")
Back to Journals » International Journal of Nanomedicine » Volume 16
New Peceol™/Span™ 60 Niosomes Coated with Chitosan for Candesartan Cilexetil: Perspective Increase in Absolute Bioavailability in Rats
Authors AbuElfadl A, Boughdady M , Meshali M
Received 10 June 2021
Accepted for publication 29 July 2021
Published 16 August 2021 Volume 2021:16 Pages 5581—5601
DOI https://doi.org/10.2147/IJN.S324171
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Israel (Rudi) Rubinstein
Aya AbuElfadl, Mariza Boughdady, Mahasen Meshali
Department of Pharmaceutics, Faculty of Pharmacy, Mansoura University, Mansoura, Egypt
Correspondence: Aya AbuElfadl
Department of Pharmaceutics, Faculty of Pharmacy, Mansoura University, Gomhoreyah St, Mansoura, 35516, Egypt
Tel +201004602870
Fax +20 50 224 7496
Email [email protected]
Purpose: Candesartan cilexetil (CC), a prodrug of candesartan (CDT), is a class II BCS drug that suffers from poor oral bioavailability because of low aqueous solubility, P-gp efflux and first-pass metabolism. The absolute bioavailability reported for CC was only 15% and the methods to increase it remain elusive, thus the aim of our work was to prepare new CC-loaded niosomes encompassing, for the first time, glycerol monooleate GMO (Peceol™), as P-gp efflux inhibitor and promoter of lymphatic transport with Span™ 60 as bioenhancer. The prepared niosomes were further coated with chitosan for augmenting the CC oral absorption.
Methods: The niosomes were prepared by thin film hydration method through quality by design approach, using two levels of each of three critical process parameters (CPPs), namely, XA (the molar ratio of surfactant mixture to cholesterol) at a ratio of 1:1 or 2:1; XB (the molar ratio of Span™ 60 to Peceol™) at a ratio of 1:1 or 2:1; and XC (the drug amount) at 15 mg or 30 mg. The investigated critical quality attributes (CQAs) were entrapment efficiency percent, particle size, and polydispersity index. The optimized uncoated and chitosan coated formulations were subjected to DSC and stability study. In vitro drug release, biocompatibility with Caco-2 cells and lastly the absolute bioavailability evaluation in rats were assessed.
Results: The physical properties of the optimized and stable niosomes were satisfactory. The ingredients were compatible with each other and biocompatible with Caco-2 cells. The synergistic combination of Peceol™ and Span™ 60 probably surmounted the P-gp efflux with an increase in oral absolute bioavailability of niosomes to five times that of CC suspension.
Conclusion: The new niosomal formulations of CC containing Peceol™ with Span™ 60 and cholesterol either uncoated or coated with chitosan were a successful paradigm in achieving high oral absolute bioavailability and increased Caco-2 cells biocompatibility.
Keywords: candesartan cilexetil, glyceryl monooleate, chitosan, niosomes, PROSOLV®
Introduction
The oral route is the most important, widespread, and convenient route of drug administration. It offers several advantages such as ease of administration, accomplished good stability, ease of manufacture, and good patient compliance. Despite all the previous advantages, most drugs possess low oral bioavailability, which is attributed to several reasons such as, poor drug solubility, inappropriate partition coefficient, first-pass metabolism, and premature degradation of the drug under the effect of either stomach pH or by enzymes. Also, the intestinal wall hampers drug absorption via drug metabolism by CYP-3A4 enzyme.1 The drug can also be subjected to some transporters inside the enterocytes such as P-glycoprotein (P-gp) efflux which carries the drug back to the intestinal lumen.2
Niosomes are colloidal carriers which are formed by self-assembly of nonionic surfactants forming lipid bilayers surrounding an aqueous core, thus they could accommodate drugs with different solubilities. Their composition as well as their characteristics such as size, lamellarity and surface charge, could be varied to fulfill a desired goal. In addition, their surface could be modified simply by different materials. They possess great stability, low cost, high effectiveness and simple preparation methodology. They showed a promising enhancement in the solubility of BCS II drugs as well as a marked increase in their oral bioavailability. They could also guard against premature degradation of drugs and sustain drug release.3,4
Glyceryl monooleate GMO (Peceol™) is an amphiphile composed of a mixture of mono and diglycerides of oleic acid which is similar to the end product of lipid digestion by the intestine. It is classified as GRAS and considered as inactive ingredient by the FDA. GMO is a lyotropic liquid which can form different crystalline phases in water (cubic, hexagonal, and lamellar), depending on its critical packing parameter (CPP) alone or in combination with other materials.5,6 It possessed a beneficial role in improving the drug absorption via lowering P-gp activity.6 Peceol™ also played a vital role in drug lymphatic transport.7 Its lamellar form could be assembled into vesicular structure when combined with Span™ 60 in the presence of cholesterol (CHOL).8 Span 60 as nonionic surfactant could provide high drug entrapment efficiency by the niosomes. It could also enhance drug permeation by facilitating paracellular transport, thus improving drug oral bioavailability.9 It was reported that Span™ 60 is a P-gp transporter inhibitor.10 CHOL could provide the rigidity to the niosomal bilayer structure.11 The surface of niosomes could be modified by coating with chitosan (CS) to increase the GIT flux of the drug via its permeation enhancing property with higher mucoadhesion to mucin compared to uncoated ones.12,13
Candesartan (CDT) is a potent angiotensin II type AT1 receptor blocker that is used orally in the treatment of hypertension and heart failure. Since it belongs to class IV according to biopharmaceutical classification system (BCS) with low solubility and low permeability, candesartan cilexetil (CC) was developed as a prodrug which is classified as class II with low solubility and high permeability. CC is bioactivated to the pharmacologically active CDT by ester hydrolysis via carboxylesterase enzymes which are present in the intestinal wall during absorption. Unfortunately, it was reported that CC showed absolute oral bioavailability of only about 15% in humans.14 This poor bioavailability was attributed to low aqueous solubility and the premature hydrolysis of CC into CDT by carboxylesterases present in the intestinal lumen, threatening the task of CC as a prodrug.15 The virtue of efflux by P-gp transporter protein in the intestinal wall16 as well as the hepatic first-pass metabolism by CYP-450,17 collectively hurt CC oral bioavailability. Several nanosystems have been implemented on CC for the sake of overcoming the previous obstacles such as self-nanoemulsifying drug delivery system, solid lipid nanoparticles, nanocrystal via solid dispersion technique, nanoparticles and proniosomes,14,17–20 but the reported absolute bioavailability did not exceed 15%, and most of these nanosystems were compared to the oral suspension of free drug.
Accordingly, the target of our study was to prepare new CC niosomes involving, for the first time, Peceol™ with the common ingredients of niosomes and to coat their surfaces by CS as cationic mucoadhesive polymer. Peceol™ was anticipated to enhance CC oral absorption via two mechanisms: the inhibition of P-gp efflux6 and stimulation of lymphatic transport,21 thus avoiding drug first-pass metabolism. CS was expected to improve CC oral absorption via adsorption to the negatively charged intestinal epithelium.12,22 In this context, silicified microcrystalline cellulose (SMCC, PROSOLV®) was used to entrap or cage the CC niosomes in the form of powder.
Materials and Methods
Materials
Candesartan cilexetil (CC) (CAS no. 145040375, Lot no. GT17090003) and valsartan (Val) (CAS no. 137862534) were obtained as gift samples from Future pharmaceutical industries company (Cairo, Egypt). Candesartan (CDT) (CAS no. 139481597) was purchased from Zhejiang Tianyu Pharmaceutical Co. Ltd (China). Span™ 60 and cholesterol were purchased from (Loba Chemie PVT, Ltd, Mumbai, India). Peceol™ (glycerol monooleate type 40) (batch no 164530) was supplied as a gift sample from Gattefossé (St Priest, France). Chitosan (CAS no. 9012764) (characterized by low M.wt and ≥ 75% degree of deacetylation) and Tween 20 (CAS no. 90005645) were purchased from Sigma-Aldrich (Saint Louis, MO, USA). PROSOLV® (SMCC) (batch no. P9B3E59), was kindly obtained from JRS Pharma (Germany). Analytical grade of ethyl alcohol 99.9%, chloroform, chromatographic grade HPLC methanol (Fischer), chromatographic grade HPLC acetonitrile (Fischer) and Millipore filter (0.45 µm) were purchased from Cornell lab (Cairo, Egypt).
Optimization of Niosomal Formulations Using 23 Full Factorial Design
The niosomes were prepared using 300 µmole of the components; the surfactant mixture (SM) consisting of Span™ 60 and Peceol™, besides CHOL as stabilizer. 23 full factorial design is a statistical tool adopted for analyzing and finding the appropriate combination of three critical process parameters (CPPs) (factors or independent variables), and their assessment at two different levels: low (−1) and high (+1). The studied CPPs are investigated in all possible combinations, in order to obtain the optimized formulation. Herein, the design of experiment (DOE) in the form of 23 full factorial has been established to explore the effects and the interactions of three CPPs, (XA) the molar ratio of the surfactant mixture (SM) to CHOL; (XB) the molar ratio of Span™ 60 to Peceol™ in the surfactant mixture; and (XC) the drug (CC) amount in mg, on three critical quality attributes (CQAs) representing (R1) entrapment efficiency percent (%EE); (R2) particle size (PS); and (R3) polydispersity index (PDI). Based on preliminary study, the ratios of the niosomal ingredients to each other were coded as (+1) and (−1) for the CPPs at high level and low level, respectively (Table 1) and the desirability constraints of each CQA was shown in Table 1.
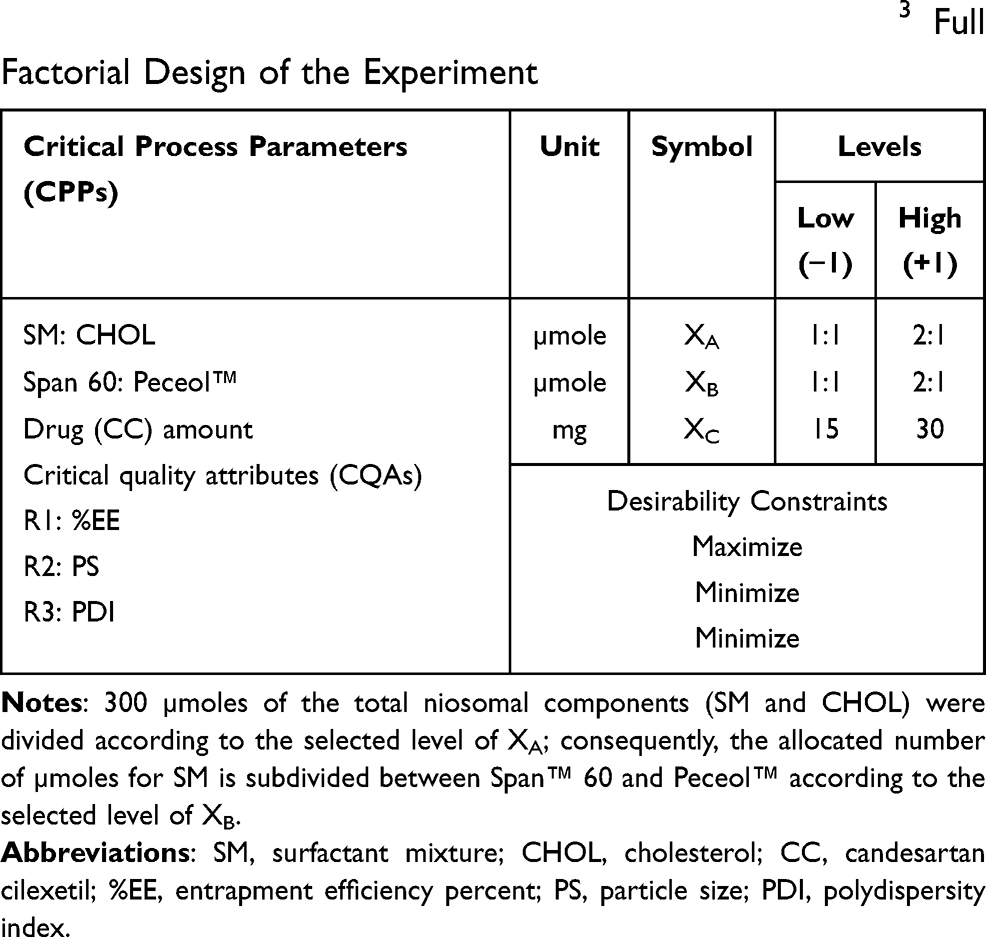 |
Table 1 Critical Process Parameters with Their Levels and Critical Quality Attributes with Their Constraints of 23 Full Factorial Design of the Experiment |
Eight niosomal formulations were prepared and characterized concerning the three CQAs, each with three runs. Finally, the DOE was precisely and accurately used to find out the optimum level of each CPP required to prepare the optimized niosomal formulation with the desired CQA.23 The DOE was employed using Design-Expert® software v12 (State-Ease Inc., Minneapolis, MN, USA). The statistical analysis was performed using the analysis of variance (ANOVA) to estimate the statistical significance of the model (P<0.05). The numerical optimization was established to find out the optimized formulation by the DOE and the checkpoint analysis was carried out by using the midpoint of the three CPPs for preparation of a checkpoint formulation and comparing it with the predicted one.
Preparation of Niosomal Formulations
The conventional thin film hydration method was used to prepare the niosomal formulations.3 Three hundred micromoles of the components consisting of surfactant mixture (Span™ 60 and Peceol™) in addition to CHOL, in different molar ratios as shown in Table 2 were dissolved in 6 mL of chloroform. The specified amount of drug as demonstrated in Table 2, was dissolved in 3 mL of methanol. The two solutions (the ratio of chloroform to methanol 2:1 v/v, was chosen based on preliminary trial) were mixed in closed tube and sonicated in an ultrasonic bath (Sonix IV, model ss101H230, USA) for 10 min; then the final mixture was transferred to 500 mL round bottom flask and evaporated by rotary evaporator (Wheaton rotary evaporator, USA) at 100 rpm under reduced pressure at 60°C for 30 min until a dry thin film was formed on the inner wall of flask. The flask was left in a desiccator overnight to allow the removal of excess organic solvent, if any. The resulting film was then hydrated with 10 mL of deionized water for 1one hour at 60°C and rotated at 150 rpm until formation of milky niosomal dispersion. The niosomal dispersion was further sonicated in an ultrasonic bath (Sonix IV, model ss101H230, USA) for 10 min at 25°C to homogenize the size of niosomes and was refrigerated at 4°C overnight.
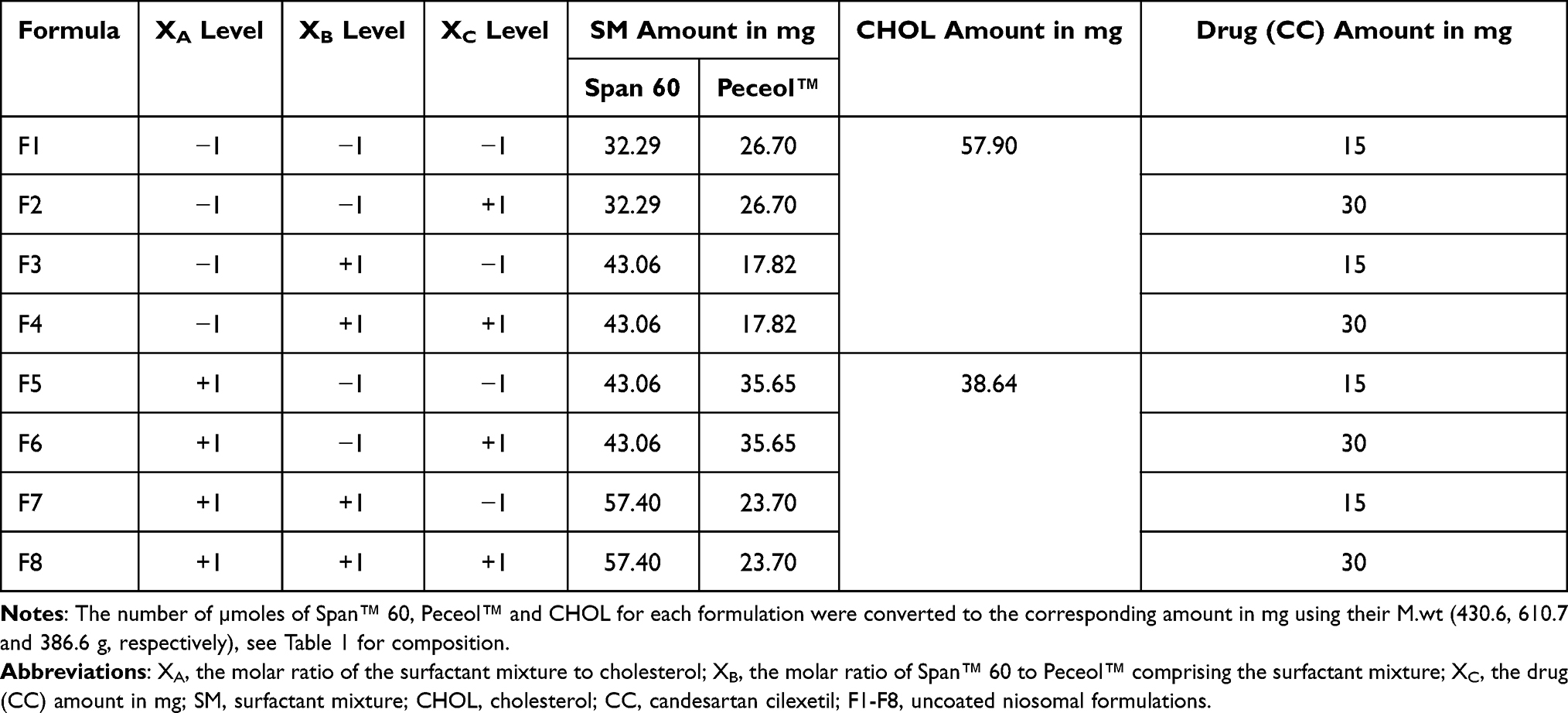 |
Table 2 Amounts of Span 60, Peceol™, Cholesterol and Candesartan Cilexetil for the Preparation of Niosomal Formulations |
Preparation of Different CS Coated Niosomal Formulations
Four CS-coated niosomal formulations (C1, C2, C3 and C4) were prepared by coating the optimized formulation (F7) with four different chitosan concentrations (0.1, 0.2, 1, and 2 mg/mL, respectively). CS was dissolved in 0.1% v/v acetic acid solution and then filtered through 0.45 µm Millipore filter before use. For coating the niosomes, CS solution was added in equivalent volume to the prepared niosomal dispersion dropwise while stirring by magnetic stirrer (model MS-300HS, Misung Scientific Co., Ltd, Korea) at 600 rpm. The resulting dispersion was left on the stirrer for two hours to ensure an efficient coating.24,25 The produced CS-coated niosomal dispersion was kept in a refrigerator for further characterization.
Characterization of Different Niosomal Formulations
Entrapment Efficiency Percent (%EE)
The prepared niosomal formulations were centrifuged at 13,000 rpm and 4°C for two hours using cooling centrifuge (Acculab CE16-4X100RD, USA). The precipitated niosomes were washed by the addition of 10 mL deionized water and centrifuged for additional one hour to remove the remaining unentrapped drug. Entrapment efficiency percent was determined by direct method via lysis of the precipitated niosomes with absolute ethanol. The precipitated vesicles were redispersed in 1 mL deionized water, then 100 µL of niosomal dispersion were transferred into a 10 mL measuring flask and filled to volume with absolute ethanol. Finally, the solution was sonicated for 10 min in ultrasonic bath (Sonix IV, model ss101H230, USA) until a clear solution was obtained to ensure complete lysis.26 The concentration of CC was measured spectrophotometrically (ultraviolet/visible [UV/VIS] spectrophotometer, JASCO, Tokyo, Japan) at λmax of 255 nm using blank of plain niosomes prepared similarly. Each measurement was carried out in triplicate and the %EE was calculated by Equation 1.26 (1)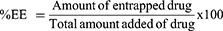
Particle Size (PS) and Polydispersity Index (PDI)
PS and PDI measurements were performed on freshly prepared formulations of CC-loaded niosomes after proper dilution with deionized water. Measurements were accomplished by dynamic light scattering using Malvern Zetasizer (Malvern Instruments Limited, UK). Each experiment was determined in triplicate.
Zeta Potential (ZP)
ZP values were determined after suitable dilution with deionized water using Malvern Zetasizer nanoseries (Malvern Instruments Limited, UK). Each measurement was determined in triplicate.
Transmission Electron Microscope (TEM) of the Optimized Uncoated Niosomes (F7)
The morphology of F7 was scrutinized by TEM (JEOL JEM-2100; JEOL Ltd, Tokyo, Japan) using a digital micrograph and soft imaging viewer software. After dilution of niosomal dispersion to 10 folds with deionized water, one drop was placed on the surface of carbon-coated copper grid for one minute to allow for some niosome adherence and the process was completed as reported.27
Scanning Electron Microscope (SEM) of the Optimized Uncoated Niosomes (F7)
The shape and the surface characteristics of niosomes in dispersion form and after freeze drying, were examined by SEM (JSM-6510LV; JOEL, Tokyo, Japan). Samples were mounted on a metalsub with double-sided adhesive carbon tapes, then coated with a gold layer and photographed at 20 KV.
Differential Scanning Calorimetry (DSC)
The niosomal dispersions of the optimized formulations F7 and C1 either containing CC or plain ones, were converted into powder by lyophilization using a freeze dryer, Labconco (LYPH.LOCK 4.5 Model, USA). To assess the compatibility of niosomal excipients and the drug, the DSC thermograms were invoked for all niosomal ingredients except Peceol™, the optimized formulations containing CC and the plain ones, using a differential scanning calorimeter (DSC 50, Shimadzu Co., Kyoto, Japan). A few milligrams of each sample were crimped in a standard aluminium pan then heated from 20°C to 300°C at heating rate of 10°C/min under constant purging of nitrogen at 20 mL/min. Similarly, an empty pan was sealed for a sample to be used as a reference.
Evaluation of the in vitro Biocompatibility of the Optimized Niosomal Formulations (Uncoated F7 and Coated C1) with Caco-2 Cells
The cytotoxicity studies of CC and the niosomal excipients were investigated on human colorectal adenocarcinoma cells (Caco-2 cells) using methyl thiazolyl tetrazolium (MTT) assay. The cells were obtained from the American Type Culture Collection (Manassas, VA, USA) and cultured as reported before.28 After cells confluency in 96-well plates, uncoated F7 and coated C1 niosomes were each incubated for 24 h with Caco-2 cells. The concentrations of CC were 5, 10, 20, 40, and 80 µg/mL in each formula. The control group was the Caco-2 cells without treatment and the blank was the corresponding plain niosomes treated similarly. For interest, the effect of free CC contact time and concentration on Caco-2 cells viability was elicited. The incubation periods arbitrary chosen were 3 and 24 h. The concentrations of free CC were the same as those of the niosomes. Each plate was incubated at 37°C in atmospheric 5% CO2 and 90% relative humidity.
After the incubation period, 20 µL of MTT solution (5 mg/mL in PBS) was added and mixed with the treated cells. Each plate was incubated for additional four hours at 37°C in a dark place. Aliquots were withdrawn and mixed with 200 µL of acidic isopropanol solution (0.1 N HCl in absolute isopropanol) with additional incubation for one hour under similar conditions. The absorbance of the purple formazan crystals formed was read by ELX 800 microplate reader (Biotek instruments, Inc., Winooski, VT, USA) at λmax of 570 nm. The percent cell viability was calculated by Equation 2.29 (2)
Preparation of Uncoated F7-SMCC and Coated C1-SMCC Powders
To obtain solid form of the optimized niosomes, a volume of niosomal dispersion (either uncoated F7 or coated C1), equivalent to 8 mg drug, was dropped on a predetermined weight (300 mg) of silicified microcrystalline cellulose (SMCC) powder. The mixture was allowed to stand for 15 min and then mixed for five minutes. Finally, the mixed powder was left overnight in a desiccator containing anhydrous calcium chloride. Plain niosomal powders were prepared similarly as blank.
Characterization of Uncoated F7-SMCC and Coated C1-SMCC Powders
Micrometric Properties
The angle of repose, compressibility index and Hausner ratio of plain SMCC, uncoated F7-SMCC and coated C1-SMCC powders were determined from Equations 3, 4, and 5 as mentioned before.30 (3) (4) (5)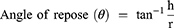
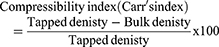
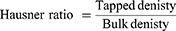
Drug Content Homogeneity
The obtained niosomal powder (either uncoated F7-SMCC or coated C1-SMCC) was equally divided into three quantities. Each quantity was sonicated with 10 mL ethanol for 30 min, then filtered and the drug concentration was measured at 255 nm by UV spectroscopy (ultraviolet/visible [UV/VIS] spectrophotometer, JASCO, Tokyo, Japan). The drug content was calculated from Equation 6.20
(6)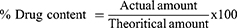
In vitro Dissolution Study of Uncoated F7-SMCC and Coated C1-SMCC Powders
In vitro dissolution study was carried out by USP apparatus II paddle method (Dissolution Apparatus USP Standards, Scientific, DA-6D, Bombay, India). The experiment was performed in different pH values of GIT, 0.1N HCl (pH 1.2), phosphate buffer (pH 6.8) and phosphate buffer (pH 7.4). Eight mg of CC and the equivalent from uncoated F7-SMCC and coated C1-SMCC powders, were subjected to the in vitro dissolution test. Each powder was transferred to 500 mL of different dissolution media,14 each containing 0.35% Tween 20 (to achieve sink condition) and stirred at 100 rpm at 37°C ±0.5. Aliquots of 5 mL were withdrawn at predetermined time intervals (0.5, 1, 2, 3, 4, 5, 6, 7, and 8 h), and replaced by same volume of fresh media. The solutions were filtered through 0.45 µm Millipore filter and the concentration of CC was measured by UV spectrophotometry spectroscopy (ultraviolet/visible [UV/VIS] spectrophotometer, JASCO, Tokyo, Japan) at predetermined λmax 254, 257, and 255 for pH values 1.2, 6.8 and 7.4,14 respectively using blank of plain formulations treated similarly. Each experiment was done in triplicate.
Kinetic Analysis of the Drug Release Data
Data of drug release from uncoated F7-SMCC and coated C1-SMCC were fitted to zero order, first order linear regression equations31 and Higuchi’s model32 to explore the kinetics of drug release. The release model with the highest coefficient of determination (R2) was considered the best to describe release kinetics of the drug. Furthermore, Korsmeyer–Peppas equation33 was utilized to inspect the drug release mechanism via determination of (n) value (diffusional exponent) from the slope of logarithmic relation between fraction of drug released and time.
Physical Stability Study
Uncoated F7-SMCC and coated C1-SMCC powders as well as uncoated F7 and coated C1 niosomal dispersions, were stored under similar conditions described in International Conference of Harmonization (ICH). The formulations were stored for three months,34 in well closed glass bottles in a refrigerator at temperature of (5°C ±3) as well as at ambient room temperature in desiccator containing anhydrous CaCl2 at temperature of (25°C ±2)/ (60% ±5%) relative humidity. The niosomal powder were tested in accordance to change in physical appearance, drug retention percent and in vitro dissolution study in phosphate buffer pH 6.8. The niosomal dispersions were examined by measuring PS, PDI, and ZP values over the period of storage. The statistical analysis of data was accomplished by one-way ANOVA, followed by Tukey–Kramer multiple comparisons tests. GraphPad Prism 5 software v5.03 (GraphPad Software Inc., San Diego, CA, USA) computer program was used for statistical analysis of data.
In vivo Study
The experimental procedure for the in vivo study was approved by the Research Ethical Committee at Mansoura University in accordance with “The principles of laboratory animal care” (NIH publication No. 85–23, revised 1985). Twenty-five male Sprague Dawley rats (weighing from 250–300 g) were adapted in environmentally controlled breeding room with free access to food and water for two weeks prior to the experiment. The rats (five groups) were fasted 12 h prior to the experiment, while allowed free access to water during the run. The selected oral dose of CC was 50 mg/kg or the equivalent from uncoated F7-SMCC and coated C1-SMCC, while that of CDT (parent drug) was 36 mg/kg orally. Each oral dose was suspended in 3 mL water containing 0.5% w/v sodium carboxy methyl cellulose and given by oral gavage to the rat under light ether anesthesia. The fifth group of rats received IV dose of 10 mg/kg of CDT via injection through the tail vein for determination of absolute bioavailability. The IV solution of CDT was prepared by dissolving CDT powder in the mixture of (1 N sodium carbonate/0.9% NaCl solution) as reported.35
Plasma Sample Preparation and Extraction Procedure
Blood samples were collected at time intervals of (0.25, 0.5, 1, 1.5, 2, 4, 6, 8, 12, and 24 h) in heparinized tubes from retro-orbital venous plexus of the rat after oral and IV administration. The plasma samples were centrifuged (Centrifuge, Hettich Micro 22 R, Germany) at 5000 rpm for 10 min and transferred to Eppendorf tubes to be stored at −20°C until analysis. After thawing, 100 µL of each sample were added to 50 µL of valsartan (Val) (internal standard solution in HPLC methanol, 10 µg/mL),36 20 µL of phosphoric acid (to adjust pH, denature and precipitate proteins)23 and 0.45 mL HPLC methanol. The mixture was vortexed by vortex tube mixer (Model VM-300, Taiwan, Gemmy Industrial Corp.) for five minutes then centrifuged (Centrifuge, Hettich Micro 22 R, Germany) at 10,000 rpm for 15 min and finally the supernatant was filtered through 0.45 µm Millipore filter.
High Performance Liquid Chromatography
The plasma concentrations of CDT were measured by HPLC–UV analysis method which was previously reported14 with slight modification. The HPLC system (Knauer, Germany) is equipped with a binary pump (Azura p 6.1 L); variable wavelength UV-VIS detector 190–750 nm (Azura UVD 2.1 L Detector) and chromatographic separation was accomplished with a C18 column (250 mm × 4.6 mm, 5 μm, Phenomenex Hyperclone ODS, USA). The sample with volume of 20 μL was injected and eluted with isocratic elution at flow rate of 1 mL/min using mobile phase consisting of a mixture of HPLC acetonitrile/potassium dihydrogen orthophosphate buffer (10 mM, pH 2.8 adjusted by few drops of phosphoric acid) (50:50% v/v). The analysis was performed at wavelength of 255 nm. The validity of the modified method was investigated in terms of selectivity/specificity, linearity, accuracy and precision according to ICH guidelines.37 Also, the detection limit (DL) and the quantification limit (QL) were calculated from Equations 7 and 8.27 (7) (8)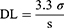
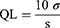
where  and
and  are the SD of y intercept and the slope of CDT plasma calibration curve, respectively.
are the SD of y intercept and the slope of CDT plasma calibration curve, respectively.
CDT plasma calibration curve was constructed by spiking 100 µL blank plasma with 50 µL of different working standard solutions over concentration range of (5–500 µg/mL) prepared by dilutions of stock CDT solution (500 µg/mL) in HPLC methanol.
Pharmacokinetic Study
CDT plasma concentrations were analyzed by non-compartmental model using GraphPad Prism 5 software computer program and the pharmacokinetic parameters were calculated as reported.23
Results and Discussion
Statistical Results of 23 Full Factorial Design
The full factorial design was applied to select the niosomal formulation with high %EE, low PS and low PDI. By the DOE, F7 was the optimized formula (Table 3), at which the CPPs levels were XA (+1), XB (+1) and XC (−1).
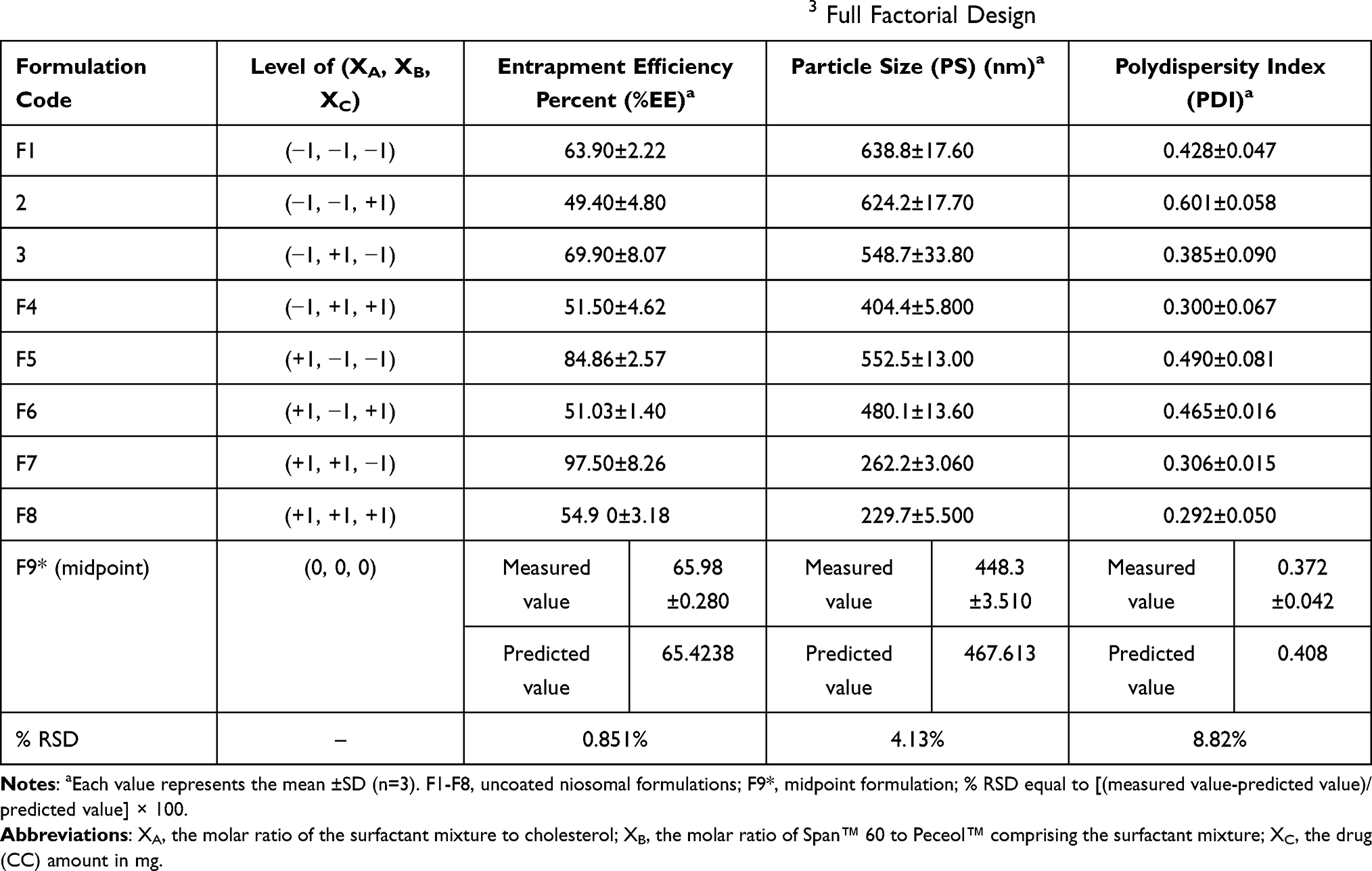 |
Table 3 Characterization of Candesartan Cilexetil Loaded Niosomes Using 23 Full Factorial Design |
The values of CQAs have been represented in Table 3. The resulted polynomial equation of each CQA was in form of terms representing the CPPs and their interactions, accompanied with regression coefficient and a sign, either positive or negative. A positive sign before the CPP indicates a positive effect, while a negative one indicates the reverse.23
Entrapment Efficiency Percent (%EE)
The %EE values of the niosomal formulations range from 49.4% to 97.5% as shown in Table 3 and the polynomial regression equation of %EE is
%EE = + 66.42 + 6.657 XA + 3.107 XB – 13.63XC + 1.02 XAB - 5.48 XAC - 1.548 XBC – 0.648 XABC.
Where P-value <0.0001 and the adjusted R2 =0.9521.
Looking carefully to the polynomial equation of %EE, it was obvious that XC (drug amount) revealed the strongest but negative influence on %EE. Even though, the interaction between XC and the other CPPs had negative values (XAC, XBC and XABC). This was explained by the fact that the formed vesicles could entrap the lipophilic drug within the niosomal membrane bilayers, until they became saturated with the drug at certain drug concentration. Beyond this concentration, the drug precipitation would occur.38 However, both the molar ratio of SM to CHOL and the molar ratio of the two surfactants Span™ 60 and Peceol™ within the SM, had positive effect on %EE (XA and XB). From Table 3, it is conspicuous that F7 complied with %EE required. Two interesting formulations are F2 and F7 having the lowest and the highest %EE, respectively and the levels of the CPPs are the reverse in both (Table 3).
It was reported that CHOL could stabilize the niosomal structure up to a certain amount, above which it could compete with the lipophilic drug for the space within the membrane bilayer. Therefore, the drug could be excluded from the bilayers structure.39 Also, one hypothesized that CHOL could disrupt the linear structure of the niosomal membrane bilayers by replacing the nonionic surfactant itself, thus the membrane permeability was increased and %EE was decreased.40
Furthermore, the molar ratio of Span™ 60 to Peceol™ influenced %EE. Span 60 has TC of 53°C and presents in the solid state at room temperature with more ordered gel phase,41 while Peceol™ (amphiphile) presents in less ordered liquid phase at room temperature with more leaky membranes.7 Hence, increasing the ratio of Span™ 60 with respect to Peceol™ (high level of XB) contributed to higher %EE.41
Particle Size (PS) and Polydispersity Index (PDI)
According to the DOE, PS and PDI with low values were required. Small values of PS are beneficial in enhancement of the drug absorption and oral bioavailability,42 while that of PDI stipulates the homogeneity of vesicle’s size distribution. The PS values of the niosomal formulations vary from 229.7 to 638.8 nm as shown in Table 3. PDI values range from 0.292 to 0.601 which are considered in the acceptable range (Table 3).
The polynomial regression equation of PS and PDI are: -
PS = +467.6–68.45 XA – 106.32 XB – 33.004 XC – 28.85 XAB +6.74 XAC-11.22 XBC+21.19 XABC.
Where P value ˂0.0001 and the adjusted R2 = 0.9875.
PDI= +0.408–0.02 XA −0.08 XB + 0.006 XC −0.001 XAB −0.015 XAC −0.035 XBC+0.033 XABC.
Where P-value ˂0.0001 and the adjusted R2 =0.7758.
It was observed that both +XA (increased SM with respect to CHOL) and +XB (increased Span™ 60 with respect to Peceol™), experienced low PS as well as low PDI. This reflected that increasing CHOL and Peceol™ had an unfriendly influence on PS and PDI. Surprisingly, the PS and PDI values of both F2 and F7 coincide with the corresponding data of the %EE as shown in Table 3. CHOL could strengthen the rigidity of niosomal membrane bilayers, hence rendering the produced vesicles more resistant to the effect of sonication on reduction of PS.11 Peceol™ encountered bent hydrocarbon chains due to the presence of double bond in its oleate moiety, thus these chains occupied more space in the bilayer structure resulting in expansion of the membrane, thus increment of PS, similar finding was reported with Tween 80.43
Figure 1 demonstrates three-dimensional (3D) surface response plots and contour plots of %EE at high level of XA (A), at low level of XA (B) and of PS at high level of XA (C), at low level of XA (D).
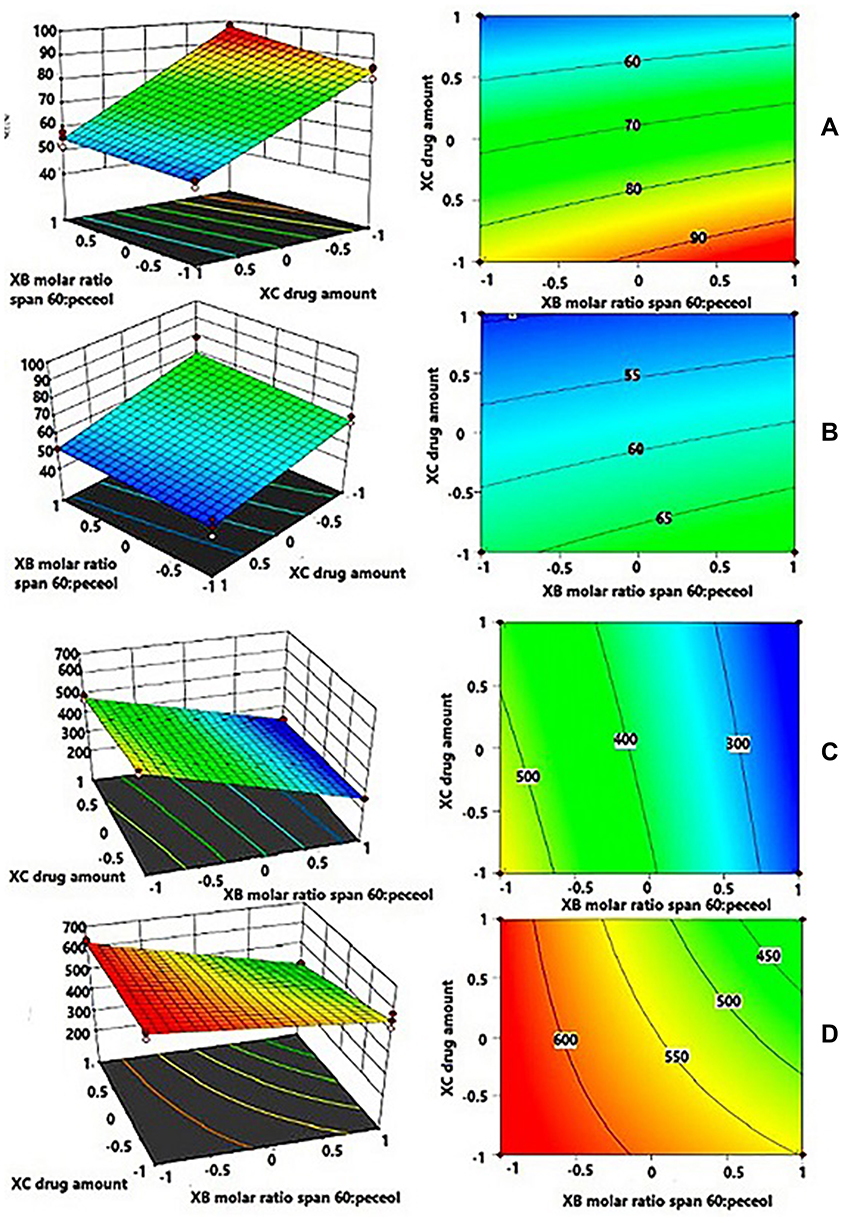 |
Figure 1 Three-dimensional (3D) surface response plots and contour plots of %EE at high level of XA (A), at low level of XA (B) and of PS at high level of XA (C), at low level of XA (D). Abbreviations: %EE, entrapment efficiency percent; PS, particle size; XA, molar ratio of surfactant mixture to cholesterol. |
For %EE, by fixing XA at the maximum level (Figure 1A), the %EE value increased at high level of XB and low level of XC. This means that the formulation with high %EE could be prepared by decreasing the utilized CHOL with respect to surfactant mixture as well as the drug amount in addition to increasing the utilized Span™ 60 within the surfactant mixture. For PS, by keeping the XA at the maximum level (Figure 1C), PS value decreased at low level of XB, reflecting the role of decreasing CHOL as well as the utilized Peceol™ in decreasing PS. The outcomes of 3D surface response plots as well as contour plots of each CQA were found to cope with the interpretation of the corresponding polynomial equation.
Check-point Analysis and Numerical Optimization by 23 Full Factorial Design
Check-point analysis was performed to verify the validity of the DOE for prediction, confirm the validity of reduced response and to investigate for any curvature in the response. Table 3 showed the predicted and the measured values of each CQA of the midpoint formulation (F9*). Percent RSD of all CQAs did not exceed 15%, confirming the validity of the DOE for prediction and absence of curvature in the response.44
Numerical optimization was applied to find out the optimized formulation with the highest desirability regarding the highest %EE and reasonable small PS along with PDI. The desirability value ranges from zero to one. If the value is closer to zero, this refers to an imprecise result, while approaching unity indicates a precise one.45 The DOE picked out the formulation prepared at XA (2:1), XB (2:1) and XC (15 mg) as the optimized one with CQAs values (Figure 2) close to those of F7 (Table 3) with the highest desirability value (0.936). Thus, F7 was chosen for further characterization.
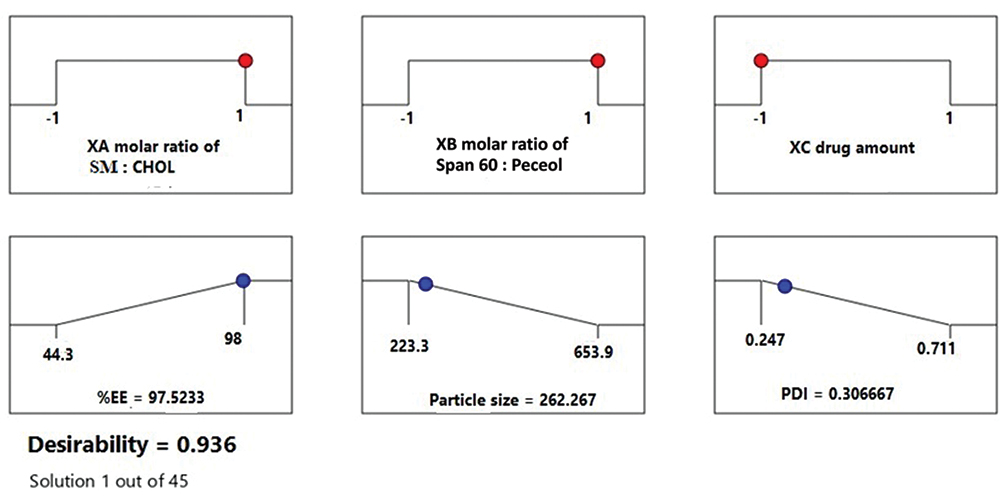 |
Figure 2 Desirability ramp of the optimized formulation by the DOE. |
Zeta Potential (ZP) of the Optimized Uncoated Niosomes (F7)
ZP is a chief parameter to judge the colloidal dispersion stability via determination of surface charge. ZP value of uncoated F7 is –39.8±0.8 mv. It is worthy to note that ZP value above –30 mv indicates good physical stability against aggregation through electrostatic repulsion.46 The imparted negative charge might be due to the adsorbed hydroxyl ion of the dispersion medium on the vesicle’s membrane47 and the adsorbed drug on niosomes surface due to the tetrazole moiety of CC with nitrogen atom bearing pair of electrons.14 In addition, it was assumed that Peceol™ could bear a negative charge due to the residuals of free oleic fatty acids with their ionized carboxylic group.6
Transmission Electron Microscope (TEM) of the Optimized Uncoated Niosomes (F7)
As depicted in Figure 3, the vesicles of F7 appear as spherical particles with dark area at the center referring to the core of vesicles which is surrounded by faint gray area corresponding to the bilayer membrane.
 |
Figure 3 Transmission electron microscope image of the optimized uncoated niosomes F7. |
Scanning Electron Microscope (SEM) of the Optimized Uncoated Niosomes (F7)
As depicted in Figure 4, niosomal particles in dispersion form (A) as well as in freeze-dried form (B), appeared as nearly spherical particles with a smooth surface, indicating that the niosomal particles could conserve their physical form after preparation and freeze-drying.48
 |
Figure 4 Scanning electron microscope of the optimized uncoated F7 niosomes in dispersion form (A) and after freeze drying (B). |
Particle Size and Zeta Potential of CS Coated Niosomal Formulations
From Table 4, it was observed that all CS-coated niosomes manifested higher PS than that of uncoated F7 ones. This emphasized that the CS coating layer around the niosomal membrane increased by increasing CS concentration with subsequent increase in PS.24,25 C1 being smaller in PS than C2, it was chosen as the optimized niosome. PS value in range of (200–500 nm) was preferred for mucoadhesion.12
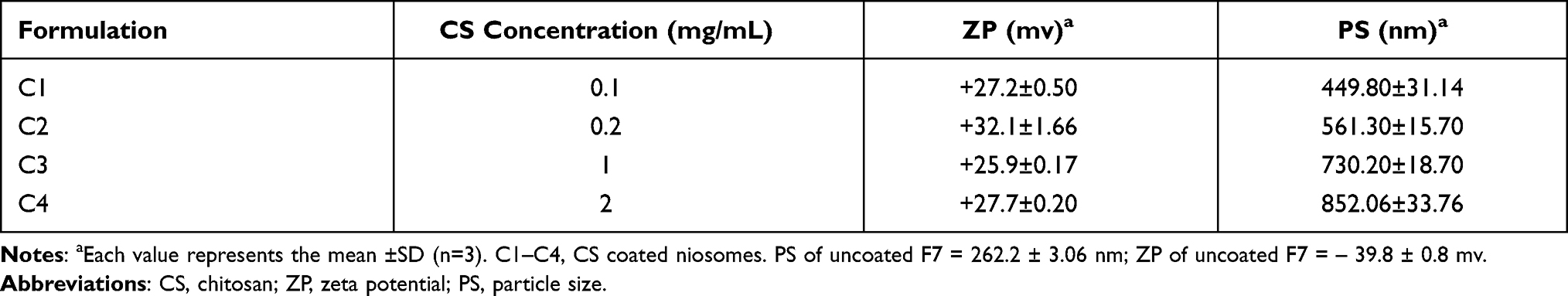 |
Table 4 Zeta Potential and Particle Size Values of Different Chitosan Coated Niosomes |
The acquired positive charge with C1 was important to fulfill interaction with mucin layer and to facilitate cellular uptake by enterocytes as well as enhancement of lymphatic transport.49 As shown in Table 4, it was noticed that ZP increased by increasing CS concentration from 0.1 mg/mL (C1) to 0.2 mg/mL (C2); while with further increase in CS concentration, ZP remained almost constant or slightly decreased as in C3 and C4.
Differential Scanning Calorimetry (DSC)
Figure 5 demonstrates the DSC thermograms of CHOL, Span™ 60, CS, CC, their physical mixture, CC-loaded F7 and C1 formulations and their corresponding plain ones. The DSC thermograms of CHOL and Span™ 60 (A and B) revealed an endothermic peak at 148.7°C and 54.2°C, respectively, representing their transition temperature (TC).50 An endothermic peak was noticed in DSC thermogram of CS (C) at 75.18°C referring to dehydration temperature, in addition a broad exothermic peak appeared around 300°C indicating CS thermal decomposition.27 The DSC thermogram of CC (D) showed sharp endothermic peak at 168.6°C representing its melting point followed by an exothermic peak at 176.9°C referring to drug decomposition.51 The physical mixture (E) (representing C1 formulation) showed only an endothermic peak of Span™ 60 at 55.35°C as its amount represented the dominant component. The disappearance of other niosomal components peaks was ascribed to their presence in small amount.52
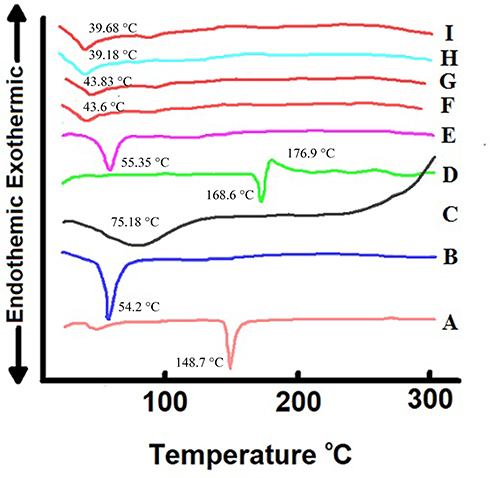 |
Figure 5 DSC thermograms of CHOL (A), Span™ 60 (B), CS (C), CC (D), physical mixture (E), lyophilized plain F7 preparation (F), lyophilized CC-loaded F7 formulation (G), lyophilized plain C1 preparation (H) and lyophilized CC-loaded C1 formulation (I). Abbreviations: DSC, differential scanning calorimetry; CHOL, cholesterol; CS, chitosan; CC, candesartan cilexetil; F7, the optimized uncoated niosomes; C1, the optimized chitosan coated niosomes. |
Interestingly, by careful examination of Figure 5, one can notice the similarity of the endothermic peaks of the plain niosomes and the corresponding medicated ones. Herein, Figure 5F and G for uncoated F7 as well as Figure 5H and I for coated C1. The peaks of CC were completely dissipated which emphasized complete drug encapsulation in niosomes and its conversion into amorphous form.26,52 They also experienced reduction in the endothermic peak of Span™ 60 (54.2°C) which designated that the assembly of Span™ 60 into the niosomal bilayers might decrease its heat of fusion.53 The results of DSC negate any incompatibility among the niosomal components.
Evaluation of the in vitro Biocompatibility of the Optimized Niosomal Formulations (Uncoated F7 and Coated C1) with Caco-2 Cells
MTT viability test was performed with uncoated F7 and coated C1 niosomes in different CC concentrations, for 24 h incubation. The concentration of CC in niosomes ranged from 5–80 µg/mL. Meanwhile, to test the effect of contact time of free CC on Caco-2 cells, the incubation was done for 3 and 24 h with equal concentrations as with that of the niosomes of uncoated F7 and coated C1. Increasing the concentration of CC resulted in a decrease in cell viability, the least effect was with the 5 µg/mL. Noticeably, the results of difference in time of exposure of Caco-2 cells was more prominent as the concentration of CC increased from 5–80 µg/mL (Figure 6).
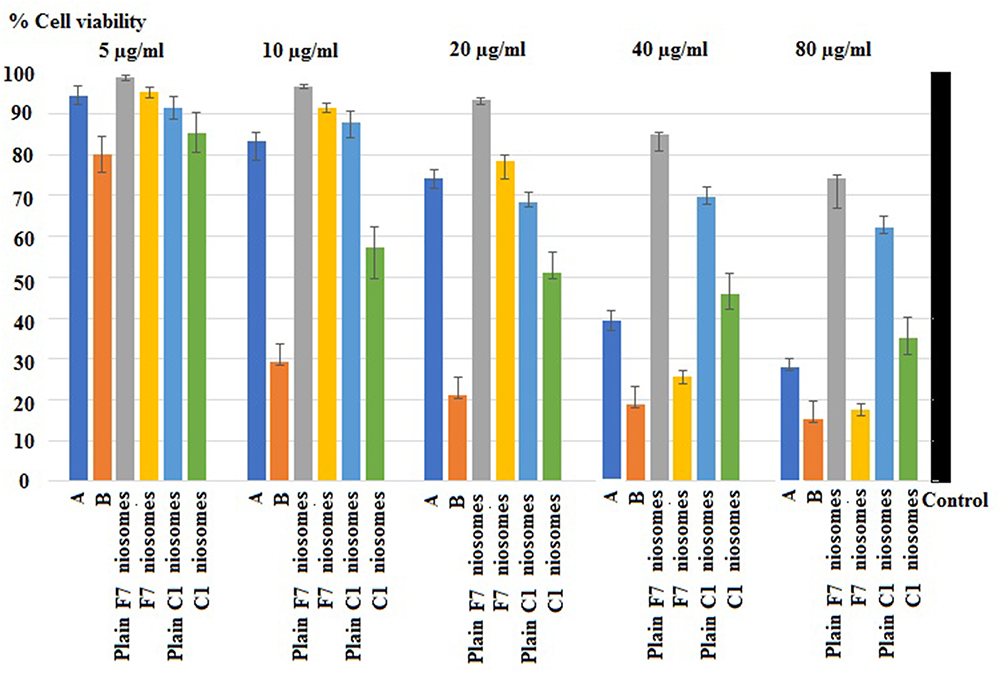 |
Figure 6 Percent cell viability of Caco-2 cells treated with free CC for three hours (A), free CC for 24 h (B), plain F7 niosomes, F7 niosomes, plain C1 niosomes and C1 niosomes. Note: Results represent the mean ±SD (n=3). Abbreviations: CC, candesartan cilexetil; Plain F7 niosomes, empty optimized uncoated niosomes; F7 niosomes, CC-loaded optimized uncoated niosomes; Plain C1 niosomes, empty optimized chitosan coated niosomes; C1 niosomes, CC-loaded optimized chitosan coated niosomes. |
As depicted in Table 5, free CC emerged the half inhibitory concentration (IC50) at 35.8 µg/mL on Caco-2 cells after contact for three hours, while IC50 decreased to 8.5 µg/mL after 24 h as the time of contact increased, the cell toxicity increased. It was reported that CC could experience certain intrinsic cell toxicity on different cell types.54
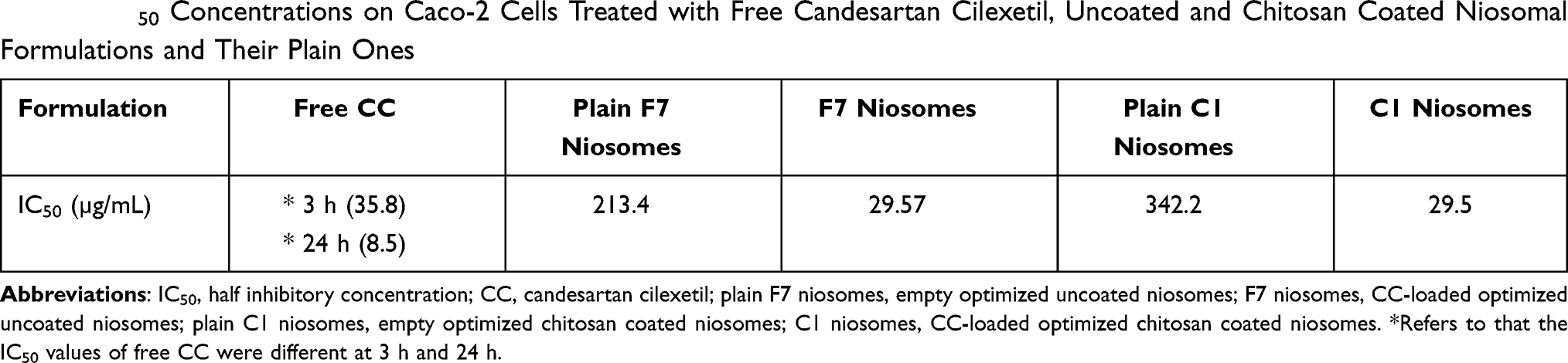 |
Table 5 IC50 Concentrations on Caco-2 Cells Treated with Free Candesartan Cilexetil, Uncoated and Chitosan Coated Niosomal Formulations and Their Plain Ones |
On incubation of niosomes of uncoated F7 and coated C1 for 24 h, both experienced the same IC50 (29.5 µg/mL) on Caco-2 cells which was higher than that of free CC (8.5 µg/mL). These findings ratified the camouflaging of the drug inside the niosomal particles with plausible reduced cell cytotoxicity.55
Plain-coated C1 revealed IC50 at 342.4 µg/mL much higher than that of plain uncoated F7 niosomes (213.4 µg/mL). CS in the plain niosomes could modulate cell toxicity.23
Undeniably, the decreased IC50 in CC-loaded formulations was attributed to the adsorbed drug on the niosomal surface or the released drug outside the niosomes during the incubation period.56 The equality of IC50 of uncoated F7 and coated C1 niosomes could be explained by the limited capacity of CS to neutralize the cytotoxicity of both CC and Peceol™ on Caco-2 cells.54,57
Conversion of the Niosomal Dispersion into Powder Form
PROSOLV® is silicified microcrystalline cellulose (SMCC) which combines microcrystalline cellulose MCC (representing 98% of its structure) and colloidal silicon dioxide (representing 2% of its structure). This combination provided more surface area than MCC alone due to the presence of SiO2 nanoparticles together with wicking effect. Also, PROSOLV® provided higher flow properties than MCC and could be directly filled into capsule. Thus, PROSOLV® (SMCC) would be utilized for conversion of niosomal dispersion into powder via high void volume that can entrap or cage the niosomal suspension within spaces of the polymer.58
Characterization of Uncoated F7-SMCC and Coated C1-SMCC Powders
Micrometric Properties
Plain SMCC powder showed angle of repose 35°±1.102 indicating good flowability, while that of uncoated F7-SMCC and coated C1-SMCC were 37.3°±1.23 and 39.3°±1.52, respectively; reflecting fair flowability. Plain SMCC, uncoated F7-SMCC, and coated C1-SMCC revealed compressibility indices (Carr’s index) equal to 18.8±0.333%, 23.3±0.306%, and 26.06±1.102%, respectively and Hausner ratio of 1.217±0.015, 1.31±0.026, and 1.35±0.02, respectively, referring to fair and passable flowability. Compressibility index between 10 and 30 indicates good flowability and angle of repose below 40° is acceptable.59
Drug Content Homogeneity
Uncoated F7-SMCC and coated C1-SMCC showed insignificant difference (P<0.05) in percent drug content of the different quantities of the powder, indicating homogenous distribution of the drug throughout the powder.59
In vitro Dissolution Study of Uncoated F7-SMCC and Coated C1-SMCC Powders
Figure 7 manifests the dissolution profiles of CC from uncoated F7-SMCC and coated C1-SMCC powders compared with free CC, in different gastrointestinal physiological pH values. Different parameters could control the drug release such as the solubility of the drug, the solubility of CS coat, pH of the dissolution medium, and PS of the niosomes. Free CC manifested a dissolution profile in acidic pH (Figure 7A) lower than those in basic pH (Figure 7B and C). This was ascribed to its acidic nature with pka=6.3.14 Uncoated F7-SMCC and coated C1-SMCC exhibited initial burst drug release in acidic and basic pH at the first hour. In pH 1.2, percent drug released at one hour (% Q1h) for uncoated F7-SMCC and coated C1-SMCC equal to 44.6% and 48.7%, respectively. This might be ascribed to the adsorbed drug on the surface of niosomal membrane which rapidly dissolved in the surrounding medium.60 The dissolution profiles of CC from uncoated F7-SMCC and coated C1-SMCC niosomes were higher than that of free CC in acidic pH and lower than those of free CC in basic pH (Figure 7). The % Q8h values of uncoated F7-SMCC and coated C1-SMCC in acidic pH were significantly (P<0.05) higher than that of free CC (93.7%, 99.9%, and 44.9%, respectively). The enhanced CC dissolution in acidic pH illuminated the ability of niosomes to motivate the CC solubility by their nanosized range. However, uncoated F7-SMCC and coated C1-SMCC niosomes in both acidic and basic pH, could release the drug over the period of eight hours as well as their dissolution profiles in basic pH, were lower than those of free CC. This was attributed to the presence of the lipid membrane bilayer of niosomal structure via which the drug was released, then dissolved in the dissolution medium.14 The role of CS could not be ignored. In pH 6.8, % Q8h of coated C1-SMCC (82.1%) was significantly (P<0.05) lower than that of uncoated F7-SMCC (97%). In basic pH, the amino groups of CS became deprotonated, so they formed a barrier against release of the drug due to H-bonds formed between these groups.
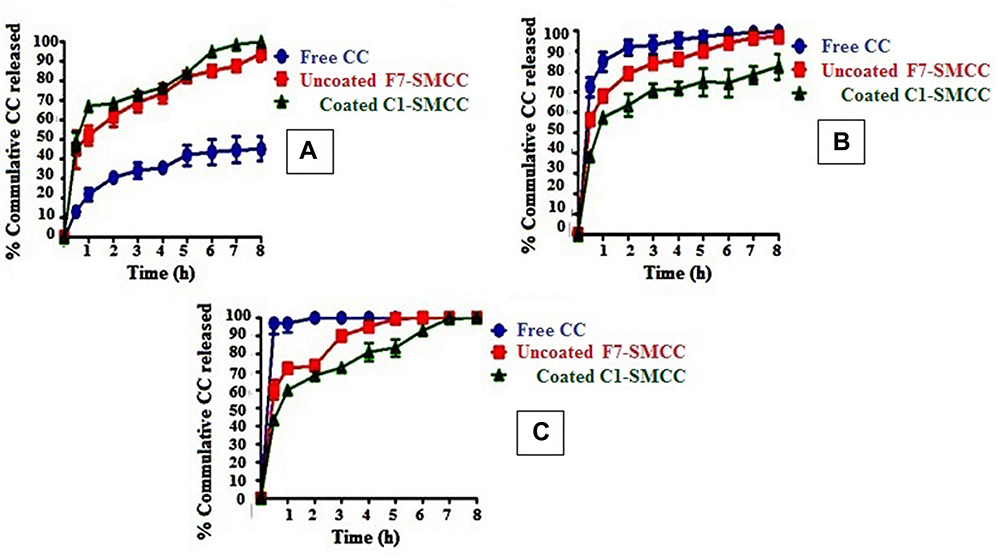 |
Figure 7 In vitro dissolution profiles of free CC, uncoated F7-SMCC and coated C1-SMCC in 0.1 N HCL, pH 1.2 (A), phosphate buffer, pH 6.8 (B) and phosphate buffer, PH 7.4 (C). Abbreviations: CC, candesartan cilexetil; Uncoated F7-SMCC, the optimized uncoated niosomes-silicified microcrystalline cellulose; Coated C1-SMCC, the optimized chitosan coated niosomes-silicified microcrystalline cellulose. |
Kinetic Analysis of the Drug Release Data
In 0.1 N HCl, uncoated F7-SMCC obeyed Higuchi’s model (Table 6). Meanwhile, in phosphate buffer, pH 6.8 and pH 7.4, it showed maximum linearity with first order kinetic. However, the drug release kinetics of coated C1-SMCC complied with Higuchi’s model either in acidic or basic media. Higuchi’s model means that the drug release occurs by diffusion in one dimension via the bilayer structure. This model hypotheses that the initial drug concentration in the matrix is greater than the solubility of the drug which creates a sink condition on the niosomal surface, the drug diffusivity is constant and the release medium maintains good sink conditions.61 First order kinetics manifests that the drug release rate is directly proportional to the amount of the drug remaining inside the niosomes which declined over time.62 Korsmeyer–Peppas model clarified the drug release mechanism from uncoated F7-SMCC and coated C1-SMCC up to 60% by Fickian diffusion in all media with n-value less than 0.5.
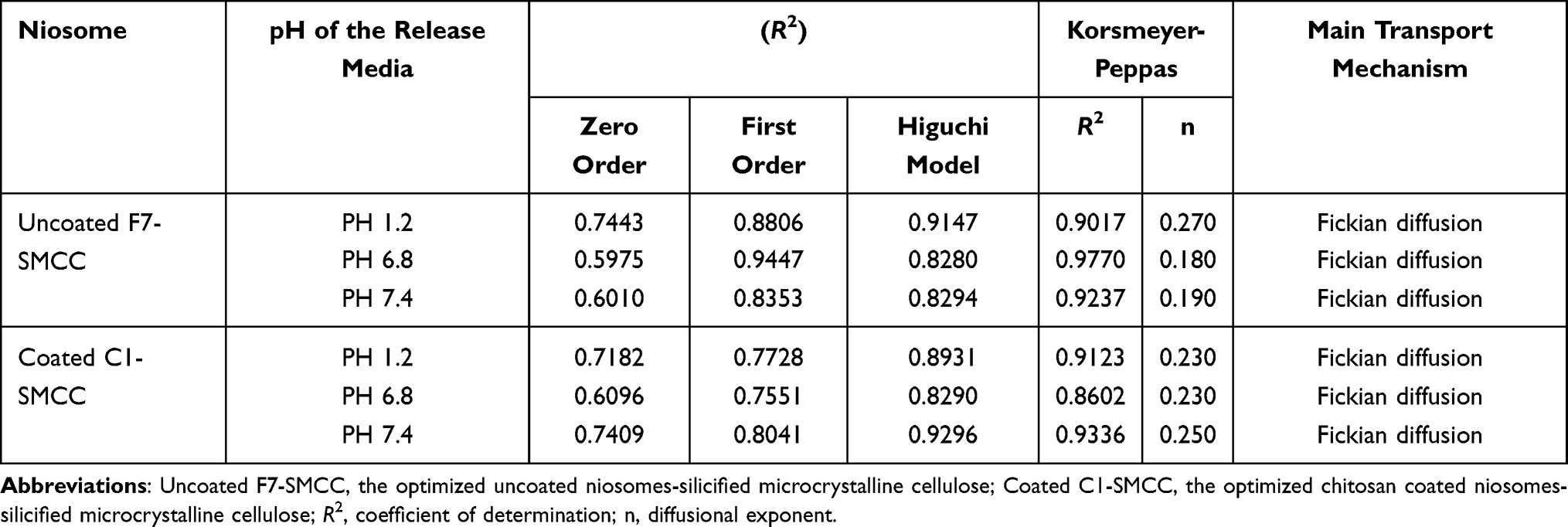 |
Table 6 Kinetic Analysis of Drug Release Data from Uncoated and Chitosan Coated Niosomal Powders |
Physical Stability Study
The powders showed no change in their color or appearance during the storage period for three months under refrigerated or ambient conditions. There was insignificant difference (P>0.001) in drug retention percent in niosomes of uncoated F7-SMCC and coated C1-SMCC for the whole storage period under both storage conditions, when compared with the corresponding drug retention percent at zero time (Table 7). Similar results were attained with the in vitro dissolution profiles which did not differ significantly (P>0.001) from the corresponding profiles at the zero time (results not shown).
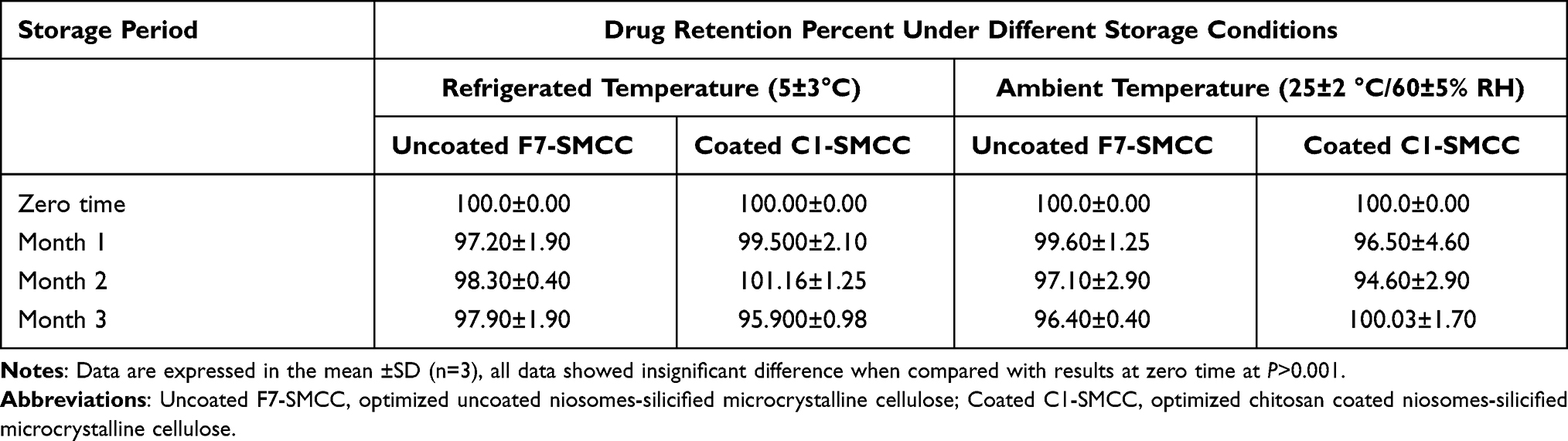 |
Table 7 Drug Retention Percent in Niosomes of Uncoated and Chitosan Coated Niosomal Powders, Stored at Refrigerated (5±3°C) and Ambient (25±2°C/60±5% RH) Conditions |
From Table 8, it was obvious that uncoated F7 niosomes preserved their stability with regard to the values of PS, PDI and ZP over the storage period. However, coated C1 niosomes manifested significant decline in ZP (+7.28±0.79 mv) accompanied with significant increase in PDI value (0.928±0.063), at the third month of storage under ambient conditions. The decreased ZP illuminated the tendency of coated niosomes to aggregate, indicating a decline in their stability over time.63
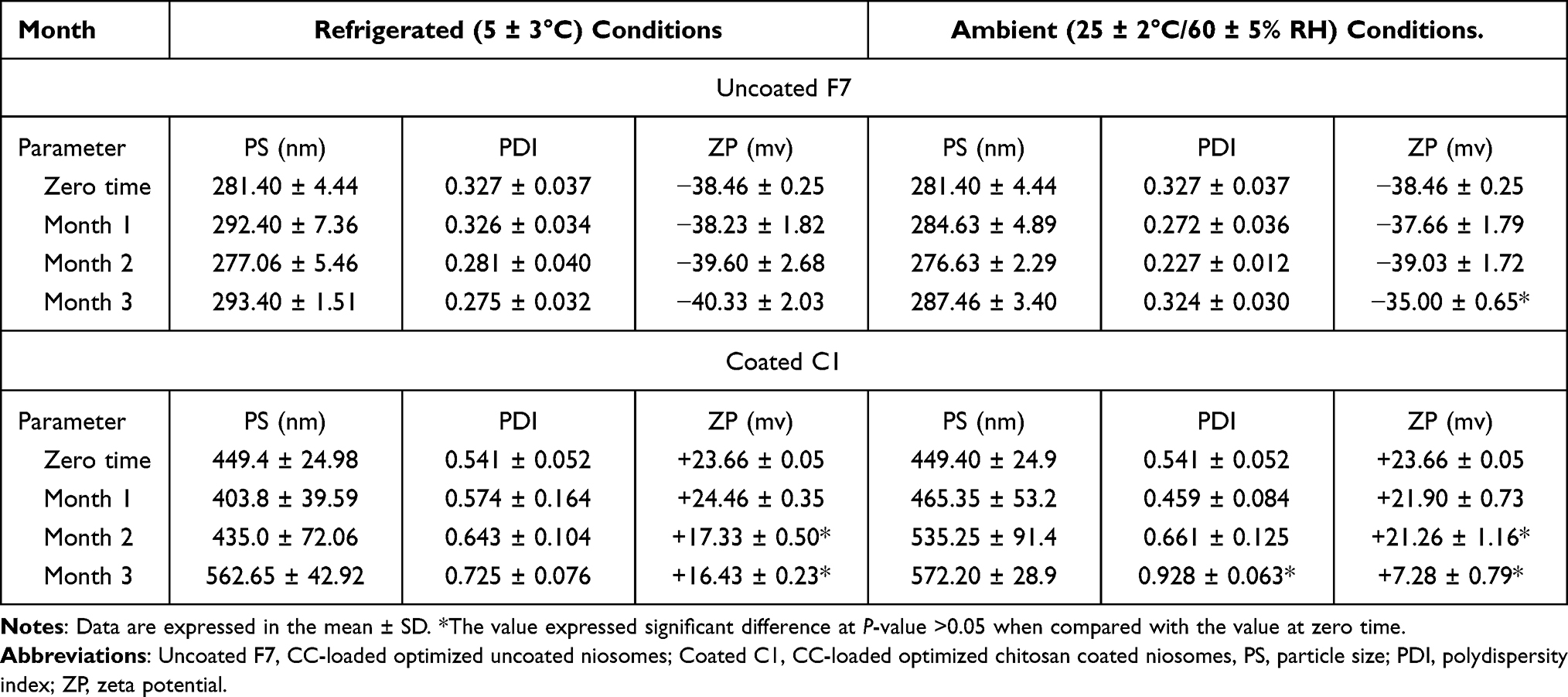 |
Table 8 PS, PDI and ZP Values of Niosomes of Uncoated and Chitosan Coated Niosomal Dispersion, Stored at Refrigerated (5 ± 3°C) and Ambient (25 ± 2°C/60 ± 5% RH) Conditions |
Pharmacokinetic Study
The modified HPLC method was validated with regard to selectivity/specificity, linearity, accuracy and precision as stated in ICH guidelines. HPLC chromatogram of blank plasma, spiked with CDT and Val (internal standard) displayed the selectivity of the method with no interferences with plasma. The retention time of CDT and Val appeared at 4.78±0.042 min and 7.17±0.068 min, respectively.
From CDT calibration curve in plasma, the linearity was achieved over concentration range of (0.5–20 µg/mL) with coefficient of determination (R2) =0.9962 and the equation of linear regression was y=(0.1320±0.003) x + (0.1894±0.005), where y was the ratio of drug peak area/IS peak area, x is the plasma concentration (µg/mL), the slope of the curve was (0.1320±0.003) and intercept was (0.1894±0.005). All values were expressed as the mean ±SEM. The calculated DL value was 0.231 µg/mL and QL was 0.646 µg/mL as SD value of y intercept was 0.009 and the slope of the plasma calibration curve was 0.1320 ± 0.003.
Finally, the intraday precision was (1.79±0.006 to 8.36±0.384) and accuracy was (92.9%±1.159 to 104.5%±3.6) while interday precision was (2.66±0 0.033 to 5.407±0.167) and accuracy was (91.5%±2.02 to 111%±3.4) Each value was expressed as the mean ±SEM.27
Figure 8A demonstrates the CDT plasma concentration–time curve of orally administered free CC and CDT suspensions in dose of 50 mg/kg and 36 mg/kg, respectively. While Figure 8B displays those of the niosomes of uncoated F7-SMCC and coated C1-SMCC, equivalent to dose of 50 mg/kg CC and Figure 8C shows the CDT plasma concentration–time curve after IV administration of 10 mg/kg of CDT solution. It was reported that the therapeutic concentration of CDT in plasma should not be less than 0.05 µg/mL.64 Further study is required to ascertain that the niosomes derived thereof can produce the therapeutic concentration in humans.
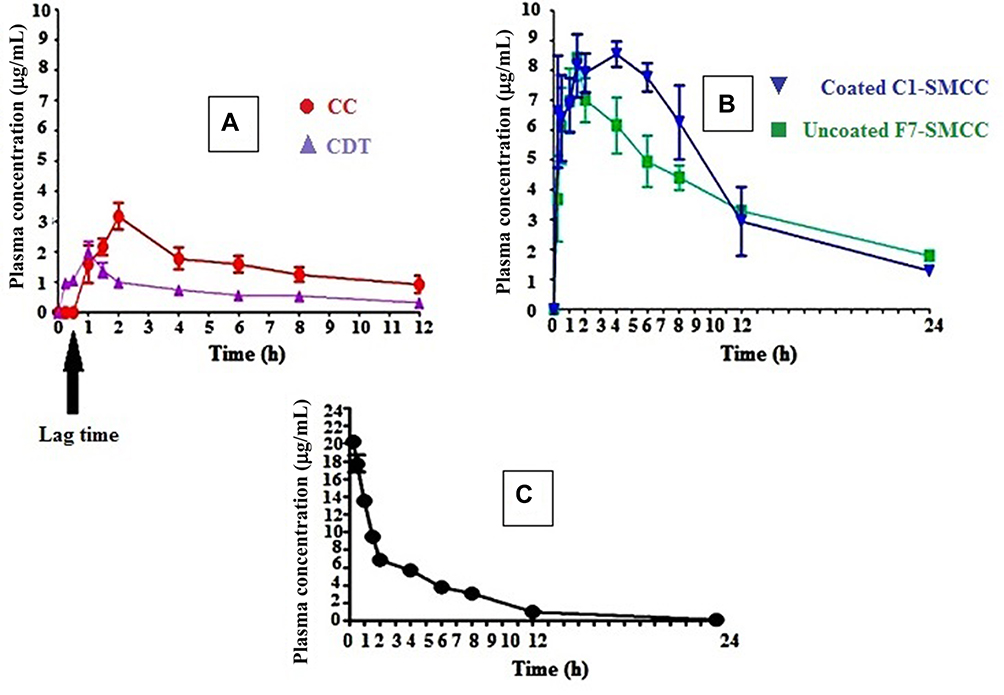 |
Figure 8 Plasma concentration–time curve of CDT after oral administration of (50 mg/kg) of CC and (36 mg/kg) CDT (A), oral administration of uncoated F7-SMCC and coated C1-SMCC each equivalent to (50 mg/kg) (B) and IV administration of CDT (10 mg/kg) (C). Abbreviations: CC, candesartan cilexetil; CDT, candesartan; Uncoated F7-SMCC, the optimized uncoated niosomes-silicified microcrystalline cellulose; Coated C1-SMCC, the optimized chitosan coated niosomes-silicified microcrystalline cellulose. Note: Each point represents the mean ±SEM (n=5). |
Surprisingly, oral free CC exhibited superior Cmax (3.543 µg/mL) and AUC (22.84±6.13 µg.h/mL) over oral CDT (2.054 µg/mL and 10.98±1.43 µg.h/mL, respectively) (Table 9). Our results were controversial to those described in previous literature.65 This literature reported that CDT as a parent drug was better in bioavailability (higher Cmax and AUC) than CC as prodrug. These results were contrary to our findings.
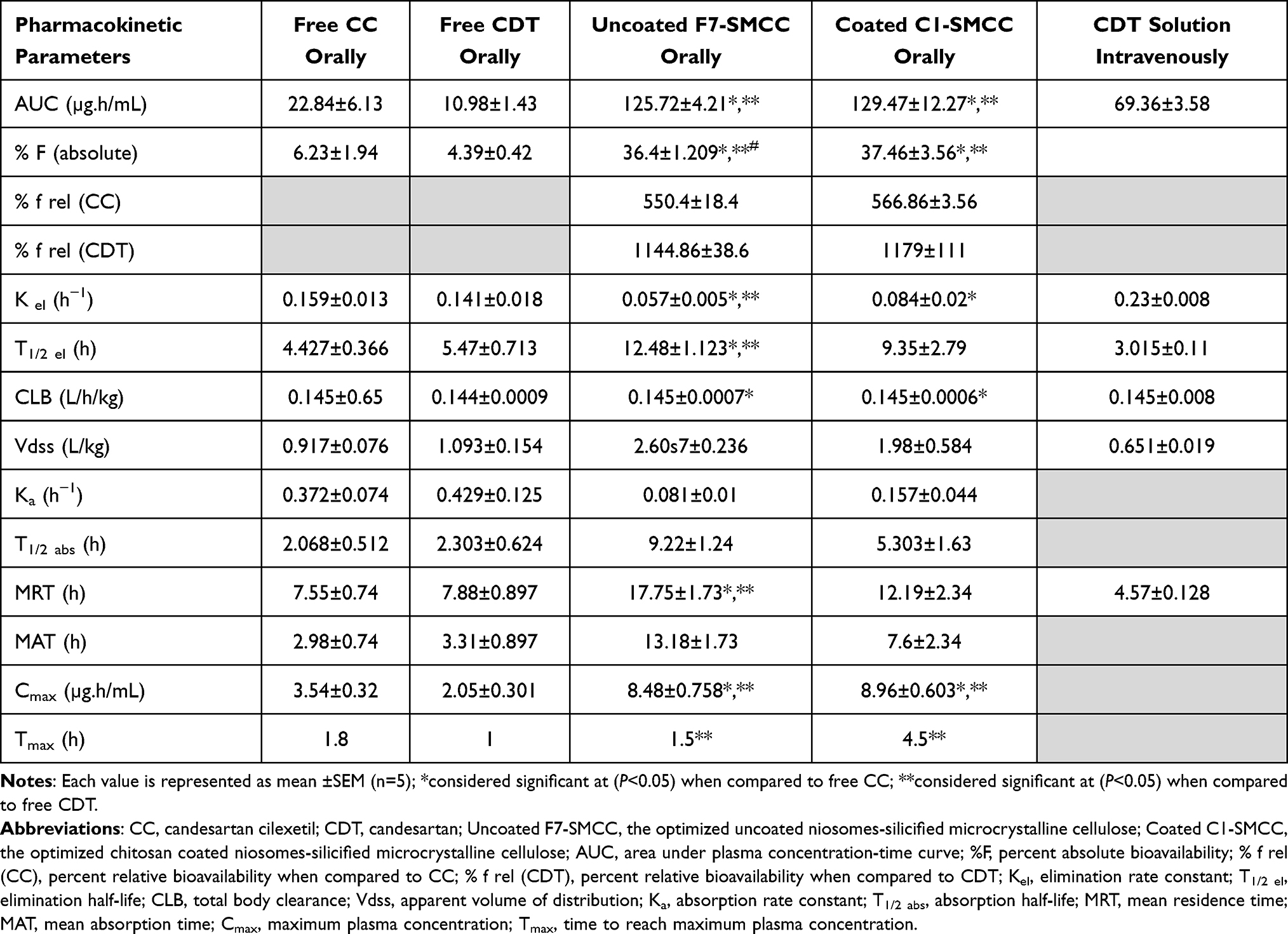 |
Table 9 Pharmacokinetic Parameters of IV Administration of CDT Solution and Oral Administration of Free Candesartan Cilexetil, Free Candesartan, Uncoated and Chitosan Coated Niosomal Powders |
The lag time in CDT plasma concentration–time curve experienced by free CC lasted for one hour, it was taken for the ester hydrolysis in the gut wall before reaching the systemic circulation in the form of CDT.19 Accordingly, there was a shift in tmax of oral free CC to 1.8 h instead of one hour in the case of oral CDT.
The attractiveness of uncoated F7-SMCC and coated C1-SMCC blood level data was the absence of lag-time period that was manifested in plasma concentration time curve of free CC suspension. The rational for that might be the membrane fluidity by Peceol™ and Span™ 60 presented in niosomes.6 Additionally, Peceol™ enhanced lymphatic transport of niosomes by M cells of Peyer’s patches of the lymphoid system as reported before.8 This also confirmed that intact niosomes endowed plentiful transfer of the drug through the epithelial cells by avoiding recognition by P-gp efflux pump and protection from hydrolysis.66 The role of CS in this respect could not be overlooked.
The high SMCC void volumes that can entrap or cage the niosomes of uncoated F7 and coated C1 resulted in promising equal increments of the drug oral absolute bioavailability (36.4±1.209% and 37.46±3.56, respectively) compared to those of orally administered free CC or CDT suspension (6.23±1.94% and 4.39±0.42%, respectively). Moreover, the relative bioavailability of uncoated F7-SMCC and coated C1-SMCC with respect to free CC suspension was much higher than that previously reported for CC-loaded proniosomes14 and as solid dispersion with PVPK-90.20
Table 9 shows higher MRT of uncoated F7-SMCC (17.75±1.73 h) which means prolonged effect compared with coated C1-SMCC with lower value (12.19±2.34 h). Meanwhile, the higher value of Ka in case of coated C1-SMCC than that of uncoated F7-SMCC reflected that CS played a pivotal role in enhancing CC absorption,12 although both niosomes experienced the same extent of bioavailability (Table 9).
From the data, it can be inferred that the niosomes acted as reservoir for CC, while SMCC ensured its sustained release. It might be speculated that SMCC in this case acted as “scaffold” for the niosomes.
Conclusion
Niosomes for CC were maneuvered successfully. For the first time, the absolute bioavailability of CC-loaded uncoated F7-SMCC and coated C1-SMCC in rats was about 37% instead of the previously reported 15% by the virtue of the “miraculous” molecules of GMO in combination with Span™ 60. The MRT of the coated C1-SMCC was lower than the corresponding uncoated one. CS probably helped rapid absorption with lower MRT and higher Ka value. IC50 (µg/mL) was the same for uncoated F7 and coated C1 niosomes but lower than the corresponding plain one on Caco-2 cells. Meanwhile, IC50 for free CC alone was much lower indicating that the excipients ameliorated the toxicity of CC. Further, it was noticed that the concentrations of free CC and the time of contact with Caco-2 cells were prominent factors in biocompatibility. The longer the time the lower the IC50. Further in vivo evaluation in humans is a prospective goal.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Vishwakarma N, Jain A, Sharma R, Mody N, Vyas S, Vyas SP. Lipid-based nanocarriers for lymphatic transportation. AAPS PharmSciTech. 2019;20(2):1–13. doi:10.1208/s12249-019-1293-3
2. Elmeliegy M, Vourvahis M, Guo C, Wang DD. Effect of P-glycoprotein (P-gp) inducers on exposure of P-gp substrates: review of clinical drug–drug interaction studies. Clin Pharmacokinet. 2020;59(6):699–714. doi:10.1007/s40262-020-00867-1
3. Bhardwaj P, Tripathi P, Gupta R, Pandey S. Niosomes: a review on niosomal research in the last decade. JDDST. 2020;56(PA):101581. doi:10.1016/j.jddst.2020.101581
4. Shilakari Asthana G, Sharma PK, Asthana A. In vitro and in vivo evaluation of niosomal formulation for controlled delivery of clarithromycin. Scientifica. 2016;2016:1–10. doi:10.1155/2016/6492953
5. Kamel AE, Fadel M, Louis D. Curcumin-loaded nanostructured lipid carriers prepared using Peceol™ and olive oil in photodynamic therapy: development and application in breast cancer cell line. Int J Nanomedicine. 2019;14:5073–5085. doi:10.2147/IJN.S210484
6. Gagliardi A, Cosco D, Udongo BP, Dini L, Viglietto G, Paolino D. Design and characterization of glyceryl monooleate-nanostructures containing doxorubicin hydrochloride. Pharmaceutics. 2020;12(11):1017. doi:10.3390/pharmaceutics12111017
7. Milak S, Zimmer A. Glycerol monooleate liquid crystalline phases used in drug delivery systems. Int J Pharm. 2015;478(2):569–587. doi:10.1016/j.ijpharm.2014.11.072
8. Sultan AA, El-Gizawy SA, Osman MA, El Maghraby GM. Peceosomes for oral delivery of glibenclamide: in vitro in situ correlation. JDDST. 2017;41:303–309. doi:10.1016/j.jddst.2017.08.003
9. Mady OY, Donia AA, Al-Shoubki AA, Qasim W. Paracellular pathway enhancement of metformin hydrochloride via molecular dispersion in span 60 microparticles. Front Pharmacol. 2019;10:713. doi:10.3389/fphar.2019.00713
10. Hoosain FG, Choonara YE, Tomar LK, et al. Bypassing P-glycoprotein drug efflux mechanisms: possible applications in pharmacoresistant schizophrenia therapy. Biomed Res Int. 2015;2015:1–21. doi:10.1155/2015/484963
11. Shah P, Goodyear B, Haq A, Puri V, Michniak-Kohn B. Evaluations of quality by design (QbD) elements impact for developing niosomes as a promising topical drug delivery platform. Pharmaceutics. 2020;12(3):246. doi:10.3390/pharmaceutics12030246
12. Shalaby TI, El-Refaie WM. Bioadhesive chitosan-coated cationic nanoliposomes with improved insulin encapsulation and prolonged oral hypoglycemic effect in diabetic mice. JPharmSci. 2018;107(8):2136–2143. doi:10.1016/j.xphs.2018.04.011
13. Khalifa A-ZM, Rasool BKA. Optimized mucoadhesive coated niosomes as a sustained oral delivery system of famotidine. AAPS PharmSciTech. 2017;18(8):3064–3075. doi:10.1208/s12249-017-0780-7
14. Yuksel N, Bayindir ZS, Aksakal E, Ozcelikay AT. In situ niosome forming maltodextrin proniosomes of candesartan cilexetil: in vitro and in vivo evaluations. Int J Biol Macromol. 2016;82:453–463. doi:10.1016/j.ijbiomac.2015.10.019
15. Fayed ND, Osman MA, Maghraby EL. Enhancement of dissolution rate and intestinal stability of candesartan cilexitil. JAPS. 2016;6(05):102–111. doi:10.7324/JAPS.2016.60516
16. Surampalli G, Nanjwade BK, Patil PA, Chilla R. Novel tablet formulation of amorphous candesartan cilexetil solid dispersions involving P-gp inhibition for optimal drug delivery: in vitro and in vivo evaluation. Drug Deliv. 2016;23(7):2124–2138. doi:10.3109/10717544.2014.945017
17. Dudhipala N, Veerabrahma K. Candesartan cilexetil loaded solid lipid nanoparticles for oral delivery: characterization, pharmacokinetic and pharmacodynamic evaluation. Drug Deliv. 2016;23(2):395–404. doi:10.3109/10717544.2014.914986
18. Zewail MB, El-Gizawy SA, Osman MA, Haggag YA. Preparation and In vitro characterization of a novel self-nano emulsifying drug delivery system for a fixed-dose combination of candesartan cilexetil and hydrochlorothiazide. JDDST. 2021;61:102320. doi:10.1016/j.jddst.2021.102320
19. Amer AM, Allam AN, Abdallah OY. Preparation, characterization and ex vivo–in vivo assessment of candesartan cilexetil nanocrystals via solid dispersion technique using an alkaline esterase activator carrier. Drug Dev Ind Pharm. 2019;45(7):1140–1148. doi:10.1080/03639045.2019.1600533
20. Aly UF, Sarhan HA-M, Ali TFS, Sharkawy HAE-B. Applying different techniques to improve the bioavailability of candesartan cilexetil antihypertensive drug. Drug Des Devel Ther. 2020;14:1851–1865. doi:10.2147/DDDT.S248511
21. Sultan AA, El-Gizawy SA, Osman MA, El maghraby GM. Niosomes for oral delivery of nateglinide: in situ–in vivo correlation. J Liposome Res. 2018;28(3):209–217. doi:10.1080/08982104.2017.1343835
22. Rinaldi F, Hanieh PN, Chan LKN, et al. Chitosan glutamate-coated niosomes: a proposal for nose-to-brain delivery. Pharmaceutics. 2018;10(2):38. doi:10.3390/pharmaceutics10020038
23. Aman RM, Abu Hashim II, Meshali MM. Novel chitosan-based solid-lipid nanoparticles to enhance the bio-residence of the miraculous phytochemical “Apocynin. Eur J Pharm Sci. 2018;124:304–318. doi:10.1016/j.ejps.2018.09.001
24. Alomrani A, Badran M, Harisa GI, et al. The use of chitosan-coated flexible liposomes as a remarkable carrier to enhance the antitumor efficacy of 5-fluorouracil against colorectal cancer. SPJ. 2019;27(5):603–611. doi:10.1016/j.jsps.2019.02.008
25. Alshraim MO, Sangi S, Harisa GI, Alomrani AH, Yusuf O, Badran MM. Chitosan-coated flexible liposomes magnify the anticancer activity and bioavailability of docetaxel: impact on composition. Molecules. 2019;24(2):250. doi:10.3390/molecules24020250
26. Abu Hashim II, Abo El-Magd NF, El-Sheakh AR, Hamed MF, Abd El-Gawad AE-GH. Pivotal role of Acitretin nanovesicular gel for effective treatment of psoriasis: ex vivo–in vivo evaluation study. Int J Nanomedicine. 2018;13:1059–1079. doi:10.2147/IJN.S156412
27. Darwesh AY, El-Dahhan MS, Meshali MM. A new dual function orodissolvable/dispersible meclizine HCL tablet to challenge patient inconvenience: in vitro evaluation and in vivo assessment in human volunteers. Drug Deliv Transl Res. 2021;1–15. doi:10.1007/s13346-020-00889-z
28. Darwesh AY, El-Dahhan MS, Meshali MM. New oral coaxial nanofibers for gadodiamide-prospective intestinal magnetic resonance imaging and theranostic. Int J Nanomedicine. 2020;15:8933–8943. doi:10.2147/IJN.S281158
29. Mady FM, Shaker MA. Enhanced anticancer activity and oral bioavailability of ellagic acid through encapsulation in biodegradable polymeric nanoparticles. Int J Nanomedicine. 2017;12:7405. doi:10.2147/IJN.S147740
30. Awadeen RH, Boughdady MF, Meshali MM. New in-situ gelling biopolymer-based matrix for bioavailability enhancement of glimepiride; in-vitro/in-vivo x-ray imaging and pharmacodynamic evaluations. Pharm Dev Technol. 2019;24(5):539–549. doi:10.1080/10837450.2018.1517366
31. Martin AN, Bustamante P, Chun AHC. Physical Pharmacy: Physical Chemical Principles in the Pharmaceutical Sciences.
32. Higuchi T. Mechanism of sustained‐action medication. Theoretical analysis of rate of release of solid drugs dispersed in solid matrices. J Pharm Sci. 1963;52(12):1145–1149. doi:10.1002/jps.2600521210
33. Korsmeyer RW, Gurny R, Doelker E, Buri P, Peppas NA. Mechanisms of solute release from porous hydrophilic polymers. Int J Pharm. 1983;15(1):25–35. doi:10.1016/0378-5173(83)90064-9
34. Khudair N, Agouni A, Elrayess MA, Najlah M, Younes HM, Elhissi A. Letrozole-loaded nonionic surfactant vesicles prepared via a slurry-based proniosome technology: formulation development and characterization. JDDST. 2020;58:101721. doi:10.1016/j.jddst.2020.101721
35. Bivalacqua TJ, Champion HC, Hyman AL, McNamara DB, Kadowitz PJ. Analysis of responses to angiotensin II in the mouse. J Renin Angiotensin Aldosterone Syst. 2001;1_suppl(2):S48–S53. doi:10.1177/14703203010020010801
36. Hamid S, Beg AE, Muhammad IN, et al. Development and validation of HPLC method for the determination of Candesartan in human plasma. Pak J Pharm Sci. 2018;31(6):2323–2327.
37. Ich I. Q2 (R1): validation of analytical procedures: text and methodology.
38. Song S, Tian B, Chen F, et al. Potentials of proniosomes for improving the oral bioavailability of poorly water-soluble drugs. Drug Dev Ind Pharm. 2015;41(1):51–62. doi:10.3109/03639045.2013.845841
39. Abdallah MH, Sabry SA, Hasan AA. Enhancing transdermal delivery of glimepiride via entrapment in proniosomal gel. J Young Pharm. 2016;8(4):335–340. doi:10.5530/jyp.2016.4.8
40. Shah A, Boldhane S, Pawar A, Bothiraja C. Advanced development of a non-ionic surfactant and cholesterol material based niosomal gel formulation for the topical delivery of anti-acne drugs. Mater Adv. 2020;1(6):1763–1774. doi:10.1039/d0ma00298d
41. Khalil RM, Abdelbary GA, Basha M, Awad GEA, El-Hashemy HA. Design and evaluation of proniosomes as a carrier for ocular delivery of lomefloxacin HCl. J Liposome Res. 2017;27(2):118–129. doi:10.3109/08982104.2016.1167737
42. Arzani G, Haeri A, Daeihamed M, Bakhtiari-Kaboutaraki H, Dadashzadeh S. Niosomal carriers enhance oral bioavailability of carvedilol: effects of bile salt-enriched vesicles and carrier surface charge. Int J Nanomed. 2015;10:4797–4813. doi:10.2147/IJN.S84703
43. Tavano L, Picci N, Loele G, Muzzalupo R. Tetracycline-niosomes versus tetracycline hydrochloride-niosomes: how to modulate encapsulation and percutaneous permeation properties. J Drug. 2017;1:1–6
44. Badawi MA, El-Khordagui LK. A quality by design approach to optimization of emulsions for electrospinning using factorial and D-optimal designs. Eur J Pharm Sci. 2014;58:44–54. doi:10.1016/j.ejps.2014.03.004
45. Aman RM, Zaghloul RA, El-Dahhan MS. Formulation, optimization and characterization of allantoin-loaded chitosan nanoparticles to alleviate ethanol-induced gastric ulcer: in-vitro and in-vivo studies. Sci Rep. 2021;11(1):1–24. doi:10.1038/s41598-021-81183-x
46. Khan DH, Bashir S, Figueiredo P, Santos HA, Khan MI, Peltonen L. Process optimization of ecological probe sonication technique for production of rifampicin loaded niosomes. JDDST. 2019;50:27–33. doi:10.1016/j.jddst.2019.01.012
47. Soliman SM, Abdelmalak NS, El-Gazayerly ON, Abdelaziz N. Novel non-ionic surfactant proniosomes for transdermal delivery of lacidipine: optimization using 23 factorial design and in vivo evaluation in rabbits. Drug Deliv. 2016;23(5):1608–1622. doi:10.3109/10717544.2015.1132797
48. Guimarães D, Noro J, Silva C, Cavaco-Paulo A, Nogueira E. Protective effect of saccharides on freeze-dried liposomes encapsulating drugs. Front Bioeng Biotechnol. 2019;7:424. doi:10.3389/fbioe.2019.00424
49. Du X-J, Wang J-L, Iqbal S, et al. The effect of surface charge on oral absorption of polymeric nanoparticles. Biomater.Sci. 2018;6(3):642–650. doi:10.1039/C7BM01096F
50. Sezgin-Bayindir Z, Antep MN, Yuksel N. Development and characterization of mixed niosomes for oral delivery using candesartan cilexetil as a model poorly water-soluble drug. AAPS PharmSciTech. 2015;16(1):108–117. doi:10.1208/s12249-014-0213-9
51. Dabhi MR, Ghodasara UK, Mori DD, Patel KA, Manek R, Sheth NR. Formulation, optimization and characterization of candesartan cilexetil nanosuspension for in vitro dissolution enhancement. Afr J Pharm Pharmacol. 2015;9(5):102–113. doi:10.5897/AJPP2013.3887
52. Mohamed HB, El-Shanawany SM, Hamad MA, Elsabahy M. Niosomes: a strategy toward prevention of clinically significant drug incompatibilities. Sci Rep. 2017;7(1):1–14. doi:10.1038/s41598-017-06955-w
53. El-Ridy MS, Abdelbary A, Essam T, Abd EL-Salam RM, Kassem AAA. Niosomes as a potential drug delivery system for increasing the efficacy and safety of nystatin. Drug Dev Ind Pharm. 2011;37(12):1491–1508. doi:10.3109/03639045.2011.587431
54. Weiss J, Sauer A, Divac N, et al. Interaction of angiotensin receptor type 1 blockers with ATP‐binding cassette transporters. Biopharm Drug Dispos. 2010;31(2‐3):150–161. doi:10.1002/bdd.699
55. Song Q, Li D, Zhou Y, et al. Enhanced uptake and transport of (+)-catechin and (−)-epigallocatechin gallate in niosomal formulation by human intestinal Caco-2 cells. Int J Nanomed. 2014;9:2157–2165. doi:10.2147/IJN.S59331
56. Li P-W, Wang G, Yang Z-M, et al. Development of drug-loaded chitosan–vanillin nanoparticles and its cytotoxicity against HT-29 cells. Drug Deliv. 2016;23(1):30–35. doi:10.3109/10717544.2014.900590
57. Tran N, Mulet X, Hawley AM, et al. Nanostructure and cytotoxicity of self-assembled monoolein–capric acid lyotropic liquid crystalline nanoparticles. RSC Adv. 2015;5(34):26785–26795. doi:10.1039/c5ra02604k
58. Mustafa O, Chaw CS. Silicified microcrystalline cellulose based pellets and their physicochemical properties. JAPS. 2016;133:33. doi:10.1002/app.43829
59. Shin H-W, Kim J-E, Park Y-J. Nanoporous silica entrapped lipid-drug complexes for the solubilization and absorption enhancement of poorly soluble drugs. Pharmaceutics. 2021;13(1):63. doi:10.3390/pharmaceutics13010063
60. Morsi NM, Aboelwafa AA, Dawoud MHS. Improved bioavailability of timolol maleate via transdermal transfersomal gel: statistical optimization, characterization, and pharmacokinetic assessment. J Adv Rse. 2016;7(5):691–701. doi:10.1016/j.jare.2016.07.003
61. Teaima MH, El Mohamady AM, El-Nabarawi MA, Mohamed AI. Formulation and evaluation of niosomal vesicles containing ondansetron HCL for trans-mucosal nasal drug delivery. Drug Dev Ind Pharm. 2020;46(5):751–761. doi:10.1080/03639045.2020.1753061
62. Costa P, Lobo JMS. Modeling and comparison of dissolution profiles. Eur J Pharm Scis. 2001;13(2):123–133. doi:10.1016/s0928-0987(01)00095-1
63. Haider M, Elsherbeny A, Jagal J, Hubatová-Vacková A, Saad Ahmed I. Optimization and evaluation of poly (Lactide-co-glycolide) nanoparticles for enhanced cellular uptake and efficacy of paclitaxel in the treatment of head and neck cancer. Pharmaceutics. 2020;12(9):828. doi:10.3390/pharmaceutics12090828
64. Anwar W, Dawaba HM, Afouna MI, Samy AM, Rashed MH, Abdelaziz AE. Enhancing the oral bioavailability of candesartan cilexetil loaded nanostructured lipid carriers: in vitro characterization and absorption in rats after oral administration. Pharmaceutics. 2020;12(11):1047. doi:10.3390/pharmaceutics12111047
65. Amer AM, Allam AN, Abdallah OY. Comparative pharmaceutical evaluation of candesartan and candesartan cilexetil: physicochemical properties, in vitro dissolution and ex vivo in vivo studies. AAPS PharmSciTech. 2018;19(2):661–667. doi:10.1208/s12249-017-0879-x
66. AboulFotouh K, Allam AA, El-Badry M, El-Sayed AM. A self-nanoemulsifying drug delivery system for enhancing the oral bioavailability of candesartan cilexetil: ex vivo and in vivo evaluation. J Pharm Sci. 2019;108(11):3599–3608. doi:10.1016/j.xphs.2019.07.004
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.