the airspaces (D), 0.1, >1; compared with the normal alveolar septum, increased thicknesses of <2×, 2–4×, >4×. The lung injury score varied from 0 to 1, based on the formula: Score=[(20×A)+(14×B)+(7×C)+(7×D)+(2×E)]/(number of fields×100).
Evans Blue Assay
Vascular permeability was evaluated using Evans blue dye, as previously described.24 In brief, mice were intravenously injected with 0.5% Evans blue dye (Sigma-Aldrich) solution. Thirty minutes later, fluorescence of formamide-treated lung was determined by a spectrometer, at excitation=620 nm and emission=680 nm. The results were referred to a standard curve and expressed as the number of micrograms of Evans blue dye per gram of organ tissue (wet weight).
Quantitative Reverse Transcription–Polymerase Chain Reaction (qRT-PCR)
Lung tissue was homogenized and dissolved in Trizol (Invitrogen, Carlsbad, CA, USA) to extract RNA. The concentration of RNA was measured by a Nanodrop 2000 spectrophotometer (Thermo Fisher, USA). Reverse transcription (RT) was performed using a cDNA Synthesis Supermix (Novoprotein, USA). The cDNA thus obtained was subjected to real-time PCR with the SYBR qPCR Supermix Plus (Novoprotein, USA). Relative mRNA levels were calculated with the ΔΔCt method using glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA as an internal control. Primer sequences are shown in Table 1.
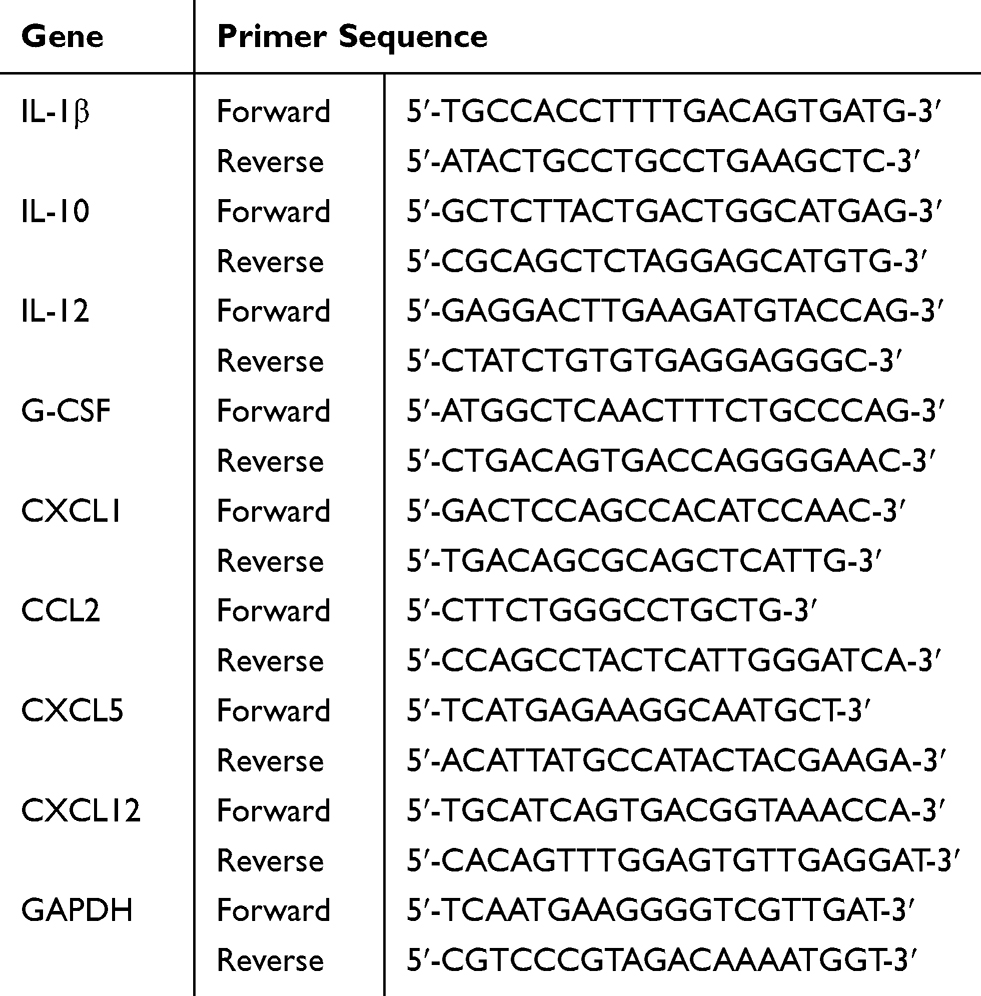 |
Table 1 Primer Sequence for Each Gene |
Multiplex Cytokine and Chemokine Analysis
Plasma samples, reagents and standards were prepared according the instructions of the Multi-Analyte Flow Assay Kit (Biolegend). In brief, standards and all diluted samples were added into 96 V-bottom plate wells in the presence of mixed beads, and shaken at 800 rpm for 2 hours at room temperature. Next, 25 µL detection antibodies was added to each well, with 1 hour incubation at room temperature. Then, 25 µL SA-PE was added to each well. The beads were resuspended and analyzed on a flow cytometer (Becton Dickinson, San Jose, USA) and the data were analyzed by LEGENDplex software (TreeStar, San Carlos, USA).
MPO Immunohistochemistry
Paraffin sections of lung tissues in all mice were rehydrated and boiled in sodium citrate buffer (pH6.0) for antigen retrieval. After blocking with 10% goat serum, rabbit anti-mouse myeloperoxidase (MPO) antibody (Abcam, UK) and goat anti-rabbit IgG (Zsbio Store, China) samples were incubated at 37°C for 30 minutes in the dark, then stained with DAB (Zsbio Store, Beijing, China). Finally, images were captured with an Olympus BX53 microscope and processed with LSM Image Examiner software (Zeiss).
Neutrophil Migration Assay
Neutrophils were purified from mouse bone marrow cells by negative selection according to the manufacturer’s protocol (Biolegend), then stained with CD11b (M1/70; Biolegend) and Ly6G antibody (1A8; Biolegend). The purity of isolated neutrophils was analyzed by a FACS Aria II (Becton Dickinson, San Jose, USA). Isolated neutrophils were placed in the upper compartment of a Transwell chamber (Corning, Corning, NY, USA), and 600 µL of the indicated N-formylmethionyl-leucyl-phenylalanine (fMLP; Sigma-Aldrich) concentration RPMI 1640 medium was added to the bottom chamber. After incubation at 37°C for 90 minutes, neutrophils on the lower surface were fixed with 5% neutral buffered formalin, stained with 1% crystal violet and counted, and the average number was recorded. The number of migrating cells per high-power field (400×) was counted in 10 random visual fields under the microscope (Olympus, Japan), and a mean estimate for individual samples was calculated.
Neutrophil Purification and Flow Cytometric Analysis
Neutrophils were purified from mouse bone marrow cells, then stained with APC/Cy7 anti-mouse Ly6G antibody (1A8), PerCP/Cyanine5.5 anti-mouse CD11b antibody (M1/70), PE/Cy7 anti-mouse CCR2 antibody (SA203G11), PE anti-mouse CXCR2 antibody (SA044G4) and APC anti-mouse CXCR4 antibody (L276F12). (All antibodies were purchased from Biolegend.) All of the above stained cells were assayed by a FACS Aria II (Becton Dickinson, San Jose, USA) and the data were analyzed by FlowJo software (TreeStar, San Carlos, USA).
CXCL12 Neutralizing Experiment In Vivo
Eight-to-twelve-week-old C57BL/6 WT mice were intraperitoneally injected with 10 mg/kg of 200 μL LPS, dissolved in sterile physiological saline, to induce ALI. Three of them received neutralizing anti-mouse CXCL12/SDF-1 antibody (79014, R&D) at a dose of 10 μg/mouse via tail intravenous injection, and the control mice were treated with the same volume of mouse isotype-matched IgG1 antibody. Twelve hours later, all mice were killed, and their lung tissue was collected and assessed.
Statistical Analysis
All data are shown as mean ± SD. Significant differences between groups were determined by unpaired t tests for neutralization assays or two-way ANOVA at different time-points (0, 6 and 12 hours), followed by multiple comparisons with Bonferroni correction, as appropriate. P<0.05 was required for statistical significance. All plots were generated with Prism software version 7 (GraphPad, San Diego, CA).
Results
NLRP3 Deficiency Increases Resistance to LPS-Induced ALI
To explore the role of NLRP3 in LPS-induced ALI, WT mice and NLRP3–/– mice were challenged with/without LPS, and lung pathology and vascular permeability were characterized and compared. On HE-stained slides, LPS administration increased the adhesive entrapment of perivascular inflammatory cells (Figure 1A, cyan arrows), interstitial infiltration (Figure 1A, blue arrows) and alveolar septal thickness (Figure 1A, red arrows) after 6 hours, and these featured injuries were further aggravated at 12 hours in both groups. However, rare inflammatory cell infiltration and proteinaceous debris were observed in the alveolar space. Compared with NLRP3–/–, WT mice presented more serious lung injury after LPS insults (Figure 1B). The Evans blue assay showed that lung vascular permeability was significantly higher after LPS treatment. (Figure 1C). BALF neutrophil counts were significantly lower in NLRP3-deficient mice (Figure 1D).
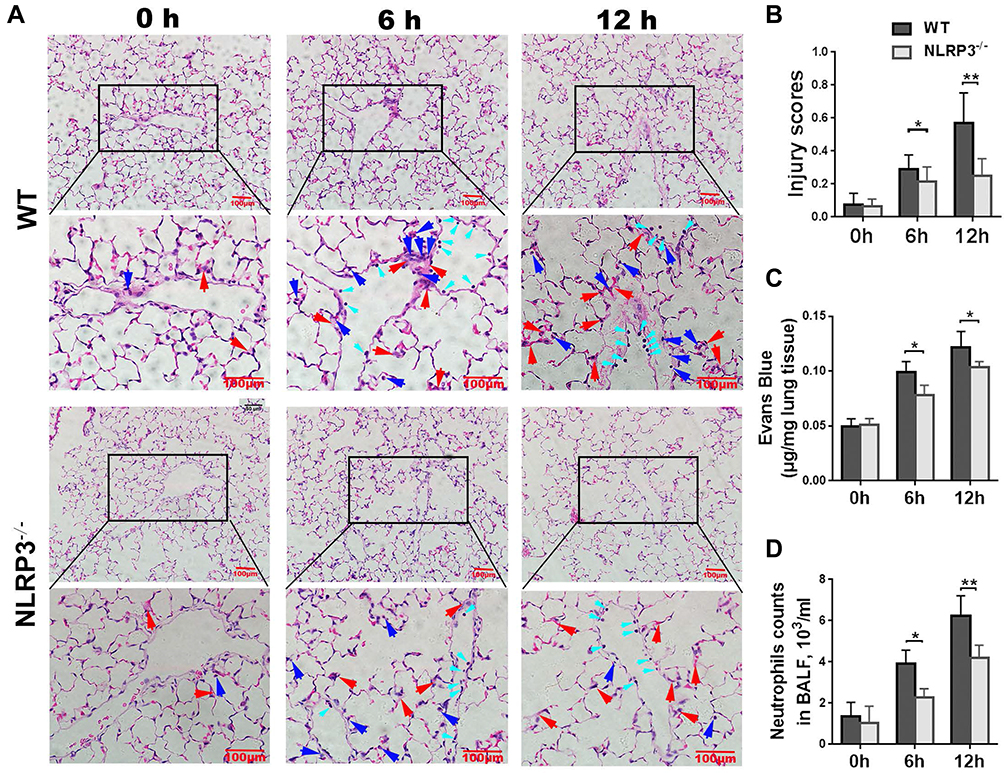 |
Figure 1 NLRP3 deficiency ameliorates LPS-induced lung injury. The wild-type (WT) mice and NLRP3–/– mice received intraperitoneal injection with 10 mg/kg LPS. Six and 12 hours later, lung injury was observed by HE staining. Compared to the WT mice, neutrophil adhesive entrapment (cyan arrows), infiltration (blue arrows) and alveolar septal thickening (red arrows) were significantly reduced in NLRP3–/– mice (A). Lung injury score was higher significantly in the WT group (B). Lung vascular permeability measured by the Evans blue assay was significantly lower in NLRP3–/– mice (C). Neutrophil counts in BALF were lower in NLRP3-deficient mice (D). Data are shown as mean±SEM. n=4 per group. Scale bars=100 μm. *P<0.05, **P<0.01, two-way ANOVA followed by multiple comparison with t test and Bonferronicorrection. |
Neutrophil Infiltration is Abrogated by NLRP3 Depletion
Neutrophil recruitment in the lung is a critical landmark in the pathogenesis in ALI. To observe the infiltration of neutrophils into the ALI lung in WT mice and NLRP3–/– mice, MPO immunohistochemical experiments were performed. The results showed that, despite the vascular contents being depleted, MPO-positive cells were still adhesively trapped in the pulmonary vessel walls (Figure 2A, red arrows), and infiltrated into stroma (Figure 2A, blue arrows). Statistical analysis revealed that 6 and 12 hours of LPS treatment strikingly increased the number of neutrophils in the WT murine lungs, whereas the NLRP3–/– groups showed much less neutrophil entrapment and infiltration (Figure 2B).
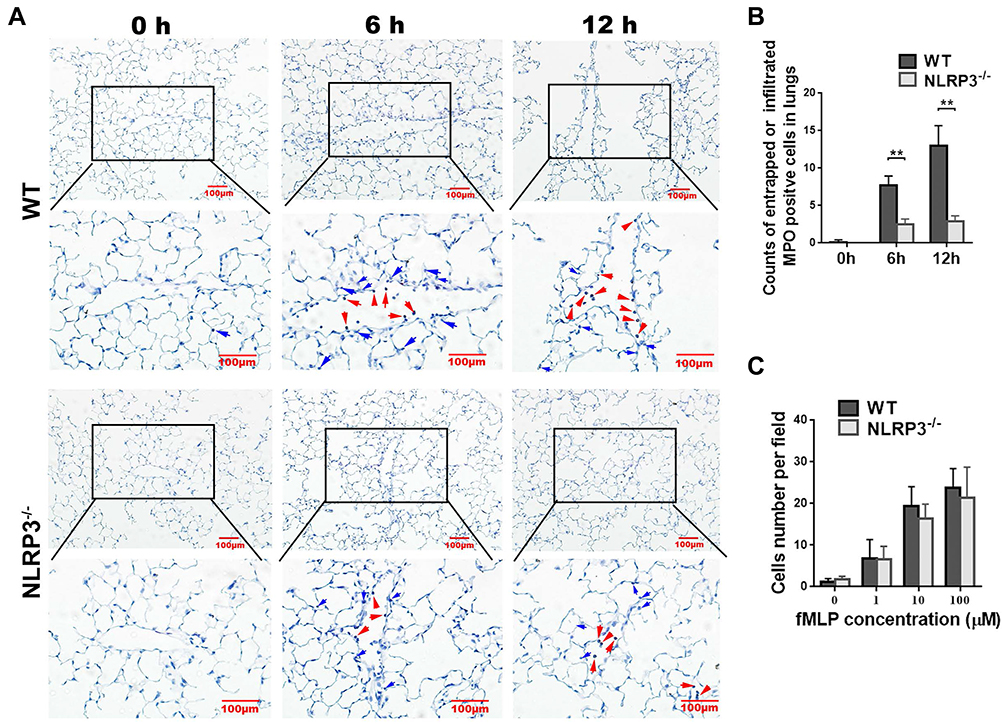 |
Figure 2 NLRP3 deficiency decreased neutrophil entrapment and infiltration into the lung. The WT mice and NLRP3–/– mice received intraperitoneal injection with 10mg/kg LPS, then the neutrophil infiltration of the lung was analyzed by MPO immunohistochemistry. Compared with the WT group, significantly fewer neutrophils were adhesively entrapped (red arrows) or infiltrated (blue arrows) in the lung of NLRP3–/– mice at both 6 and 12 hours (A, B). Isolated neutrophils were incubated at 0.1, 1 and 10 μM fMLP in transwell cultrue system. Migrated cells were counted. Results showed that deletion of NLRP3 did not affect neutrophil migration in response to fMLP, regardless its concentrations (C). MPO immunohistochemistry is shown at 40× and 100× magnification. Scale bar=100 μm. Images are representative of four independent experiments. MPO-positive cell counts were analyzed in 8–10 fields (100×) for each slide. Scale bars=100μm. Data are shown as mean±SEM. n=4 per group. **P<0.01, two-way ANOVA followed by unpaired multiple comparison with t test and Bonferroni correction. |
To investigate whether the lower neutrophil infiltration in the NLRP3–/– ALI mice was caused by an impaired migration capability, which was probably affected by NLRP3 gene knockout, we compared the neutrophil migration abilities between WT mice and NLRP3–/– mice in vitro. As shown in Figure 2C, the deletion of NLRP3 did not affect neutrophil migration in response to fMLP; there were no significant differences between two genotypes’ neutrophils at 0.1, 1 and 10 μM fMLP.
Collectively, these results proved that the deletion of NLRP3 had no effect on neutrophil migration, but strikingly reduced the infiltration of neutrophils and attenuated injury in LPS-induced ALI.
Deficiency of NLRP3 Downregulates CXCL12 Levels in Both Serum and Lung
Recruitment of neutrophils in the lung is conducted by chemotaxis. Hence, the serum levels of cytokines, such as IL-1β, IL-10, IL-12 and G-CSF, and chemokines, including CXCL1, CCL2, CXCL5 and CXCL12, in all mouse groups were measured. As expected, the concentrations of serum IL-1β, IL-10, IL-12, G-CSF, CCL2, CXCL1 and CXCL5 were increased significantly in the WT mice after LPS challenge, and their concentrations were significantly lower in the NLRP–/– mice (Figure 3A). In contrast, LPS treatment decreased the serum levels of CXCL12 in WT mice surprisingly; NLRP3 depletion suppressed serum CXCL12 to baseline levels and LPS stimulation did not change the serum concentration. Strikingly, compared to decreased or intact lung, IL-10, IL-12, G-CSF, CCL2, CXCL1, CXCL5 and lung CXCL12 concentrations were elevated by LPS administration in WT mice, and NLRP–/– ALI mice showed a low amount of lung CXCL12 which remained intact after LPS treatment (Figure 3B).
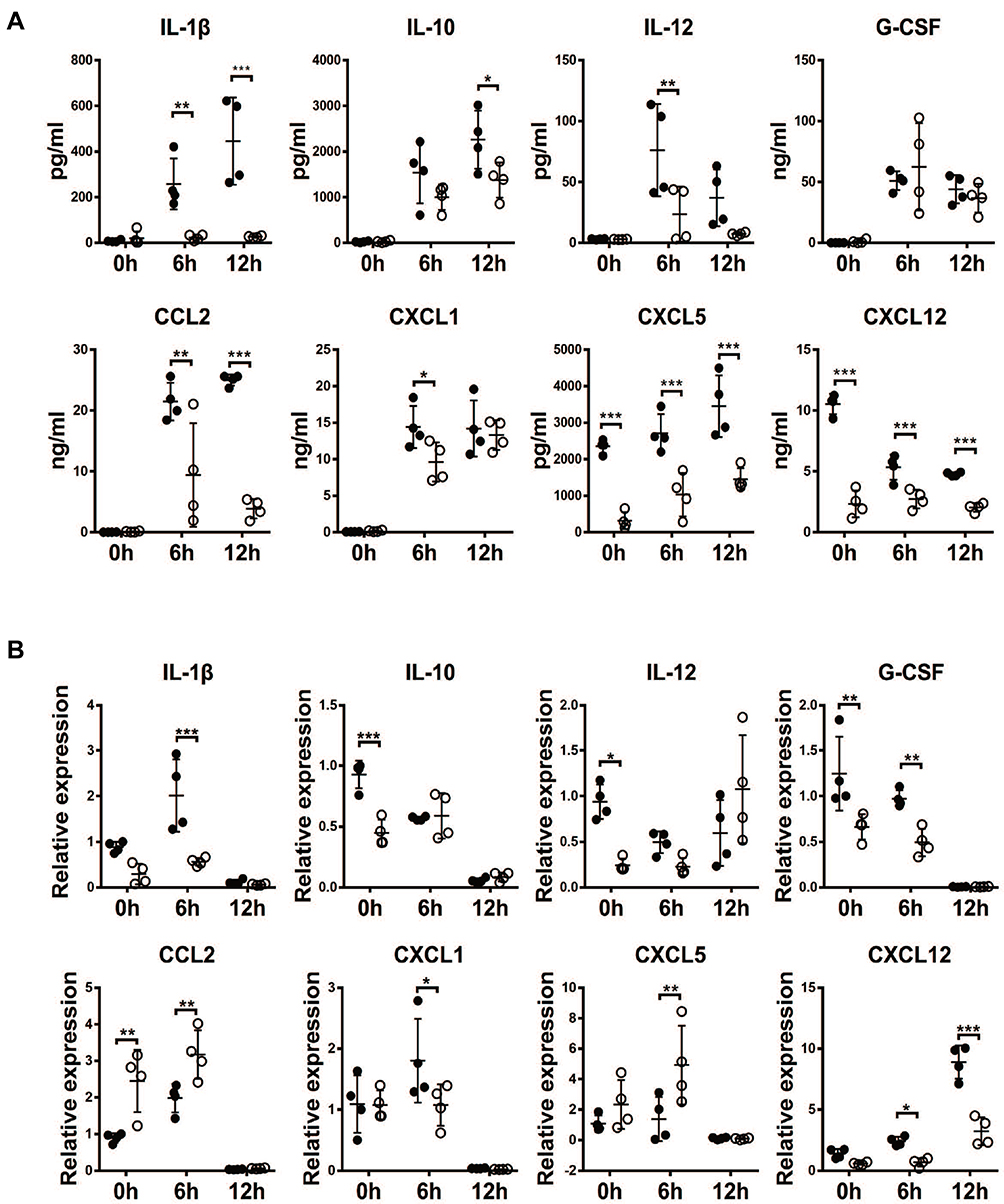 |
Figure 3 NLRP3 deficiency resulted in reduced cytokine and chemokine levels in serum and lung tissue. Serum or lungs were collected at 6 and 12 hours after LPS challenge in WT mice and NLRP3–/– mice. IL-1β, IL-10, IL-12, G-CSF, CCL2, CXCL1, CXCL5 and CXCL12 were analyzed by Multiplex test in serum samples (A) or by qPCR in lung tissue (B). Data are shown as mean±SEM. n=4 per group. *P<0.05, **P<0.01, ***P<0.001, two-way ANOVA followed by unpaired multiple comparison with t test and Bonferroni correction. |
Both chemokines and their receptors regulate the migratory patterns of neutrophils. To explore the key factors in the neutrophil migration to the lung, we studied the surface expression of some chemokine receptors, such as CCR2, CXCR1 and CXCR4, by flow cytometry. In general, flow cytometric data showed that there were no significant differences in the expression of CCR2 (Figure 4A) and CXCR2 (Figure 4B), while there was an increase in the expression of CXCR4 (Figure 4C) on neutrophils from the NLRP3–/– mice compared to those from the WT mice.
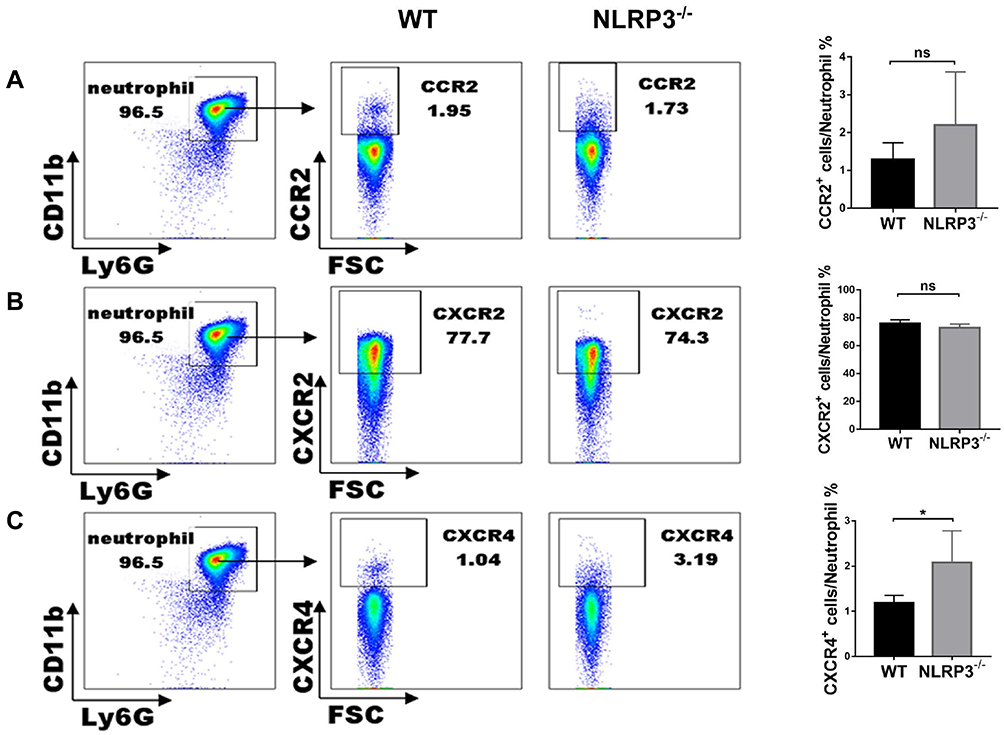 |
Figure 4 Chemokine receptors in NLRP3-deficient neutrophils. In Ly6GhiCD11bhi bone marrow cells, CCR2 (A) and CXCR2 (B) expression were similar in the two genotypes. CXCR4 expression (C) was significantly higher in the NLRP3–/– group. Data are shown as mean±SEM. n=3 per group. *P<0.05, unpaired Student’s t test. |
These data strongly highlighted the critical contribution of CXCL12 in neutrophil recruitment into the lung during LPS-induced ALI.
Blocking CXCL12 Moderates ALI
To verify the role of CXCL12 during neutrophil recruitment, neutralizing CXCL12 antibody was administered, then lung injury and neutrophil infiltration were assessed at 12 hours after LPS injection. HE staining of the lung revealed less destruction of the alveolar structure and subalveolar edema in the WT mice with CXCL12 blockage than in the control group (Figure 5A and C), and less neutrophil accumulation was observed in the mice with neutralization of CXCL12 than in those without this neutralization (Figure 5B and D).
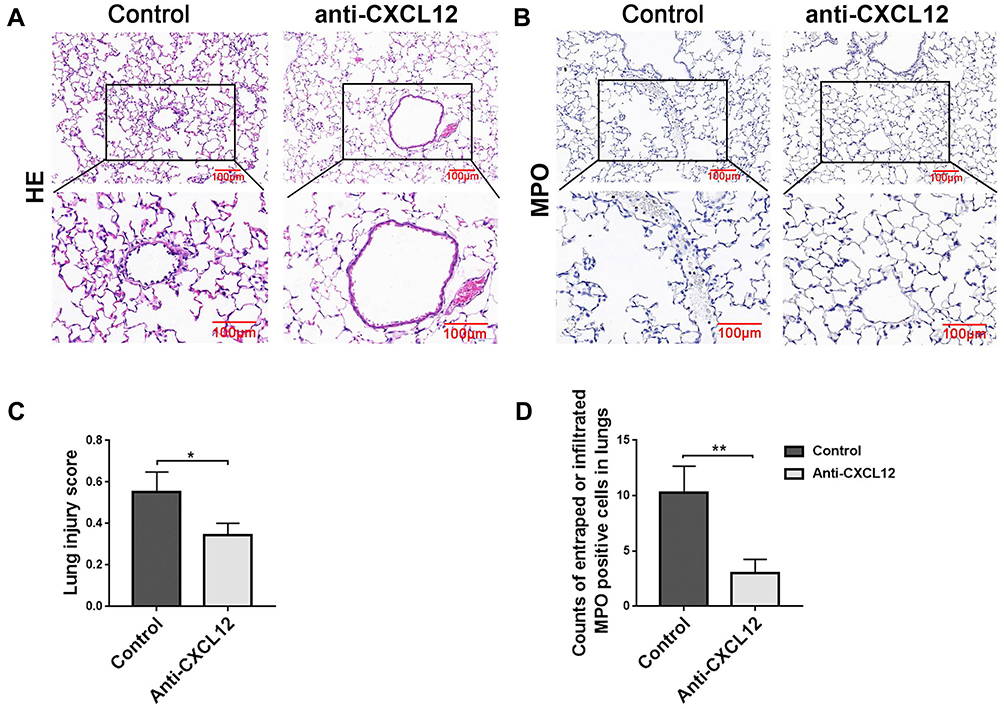 |
Figure 5 CXCL12 recruited neutrophils to the lung in acute lung injury (ALI). Compared with the isotype IgG1-matched control group, neutralizing anti-CXCL12 antibody alleviated ALI (A, C), and dramatically reduced neutrophil number (B, D) in lungs from the WT mice after exposure to LPS for 12 hours. The photographs are shown as 40× and 100× magnification. Scale bars=100 μm. Images are representative of three independent experiments. MPO-positive cells were counted and analyzed in 8–10 fields (100×) for each slide. Data are shown as mean±SEM. n=6 per group. *P<0.05, **P<0.01, unpaired Student’s t test. |
Collectively, these data imply that deletion of NLRP3 strikingly attenuated lung injury and neutrophil accumulation in LPS-induced ALI by reducing the expression of CXCL12 chemokines.
Discussion
It is well known that the NLRP3 inflammasome is required for the development of LPS-induced ALI,19 but the molecular and cellular mechanism is still elusive. In this study, we observed that mice with NLRP3 depletion were strongly resistant to LPS-induced ALI. NLRP3 deficiency decreases neutrophil recruitment in injured lungs, which is partially mediated by CXCL12 downregulation.
In ALI, neutrophils are recruited to and infiltrated the lung, which is critical in the pathogenesis of lung injury. Activated neutrophils can secrete a variety of pro-inflammatory cytokines.4,25 With NLRP3 inflammasome deletion, IL-1β increased significantly more in both serum and lung from the WT mice than the NLRP–/– mice, which is consistent with previous studies.26–28 Although it has been reported that IL-10 could play a protective role in ALI,29 there were higher concentrations of IL-10 in the serum from the WT mice than in that from the NLRP3–/– mice in this study. The higher IL-10 level did not alleviate lung injury compared to the NLRP3–/– mice, which was probably related to the expression in the lung having no significant difference between the two groups.
Chemokines are a family of chemotactic cytokines, which play a crucial role in leukocyte migration to inflammatory sites. They are also pivotal mediators that recruit neutrophils to the lung in ALI, and neutralization of chemokines could attenuate the lung injury.13,30 After LPS treatment, obviously higher levels of CCL2, CXCL1, CXCL5 and CXCL12 were found in serum from the WT group at the 6-hour time-point, compared to the NLRP3–/– group. Further investigation of chemokine expression in the lungs revealed that there were significantly higher levels of CXCL12 in the WT ALI group than in the NLRP3–/– ALI group at 6 and 12 hours, in line with the serum results. The accumulated evidence suggests that both the chemokine CXCL12 and its receptor CXCR4 contribute to the control of neutrophil migration.16,18,31 Although our data showed that the surface CXCR4 expression levels on neutrophils from the NLRP3–/– mice were higher than on those from the WT mice, there was less neutrophil infiltration in the NLRP3–/– mice compared with the WT mice during LPS-induced lung injury. Thus, the data suggest that NLRP3 deficiency markedly weakened neutrophil accumulation in LPS-induced ALI by reducing CXCL12 expression.
To further investigate the effect of CXCL12, we used anti-CXCL12 neutralizing antibody to block CXCL12 chemotaxis, and the experiments confirmed the important role of CXCL12 in LPS-induced ALI. Although the mechanisms by which bone-marrow derived stem cells are recruited to the lungs have not been completely elucidated, there is significant evidence that the injured lung expresses stromal-derived factor-1 (CXCL12), which interacts with the CXCR4 receptor on the surface of bone-marrow derived stem cells.32–34 Thus, it is likely that the deletion of the NLRP3 molecule leads to a reduction in CXCL12 gene expression in the lung, and protects NLRP3–/– mice from LPS-induced ALI.
Conclusion
Our study suggests that CXCL12 is one of the pivotal drivers of neutrophil infiltration during ALI development by LPS challenge; the deletion of NLRP3 suppressed neutrophil migration and lung injury, which may be associated with the suppression of CXCL12 expression by NKRP3 depletion.
Disclosure
The authors declare that no conflict of interest exists.
References
1. Thompson BT, Chambers RC, Liu KD. Acute respiratory distress syndrome. N Engl J Med. 2017;377(6):562–572. doi:10.1056/NEJMra1608077
2. Wong JJM, Leong JY, Lee JH, Albani S, Yeo JG. Insights into the immuno-pathogenesis of acute respiratory distress syndrome. Ann Transl Med. 2019;7(19):504. doi:10.21037/atm.2019.09.28
3. Williams AE, Chambers RC. The mercurial nature of neutrophils: still an enigma in ARDS? Am J Physiol Lung Cell Mol Physiol. 2014;306(3):L217–L230. doi:10.1152/ajplung.00311.2013
4. Potey PMD, Rossi AG, Lucas CD, Dorward DA. Neutrophils in the initiation and resolution of acute pulmonary inflammation: understanding biological function and therapeutic potential. J Pathol. 2019;247(5):672–685. doi:10.1002/path.5221
5. Giacalone VD, Margaroli C, Mall MA, Tirouvanziam R. Neutrophil adaptations upon recruitment to the lung: new concepts and implications for homeostasis and disease. Int J Mol Sci. 2020;21:3. doi:10.3390/ijms21030851
6. Steinberg KP, Milberg JA, Martin TR, Maunder RJ, Cockrill BA, Hudson LD. Evolution of bronchoalveolar cell populations in the adult respiratory distress syndrome. Am J Respir Crit Care Med. 1994;150(1):113. doi:10.1164/ajrccm.150.1.8025736
7. Summers C, Singh NR, White JF, et al. Pulmonary retention of primed neutrophils: a novel protective host response, which is impaired in the acute respiratory distress syndrome. Thorax. 2014;69(7):623–629. doi:10.1136/thoraxjnl-2013-204742
8. Narasaraju T, Yang E, Samy RP, et al. Excessive neutrophils and neutrophil extracellular traps contribute to acute lung injury of influenza pneumonitis. Am J Pathol. 2011;179(1):199–210. doi:10.1016/j.ajpath.2011.03.013
9. Park I, Kim M, Choe K, et al. Neutrophils disturb pulmonary microcirculation in sepsis-induced acute lung injury. Eur Respir J. 2019;53:3. doi:10.1183/13993003.00786-2018
10. Williams AE, Jose RJ, Mercer PF, et al. Evidence for chemokine synergy during neutrophil migration in ARDS. Thorax. 2017;72(1):66–73. doi:10.1136/thoraxjnl-2016-208597
11. Zemans RL, Matthay MA. What drives neutrophils to the alveoli in ARDS? Thorax. 2017;72(1):1–3. doi:10.1136/thoraxjnl-2016-209170
12. Bhatia M, Zemans RL, Jeyaseelan S. Role of chemokines in the pathogenesis of acute lung injury. Am J Respir Cell Mol Biol. 2012;46(5):566–572. doi:10.1165/rcmb.2011-0392TR
13. Rudd JM, Pulavendran S, Ashar HK, et al. Neutrophils induce a novel chemokine receptors repertoire during influenza pneumonia. Front Cell Infect Microbiol. 2019;9:108. doi:10.3389/fcimb.2019.00108
14. Wang CY, Shang M, Zhou CL, Feng LZ, Zhou QS, Hu K. Mechanism of Cxc Chemokine Ligand 5 (CXCL5)/Cxc Chemokine Receptor 2 (CXCR2) bio-axis in mice with acute respiratory distress syndrome. Med Sci Monit. 2019;25:5299–5305. doi:10.12659/MSM.915835
15. Sawant KV, Xu R, Cox R, et al. Chemokine CXCL1-mediated neutrophil trafficking in the lung: role of CXCR2 activation. J Innate Immun. 2015;7(6):647–658. doi:10.1159/000430914
16. Isles HM, Herman KD, Robertson AL, et al. The CXCL12/CXCR4 signaling axis retains neutrophils at inflammatory sites in zebrafish. Front Immunol. 2019;10:1784. doi:10.3389/fimmu.2019.01784
17. Mousavi A. CXCL12/CXCR4 signal transduction in diseases and its molecular approaches in targeted-therapy. Immunol Lett. 2020;217:91–115. doi:10.1016/j.imlet.2019.11.007
18. Konrad FM, Meichssner N, Bury A, Ngamsri KC, Reutershan J. Inhibition of SDF-1 receptors CXCR4 and CXCR7 attenuates acute pulmonary inflammation via the adenosine A2B-receptor on blood cells. Cell Death Dis. 2017;8(5):e2832. doi:10.1038/cddis.2016.482
19. Hosseinian N, Cho Y, Lockey RF, Kolliputi N. The role of the NLRP3 inflammasome in pulmonary diseases. Ther Adv Respir Dis. 2015;9(4):188–197. doi:10.1177/1753465815586335
20. Tian X, Sun H, Casbon AJ, et al. NLRP3 inflammasome mediates dormant neutrophil recruitment following sterile lung injury and protects against subsequent bacterial pneumonia in mice. Front Immunol. 2017;8:1337. doi:10.3389/fimmu.2017.01337
21. Chen J, Wang S, Fu R, et al. RIP3 dependent NLRP3 inflammasome activation is implicated in acute lung injury in mice. J Transl Med. 2018;16(1):233. doi:10.1186/s12967-018-1606-4
22. Fukumoto J, Fukumoto I, Parthasarathy PT, et al. NLRP3 deletion protects from hyperoxia-induced acute lung injury. Am J Physiol Cell Physiol. 2013;305(2):C182–C189.
23. Boro M, Balaji KN. CXCL1 and CXCL2 regulate NLRP3 inflammasome activation via G-protein-coupled receptor CXCR2. J Immunol. 2017;199(5):1660–1671. doi:10.4049/jimmunol.1700129
24. Matute-Bello G, Downey G, Moore BB, et al. An official American Thoracic Society workshop report: features and measurements of experimental acute lung injury in animals. Am J Respir Cell Mol Biol. 2011;44(5):725–738. doi:10.1165/rcmb.2009-0210ST
25. Grommes J, Soehnlein O. Contribution of neutrophils to acute lung injury. Mol Med. 2011;17(3–4):293–307. doi:10.2119/molmed.2010.00138
26. Grailer JJ, Canning BA, Kalbitz M, et al. Critical role for the NLRP3 inflammasome during acute lung injury. J Immunol. 2014;192(12):5974–5983. doi:10.4049/jimmunol.1400368
27. Fukumoto J, Fukumoto I, Parthasarathy PT, et al. NLRP3 deletion protects from hyperoxia-induced acute lung injury. Am J Physiol Cell Physiol. 2013;305(2):C182–189.
28. Jones HD, Crother TR, Gonzalez-Villalobos RA, et al. The NLRP3 inflammasome is required for the development of hypoxemia in LPS/mechanical ventilation acute lung injury. Am J Respir Cell Mol Biol. 2014;50(2):270–280. doi:10.1165/rcmb.2013-0087OC
29. Inoue G. Effect of interleukin-10 (IL-10) on experimental LPS-induced acute lung injury. J Infect Chemother. 2000;6(1):51–60. doi:10.1007/s101560050050
30. Griffith JW, Sokol CL, Luster AD. Chemokines and chemokine receptors: positioning cells for host defense and immunity. Annu Rev Immunol. 2014;32:659–702. doi:10.1146/annurev-immunol-032713-120145
31. Tulotta C, Stefanescu C, Chen Q, Torraca V, Meijer AH, Snaar-Jagalska BE. CXCR4 signaling regulates metastatic onset by controlling neutrophil motility and response to malignant cells. Sci Rep. 2019;9(1):2399. doi:10.1038/s41598-019-38643-2
32. Hannoush EJ, Sifri ZC, Elhassan IO, et al. Impact of enhanced mobilization of bone marrow derived cells to site of injury. J Trauma. 2011;71(2):283–291. doi:10.1097/TA.0b013e318222f380
33. Carevic M, Singh A, Rieber N, et al. CXCR4+ granulocytes reflect fungal cystic fibrosis lung disease. Eur Respir J. 2015;46(2):395–404. doi:10.1183/09031936.00173514
34. Yamada M, Kubo H, Kobayashi S, et al. The increase in surface CXCR4 expression on lung extravascular neutrophils and its effects on neutrophils during endotoxin-induced lung injury. Cell Mol Immunol. 2011;8(4):305–314. doi:10.1038/cmi.2011.8
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.