")
Back to Journals » OncoTargets and Therapy » Volume 12
Nicotine promotes cervical metastasis of oral cancer by regulating peroxiredoxin 1 and epithelial–mesenchymal transition in mice
Authors Wang M, Niu W, Qi M, Chen H , Zhang M , Wang C, Ge L, Yang J, Miao C, Shi N , Chen T, Tang X
Received 10 November 2018
Accepted for publication 13 February 2019
Published 1 May 2019 Volume 2019:12 Pages 3327—3338
DOI https://doi.org/10.2147/OTT.S194129
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Federico Perche
Min Wang,1 Wenwen Niu,2 Moci Qi,1 Hui Chen,1 Min Zhang,1 Chunxiao Wang,1 Lihua Ge,1 Jing Yang,1 Congcong Miao,1 Ni Shi,3 Tong Chen,3 Xiaofei Tang1
1Beijing Institute of Dental Research, Beijing Stomatological Hospital and School of Stomatology, Capital Medical University, Beijing 100050, People’s Republic of China; 2Beijing Shibalidian Community Hospital, Beijing, People’s Republic of China; 3Division of Medical Oncology, Department of Internal Medicine, The Arthur G. James Cancer Hospital and Richard J. Solove Research Institute, The Ohio State University, Columbus, OH 43210, USA
Background: Tobacco is a major risk factor for oral squamous cell carcinoma (OSCC). However, the role of nicotine in OSCC is not completely understood.
Materials and methods: To analyze the mechanisms of nicotine-induced cervical metastasis, we investigated whether nicotine induced invasion, migration, and epithelial–mesenchymal transition (EMT) via regulating peroxiredoxin 1 (Prx1) in CAL 27 cells. In addition, we established a mouse model to confirm the roles of nicotine in regulating Ets1/Prx1/EMT signaling in OSCC metastasis.
Results: We showed that nicotine induced CAL 27 cell invasion, migration, EMT, and Prx1 and Ets1 expression. Prx1 knockdown inhibited cell invasion, migration, and EMT. Ets1 silencing downregulated Prx1 expression and EMT. Prx1 and Ets1 were shown to interact in CAL 27 cells treated with nicotine, and nicotine could significantly upregulate the binding of the transcription factor Ets1 to the Prx1 gene promoter region. Additionally, an in vivo study showed that nicotine induced tumor metastasis and EMT. Prx1 knockdown inhibited cervical metastasis rates and EMT progression. No significant differences in metastasis rates and EMT-related marker expression levels were observed between vehicle- and nicotine-treated mice.
Conclusion: The results indicate that nicotine promotes cervical lymph node metastasis through regulating Ets1/Prx1/EMT signaling during OSCC pathogenesis; consequently, Prx1 may represent a potential target for the prevention and treatment of OSCC.
Keywords: oral squamous cell carcinoma, peroxiredoxin 1, metastasis, epithelial–mesenchymal transition, mouse model
Introduction
Oral squamous cell carcinoma (OSCC) is among the most common head and neck cancers worldwide.1 Despite the advances in the prevention and treatment of OSCC, the 5-year survival rate has not improved in 30 years.2 Cancer recurrence and metastases are the main causes of mortality, and OSCC metastasizes mainly to the lymph nodes in the cervical regions.
Tobacco use has been considered as the most important risk factor for OSCC.3 Nicotine is a major component of cigarette smoke and an exogenous source of oxidative stress, and it has been suggested that it may contribute to cancer development and induce epithelial–mesenchymal transition (EMT) and metastases.4 As an antioxidant protein family member, peroxiredoxin (Prx) can be found in a wide variety of species.5 Prx1 is a key member of this family and represents an oxidative stress-inducible protein. Prx1 is overexpressed in several human cancers, including OSCC, and has been reported to be associated with cell proliferation, apoptosis, cancer development, and lymph node metastasis.6–8 Ets1 is a member of the Ets family of transcription factors, and studies have demonstrated that Ets1 may be overexpressed in several tumors; it also promotes invasive behavior and may affect ROS sensitivity through the expression of Prx1.9–12 Previously, we showed that nicotine induced cell proliferation and Prx1 overexpression in OSCC cells in vitro, and Prx1 overexpression promoted tumor growth in OSCC xenografts; these findings indicate that Prx1 plays a key role in the pathogenesis of tobacco-related OSCC.13 However, the molecular mechanisms underlying the potential role of nicotine, Prx1, and Ets1 in OSCC metastasis have not been completely elucidated.
EMT is considered to be necessary for metastasis formation and responsible for the increase in invasiveness.14,15 Various biomarkers have been found for different types of EMT.16 E-cadherin, vimentin, and Snail have been shown to be EMT-related proteins involved in type 3 EMT, which is related to cancer metastasis and progression.17,18 In this process, E-cadherin represents an attenuated epithelial marker, vimentin is an acquired mesenchymal marker, and Snail mediates the transition.19
In this study, to analyze the roles and mechanisms of nicotine-induced cervical metastasis, we investigated whether nicotine induced invasion and cervical metastasis via regulating Prx1 and EMT in oral cancer CAL 27 cells. Next, we established a mouse cancer model originating in the tongue with cervical metastasis by the orthotopic transplantation of control CAL 27 cells and siPrx1 CAL 27 cells labeled with green fluorescent protein (GFP); this step served to confirm the roles of nicotine in regulating Ets1/Prx1/EMT signaling in OSCC metastasis.
Materials and methods
Cells
Human CAL 27 cells (American Type Culture Collection [ATCC], Manassas, VA, USA) were cultured in DMEM/high glucose (HyClone, Logan, UT, USA) containing 10% (v/v) FBS (Thermo Fisher Scientific, Waltham, MA, USA) with 100 U/mL penicillin and 100 μg/mL streptomycin (Thermo Fisher Scientific). The cells were grown at 37°C with 5% CO2 in a humidified atmosphere.
Lentivirus-mediated transfection
Prx1-shRNA and GFP-expressing lentiviral particles were prepared by Shanghai GenePharma (Shanghai, People’s Republic of China). The Prx1-targeting sequence was 5′-AGGGTATTCTTCGGCAGATCA-3′. CAL 27 cells were seeded at a density of 1.5×105 cells/well in six-well plates and cultured overnight. Negative control or Prx1-shRNA-expressing lentiviruses were transfected at a multiplicity of infection (MOI) of 10 with 5 μg/mL polybrene in serum-free media for 48 h. Control CAL 27 cells (siControl) and stable Prx1 knockdown (siPrx1) cells were selected with puromycin. Prx1 protein expression was detected by Western blot with an anti-Prx1 antibody (Abcam, Cambridge, UK).
Transient transfection
For Ets1 knockdown, siRNAs were commercially generated (Sangon Biotech, Shanghai, People’s Republic of China): 5′-GCAAAGAAAUGAUGUCUCAAGCAUU-3′ was used for Ets1 targeting, while 5′-UUCUCCGAACGUGUCACGUTT-3′ was used as a negative control. Lipofectamine 2000 (Thermo Fisher Scientific) was used to perform siRNA transfections according to the manufacturer’s instructions. Total protein was collected 72 h after transfection for Western blot analysis.
Nicotine treatment in vitro
CAL 27 cells were treated with 1 μmol/L nicotine for 7 days for the following in vitro experiments involving nicotine treatment. Cells were subcultured every 3 days and treated with 1 μmol/L nicotine for 48 h after every passage. The regimen of 7-day nicotine treatment was selected based on the nicotine dose–response experiments.
Invasion assay
Cell invasion assays were performed in Transwell chambers (24-well format; 8 mm pore size; Falcon, Austin, TX, USA) coated with Matrigel (1 mg/mL; Corning Incorporated, Corning, NY, USA). CAL 27 cells (2.5×105/well) were plated in the upper chambers filled with medium containing 0.2% FBS, while the lower chambers were filled with medium containing 10% FBS. After a 24 h incubation, the cells inside the chamber were removed, and the invaded cells were stained with hematoxylin (Solarbio, Beijing, People’s Republic of China) and examined with an inverted microscope (Olympus Corporation, Tokyo, Japan).
In vitro scratch assay
Cells (5×105 cells/well) were seeded onto six-well plates and incubated in complete medium for 12 h at 37°C. Scratch wounds were applied to each well with a sterilized 200 μL pipette tip, and the non-adherent cells were washed off three times with warm PBS. Fresh serum-free medium was added to the wells, and the cells were incubated for an additional 48 h. All experiments were conducted independently in triplicate.
Co-immunoprecipitation (Co-IP) assay
Following 7-day treatment with 1 μmol/L nicotine, CAL 27 cells were harvested. The cell lysates were incubated with antibodies against Prx1 (0.5 μg; Abcam) or negative control IgG (0.5 μg; Santa Cruz Biotechnology Inc., Dallas, TX, USA) overnight at 4°C, followed by the addition of 40 μL of protein A plus Sepharose (Sigma-Aldrich Co., St Louis, MO, USA) and incubation for 4 h at 4°C. After centrifugation for 1 min at 4°C, the pellets were washed three times with cell lysis buffer. Protein complexes were eluted and then separated by SDS-PAGE; Western blot analyses were performed to detect Ets1 (1:1,000; Cell Signaling Technology, Danvers, MA, USA) and Prx1 (1:5,000; Abcam). In addition, whole cell extracts were separated and analyzed by Western blot.
Chromatin immunoprecipitation (ChIP)
ChIP assays were performed as reported previously.20 Precipitated chromatin was purified, and DNA was quantified by quantitative real-time PCR (qRT-PCR). A ChIP-grade antibody against Ets1 was used in this study. Primers for the Prx1 promoter region were as follows: forward, CTGACTAAACCATGTCACAGAC; reverse, AGCCTGGCAACAGAGCAAG.
Cervical metastasis mouse model of OSCC
Five-week-old BALB/c nude mice (Vital River Laboratory Animal Technology, Beijing, People’s Republic of China) were bred under specific pathogen-free conditions. Experiments were conducted in accordance with the Beijing Municipality on the Review of Welfare and Ethics of Laboratory Animals guidelines approved by the Beijing Municipality Administration Office of Laboratory Animals (BAOLA). Furthermore, the study was approved by the Animal Ethical and Welfare Committee of Beijing Stomatological Hospital (Approval No KQYY-201503-010).
The animals were separated randomly into five groups: 1) control group: siControl CAL 27 cell transplantation with vehicle treatment (corn oil; Sigma-Aldrich Co.); 2) nicotine group: siControl cell transplantation with nicotine treatment (Wako Pure Chemical Industries, Ltd., Osaka, Japan); 3) Prx1 knockdown group: siPrx1 cell transplantation with vehicle treatment; 4) Prx1 knockdown + nicotine group: siPrx1 cell transplantation with nicotine treatment; and 5) no treatment group. Each group contained 10 male and 10 female mice.
The mice were anesthetized using sodium pentobarbital (50 mg/kg body weight), and they received a submucosal injection of 5×106 cells (in a 25 μL volume) into their tongue using a sterile insulin syringe (BD, Franklin Lakes, NJ, USA). The mice in the nicotine and Prx1 knockdown + nicotine groups received 10 μL of a nicotine solution (6% in corn oil) on their tongue three times a week. The development of tongue cancer, overall health condition, and total body weight of the mice were examined daily. The tongues and cervical lymph nodes were observed under a dissecting fluorescence microscope (Olympus Corporation). The number of mice with lymph node metastases was counted, and the metastasis rates were calculated. Metastasis to the lymph nodes was observed by fluorescence stereomicroscopy from 11 days after cell implantation. At day 13 after siControl cell or siPrx1 cell implantation, the tongues and cervical lymph nodes with green fluorescence were removed and immediately stored in liquid nitrogen for further qRT-PCR and Western blot analyses or placed in formalin solution for histological examination.
H&E staining
Tongue and cervical lymph node tissues were fixed in formalin and embedded in paraffin; 5μm sections were cut using a microtome and stained with H&E to examine tumor morphology as described previously.21
Immunohistochemistry
Tissue sections were deparaffinized and rehydrated, and antigen retrieval was performed by boiling the samples in 0.01 M citrate buffer (pH 6.0) for 10 min. The sections were incubated with a mouse antihuman cytokeratin antibody (Fuzhou Maixin Biotech. Co., Ltd., Fujian, People’s Republic of China) overnight at 4°C. Afterward, the sections were incubated with a biotinylated secondary antibody for 30 min at 37°C, and the signals were visualized using 3,3′-diaminobenzidine (Solarbio) for 3–5 min. PBS was used as a negative control.
Quantitative PCR (qPCR) analysis
TRIzol (Thermo Fisher Scientific) was used to extract the total RNA from tongue samples and cervical lymph nodes. Reverse transcription was conducted using HiFi-MMLV Reverse Transcriptase (CW Biotech, Beijing, People’s Republic of China). Gapdh was used as an internal control. qPCR was performed using a CFX Connect Real-Time PCR Detection System (Bio-Rad Laboratories Inc., Hercules, CA, USA) with the primer pairs listed in Table 1. The comparative cycle threshold method (2−ΔΔCT) was used to analyze the data. All samples were analyzed in triplicate.
 | Table 1 Primers used for qPCR |
Western blot
Tumor cells and tissues were lysed using immunoprecipitation lysis buffer. Equal amounts of protein were separated by SDS-PAGE and transferred onto polyvinylidene fluoride (PVDF) membranes (0.45 μm; EMD Millipore, Billerica, MA, USA). Following blocking with 5% fat-free milk in Tris-buffered saline with Tween-20 (TBST) for 1 h at 37°C, the membranes were incubated with anti-Prx1 (1:5,000; Abcam), anti-Ets1 (1:1,000; Cell Signaling Technology), anti-E-cadherin (1:1,000; Cell Signaling Technology), anti-vimentin (1:1,000; Bioss Inc., Beijing, People’s Republic of China), anti-Snail (1:1,000; Abcam), and anti-GAPDH antibodies (1:20,000; Immunoway, Plano, TX, USA) overnight at 4°C. Immunoreactive bands were detected using horseradish peroxidase-conjugated secondary antibodies and enhanced with chemiluminescence reagents (Amersham Biosciences, Piscataway, NJ, USA).
Statistical analyses
All assays were independently performed in triplicate. SPSS software Version 17.0 (SPSS Inc., Chicago, IL, USA) was used for statistical analyses. The results are expressed as mean ± SD. Statistical significance was assessed using unpaired two-tailed Student’s t-tests. Differences were considered significant when p<0.05.
Results
Nicotine induces cell invasion, migration, and EMT in CAL 27 cells
We investigated the effect of nicotine on cell invasion and migration in CAL 27 cells. Matrigel invasion assay results demonstrated that a higher number of nicotine-treated cells than control cells penetrated through the filters after 12 h (Figure 1A and B). Consistently, higher rates of wound healing and cell migration were observed in the nicotine-treated cells than in the controls (Figure 1C). These results suggest that nicotine promotes cell invasion and migration in OSCC cells.
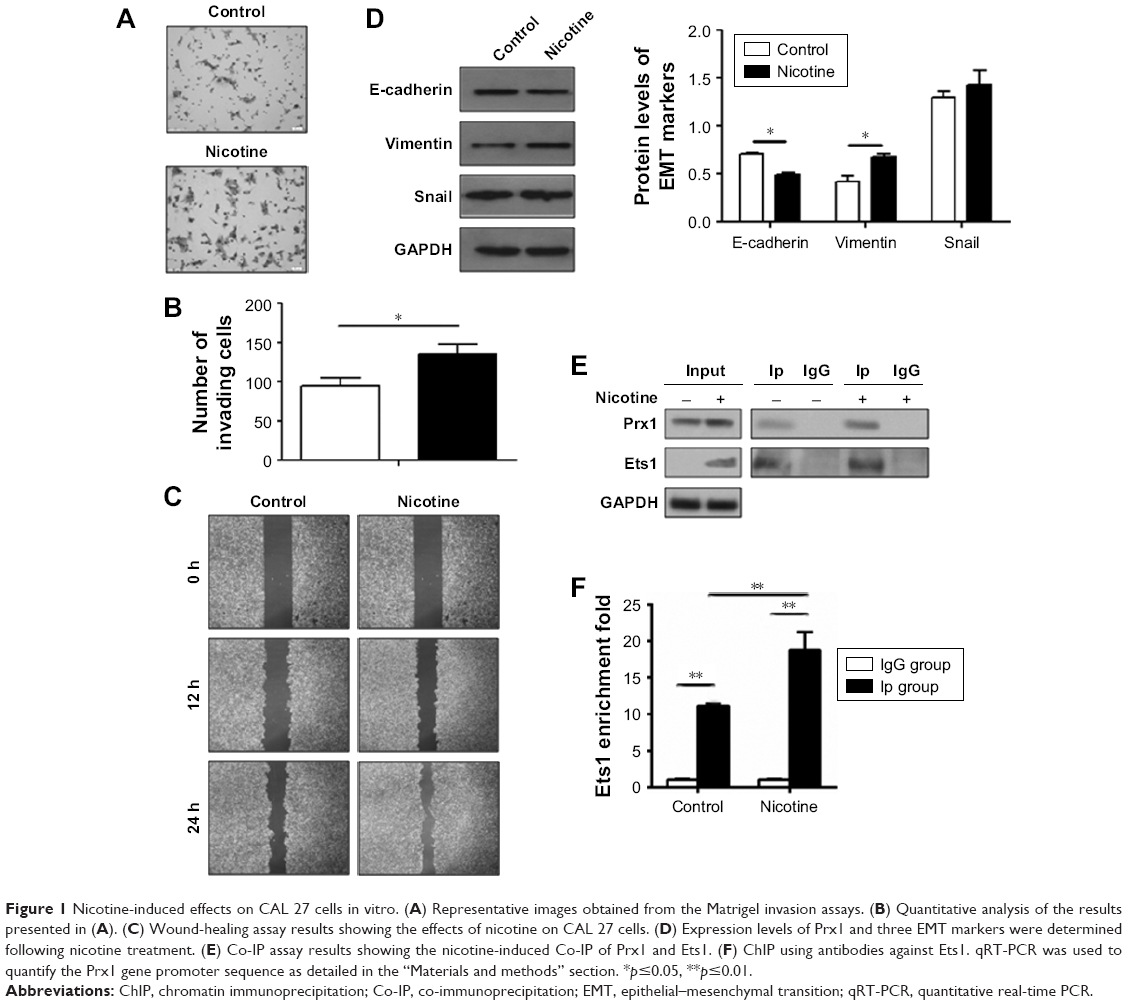 | Figure 1 Nicotine-induced effects on CAL 27 cells in vitro. (A) Representative images obtained from the Matrigel invasion assays. (B) Quantitative analysis of the results presented in (A). (C) Wound-healing assay results showing the effects of nicotine on CAL 27 cells. (D) Expression levels of Prx1 and three EMT markers were determined following nicotine treatment. (E) Co-IP assay results showing the nicotine-induced Co-IP of Prx1 and Ets1. (F) ChIP using antibodies against Ets1. qRT-PCR was used to quantify the Prx1 gene promoter sequence as detailed in the “Materials and methods” section. *p≤0.05, **p≤0.01. |
Furthermore, the protein expression levels of E-cadherin were decreased, and the expression levels of vimentin were increased in CAL 27 cells treated with nicotine compared to control cells. Additionally, Snail expression levels were higher, but no significant difference was observed (Figure 1D).
Nicotine induces the expression of Prx1 and Ets1 in CAL 27 cells
To determine the potential interaction between Prx1 and the nuclear transcriptional factor Ets1, Co-IP assays were performed. As shown in Figure 1E, Prx1 and Ets1 were detected in total cell lysate samples, and their expression levels were higher in CAL 27 cells treated with nicotine for 7 days than in control cells. ChIP assays were conducted to observe the effect of nicotine on the binding activity of the transcriptional factor Ets1 to the promoter region of the Prx1 gene.
As shown in Figure 1F, compared to the control group, the combined activity of Ets1 and the promoter region of the Prx1 gene was significantly increased in the nicotine group (p<0.01). It is suggested that Ets1 in CAL 27 cells can be directly combined with the Prx1 gene promoter region to regulate Prx1 gene transcription. Nicotine can increase the binding activity of Ets1 and promote the transcription of the Prx1 gene.
Prx1 and Ets1 promote cell invasion, migration, and EMT in CAL 27 cells
To confirm the roles of Prx1 and Ets1 in EMT, cell invasion, and migration, we performed stable Prx1 knockdown and transient Ets1 knockdown in CAL 27 cells. Western blots were performed to detect the expression levels of EMT markers; Prx1 knockdown increased E-cadherin expression levels and decreased vimentin and Snail expression levels (Figure 2A and B). Ets1 knockdown decreased Prx1 and Snail expression levels and increased E-cadherin expression levels, while vimentin expression levels were not significantly different (Figure 2C and D). These results indicate that Prx1 promotes the EMT process, which may be regulated by Ets1.
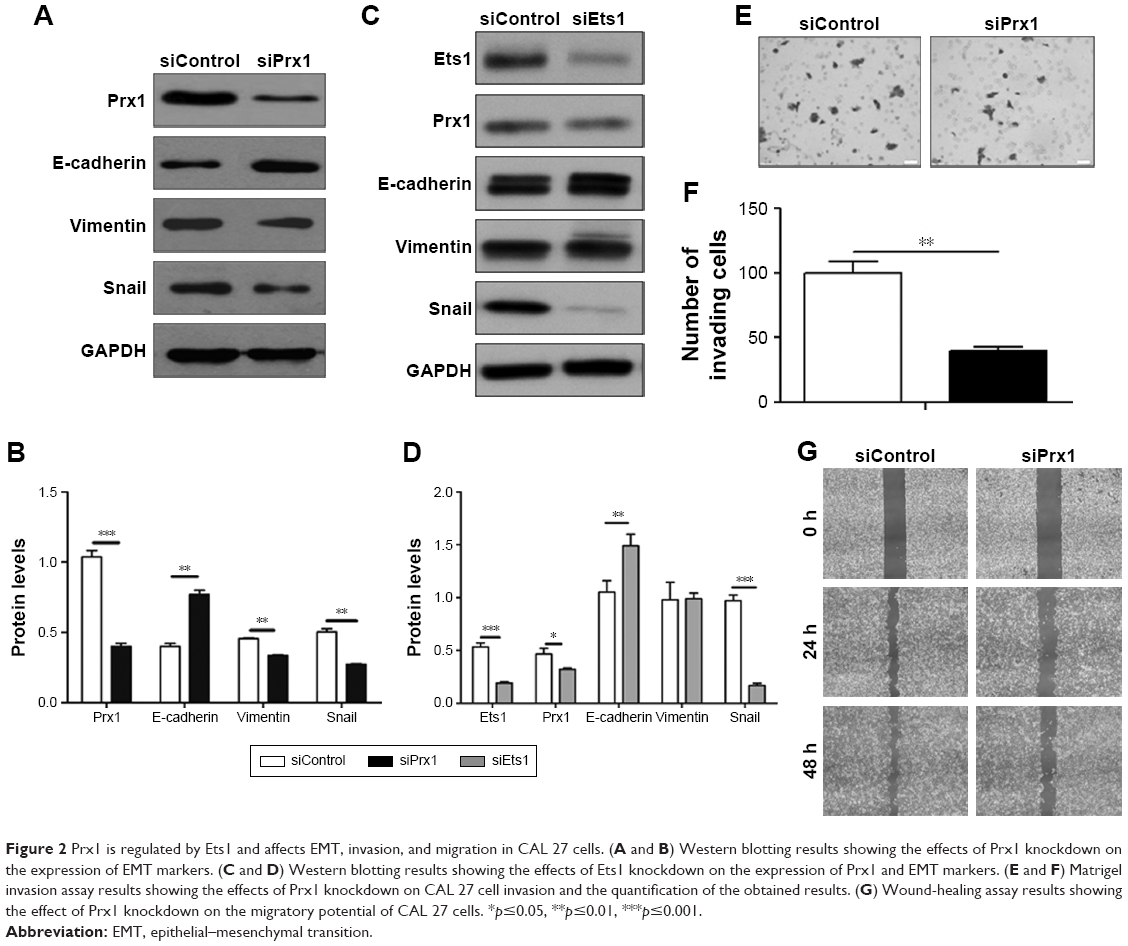 | Figure 2 Prx1 is regulated by Ets1 and affects EMT, invasion, and migration in CAL 27 cells. (A and B) Western blotting results showing the effects of Prx1 knockdown on the expression of EMT markers. (C and D) Western blotting results showing the effects of Ets1 knockdown on the expression of Prx1 and EMT markers. (E and F) Matrigel invasion assay results showing the effects of Prx1 knockdown on CAL 27 cell invasion and the quantification of the obtained results. (G) Wound-healing assay results showing the effect of Prx1 knockdown on the migratory potential of CAL 27 cells. *p≤0.05, **p≤0.01, ***p≤0.001. |
Matrigel invasion assay results demonstrated that after 24 h, a lower number of siPrx1 CAL 27 cells than siControl cells could penetrate through the Transwell membranes (Figure 2E and F). Consistently, after 24 h, the wound healing and migration rates were lower in siPrx1 cells than in siControl cells (Figure 2G).
Cervical lymph node metastasis model of tongue cancer establishment in mice
CAL 27 cells, siPrx1 and siControl, were orthotopically implanted into mouse tongues. Approximately 1 week later, white plaque-like lesions were observed on the tongue surfaces, and the mice started losing body weight. Tongue tumor sizes increased, which was accompanied by anabrosis, cachexia, and emaciation (Figure 3A[a]). The tongues and lymph nodes were much larger in the mice with tumors than in the negative controls (Figure 3A[b] and [c]). Fluorescence stereomicroscopy was used to confirm tumor formation and cervical metastases since the tumors in the mouse tongues and lymph nodes showed distinct green fluorescence (Figure 3B). At day 13 after CAL 27 cell implantation, H&E staining was used to confirm tumor formation and lymph node metastases (Figure 3C and D). We found that all tumors were moderately differentiated according to histological grades confirmed by H&E staining. Immunohistochemical staining with an anti-keratin antibody showed many positive cells in both tongue tissues and lymph nodes (Figure 3E and F), further demonstrating the successful establishment of a cervical metastasis mouse model of OSCC.
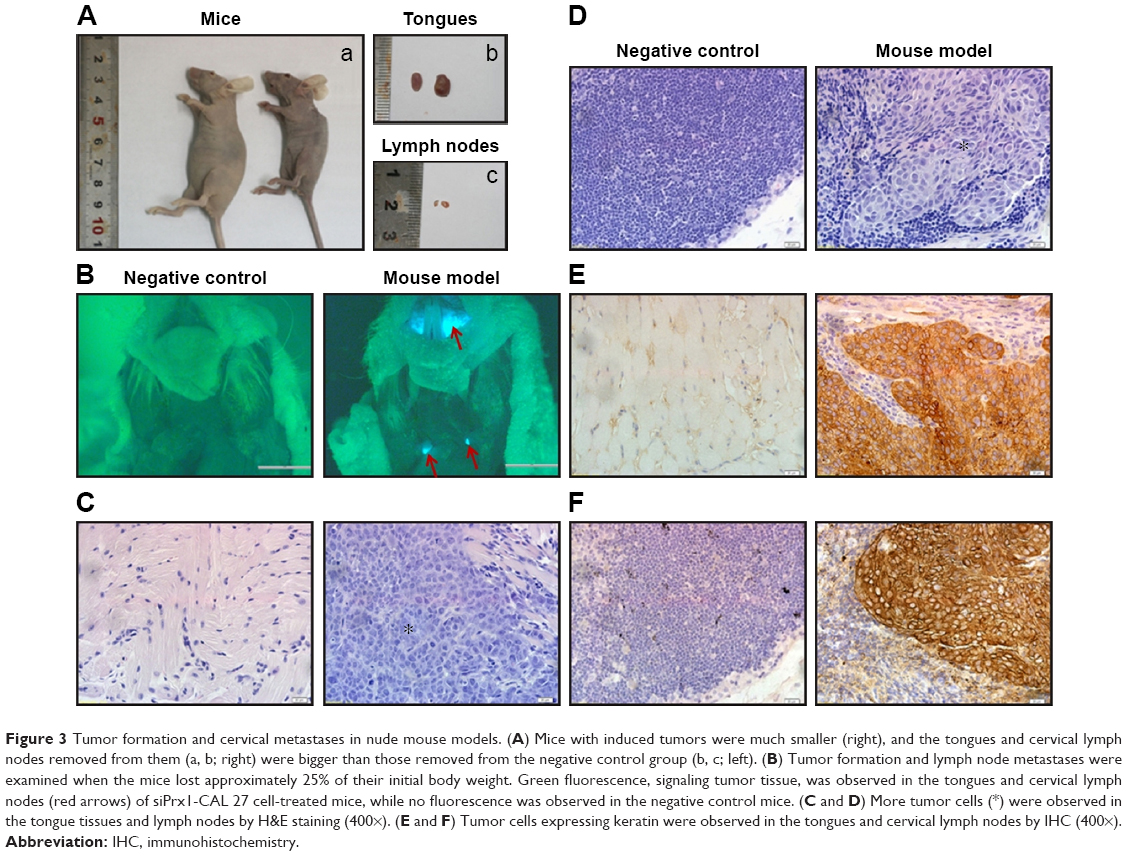 | Figure 3 Tumor formation and cervical metastases in nude mouse models. (A) Mice with induced tumors were much smaller (right), and the tongues and cervical lymph nodes removed from them (a, b; right) were bigger than those removed from the negative control group (b, c; left). (B) Tumor formation and lymph node metastases were examined when the mice lost approximately 25% of their initial body weight. Green fluorescence, signaling tumor tissue, was observed in the tongues and cervical lymph nodes (red arrows) of siPrx1-CAL 27 cell-treated mice, while no fluorescence was observed in the negative control mice. (C and D) More tumor cells (*) were observed in the tongue tissues and lymph nodes by H&E staining (400×). (E and F) Tumor cells expressing keratin were observed in the tongues and cervical lymph nodes by IHC (400×). |
Nicotine induces tumor metastasis and EMT in vivo
Cervical lymph node metastasis rates were analyzed in mice after CAL 27 cell implantation at day 13. Nicotine treatment, in comparison to control treatment, promoted cervical lymph node metastasis (Figure 4A). We examined the mRNA and protein expression levels of E-cadherin, vimentin, and Snail in the mouse tongues and lymph nodes. The results showed that compared to the control treatment, nicotine treatment downregulated E-cadherin mRNA expression and upregulated vimentin and Snail expression in tongues and lymph nodes (Figure 4B and C). Protein expression levels showed almost the same tendency, except for Snail protein expression, which did not significantly differ among different tongue samples (Figure 4D–F).
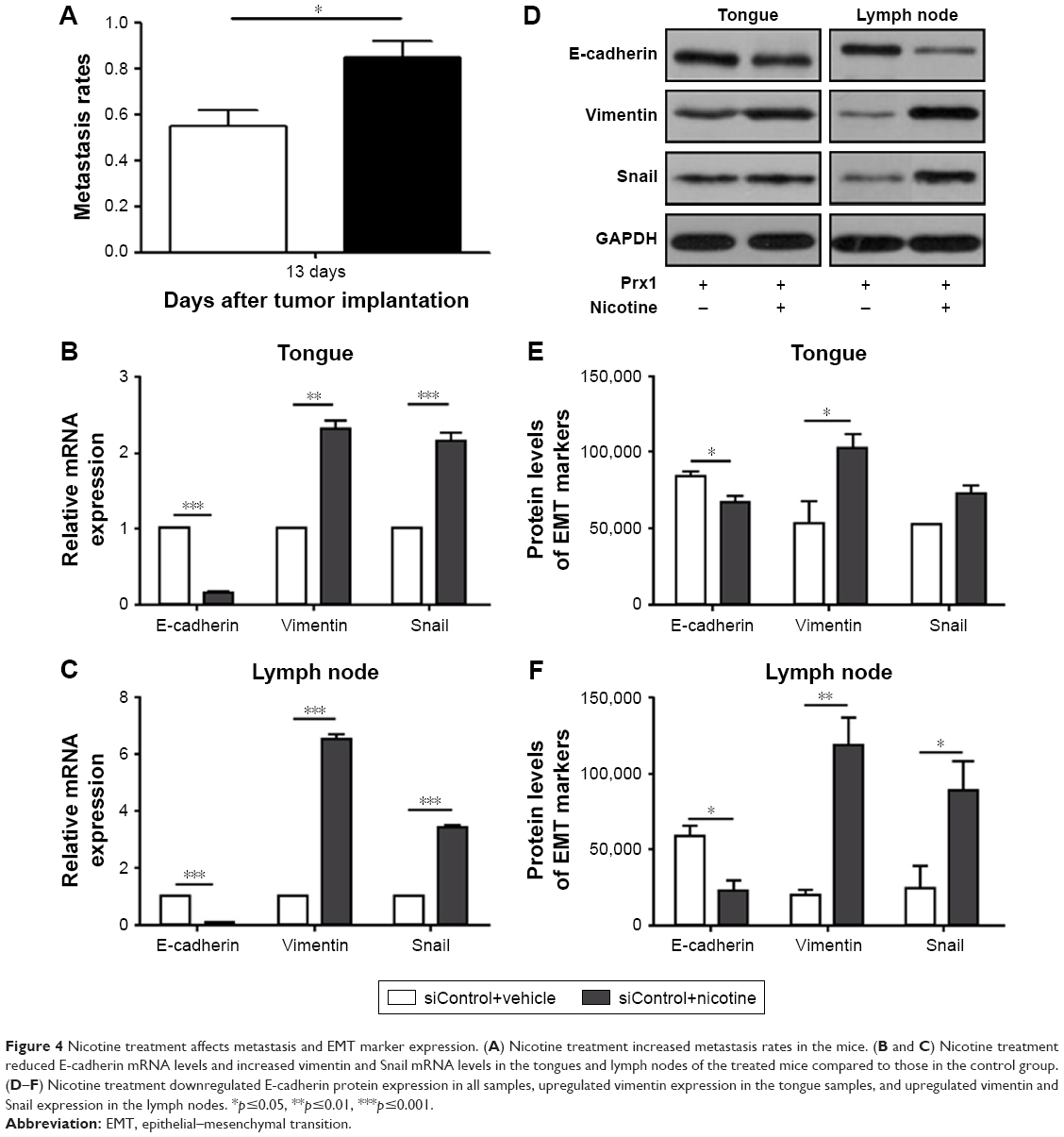 | Figure 4 Nicotine treatment affects metastasis and EMT marker expression. (A) Nicotine treatment increased metastasis rates in the mice. (B and C) Nicotine treatment reduced E-cadherin mRNA levels and increased vimentin and Snail mRNA levels in the tongues and lymph nodes of the treated mice compared to those in the control group. (D–F) Nicotine treatment downregulated E-cadherin protein expression in all samples, upregulated vimentin expression in the tongue samples, and upregulated vimentin and Snail expression in the lymph nodes. *p≤0.05, **p≤0.01, ***p≤0.001. |
Prx1 knockdown decreases cervical metastasis rates and inhibits EMT progression in vivo
Compared to the control group, Prx1 knockdown reduced metastasis rates (Figure 5A), upregulated E-cadherin mRNA, and downregulated vimentin and Snail mRNA expression in tongues and lymph nodes (Figure 5B and C). Protein expression levels showed the same tendency, except for vimentin expression in the tongue samples and E-cadherin levels in the lymph nodes, which were not significantly different between the groups (Figure 5D–F).
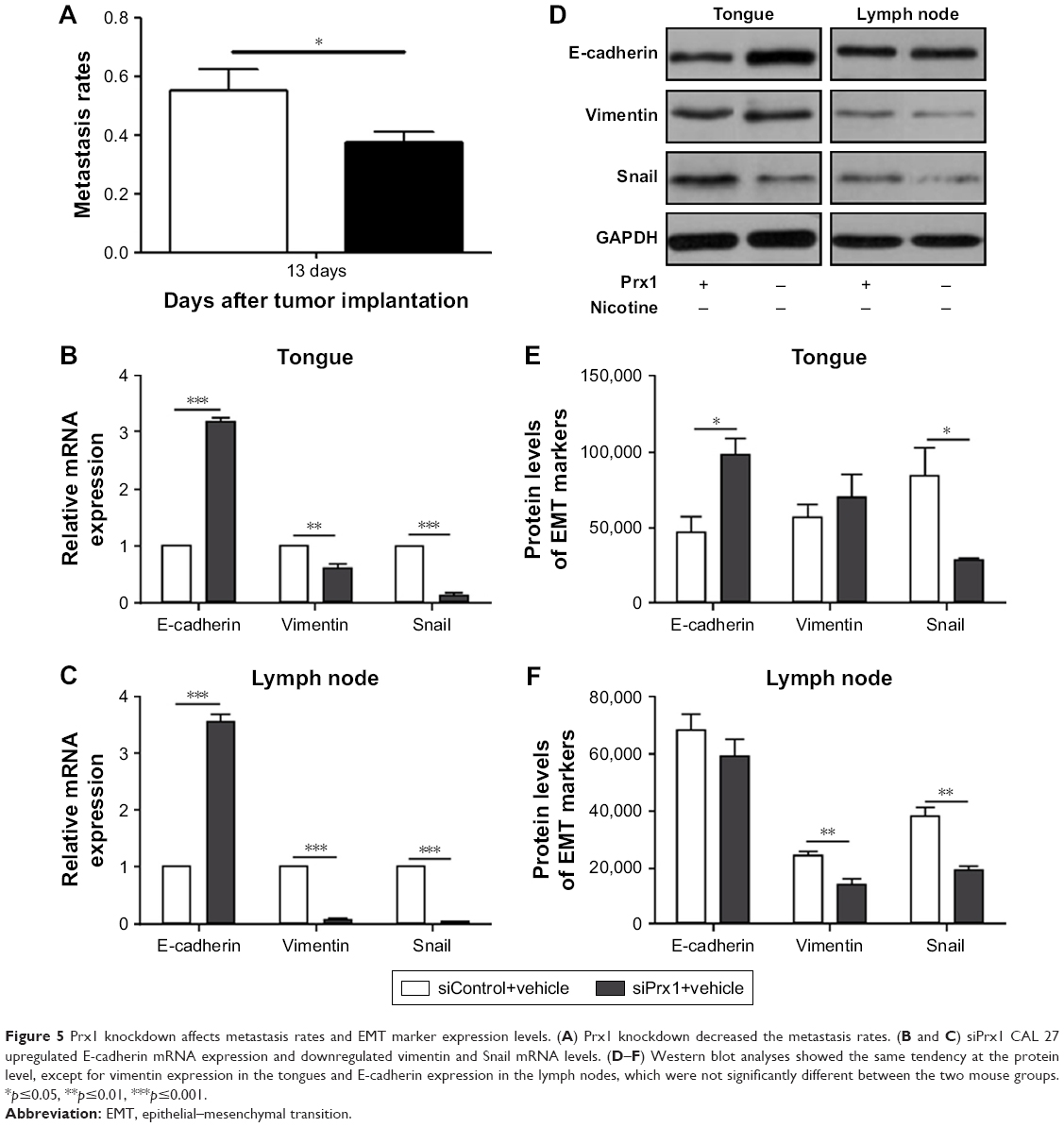 | Figure 5 Prx1 knockdown affects metastasis rates and EMT marker expression levels. (A) Prx1 knockdown decreased the metastasis rates. (B and C) siPrx1 CAL 27 upregulated E-cadherin mRNA expression and downregulated vimentin and Snail mRNA levels. (D–F) Western blot analyses showed the same tendency at the protein level, except for vimentin expression in the tongues and E-cadherin expression in the lymph nodes, which were not significantly different between the two mouse groups. *p≤0.05, **p≤0.01, ***p≤0.001. |
Nicotine induces lymph node metastasis and EMT through regulating Prx1 expression in vivo
To further explore the role of Prx1 in nicotine-induced metastasis, we investigated the effects of nicotine on lymph node metastasis rates and EMT-related marker expression in the investigated animals. The data obtained by analyzing the tongue and lymph node samples showed no significant differences between nicotine-treated and vehicle-treated mice after Prx1 knockdown (Figure 6). These results suggest that Prx1 plays a critical role in nicotine-induced metastasis in OSCC.
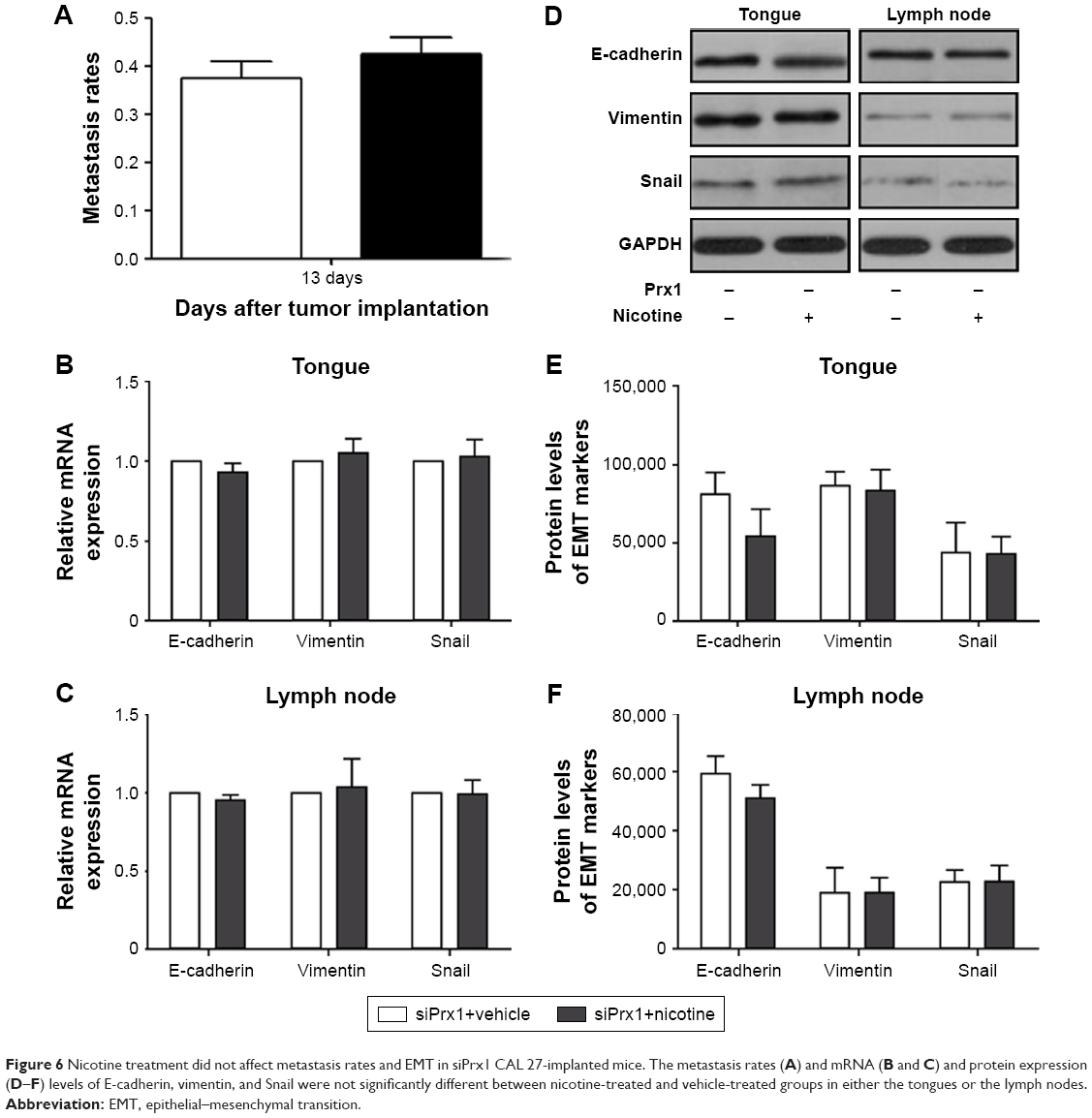 | Figure 6 Nicotine treatment did not affect metastasis rates and EMT in siPrx1 CAL 27-implanted mice. The metastasis rates (A) and mRNA (B and C) and protein expression (D–F) levels of E-cadherin, vimentin, and Snail were not significantly different between nicotine-treated and vehicle-treated groups in either the tongues or the lymph nodes. |
Discussion
Treatment failure in OSCC is primarily a consequence of tumor recurrence and metastasis.22 Nicotine is a source of exogenous oxidative stress, which is related to the pathogenesis of OSCC. Although some studies showed that nicotine is associated with cell proliferation, tumor growth, invasion, and metastasis in OSCC, the specific mechanisms underlying OSCC metastasis remain unclear.23–25
Previous studies have reported OSCC cervical metastasis models to investigate the cervical lymph node metastasis of oral cancer. Sun et al26 used H&E staining to observe that cancer in mouse tongues caused by implanting OSCC cell line Tca83 cells metastasized to the cervical lymph nodes at day 49. Here, we present an improved animal model of OSCC metastasis that enables us to identify and analyze lymph node metastases in vivo. This model was established by the orthotopic transplantation of GFP-labeled CAL 27 cells into mouse tongues; this process facilitated the identification of the development of metastases. Fluorescence stereomicroscopy was used for monitoring lingual tumor and cervical metastasis development in vivo. Following the development of tongue tumors at day 13 after CAL 27 cell implantation, our results showed rapid body weight loss and lymph node metastases due to nicotine treatment and the invasive and metastatic nature of CAL 27 cells. Using H&E and cytokeratin immunohistochemical staining, we confirmed that the GFP-positive tissues in mouse tongues and cervical lymph nodes were OSCCs. Our data suggest that examination performed by using fluorescence stereomicroscopy is more convenient, quicker, and more intuitive for the detection of tumor formation and early lymphatic metastases before animal sacrifice and biopsy.
Nicotine has been reported to induce EMT and promote the progression and metastasis of several malignant tumors.25 During the process of EMT, cell phenotypic changes are associated with alterations in the expression of many molecules, such as cadherins, vimentin, and transcription factors.16,27,28 E-cadherin plays a key role in cell adhesion. In addition, E-cadherin-regulated intercellular adhesion has been related to tumor cell invasion, metastasis, and oral cancer prognosis.29 Vimentin was shown to be upregulated during EMT, and its expression was associated with the acquisition of a migratory phenotype and invasion.30 As a zinc-finger transcription factor, Snail can promote EMT progression via inhibiting E-cadherin expression in the pathogenesis of cancer.15 EMT regulators are being investigated as potential molecular biomarkers and therapeutic targets for cancer therapies.31–33 Some studies have demonstrated that tumor cells treated with nicotine expressed significantly higher levels of mesenchymal markers and lower levels of epithelial markers, suggesting that nicotine can induce EMT in vitro.34–36 The previously obtained results are consistent with the results of our in vitro experiments, demonstrating that nicotine promotes OSCC cell invasion and migration accompanied by increased EMT progression and Prx1 upregulation in CAL 27 cells. To investigate the roles and molecular mechanisms underlying the effects of nicotine on OSCC metastases, a 6% nicotine solution was applied topically to mouse tongues. We observed that nicotine increased OSCC metastasis rates, downregulated E-cadherin expression, and upregulated vimentin and Snail expression in tongue and lymph node samples. These data indicate that nicotine may induce cervical lymph node metastases through regulating EMT progression during OSCC pathogenesis.
Our previous study demonstrated that nicotine treatment can increase ROS levels and induce oxidative stress in OSCC cells, and Prx1 upregulation is associated with nicotine-induced oxidative stress.13 In another previous report, Prx1 overexpression has been shown to enhance the growth of OSCC tumor xenografts.37 However, to date, the relationship between Prx1 and OSCC metastasis in vivo has not been investigated. In this study, we found that nicotine treatment upregulated the expression of Prx1. To investigate the role of Prx1, we generated stable Prx1 knockdown CAL 27 cells. Prx1 knockdown inhibited cell invasion, migration, and EMT progression, which led us to establish an additional model of OSCC cervical metastasis using siPrx1 CAL 27 cells. We demonstrated that Prx1 knockdown significantly inhibited tongue cancer metastasis rates and EMT progression in vivo and that it upregulated E-cadherin expression and downregulated vimentin and Snail expression in both tongue and lymph node samples. These findings indicate that Prx1 plays a key role in the nicotine-induced invasion and metastasis of OSCC. However, a study conducted by Yanagawa et al8 provided a different perspective regarding the role of Prx1 in OSCC. They showed that low Prx1 expression levels correlated with increased lymph node metastasis risk.
Ets1 has been shown to be a key transcription factor involved in oxidative stress, and its expression was shown to be upregulated in invasive high-grade tumors and correlated with a higher incidence of lymph node metastasis.9,38 Shiota et al12 showed that inhibiting Ets1 expression significantly decreased Prx1 expression in prostate cancer PC3 cells. To further explore the mechanism of Prx1 in nicotine-induced OSCC, we examined the relationship between Prx1 and Ets1 in OSCC cells. We found that nicotine induced Ets1 expression in CAL 27 cells, while Ets1 knockdown inhibited Prx1 expression and EMT progression. Furthermore, Co-IP and ChIP results confirmed that nicotine could significantly upregulate the binding of the transcription factor Ets1 to the Prx1 gene promoter region to regulate Prx1 gene transcription and expression. Taken together, the data indicate that nicotine promotes cell invasion and lymph node metastasis development through regulating the Ets1/Prx1/EMT signaling pathway during the pathogenesis of OSCC.
Conclusion
Our results demonstrate for the first time that nicotine promotes invasion, migration, and lymph node metastasis through regulating Prx1/EMT signaling in tongue cancer. Our findings provide significant mechanistic and functional insights into OSCC pathogenesis, and Prx1 may represent a potential diagnostic marker and a therapeutic target for OSCC treatment.
Acknowledgments
This work was supported by the Beijing Natural Science Foundation of China (Project No 7152066) and the National Natural Science Foundation of China (Project No 81470752). This paper was presented at the 2017 AACR Special Conference: Advances in Modeling Cancer in Mice: Technology, Biology, and Beyond as a poster presentation with interim findings. The poster’s abstract was published in “Poster Abstracts” in Cancer Research Volume 78, Issue 10 Supplement, pp. A51: https://doi.org/10.1158/1538-7445.DOI:10.1158/1538-7445.MOUSEMODELS17-A51.
Disclosure
The authors report no conflicts of interest in this work.
References
Greenlee RT, Taylor M, Sherry B, et al. Cancer statistics 2000. CA Cancer J Clin. 2000;50:7–33. | ||
Bagan JV, Scully C. Recent advances in Oral Oncology 2007: epidemiology, aetiopathogenesis, diagnosis and prognostication. Oral Oncol. 2008;44:103–108. doi:10.1016/j.oraloncology.2008.01.008 | ||
Schottenfeld D. The etiology and prevention of aerodigestive tract cancers. Adv Exp Med Biol. 1992;320:1–19. | ||
Sanner T, Grimsrud TK. Nicotine: carcinogenicity and effects on response to cancer treatment – a review. Front Oncol. 2015;5:196. doi:10.3389/fonc.2015.00196 | ||
Ishii T, Yamada M, Sato H, et al. Cloning and characterization of a 23-kDa stress-induced mouse peritoneal macrophage protein. J Biol Chem. 1993;268:18633–18636. | ||
Yanagawa T, Ishikawa T, Ishii T, et al. Peroxiredoxin I expression in human thyroid tumors. Cancer Lett. 1999;145:127–132. doi:10.1016/S0304-3835(99)00243-8 | ||
Noh DY, Ahn SJ, Lee RA, et al. Overexpression of peroxiredoxin in human breast cancer. Anticancer Res. 2001;21:2085–2090. | ||
Yanagawa T, Iwasa S, Ishii T, et al. Peroxiredoxin I expression in oral cancer: a potential new tumor marker. Cancer Lett. 2000;156:27–35. doi:10.1016/S0304-3835(00)00434-1 | ||
Pande P, Mathur M, Shukla NK, Ralhan R. Ets-1: a plausible marker of invasive potential and lymph node metastasis in human oral squamous cell carcinomas. J Pathol. 1999;189:40–45. doi:10.1002/(SICI)1096-9896(199909)189:1<40:AID-PATH405>3.0.CO;2-# | ||
Cao L, Xie B, Yang X, et al. MiR-324-5p suppresses hepatocellular carcinoma cell invasion by counteracting ECM degradation through post-transcriptionally downregulating ETS1 and SP1. PLoS One. 2015;10:e0133074. doi:10.1371/journal.pone.0133074 | ||
Chang XZ, Li DQ, Hou YF, et al. Identification of the functional role of peroxiredoxin 6 in the progression of breast cancer. Breast Cancer Res. 2007;9:R76. doi:10.1186/bcr1789 | ||
Shiota M, Izumi H, Miyamoto N, et al. Ets regulates peroxiredoxin1 and 5 expressions through their interaction with the high-mobility group protein B1. Cancer Sci. 2008;99:1950. | ||
Zhao YH, Zhang M, Yan F, et al. Nicotine-induced upregulation of antioxidant protein Prx1 in oral squamous cell carcinoma. Chin Sci Bull. 2013;58:1912–1918. | ||
Mathias RA, Gopal SK, Simpson RJ. Contribution of cells undergoing epithelial–mesenchymal transition to the tumour microenvironment. J Proteomics. 2013;78:545–557. doi:10.1016/j.jprot.2012.10.016 | ||
Brzozowa M, Michalski M, Wyrobiec G, et al. The role of Snail1 transcription factor in colorectal cancer progression and metastasis. Contemp Oncol (Pozn). 2015;19:265–270. doi:10.5114/wo.2014.42173 | ||
Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119:1420–1428. doi:10.1172/JCI39104 | ||
Nieto MA. The ins and outs of the epithelial to mesenchymal transition in health and disease. Annu Rev Cell Dev Biol. 2011;27:347–376. doi:10.1146/annurev-cellbio-092910-154036 | ||
Tam WL, Weinberg RA. The epigenetics of epithelial-mesenchymal plasticity in cancer. Nat Med. 2013;19:1438–1449. doi:10.1038/nm.3336 | ||
Zeisberg M, Neilson EG. Biomarkers for epithelial-mesenchymal transitions. J Clin Invest. 2009;119:1429–1437. doi:10.1172/JCI36183 | ||
Nelson JD, Denisenko O, Bomsztyk K. Protocol for the fast chromatin immunoprecipitation (ChIP) method. Nat Protoc. 2006;1:179–185. doi:10.1038/nprot.2006.27 | ||
Yu J, Hui AY, Chu ESH, et al. Expression of a cyclo-oxygenase-2 transgene in murine liver causes hepatitis. Gut. 2007;56:991–999. doi:10.1136/gut.2006.097923 | ||
Myers JN. Holsinger FC, Jasser SA, et al. An orthotopic nude mouse model of oral tongue squamous cell carcinoma. Clin Cancer Res. 2002;8:293–298. | ||
Heeschen C, Jang JJ, Weis M, et al. Nicotine stimulates angiogenesis and promotes tumor growth and atherosclerosis. Nat Med. 2001;7:833–839. doi:10.1038/89961 | ||
Dasgupta P, Chellappan SP. Nicotine-mediated cell proliferation and angiogenesis: new twists to an old story. Cell Cycle. 2006;5:2324–2328. doi:10.4161/cc.5.20.3366 | ||
Davis R, Wasia R, Sarmistha B, et al. Nicotine promotes tumor growth and metastasis in mouse models of lung cancer. PLoS One. 2009;4:e7524. doi:10.1371/journal.pone.0007524 | ||
Sun R, Zhang JG, Guo CB. Establishment of cervical lymph node metastasis model of squamous cell carcinoma in the oral cavity in mice. Chin Med J (Engl). 2008;121:1891–1895. | ||
Thiery JP. Epithelial–mesenchymal transitions in development and pathologies. Curr Opin Cell Biol. 2003;15:740–746. | ||
Pasquier J, Abu-Kaoud N, Thani HA, Rafii A. Epithelial to mesenchymal transition in a clinical perspective. J Oncol. 2015;2015:792182. doi:10.1155/2015/792182 | ||
Diniz-Freitas M, García-Caballero T, Antúnez-López J, Gándara-Rey JM, García-García A. Reduced E-cadherin expression is an indicator of unfavourable prognosis in oral squamous cell carcinoma. Oral Oncol. 2006;42:190–200. doi:10.1016/j.oraloncology.2005.07.010 | ||
Gilles C, Polette M, Zahm JM, et al. Vimentin contributes to human mammary epithelial cell migration. J Cell Sci. 1999;112:4615–4625. | ||
Pirozzi G, Virginia T, Rosa C, et al. Epithelial to mesenchymal transition by TGFβ-1 induction increases stemness characteristics in primary non small cell lung cancer cell line. PLoS One. 2011;6:e21548. doi:10.1371/journal.pone.0021548 | ||
Nagaraj NS, Datta PK. Targeting the transforming growth factor-beta signaling pathway in human cancer. Expert Opin Investig Drugs. 2010;19:77–91. doi:10.1517/13543780903382609 | ||
Park CY, Min KN, Son JY, et al. An novel inhibitor of TGF-β type I receptor, IN-1130, blocks breast cancer lung metastasis through inhibition of epithelial-mesenchymal transition. Cancer Lett. 2014;351:72–80. doi:10.1016/j.canlet.2014.05.006 | ||
Yu MA, Kiang A, Wang-Rodriguez J, et al. Nicotine promotes acquisition of stem cell and epithelial-to-mesenchymal properties in head and neck squamous cell carcinoma. PLoS One. 2012;7:e51967. doi:10.1371/journal.pone.0051967 | ||
Wu SQ, Lv YE, Lin BH, et al. Silencing of periostin inhibits nicotine-mediated tumor cell growth and epithelial-mesenchymal transition in lung cancer cells. Mol Med Rep. 2013;7:875–880. doi:10.3892/mmr.2013.1267 | ||
Dasgupta P, Rizwani W, Pillai S, et al. Nicotine induces cell proliferation, invasion and epithelial-mesenchymal transition in a variety of human cancer cell lines. Int J Cancer. 2009;124:36–45. doi:10.1002/ijc.23894 | ||
Zhang M, Hou M, Ge L, et al. Induction of peroxiredoxin 1 by hypoxia regulates heme oxygenase-1 via NF-κB in oral cancer. PLoS One. 2014;9:e105994. doi:10.1371/journal.pone.0105994 | ||
Calli AO, Sari A, Cakalagaoglu F, Altinboga AA, Oncel S. ETS-1 proto-oncogene as a key newcomer molecule to predict invasiveness in laryngeal carcinoma. Pathol Res Pract. 2011;207:628–633. doi:10.1016/j.prp.2011.07.010 |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.