")
Back to Journals » OncoTargets and Therapy » Volume 13
NHE1 Mediates 5-Fu Resistance in Gastric Cancer via STAT3 Signaling Pathway
Authors Sun Z, Luan S, Yao Y, Qin T, Xu X, Shen Z, Yao R, Yue L
Received 3 April 2020
Accepted for publication 5 August 2020
Published 24 August 2020 Volume 2020:13 Pages 8521—8532
DOI https://doi.org/10.2147/OTT.S256274
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Carlos E Vigil
Zhenni Sun,1 Shufang Luan,1 Yasai Yao,1 Tao Qin,1 Xiaomei Xu,1 Zan Shen,2 Ruyong Yao,3 Lu Yue1
1Department of Oncology, Qingdao Municipal Hospital, School of Medicine, Qingdao University, Qingdao, Shandong Province 266071, People’s Republic of China; 2Department of Oncology, The Sixth People’s Hospital, Medical College of Shanghai Jiao Tong University, Shanghai 200233, People’s Republic of China; 3Central Laboratory, Qingdao Municipal Hospital, School of Medicine, Qingdao University, Qingdao, Shandong Province 266071, People’s Republic of China
Correspondence: Lu Yue
Department of Oncology, Qingdao Municipal Hospital, School of Medicine, Qingdao University, Shandong Province 266071, People’s Republic of China
Tel + 86 18887112038
Fax +86 871-68250639
Email [email protected]
Background: Several recent studies have addressed the role of Na+/H+ exchanger isoform 1 (NHE1) in tumor cell growth and apoptosis, including in gastric cancer. However, the role of NHE1 expression related to the 5-Fu resistance in gastric cancer has not been investigated.
Methods: The expression of NHE1 was examined by qPCR in the SGC7901/5-FU cell line and its parental cell line. pcDNA3.1-NHE1 and NHE1-siRNA were transfected to SGC7901/5-FU resistance cells and cell apoptosis was detected via TUNEL assay. The upstream activators in NHE1 mediated 5-Fu resistant gastric cancer cells were detected by Western blot and immunofluorescent.
Results: A significant increase of the expression of NHE1 was observed in SGC7901 5-FU resistance cells compared to the GES-1 and SGC7901 cell line. NHE1 can suppress the cell apoptosis of SGC7901 5-FU resistance cells and involved in cell cycle. Also, the migration and invasion of SGC7901 5-FU resistance cells were promoted by NHE1. NHE1 also increases the intracellular pH. The results of Western blot analysis showed that NHE1 overexpression induced an increase in the expression of phosphorylated activator transcription factor 3 (pSTAT3). The more obvious phosphorylated level was shown in the phosphorylated STAT3 at pSTAT3tyr705. Further investigations revealed that the constitutive activation of STAT3 may be induced by JAK1 and JAK2, and thus effect the 5-FU resistance by regulating NHE1.
Discussion: In summary, our findings provided evidence that NHE1 contributed to 5-Fu resistance in gastric cancer cells by regulating the JAK/STAT3 pathway. Therefore, NHE1 can be a useful marker for predicting and monitoring 5-Fu resistance.
Keywords: NHE1, 5-FU resistance, JAK/STAT3 pathway, gastric cancer
Introduction
Despite a considerable decrease in its incidence rate in many developed countries, gastric cancer (GC) remains the fifth most commonly diagnosed malignancy and the third most lethal cancer worldwide.1 More than two-thirds of gastric cancer patients are diagnosed in advanced stages clinically. Surgery is the only treatment modality with curative intention, and the first choice for localized gastric cancer, but recurrence is common even with complete resection.2–4 Therefore, a multidisciplinary approach is required for optimal treatment. Systemic chemotherapy is one of the common palliative treatments clinically.5–7 Recently, intravenous 5-fluorouracil (5-FU) and its derivatives (S-1 and Xeloda etc.) remain the most widely used drugs in the treatment of gastrointestinal cancers.8 However, many patients still result in recurrence after several courses of 5-Fu-based chemotherapy due to the rapid emergence of drug resistance, which has become a major clinical problem. A previous study showed that the response rate of 5-FU monotherapy is only 10–30%, and its efficacy is unsatisfactory.9 Therefore, the molecular mechanisms underlying the development of 5-FU chemoresistance in gastric cancer have drawn attention.
The interstitial environment in malignant tumors is acidic as a result of accumulation of lactic acid and other acidic metabolites. The maintenance of pH homeostasis is chiefly regulated by Na+/H+ exchanger isoform 1 (NHE1), a transmembrane protein that mediates the 1:1 exchange of extracellular sodium for intracellular protons across the cell membrane.10,11 In mammals, aside from its pH regulatory role, NHE1 is also important in regulation of cell volume, proliferation, and differentiation and in metastasis of some types of tumor cells.12,13 NHE1 has also been detected in various human tumor samples to facilitate the development of resistance to chemotherapy drugs because of disruption of the absorption of weakly basic chemotherapy agents.14 However, the mechanistic contributions and pathologic significance of NHE1 activation in elaborating 5-Fu chemoresistance of GC have little been demonstrated.
In this study, we sought to determine the relationship between NHE1 expression and chemotherapy response to 5-Fu in gastric cancer cells and further investigate its possible mechanism.
Materials and Methods
Cell Culture and the Establishment of 5-FU Resistance Cell
Human gastric cancer cell lines AGS, BGC823, SGC7901 were obtained from the central laboratory of Affiliated Hospital of Qingdao University. The use of the cell lines was approved by the ethics committee of the Affiliated Hospital of Qingdao University. BGC823 and SGC7901 cells were authenticated by STR profile. All the cells were cultured in RPMI 1640 (Hyclone, USA) containing 10% fetal bovine serum (FBS) (Hangzhou Sijiqing Biotech, Co. Ltd. China) and 1% Penicillin/Streptomycin in a humidified 5% CO2 at 37°C. The 5-Fu (Amersham Biosciences, England) was diluted to a final concentration of 10 mg/L. EIPA were obtained from Sigma (St. Louis, MO, USA).
The variants of SGC7901 cells, SGC7901/FU, which are 5-Fluorouracil resistant cells, were established by step-wise exposure as described before.15 In brief, parental cells were exposed to escalating concentrations of 5-Fluorouracil, ranging from 1 μM to 10 mM for more than 6 months.
Construction of NHE1 Expression Vector and Transfection
NHE1 expression vectors pcDNA3.1-NHE1 and siRNA-NHE1 were purchased from Genepharma (Shanghai). The pcDNA3.1-NHE1, siRNA-NHE1, pcDNA3.1 and non-specific siRNA were transfected to SGC7901 cells via Lipofectamine 2000 agent (Invitrogen, Carlsbad, CA, USA) in accordance with the manufacturer’s protocol. Forty-eight hours after transfection, fresh RPMI 1640 medium with 10% FBS was added. NHE1 inhibitor EIPA (25 μM, GLPBIO, CA, USA) was dissolved and stocked in DMSO and applied to the cells in the culture medium.
Cytotoxicity Assay
5-Fu resistance of cell lines was examined using MTT assay. Cells were seeded at 5000–8000 cells/well in 96-well plates in 100 μL fresh medium with the presence of different concentrations of 5-Fu, and each concentration was repeated in triplicate. After a 24 hour incubation, 10 μL MTT (3-(4,5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium) (5 mg/mL) (Sigma, St. Louis, MO, USA) was added to each well and incubated for 4 hours at 37°C. Dimethyl sulfoxide (DMSO) (100 μL/well) was added to dissolve the blue formazan crystals converted by cells. The absorbance at 570 nm was measured with a microplate reader (BioTek, USA).
TUNEL Assay
The cell lines were treated with 2.5 μg/mL 5-FU for 48 hours according to the previous research and the results obtained in the cytotoxicity assay.16 The number of apoptotic cell deaths was evaluated by transferase (TdT)-mediated dUTP nick-end labeling (TUNEL) assay. The TUNEL assay was performed according to the manufacturer’s instructions. In brief, cells were fixed with 4% formaldehyde in PBS and then incubated in a TUNEL reaction mixture in the dark for 1 hour at 37°C. Labeled samples were visualized with a fluorescence microscope (Olympus, Tokyo, Japan) and examined by Zen 2011 software (Carl Zeiss, Weimar, Germany).
Western Blot
Cells were collected and lysed in RIPA lysis buffer (containing 0.01% PMSF, 150 nmol/L Tris (pH=8), 0.1% SDS, 0.2% EDTA, 1% Triton X-100, 1% sodium deoxycholate). Protein concentration of cellular extracts was determined by Bicinchoninic Acid (BCA) Protein Assay (KeyGen Biotech, China) following the manufacturer’s instructions. Equal amounts (30–50 μg) of proteins were applied to a 8–12% SDS-polyacrylamide separating gel and transferred to a polyvinylidene fluoride (PVDF) membrane (Millipore, USA). The membrane was blocked with 5% skim milk powder and then probed with indicated primary antibodies with gentle shaking at 4°C overnight, followed by appropriate HRP-conjugated secondary antibodies. Antibody staining was visualized by enhanced chemiluminescence (Millipore, USA) and the images were analyzed by Quantity One V4.31 (Bio-Rad, USA). β-actin was used as the loading control. The information of primary antibodies was listed in Table 1.
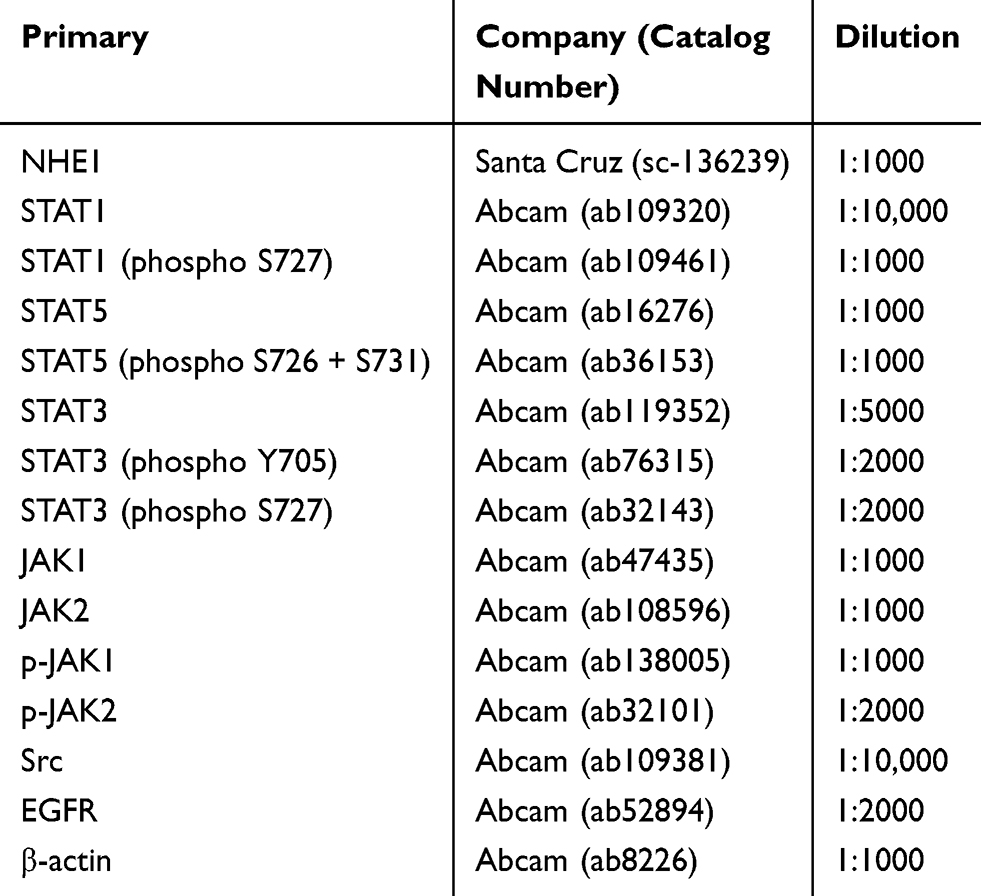 |
Table 1 Primary Antibodies |
pHi Assay
The intracellular pH (pHi) was measured as described earlier.13 GC cells were loaded with the acetoxy-methyl ester derivative of the pH-sensitive dye 2,7-biscarboxyethl-5(6)-carboxyfluorescein (BCECF) (Eugene, OR, USA) for 30 minutes. The acetoxymethyl ester form of BCECF enters the cell and is rapidly converted to the anionic-free acid form by intracellular esterizes. Then cells were washed with HEPES buffered solution to remove the extracellular. Fluorescence of intracellular BCECF was detected by Nikon A1-Rsi confocal microscopy at the excitation wavelength of 530 nm.
Co-Immunoprecipitation (Co-IP)
Cells were lysed in 20 mM Tris-HCl buffer (pH=7.4) containing 150 mM NaCl, 2 mM EDTA, 1% Nonidet P-40 (NP-40), 50 mM NaF, 1 mM Na3VO4, 1 mM Na2MoO4, 10 mM/mL aprotinin, and 10 mM/mL leupeptin. Protein concentration of cellular extracts was determined as described in Western blot assay. The extracts were incubated with antibodies to NHE1, JAK1, or JAK2 immobilized on protein A-sepharose (Santa Cruz Biotechnology, CA, USA) at 4°C overnight with constant rotation. The immunoprecipitates were extensively washed with lysis buffer and subjected to electrophoresis through sodium dodecyl sulfate (SDS)-polyacrylamide gel. The separated proteins were then analyzed by Western blot assay.
Enzyme-Linked Immunosorbent Assay (ELISA)
The cells were seeded in 6-well plates in duplicate wells before the indicated treatment. The supernatants were collected and centrifugated at 5000 rpm. The concentration of caspase 3 was measured using ELISA kits (eBioscience, Austria) according to the manufacturer’s instructions.
Real-Time Reverse Transcription Polymerase Chain Reaction (RT-PCR)
Total cellular RNA was extracted with Trizol Reagent (Invitrogen, USA) and quantified with a spectrophotometer. Then the samples were reverse-transcribed using random hexamers and reverse transcriptase (Invitrogen, USA) to obtain cDNA. A PCR reaction system was prepared using cDNA and SYBR Green Real-Time PCR Master Mixes (Thermo Fisher Scientific, USA). Quantitative real-time PCR was performed using the UltraSYBR (with ROX, CWbio. Co. Ltd., Beijing, China). The samples were processed using an ABI StepOne (ABI, Foster City, CA, USA). The PCR amplification included an initial denaturation at 95°C for 1 minute, 35 cycles of denaturation at 95°C for 1 minute, annealing at 60°C for 2 minutes, and extension for 30 seconds, at 72°C. Primers were designed and validated by Invitrogen Biotechnology Co. Ltd. The sequences of primers used in reverse transcription PCR are listed in Table 2. Results of the log-linear phase of the growth curve were analyzed and relative quantification was performed using the 2-ΔΔCt method with β-actin as a house-keeping gene. Each sample was tested in triplicate.
 |
Table 2 The Sequences of Primers Used in Reverse Transcription PCR |
Immunofluorescent
The cells were inoculated in 24-well slides. They were then grown to about 80% and fixed by 4% polyformaldehyde for 15 minutes at room temperature and washed with PBS. After blocking with normal goat serum (Thermo Fisher Scientific, USA), cells were stained with the primary antibody for STAT3 (Cell Signaling Technology, USA), p-STAT3 (Ser 727) (omnimabs, USA), and p-STAT3 (Tyr 705) (SAB, USA). Then the primary antibody was visualized with Alexa Fluor® 594-conjugated goat anti-rabbit IgG (abcam, USA). Cell nuclei were stained with DAPI. Fluorescence images were taken using a fluorescence microscope.
Statistical Analysis
Data were expressed as mean±SD, and analyzed with Graph-Pad Prism 5.0 software (GraphPad Software, San Diego, CA). Statistical analyses were performed using one-way ANOVA, followed by Tukey’s post-test. Differences with P<0.05 were considered statistically significant.
Results
Verification of SCG-7901/5‑FU Cells Establishment
The expression of NHE1 in three different human gastric cancer cell lines, ie, AGS, BGC823, SGC7901, was detected by Western blot. The AGS cell line owed the highest expression of NHE1, while the lowest expression of NHE1 appeared in BGC823 cell lines (P<0.05, Figure 1A and B). Moreover, among three human gastric cancer cell lines, BGC823 was the most sensitive cell line to the 5-FU (IC50=7.502±0.871), while the AGS cell line was the least sensitive one (IC50=11.211±0.871) (P<0.05, Figure 1C and D). Collectively, the SGC7901 cell line was more suitable to transfected pcDNA3.1-NHE1/NHE1-siRNA to overexpress/knockdown NHE1 in cell lines and was therefore selected. To estimate the resistance of SCG-7901/5-FU cells to 5-FU, the IC50 of 5-FU in SGC7901/5-FU and its parental cell was evaluated by MTT assay. SCG-7901/5-FU showed poorer response to 5-FU compared with SCG-7901 cells, as evidenced by the increased IC50 (Resistance index=11.995±0.314) (Figure 1E and F). Also, the expression of drug resistance related protein P-glycoprotein (P-gp), MDR-1, and GST-π were detected via Western blot (Figure 1G and H). The results showed that the expressions were increased in SCG-7901/5‑FU cells compared with SCG-7901 cells, which verified the successful establishment of SCG-7901/5‑FU cells.
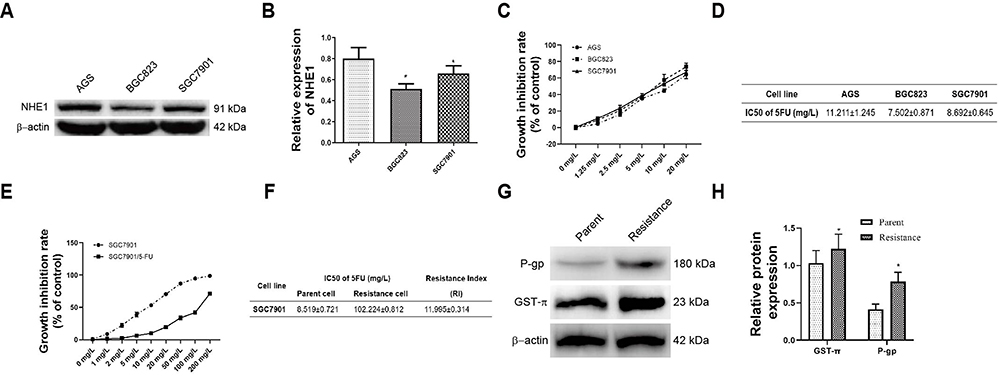 |
Figure 1 Verification of SCG-7901/5‑FU cells establishment. (A and B) The NHE1 expression in three different gastric cell lines. (C) Growth inhibition rate of three different gastric cell lines in response to 5-FU. (D) IC50 value of the parental gastric cancer (SGC7901) and 5-fluorouracil resistant gastric cancer cell lines SGC7901/FU. (E) Growth inhibition rate of the parental gastric cancer (SGC7901) and 5-fluorouracil resistant gastric cancer cell lines SGC7901/FU in response to 5-FU. (F) Resistance Index of the parental gastric cancer line SGC7901 and its 5-fluorouracil resistant cell lines to 5-FU. (G and H) Drug resistance related proteins (GST-π and P-gp) were up-regulated in the 5-fluorouracil resistant gastric cancer cell SGC7901/FU contrast to the parental cell lines. *P<0.05, compared to the control group. |
NHE1 Attenuate the Cell Apoptosis Mediated by 5-FU and Involved in the Cell Cycle
Next, the expression of NHE1 in SCG-7901/5‑FU cells was detected. The expression of NHE1 in SGC7901 and SGC7901/5-FU was obviously increased compared to GES-1 cells (Figure 2A). Also, when the pcDNA3.1-NHE1 was transfected, the expression of NHE1 was increased compared with the vector transfection, while the expression was decreased when NHE1-siRNA was transfected, which indicated the successful transfection of the plasmids (P<0.05, Figure 2B and C). We then detected the cell apoptosis by TUNEL assay. EIPA was identified as a selective inhibitor of NHE1 isoform that does not interfere with other pH-regulating transporters.17 Therefore, EIPA was used as a NHE1 inhibitor in the subsequent researches. 5-FU was applied at a concentration of 80 μg/mL. After the pcDNA3.1-NHE1 was transfected, and the percentage of cell apoptosis was decreased significantly. When the NHE1-siRNA was transfected, the percentage of cell apoptosis was increased (Figure 2D, P<0.01). The result of caspase 3 concentration showed the same trend (Figure 2E, P<0.01). Also, Western blot was used to detect the expression of downstream protein Bcl-xl and Bcl-2. Compared to the control group, the expression of Bcl-xl and Bcl-2 was increased after pcDNA3.1-NHE1 transfection, while when the NHE1-siRNA was transfected, the expression of Bcl-xl and Bcl-2 was significantly decreased (Figure 2F and G, P<0.01). This indicated that 5-FU could mediate the cell apoptosis of SGC7901 cell lines, while NHE1 overexpression attenuates the cell apoptosis mediated by 5-FU. Flow cytometry was also applied to find the effect of NHE1 on the cell cycle of SGC7901. We found that the percentage of cell numbers in the G1 phase was decreased (48.830±2.826% vs 62.157±2.386%, P<0.01) and the percentage in the S phase was increased (45.353±1.744% vs 35.983±1.876%, P<0.01), when NHE1 was overexpressed, the NHE1 inhibitor EIPA reversed the results (56.803±1.978% vs 48.830±2.826% in G1 phase, P<0.01 and 31.383±1.752% vs 45.353±1.744% in S phase, P<0.01). When NHE1 expression was suppressed by NHE1-siRNA, the percentage in G1 phase was increased (76.327±3.385% vs (62.157±2.386%, P<0.01) while the percentage in S phase was decreased compared with the control group (15.78±3.232% vs 35.983±1.876%, P<0.01) (Figure 3A). Also, the cell cycle related proteins CDK2, Cyclin A, CDK4, Cyclin D1, and C-Myc were increased when NHE1 was overexpressed and decreased when the NHE1 expression was suppressed (Figure 3B, P<0.01). These results showed that NHE1 can suppress the cell apoptosis of SGC7901 cells mediated by 5-FU and involved in the cell cycle.
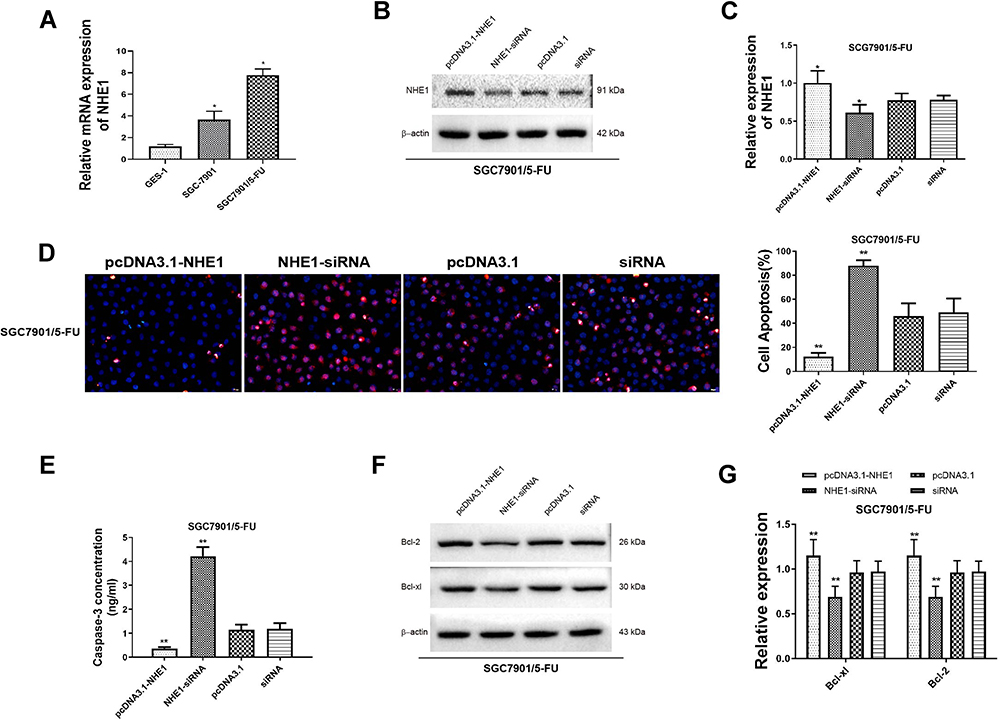 |
Figure 2 NHE1 attenuate the cell apoptosis mediated by 5-FU (A). Relative mRNA expression of NHE1 in the GES-1 cell line, the parental gastric cancer (SGC7901), and 5-fluorouracil resistant gastric cancer cell lines SGC7901/FU. (B and C) The relative protein expression of NHE1 after plasmids transfection. (D) Cell apoptosis percentage of SGC7901 cell line. Blue represents Dapi stain, Red represents the apoptosis cells. (E) The concentration of caspase 3. (F and G) The expression of anti-apoptosis proteins (Bcl-xl and Bcl-2). Error bars denote SD. *P<0.05, compared to the control group. **P<0.01, compared to the control group. |
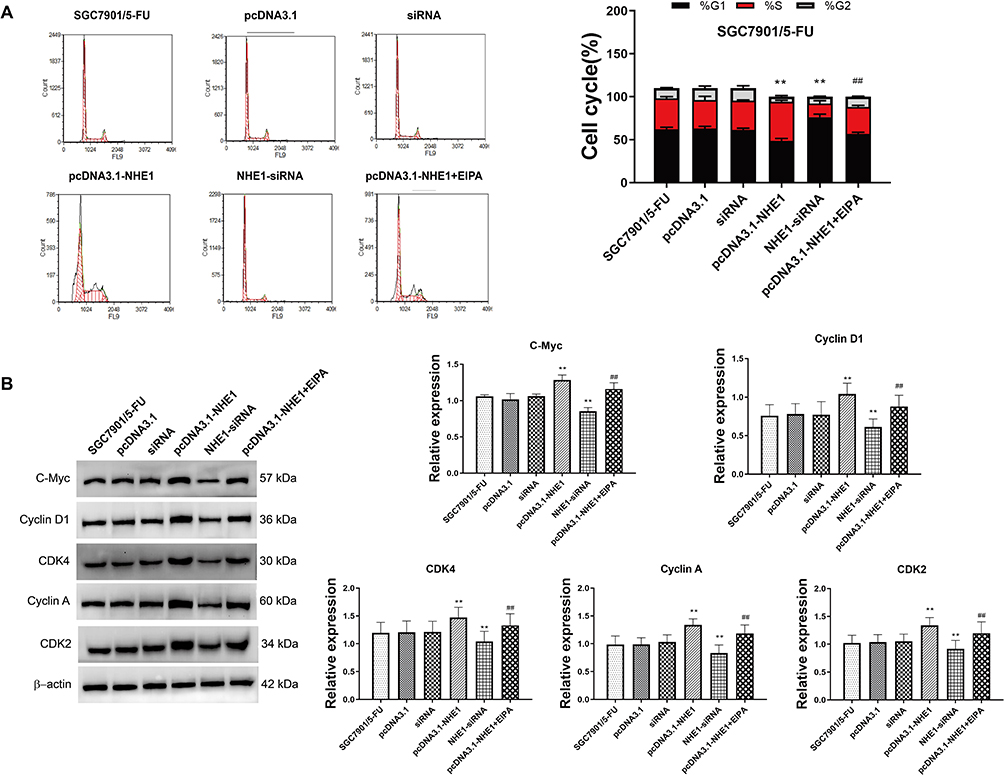 |
Figure 3 NHE1 overexpression involved in the cell cycle. (A) The change of cell cycle. (B) The relative expression of cell cycle related proteins C-Myc, Cyclin D1, CDK4, Cyclin A, and CDK2. Error bars denote SD. **P<0.01, compared to the control group. ##P<0.01, compared to the pcDNA3.1-NHE1 group. |
NHE1 Promotes the Migration and Invasion of SGC7901 Cells
We further investigated whether NHE1 can affect the migration and invasion of SCG-7901/5‑FU cell lines. Wound scratch assay showed that the migration ability was significantly increased when NHE1 was overexpressed compared to the control group ((0.866±0.014) vs (0.505±0.026), P<0.01). Then NHE1-siRNA was transfected, and the migration ability was decreased ((0.291±0.015) vs (0.505±0.026), P<0.01). The NHE1 inhibitor EIPA reversed the increase of migration ability ((0.650±0.015) vs (0.866±0.014), P<0.01) (Figure 4A). Also, the result of Transwell assay revealed that NHE1 overexpression obviously increased the number of invasion cell. The number of invasion cells was increased significantly when the expression of NHE1 was increased by pcDNA3.1-NHE1 transfection (282.33±21.825 vs 124.333±20.404, P<0.01). The NHE1 inhibitor EIPA suppressed the increase of the number of invasion cells (159.333±12.055 vs 282.33±21.825, P<0.01). NHE1-siRNA transfection also decreased the number of invasion cells (56.333±9.452 vs 124.333±20.404, P<0.01) (Figure 4B). The expression of migration and invasion associated gene ROCK1 and Rac also showed the same trend (Figure 4C and D, P<0.01).
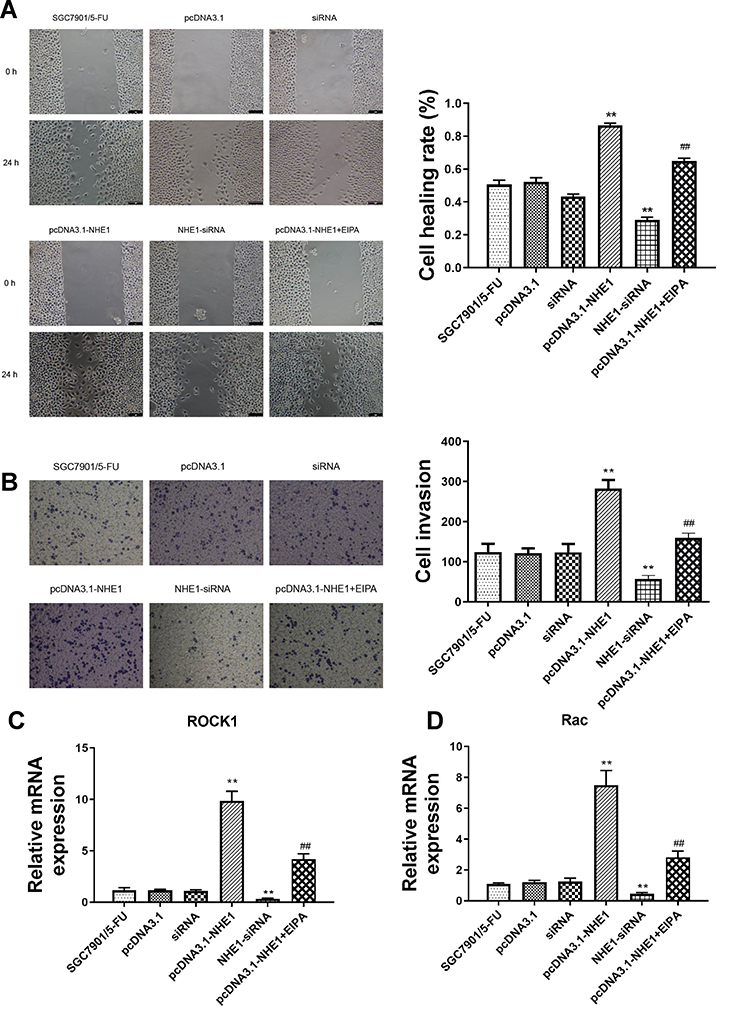 |
Figure 4 NHE1 promotes the migration and invasion of SGC7901 cells. (A) Cell wound scratch assay at 0 and 48 hours after the scratch. (B) Cells invasion to the bottom of membranes. (C and D) The relative expression of migration and invasion associated gene ROCK1 and Rac. Error bars denote SD. **P<0.01, compared to the control group. ##P<0.01, compared to the pcDNA3.1-NHE1 group. |
NHE1 Prompt 5-Fu Resistance by Decreasing the Intracellular pH
To analyze the effect of NHE1 transfection on intracellular pH, the pHi analysis was applied. As shown in Table 3, when NHE1 was overexpressed in the 5-FU treated SGC7901 cell lines, the pHi value was increased (7.35±0.078 vs 7.26± 0.04, P<0.05). While, when the NHE1 expression was suppressed, the pHi value was decreased (7.15±0.071 vs 7.26±0.04, P<0.05). The results suggested that an acidic microenvironment may play a crucial role in 5-Fu resistance in gastric cancer cells, and NHE1 prompted 5-Fu resistance by increasing the intracellular pH.
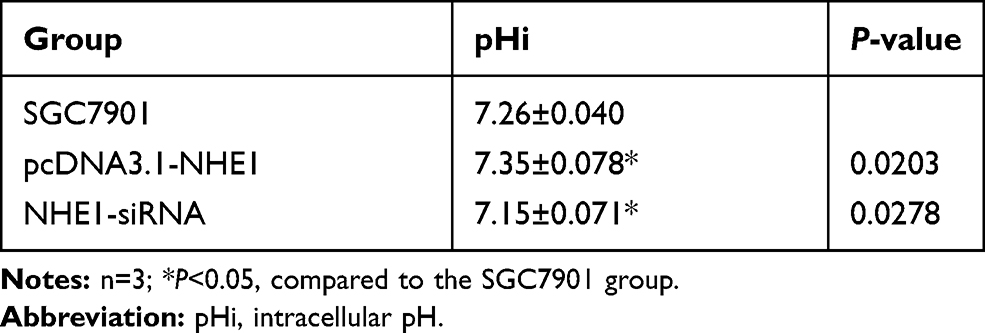 |
Table 3 Intracellular pH Value |
STAT3 is Constitutively Phosphorylated in NHE1 Overexpressed Gastric Cancer Cells
Constitutive activation of STAT3 has been reported in several primary cancers and tumor cell lines. STAT3 induces cell transformation through a combined inhibition of apoptosis and cell-cycle activation, which highlights a likely role for STAT3 signaling in regulation of drug resistance.18 Therefore, the expression of STAT3 was detected. NHE1 overexpression induced the increase in the expression of pSTAT3, EIPA reverses the increase. Also, NHE1-siRNA transfection decrease the expression of pSTAT3 (Figure 5A, P<0.01). The expressions of pSTAT1 and pSTAT5 showed the same trend. Also, the expression of STAT3, pSTAT3ser727, and pSTAT3tyr705 was detected by immunofluorescence. Compared with the SGC7901/5-FU group, the elevated levels of phosphorylated STAT3 was detected both at pSTAT3tyr705 and pSTAT3ser727, while the more obvious phosphorylated level was shown in the phosphorylated STAT3 at pSTAT3tyr705. The NHE1 inhibitor EIPA reversed the increase. The expression levels of phosphorylated STAT3 both at pSTAT3tyr705 and pSTAT3ser727 were reduced when the NHE1-siRNA transfection compared with the SGC7901/5-FU group (Figure 5B).
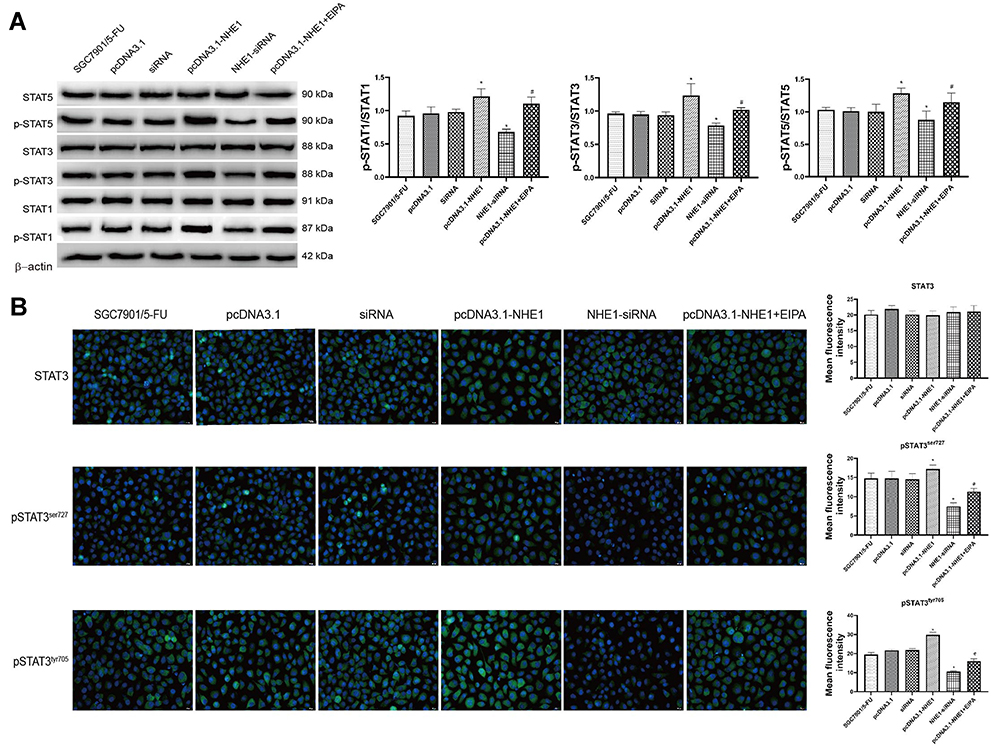 |
Figure 5 STAT3 is constitutively phosphorylated in NHE1 overexpressed gastric cancer cells. (A) The relative expression of STATs and their phosphorylation. (B) Immunofluorescent of STAT3 and phosphorylated STAT3 at pSTAT3tyr705 and pSTAT3ser727 (40x). Green represents the STAT 3 or its phosphorylation, blue represents the Dapi stain. Error bars denote SD. *P<0.05, compared to the control group. #P<0.05, compared to the pcDNA3.1-NHE1 group. |
STAT3 Phosphorylation is Triggered by JAK but Not Src or EGFR in NHE1 Overexpressed Gastric Cells
We next examine the upstream activators of STAT3 in NHE1 mediated 5-Fu resistant gastric cancer cells. As revealed by Western blot assay, compared with the control group, NHE1 overexpressed cells showed elevated expression of p-JAK1 and p-JAK2, the NHE1 inhibitor reversed the increase. The NHE1-siRNA transfection decreased the expression. Interestingly, no statistical significance appeared in the expression of epidermal growth factor receptor (EGFR) and src (P<0.01, Figure 6A). We subsequently conducted the Co-IP assay to investigate whether NHE1 could directly interact with JAKs. As showed Figure 6C, JAK1/2 could form a complex with NHE1. Conversely, no obvious complex was detected after the NHE1-siRNA transfection (P<0.01, Figure 6B). Also, when the JAK inhibitor was applied, the cell apoptosis suppressed by NHE1 was aggravated (P<0.01, Figure 6C and D). These findings demonstrated that the constitutive activation of STAT3 may be induced by JAK1 and JAK2, and thus affected the 5-FU resistance by regulating NHE1.
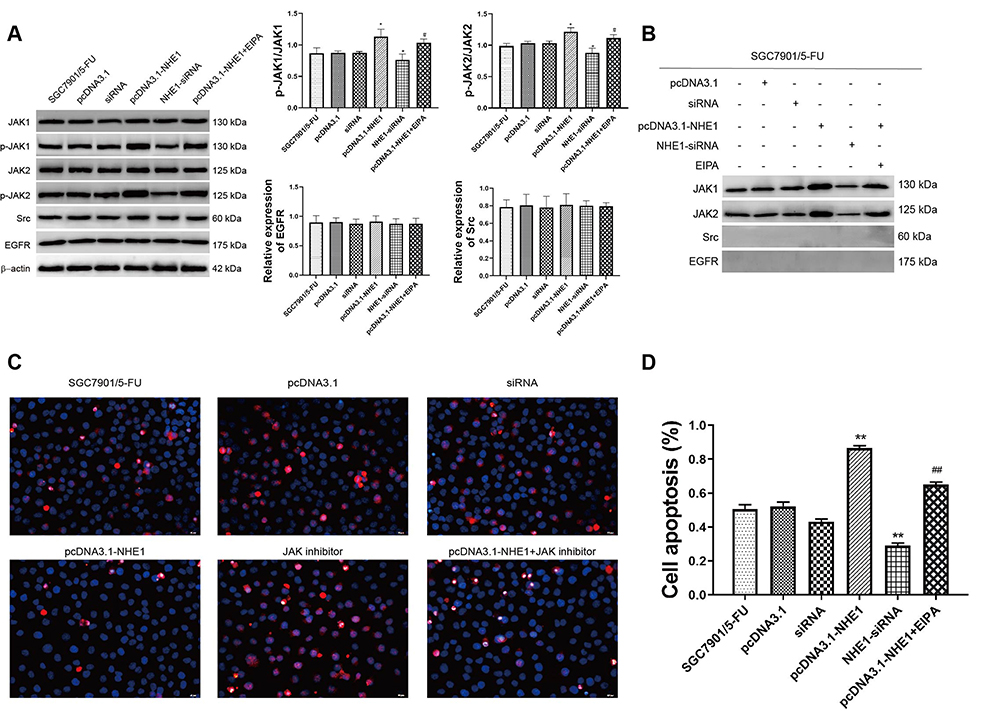 |
Figure 6 STAT3 phosphorylation is triggered by JAK but not Src or EGFR in NHE1 overexpressed gastric cells. (A) The relative expression of JAKs, Src, and EGFR. (B) Co-IP assay revealed that NHE1 could directly interact with JAKs and their phosphorylation. (C and D) Cell apoptosis percentage of SGC7901 cell line (40x). Blue represents Dapi stain, Red represents the apoptosis cells. Error bars denote SD. *P<0.05, compared to the control group. **P<0.01, compared to the control group. #P<0.05, compared to the pcDNA3.1-NHE1 group. ##P<0.01, compared to the pcDNA3.1-NHE1 group. |
Discussion
Several recent studies have addressed the role of NHE1 in tumor cell growth and apoptosis, including in gastric cancer.19 However, the role of NHE1 expression related to the 5-Fu resistance in gastric cancer has not been investigated. The results presented herein provide provocative evidence for a functional role of the NHE1 in 5-Fu resistance in human gastric cancer cells and clarify the underlying molecular mechanism, which has little been described. In the current study, we found that NHE1 overexpression aggravated the 5-FU resistance through the JAK/STAT3 pathway.
The 5-fluorouracil (5-FU) based chemotherapy is a standard treatment for patients with advanced GC.4,8,20 Although combining 5-FU with cisplatin, docetaxel, or oxaliplatin can improve treatment response, resistance to 5-FU in GC is still an intractable issue in the clinic. Recent research has shown that enzymes upregulation associated with 5-FU metabolism such as thymidylate synthase (TS), orotate phosphoribosyl transferase (OPRT), and dihydropyrimidine dehydrogenase (DPD) were involved in the 5-FU-resistance. Besides, alterations of the apoptosis pathway or proteins in response to DNA damage can lead to resistance to 5-FU.4,21 It was recently shown that acidification of intracellular milieu is critical to induce efficient caspase activation and cell death following drug-induced apoptosis. However, cancer cells survive by manipulating and exploiting a host of ion exchangers including NHE.22,23 High expression of NHE1 has been observed resistant to chemotherapy treatment in many different tumor types, including pancreas,23 esophageal,24 lung,25 and breast cancers.13 Accordingly, NHE1 has been proposed as a promising molecular target for cancer chemotherapy.25,26 In the current research, NHE1 was overexpressed when the resistance of SCG-7901/5-FU cells to 5-FU increased, and the overexpression of NHE1 obviously alleviated the cell apoptosis of SGC7901/5-FU cells. It indicated that NHE1 overexpression would alleviate cell apoptosis, thus promotes the 5-FU resistant to the SGC7901 cell line. Moreover, NHE1 overexpression also elevated the migration and invasion, and was involved in the cell cycle of SGC7901 cells.
Also, we found that when NHE1 was overexpressed in the SGC7901/5-FU cell lines, the pHi value was increased. Also, when the NHE1 expression was suppressed, the pHi value was decreased. The NHE1, which is highly conserved across vertebrate species, is activated by acidic deviations from steady-state pHi and by osmotic cell shrinkage, and is, in this capacity, abundantly characterized as a major membrane transport mechanism in the regulation of pHi and cell volume.20 It is been reported that the mechanism that NHE1 modulate cell proliferation and cell death appeared to be changes in pHi, [Na+]i, and/or cell volume, directly affecting components of metabolic, cell cycle, and death effector pathways.12 The results suggested that an acidic microenvironment may play a crucial role in 5-Fu resistance in gastric cancer cells, and NHE1 promotes 5-Fu resistance by increasing the intracellular pH.
STAT3, a cytoplasmic transcription factor that translocates into the nucleus following cytokine activation, has important roles in various biological processes such as inflammation, metabolism, and tumorigenesis.27,28 The pervious researches showed that the activation of STAT3 is closely related to the malignant tumors. The abnormal activation of STAT3 could promote the gene transcription of CyclinD1, Bcl-xl, c-Myc, interleukin (IL)-10, and vascular endothelial growth factor (VEGF) etc., and therefore involves the cell survival, proliferation, angiogenesis, migration, inflammation, immunity, and mitochondrial respiratory chain transmission.29,30 We found that NEH1 overexpression could increase the expression of pSTAT3. As naturally occurring mutations of STAT3 have not been observed, constitutive activation of STAT3 seems to be mediated by aberrant growth factor signaling. Tyrosine phosphorylation of STAT3 (Tyrosine 705) is mediated by a wide variety of polypeptides and is essential for STAT3 dimerization and nuclear translocation. STAT3 also has a conserved serine727 residue, which is a target for phosphorylation.31 Evidence indicates that cooperation of both tyrosine and serine phosphorylation is necessary for full activation of STAT3.30 Compared with the SGC7901/5-FU group, phosphorylated STAT3 had elevated levels of both at pSTAT3tyr705 and pSTAT3ser727, while the more obvious phosphorylated level was shown in the phosphorylated STAT3 at pSTAT3tyr705. The results indicated that NHE1 promote the 5-FU resistance by constitutive phosphorylation of STAT3. A number of different tyrosine kinases, including the JAKs, Src, and the epidermal growth factor receptor (EGFR) family of tyrosine kinases, have been described as mediators of STAT3 phosphorylation in various primary tumors.32–34 Further studies clearly demonstrate that the NHE1 mediated pSTAT3 elevation was triggered by JAKs instead of Src or EGFR, indicating a tight link between NHE1 and JAK/STAT3 signaling pathways in 5-Fu resistance of gastric cancer cells.
STAT3 is activated for a few seconds or hours and then is deactivated to maintain homeostasis under normal circumstance. However, STAT3 activation continues, which triggers oncogene transcription under abnormal conditions.35 Following activation, STAT3 homodimerizes, rapidly translocates into the nucleus, and induces uncontrolled tumor cell growth and survival through multiple mechanisms.36 In our study, the downstream targets of STAT3, anti-apoptosis protein Bcl-2 and Bcl-xl, as well as proliferation genes c-Myc and cyclin D1 were elevated in NHE1 overexpressed gastric cancer cells. Additionally, the apoptosis rates of cells with different expression of NHE1 were in accordance with the expression of anti-apoptosis protein. These findings were consistent with the previous findings in breast cancer and myeloma, in which constitutive activation of STAT3 induced antiapoptotic genes and confers resistance to apoptosis.37
The results revealed the mechanism of NHE1 mediated 5-Fu resistant in gastric cancer cell line SGC7901 is associated with the JAK/STAT3 pathway. However, the accurate binding sites at gene level were not revealed yet. Moreover, considering that NHE1 was involved in various aspects of physiological progression of tumorigenesis, some other mechanism such as regulating the expression of DNA-repaired genes may also correlate with the occurrence of 5-Fu resistance, which need further investigation.
In summary, our findings provided evidence that NHE1 contributed to 5-Fu resistance in gastric cancer cells by upregulating the JAK/STAT3 pathway. Effective inhibition of NHE1 remarkably sensitized gastric cancer cells to 5-Fu chemotherapy. Therefore, NHE1 can be a useful marker for predicting and monitoring 5-Fu response. The present study provided a molecular basis for potential application of NHE1 inhibition in combination with 5-Fu in drug resistance cases.
Author Contributions
All authors made substantial contributions to the conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Disclosure
There is no conflict of interest because of the role of the post which affects the opinions of the article and reports on the results of the data.
References
1. Torre LA, Bray F, Siegel RL, et al. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87–108.
2. Lei Z, Tan IB, Das K, et al. Identification of molecular subtypes of gastric cancer with different responses to PI3-kinase inhibitors and 5-fluorouracil. Gastroenterology. 2013;145(3):554–565. doi:10.1053/j.gastro.2013.05.010
3. Bang YJ, Van Cutsem E, Feyereislova A. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junc tion cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet. 2010;376(9742):687–697. doi:10.1016/S0140-6736(10)61121-X
4. Matsuhashi N, Saio M, Matsuo A, et al. The evaluation of gastric cancer sensitivity to 5-FU/CDDP in terms of induction of apoptosis: time- and p53 expression-dependency of anti-cancer drugs. Oncol Rep. 2005;14(3):609–615.
5. Kim HS, Kim JH, Kim JW, et al. Chemotherapy in elderly patients with gastric cancer. J Cancer. 2016;7(1):88–94. doi:10.7150/jca.13248
6. Farran B, Müller S, Montenegro RC. Gastric cancer management: kinases as a target therapy. Clin Exp Pharmacol Physiol. 2017;44(6):613–622. doi:10.1111/1440-1681.12743
7. Johnston FM, Beckman M. Updates on management of gastric cancer. Curr Oncol Rep. 2019;21(8):67. doi:10.1007/s11912-019-0820-4
8. Shen L, Shan YS, Hu HM, et al. Management of gastric cancer in Asia: resource-stratified guidelines. Lancet Oncol. 2013;14(12):e535–e547.
9. Rougier P, Ducreux M, Mahjoubi M, et al. Efficacy of combined 5-fluorouracil and cisplatinum in advanced gastric carcinomas. A phase II trial with prognostic factor analysis. Eur J Cancer. 1994;30(9):1263–1269. doi:10.1016/0959-8049(94)90170-8
10. Stock C, Pedersen SF. Roles of pH and the Na(+)/H(+) exchanger NHE1 in cancer: from cell biology and animal models to an emerging translational perspective? Semin Cancer Biol. 2017;43:5. doi:10.1016/j.semcancer.2016.12.001
11. Tomoda A, Marunaka Y, Eaton DC, et al. Membrane transport: ionic environments, signal transduction, and development of therapeutic targets. Biomed Res Int. 2015;2015:1–2.
12. Pedersen SF. The Na+/H+ exchanger NHE1 in stress-induced signal transduction: implications for cell proliferation and cell death. Pflugers Arch. 2006;452(3):249–259. doi:10.1007/s00424-006-0044-y
13. Amith SR, Fliegel L. Regulation of the Na+/H+ exchanger (NHE1) in breast cancer metastasis. Cancer Res. 2013;73(4):1259–1264. doi:10.1158/0008-5472.CAN-12-4031
14. Webb BA, Chimenti M, Jacobson MP, et al. Dysregulated pH: a perfect storm for cancer progression. Nat Rev Cancer. 2011;11(9):671–677. doi:10.1038/nrc3110
15. Tang H, Zeng L, Wang J, et al. Reversal of 5-fluorouracil resistance by EGCG is mediate by inactivation of TFAP2A/VEGF signaling pathway and down-regulation of MDR-1 and P-gp expression in gastric cancer. Oncotarget. 2017;8(47):82842–82853. doi:10.18632/oncotarget.20666
16. Jiang C-P, Wu B-H, Wang B-Q, et al. Overexpression of ECRG4 enhances chemosensitivity to 5-fluorouracil in the human gastric cancer SGC-7901 cell line. Tumour Biol. 2013;34(4):2269–2273. doi:10.1007/s13277-013-0768-1
17. Hosogi S, Miyazaki H, Nakajima K-I, et al. An inhibitor of Na+/H+ exchanger (NHE), ethyl-isopropyl amiloride (EIPA), diminishes proliferation of MKN28 human gastric cancer cells by decreasing the cytosolic Cl- concentration via DIDS-sensitive pathways. Cell Physiol Biochem. 2012;30(5):1241–1253. doi:10.1159/000343315
18. Zhao C, Li H, Lin H-J, et al. Feedback activation of STAT3 as a cancer drug-resistance mechanism. Trends Pharmacol Sci. 2016;37(1):47–61. doi:10.1016/j.tips.2015.10.001
19. Xie R, Wang H, Jin H, et al. NHE1 is upregulated in gastric cancer and regulates gastric cancer cell proliferation, migration and invasion. Oncol Rep. 2017;37(1):333–340. doi:10.3892/or.2017.5386
20. Li Q-F, Yao R-Y, Liu K-W, et al. Genetic polymorphism of GSTP1: prediction of clinical outcome to oxaliplatin/5-FU-based chemotherapy in advanced gastric cancer. J Korean Med Sci. 2010;25(6):846–852. doi:10.3346/jkms.2010.25.6.846
21. Leong T, Michael M, Joon DL, et al. Adjuvant chemoradiation for gastric cancer using epirubicin, cisplatin, and 5-FU (ECF) before and after 3D- conformal radiotherapy with continuous infusional 5-FU: a multicentre study fo the trans-tasman radiation oncology group (TROG). Int J Radiat Oncol Biol Phys. 2007;69:S107. doi:10.1016/j.ijrobp.2007.07.197
22. Hirpara JL, Clément MV, Pervaiz, S. Intracellular acidification triggered by mitochondrial-derived hydrogen peroxide is an effector mechanism for drug-induced apoptosis in tumor cells. J Biol Chem. 2001;276(1):514. doi:10.1074/jbc.M004687200
23. Cardone RA, Greco MR, Zeeberg K, et al. A novel NHE1-centered signaling cassette drives epidermal growth factor receptor–dependent pancreatic tumor metastasis and is a target for combination therapy 1. Neoplasia. 2015;17(2):155–166. doi:10.1016/j.neo.2014.12.003
24. Park SY, Lee YJ, Cho EJ, et al. Intrinsic resistance triggered under acid loading within normal esophageal epithelial cells: NHE1- and ROS-mediated survival. J Cell Physiol. 2015;230(7):1503–1514. doi:10.1002/jcp.24896
25. Provost JJ, Rastedt D, Canine J, et al. Urokinase plasminogen activator receptor induced non-small cell lung cancer invasion and metastasis requires NHE1 transporter expression and transport activity. Cell Oncol. 2012;35(2):95–110. doi:10.1007/s13402-011-0068-y
26. Pedersen SF, Darborg BV, Rentsch ML, et al. Regulation of mitogen-activated protein kinase pathways by the plasma membrane Na+/H+ exchanger, NHE1. Arch Biochem Biophys. 2007;462(2):195–201. doi:10.1016/j.abb.2006.12.001
27. Yu H, Lee H, Herrmann A, et al. Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nat Rev Cancer. 2014;14(11):736–746. doi:10.1038/nrc3818
28. Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. 2009;9(11):798–809. doi:10.1038/nrc2734
29. Aziz MH, Hafeez BB, Sand JM, et al. Protein kinase Cvarepsilon mediates Stat3Ser727 phosphorylation, Stat3-regulated gene expression, and cell invasion in various human cancer cell lines through integration with MAPK cascade (RAF-1, MEK1/2, and ERK1/2). Oncogene. 2010;29(21):3100–3109. doi:10.1038/onc.2010.63
30. Hsu KW, Hsieh R-H, Huang K-H, et al. Activation of the Notch1/STAT3/Twist signaling axis promotes gastric cancer progression. Carcinogenesis. 2012;33(8):1459–1467. doi:10.1093/carcin/bgs165
31. Decker T, Kovarik P. Serine phosphorylation of STATs. Oncogene. 2000;19(21):2628–2637. doi:10.1038/sj.onc.1203481
32. Murone M, Vaslin Chessex A, Attinger A, et al. Debio 0617B inhibits growth of STAT3-driven solid tumors through combined inhibition of JAK, SRC, and class III/V receptor tyrosine kinases. Mol Cancer Ther. 2016;15(10):1535–7163. doi:10.1158/1535-7163.MCT-15-0974
33. Wen W, Wu J, Liu L, et al. Synergistic anti-tumor effect of combined inhibition of EGFR and JAK/STAT3 pathways in human ovarian cancer. Mol Cancer. 2015;14(1):100. doi:10.1186/s12943-015-0366-5
34. Nam S, Wen W, Schroeder A, et al. Dual inhibition of Janus and Src family kinases by novel indirubin derivative blocks constitutively-activated Stat3 signaling associated with apoptosis of human pancreatic cancer cells. Mol Oncol. 2013;7(3):369–378. doi:10.1016/j.molonc.2012.10.013
35. Yu H, Jove R. The STATs of cancer — new molecular targets come of age. Nat Rev Cancer. 2004;4(2):97–105. doi:10.1038/nrc1275
36. Shou J, You L, Yao J, et al. Cyclosporine A sensitizes human non-small cell lung cancer cells to gefitinib through inhibition of STAT3. Cancer Lett. 2016;379(1):124–133. doi:10.1016/j.canlet.2016.06.002
37. Zhang J, Du J, Liu Q, et al. Down-regulation of STAT3 expression using vector-based RNA interference promotes apoptosis in Hepatocarcinoma cells. Artif Cells Nanomed Biotechnol. 2015;44(5):1201–1205.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.