")
Back to Journals » Lung Cancer: Targets and Therapy » Volume 5
New options in the management of intractable ALK(+) metastatic non-small-cell lung cancer
Received 17 April 2014
Accepted for publication 28 April 2014
Published 6 August 2014 Volume 2014:5 Pages 35—42
DOI https://doi.org/10.2147/LCTT.S50414
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Amy M Weise, Shirish Gadgeel
Department of Oncology, Karmanos Cancer Institute, Wayne State University, Detroit, MI, USA
Abstract: Non-small-cell lung cancer (NSCLC) is a heterogeneous disease and a challenging malignancy to treat, as many patients have advanced disease at the time of diagnosis. Recent advances have led to the identification of molecularly defined subtypes of NSCLC, namely for patients with adenocarcinoma histology. The most recently identified molecular target is the anaplastic lymphoma kinase (ALK) gene rearrangement, and patient responses to the ALK inhibitor crizotinib have led to its approval in this selected patient population. Like other tyrosine kinase inhibitors, resistance to crizotinib ultimately develops by various mechanisms requiring alternative therapeutic options. This review article discusses the management of patients with the ALK gene rearrangement, mechanisms of crizotinib resistance, and future potential therapeutic options.
Keywords: non-small-cell lung cancer, disease management, cancer therapy
Introduction
Over one million deaths occur worldwide each year as a result of lung cancer, and approximately 85% of these are from non-small-cell lung cancer (NSCLC).1 Cytotoxic chemotherapy remains the mainstay of treatment for the majority of patients with advanced disease; however, recent pivotal discoveries have led to molecularly defined subtypes of NSCLC, particularly in adenocarcinoma. Additionally, safe and effective targeted therapies for these specific subgroups have emerged, resulting in a paradigm shift in how clinicians diagnose and treat patients with this disease.
The identification of epidermal growth factor receptor (EGFR) activating mutations and EGFR-tyrosine kinase inhibitors (TKIs) led to the 2004 United States Food and Drug Administration (FDA) approval of erlotinib as first-line therapy for EGFR mutation-positive patients.2,3 More recently, a fusion gene has been discovered between echinoderm microtubule-associated protein-like 4 (EML4) and anaplastic lymphoma kinase (ALK) in a small subset of NSCLC patients.4 Crizotinib, a multitarget TKI, has demonstrated overall response rates in approximately 60% of patients with tumors containing the ALK gene rearrangement.5
ALK is a member of the insulin receptor superfamily and is a transmembrane receptor tyrosine kinase found on the small intestines, testes, and selected neural tissue of adult humans.6 Expression of ALK during development is tightly regulated and has been implicated to have an important role in differentiation and cell survival. Aberrant ALK has been described in several neoplasms, including NSCLC, rhabdomyosarcoma, neuroblastomas, glioblastoma, melanoma, breast cancer, and neuroectodermal tumors.3
Constitutive ALK activation in NSCLC was first identified by screening a cDNA library for genes with transforming capacity from the tumor of a Japanese male smoker with adenocarcinoma.4 This screening identified a fusion gene resulting from an inversion of the short arm of chromosome 2 producing the joining of exons 20–29 of the ALK gene to exons 1–13 of the EML4 gene. The product of this unique fusion gene is a chimeric protein containing the C-terminus of the ALK gene and the N-terminus of the EML4 gene, which ultimately leads to the constitutive activation of ALK tyrosine kinase resulting in the activation of multiple downstream signaling pathways, including Akt, STAT3, and ERK (Figure 1).7 Multiple distinct variants have been identified since the initial discovery of ALK-EMLA resulting from different breakpoints in the EML4 gene.8 The most frequent fusion gene variants of EML4-ALK include; E13;A20 (34%), E6ab;A20 (30%), and E20;A20 (10%).9 ALK has also been shown to partner with additional genes such as KIF5B and TFG to form other aberrant fusion proteins.10,11 This review article will discuss current diagnostic techniques, development of resistance to ALK inhibitors, impact of therapy on quality of life, and future therapeutic directions.
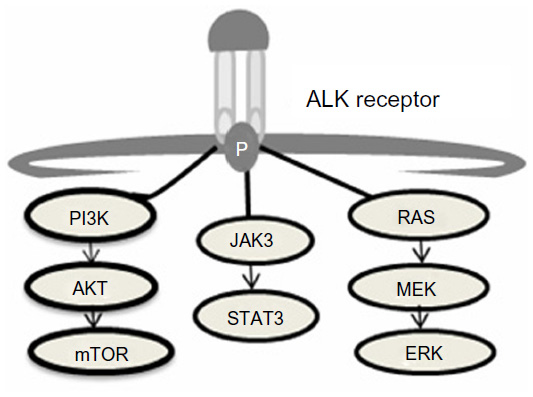 | Figure 1 ALK downstream signaling cascade.7 |
Management of ALK(+) metastatic NSCLC
Detection of the ALK gene rearrangement is critical for managing the treatment of NSCLC patients with ALK-rearranged tumors. A variety of methods are currently available to test for the ALK rearrangement, including fluorescence in situ hybridization (FISH), immunohistochemistry (IHC), and reverse transcriptase polymerase chain reaction (RT-PCR). RT-PCR has thus far been limited to clinical trials evaluating the sensitivities of IHC and FISH. Although RT-PCR can identify ALK gene rearrangements at the mRNA level, its limits include mRNA degradation in formalin-fixed, paraffin-embedded tissues; inability to detect unknown genetic alterations; and the need for highly specific primers.12 Multiplexed RT-PCR may overcome some of these limitations, but its current availability is quite limited.
The Vysis ALK Break Apart FISH Probe Kit (Abbott Molecular Inc., Des Plaines, IL, USA) was utilized to detect the ALK gene rearrangement in studies leading to the approval of the ALK inhibitor, crizotinib.13 This test consists of a red-labeled fluorescent probe at the 3′ end of ALK and a green-labeled fluorescent probe at the 5′ end of ALK. In tumors with rearranged ALK, the red and green probes are split apart (“break-apart test”). In tumors with a normal ALK gene, the two probes stay in close proximity and appear as a single yellow signal. It is recommended that at least four fields (about 60 cells) are evaluated and if ≥15% of tumor cells have split ALK 5′ and 3′ probe signals or have isolated 3′ signals, the tumor is considered positive for the ALK gene rearrangement. Detection of the break-apart signal can be challenging, as EML4 and ALK normally reside in close proximity to each other on the short arm of chromosome 2. Additionally, although FISH detects the disruption of the ALK locus, it does not identify the resulting partner fusion gene. Further, the instrumentation and expertise required for FISH are not readily available in all pathology laboratories, thus limiting the applicability of this test in routine care.
The use of IHC for detecting ALK protein expression has emerged as a conceivable alternative to FISH.14 Early IHC reports found a large number of false negatives, likely resulting from low-level mRNA expression in NSCLC versus (vs) anaplastic large cell lymphoma and thus requiring a greater dependence on the primary antibody used in the test. Because ALK is not normally expressed in lung tissue, any expression of ALK in the lung cancer tissue represents an altered status of the ALK gene. Multiple researchers have demonstrated a strong correlation between IHC and FISH.15–20 Conklin et al examined 377 tumors by IHC, 273 of which were also evaluated by FISH.16 They concluded that the sensitivity and specificity of IHC is antibody dependent, with 5A4 (Novocastra with ADVANCE detection) and D5F3 (Cell Signaling with ADVANCE detection) clones proving to be the most accurate and consistent among the antibodies tested. Additionally, the presence or absence of staining by IHC better correlated with FISH than the degree of staining (0 to 3+) as has been observed with HER2 testing. Keep in mind, however, that false positives and negatives have been reported.15,18 Further optimization of the antibodies and methodology applied is necessary before IHC can be used in routine patient care; however, IHC may become the preferred test, as it is routinely used in pathology laboratories and is generally simpler and less expensive to perform. The United States FDA approved crizotinib in conjunction with the break-apart FISH-based assay in August 2011. A year later, the European Commission approved crizotinib in conjunction with IHC using the D5F3 rabbit monoclonal primary antibody. Trials are ongoing to clarify the ALK diagnostic issues, but to date FISH remains the standard of care in the United States.
Another critical question in the management of advanced NSCLC involves determining which patient population should be tested for the ALK gene rearrangement and in what sequence testing should occur. The frequency of ALK rearrangements has been reported to range from 1% to up to 33% of the population.21 This wide range in the prevalence of ALK rearrangement can be attributed to the clinical and molecular characteristics of the populations being tested. Clinicopathologic and molecular features linked with a higher occurrence of ALK rearrangement include adenocarcinoma, light or never smokers, younger age, EGFR wild type, and KRAS wild type.21–23 In a report by Shaw et al, the frequency of ALK in never/light smokers was 22%, in never/light smokers not harboring an EGFR mutation, the frequency was 33%.21 For the most part, the ALK gene rearrangement is thought to be mutually exclusive of EGFR and KRAS mutations; however, tumors containing more than one mutation have been reported.22,24 In one series, 8% of ALK-rearranged tumors also contained either an EGFR or a KRAS mutation.24 Some investigators have suggested an algorithmic approach to testing, but to date a consensus on an algorithm has not been reached.22,25 Currently, the International Association for the Study of Lung Cancer recommends EGFR and ALK testing on adenocarcinomas or tumors with an adenocarcinoma component.26 Squamous cell tumors may be considered for testing if the abovementioned clinical features are present. In comparison, the European Society for Medical Oncology guidelines recommends testing in patients with a diagnosis of non-squamous advanced lung cancer or on any type of histology in never smokers who have been screened first for the EGFR mutation.27 Given the dramatic and rapid impact of ALK-targeted therapy, the authors favor erring on the side of testing rather than potentially missing a patient who may derive meaningful benefit from ALK inhibition.
Clinical benefit of the ALK inhibitor crizotinib
Crizotinib (Pfizer Inc., New York, NY, USA) is an oral small molecule inhibitor of ALK, c-Met, ROS1, and RON tyrosine kinases.28,29 Crizotinib was shown to be a potent ATP-competitive inhibitor of recombinant human c-Met kinase activity resulting in regression of tumors expressing constitutive activity of c-Met in preclinical studies. In fact, the drug was initially developed as a c-Met inhibitor, but was later found to inhibit tyrosine phosphorylation of ALK with a mean half maximal inhibitory concentration (IC50) of 24 nmol/L.
The initial Phase I dose escalation trial of crizotinib (PF-02341066) included 37 patients with various solid tumors; dose-limiting toxicities included grade 3 alanine aminotransferase elevation and grade 3 fatigue, and the recommended Phase II dose was established as 250 mg twice daily.5 Of the ten patients in the Phase I trial with ALK-positive NSCLC, one had a confirmed partial response (PR), two had unconfirmed PRs, and four had stable disease. These results led to a Phase I expansion cohort that included 149 ALK-rearranged NSCLC patients.30 Investigators reported a 61% overall response rate (ORR), a median progression-free survival of 9.7 months, and a median duration of response of 49.1 weeks. This promising activity gave rise to a Phase II trial (PROFILE 1005) conducted in 901 pretreated patients, the majority of whom had at least two prior therapies.31 Mature data is available on 261 patients, 259 of whom are considered response evaluable. The ORR was approximately 60%, the median duration of response was 11 months, and the median time to response was 6 weeks. Eighteen of the patients had untreated brain metastasis, two patients achieved a CR, two had a PR, and 12 had stable disease. Adverse events that were reported in more than 25% of patients included visual disorder, nausea, vomiting, diarrhea, constipation, and peripheral edema. Unfortunately, investigators also reported fatalities due to presumed treatment related pneumonitis and hepatotoxicity, occurring in less than 3% of patients. Crizotinib was approved by the FDA based on data from the expansion cohort of the Phase I study and the Phase II study.
This approval led to a series of Phase III trials beginning with PROFILE 1007, in which patients were randomly assigned to receive either crizotinib or chemotherapy with pemetrexed or docetaxel.14 Three hundred and forty-seven patients with locally advanced or metastatic ALK-positive lung cancer who had received one prior platinum-based regimen were enrolled; crossover was permitted at disease progression. The median progression-free survival more than doubled (7.7 months vs 3 months, P<0.001) and the response rates more than tripled (65% vs 20%, P<0.001) in the crizotinib group. A survival benefit was not seen, perhaps because crossover was permitted. The most common side effect reported with crizotinib was visual disorder, nausea, and diarrhea. Approximately 16% of patients experienced grade 3 elevation in transaminases. Three patient deaths were thought to be treatment related in the crizotinib group, one due to a ventricular arrhythmia, and two due to pneumonitis. The median time to response was 6.3 weeks, and the median duration of response was 32.1 months. Although the duration of response is highly significant, the majority of patients will ultimately develop resistance.
Resistance to crizotinib in ALK-positive NSCLC
It is well documented that tumors with kinase driver genetic alterations eventually develop resistance to kinase inhibitors. A common mechanism of resistance is the development of secondary mutations that alter inhibitor sensitivity. For instance, many NSCLC patients with EGFR mutations who develop resistance to EGFR TKIs demonstrate T790M mutation in the kinase domain of EGFR at the time of tumor progression.32 Tumors with this mutation are resistant to currently available EGFR TKIs. Similar resistant mutations have been observed in patients with imatinib-resistant chronic myeloid leukemia.33 Many other mechanisms of secondary resistance in tumors with driver genetic alterations have been identified apart from mutations, including amplification of the target gene and activation of alternate pathways of signaling.34
Almost all ALK-positive NSCLC patients will eventually develop resistance to crizotinib. An effort has been made to assess whether such patients demonstrate resistant mutations in their tumors based on previous experience with kinase inhibitors. Choi et al analyzed tumor cells in pleural effusion obtained from a 28-year-old patient with ALK-positive NSCLC who showed clinical benefit from crizotinib for 5 months followed by disease progression.35 Molecular analysis conducted on the tumor cells at progression revealed two mutations within the kinase domain of EML4-ALK. These mutations resulted from a G to A transition mutation at nucleotide 4374 and a C to A transversion mutation at nucleotide 4493 of the ALK cDNA. The 4374 G to A transition resulted in a cysteine to tyrosine mutation at amino acid 1156 (C1156Y). The 4493 C to A transversion resulted in a leucine to methionine mutation at amino acid 1196 (L1196M) in the ALK protein. Among the various clones isolated from the patient’s tumor cells, they found the C1156Y mutation at a rate of 47% and the L1196M mutation at a rate of 15%. Both mutations were not present in the same clone, and some clones did not have either of these mutations.
Interestingly, leucine 1196 in the kinase domain of ALK corresponds to the threonines in positions 315 in ABL and 790 in EGFR, each of which are the most frequent site of resistant mutations in chronic myeloid leukemia and EGFR mutation-positive NSCLC, respectively. These amino acids are each located at the bottom of the ATP binding pocket; thus the presence of an amino acid with a bulky side chain such as methionine may prevent the binding of the kinase inhibitor molecules. Recently, a number of investigators have examined the activities of other ALK inhibitors in tumors with this mutation in preclinical models.36,37
As with other tumors with genetic alterations, mechanisms other than resistant phenotypes have been identified. Doebele et al analyzed the tumors of 14 ALK-positive NSCLC patients who had developed resistance to crizotinib.38 Four patients had evidence of mutations in the kinase domain of the ALK receptor: two had tumors with a copy number gain in the ALK-rearranged gene, and one of these also had the G1269A mutation. Interestingly, these investigators found that one patient had an EGFR mutation and two patients had KRAS mutations in their tumors. One of the patients with the KRAS mutation was shown to have this mutation in the precrizotinib specimen.
In order to gain a greater understanding of the possible mechanisms of crizotinib resistance caused by ALK-rearranged NSCLCs, Katayama et al developed an in vitro acquired resistance model by exposing NSCLC cell line 3122, known to harbor the EML4-ALK translocation, to increasing doses of crizotinib.36 They found that cells resistant to intermediate doses of crizotinib developed amplification of the EML4-ALK gene, whereas cells resistant to higher doses of crizotinib additionally developed the L1196M mutation. Based on this cell line data, the authors concluded that resistance to crizotinib may evolve in a stepwise manner, first involving amplification of EML4-ALK followed by point mutations. Additionally, two structurally distinct ALK inhibitors, AP26113 and NV-TAE684, as well as an HSP 90 inhibitor, 17-AAG, were shown to have activity in these crizotinib-resistant 3122 cells.
Taken together, these data show that the mechanisms of resistance to crizotinib in ALK-positive NSCLCs are diverse and variable, even within a single patient. The presence of these multiple mechanisms of resistance and EGFR and KRAS mutations suggest that some tumors have multiple clones with varying genetic alterations; therefore, appropriate management of this patient population may require several biopsies to identify all the mechanisms of resistance and treatment with multiple agents to target all the genetic alterations in the tumor. Further studies of patients with crizotinib resistance are required in order to properly define all of the possible mechanisms of resistance and disease management.
Current and emerging treatment options
Multiple ALK inhibitors are currently in development. In March of 2013, the selective ALK inhibitor, ceritinib or LDK378 (Novartis International AG, Basel, Switzerland) received Breakthrough Therapy designation by the FDA for patients with metastatic NSCLC containing the ALK gene rearrangement and prior exposure to crizotinib. A Phase I study included 26 NSCLC patients with ALK gene rearrangement, gene amplification or another mutation, with and without prior exposure to other ALK inhibitors, as well as five patients with other ALK-positive malignancies.39 Responses were reported in both the crizotinib-naïve and crizotinib-resistant populations. Toxicities were similar to those reported with crizotinib, mainly nausea, vomiting, diarrhea, dyspnea, and transaminase elevation. In a separate Phase I trial that accrued a total of 131 patients with advanced cancer harboring aberrant ALK, patients received LDK378 at a dose range of 50–750 mg daily.40 This trial showed that a minimum daily dose of 400 mg was necessary to achieve a response. Investigators reported a 70% ORR in NSCLC patients (receiving 400–750 mg) and a 73% ORR in the subset of patients who had prior crizotinib exposure. The recommended Phase II dose was determined to be 750 mg daily. Interestingly, responses were also noted in patients with untreated brain metastasis. The most recently reported Phase I study included 114 patients with NSCLC, either crizotinib-resistant or -naïve.41 In patients with prior crizotinib exposure, the ORR was 56%. Nineteen crizotinib-resistant patients underwent repeat tumor biopsy prior to therapy with LDK378; two revealed ALK gene amplification, five had secondary mutations in the ALK TKI domain, and the remaining 12 did not demonstrate any additional alteration. This suggests that a higher potency drug may be able to overcome resistance regardless of its mechanism. Given the activity of LDK378 in patients with prior crizotinib exposure, a Phase II trial is being conducted in patients who have received prior chemotherapy and prior crizotinib.42 In addition, trials are also being conducted in patients who are naïve to prior ALK inhibitors.
Another second-generation selective ALK inhibitor receiving Breakthrough Therapy designation by the FDA is alectinib (CH5424802/RO5424802). Alectinib has demonstrated activity against the L1196M gatekeeper mutation, as well as wild-type ALK and C1156Y mutation in preclinical models.37 The increased potency of alectinib is believed to be related to a unique hydrogen-bonding network in the ATP binding site. The first in human Phase I/II trial in ALK-positive NSCLC patients demonstrated tumor regression at all dose levels.43 Toxicities included myalgia, hypophosphatemia, neutropenia, hypermagnesemia, and elevation in CPK. The Phase II portion of this study included 46 patients and revealed an ORR of 93.5%, including two complete responses.44 It is important to note that patients treated in this study were crizotinib naïve. Significant decreases in tumor sizes were noted in 65% of patients by 3 weeks and in 87% of patients by 6 weeks, with a response duration of more than 10 months. Additional toxicities included rash, dysgeusia, constipation, and elevations in AST, bilirubin, and creatinine. Visual disorders and gastrointestinal toxicities were mild and rare. Alectinib has also been studied in a Phase I trial of 37 crizotinib-resistant NSCLC patients that demonstrated an ORR of 59.5% at therapeutic doses defined as >460 mg twice daily.45 The population included 21 patients with CNS metastases with an ORR within the brain of 53%.46
A dual ALK/EGFR inhibitor, AP26113 (ARIAD Pharmaceuticals Inc., Cambridge, MA, USA), has been developed with TKI activity on L1196M (ALK variant) and T790M (EGFR variant) mutations, but not native EGFR.47 The updated results of the dose-finding study included 44 patients with advanced malignancies excluding leukemia. The ORR in the evaluable ALK-positive population was 56%, and 67% in the group with documented exposure to crizotinib. Additionally, four of five patients with active CNS disease demonstrated radiographic improvement. Fatigue, nausea, and diarrhea of any grade were reported in over a quarter of patients. Grade 3 or 4 toxicities in 4%–5% of patients included fatigue, diarrhea, pneumonia, and hypoxia.
Other emerging strategies against the ALK driver mutation include heat shock protein 90 (HSP90) inhibitors. HSP90 is a molecular chaperone that can be present in an abundant amount in tumor cells.48 ALK and ALK fusion proteins are chaperone proteins of HSP90, and binding of ALK to HSP90 inhibits its degradation. Thus inhibition of HSP90 will limit the binding of ALK to HSP90 and enhance the degradation of ALK. In preclinical studies, HSP90 inhibition has led to antitumor activity in ALK-positive NSCLCs, including tumors with crizotinib resistance.49 IPI-504 and STA9090, both HSP90 inhibitors in development, have shown activity in a limited number of ALK-positive NSCLC patients. Because mechanisms of resistance are varied, it is unclear if these drugs will be effective in all patients or merely in a subset of patients with crizotinib resistance.50 Table 1 represents a sample of trials in the ALK (+) population available through http://www.clinicaltrials.gov.
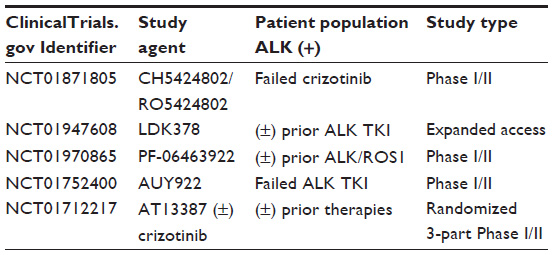 | Table 1 Sample of currently active clinical trials |
Implications for enhanced quality of life
This process of discovery of ALK gene rearrangement, rapid movement of targeted treatment through clinical trials, and early investigation of resistance mechanisms has established a new model for personalized medicine. Investigators not only simultaneously gained FDA approval and explored resistance mechanisms, they also elucidated the effect crizotinib would have on quality of life. Both the Phase II and Phase III trials used the European Organization for Research and Treatment of Cancer (EORTC) Quality of Life Questionnaire-Core 30 (QLQ-C30) and the lung cancer module (QLQ-LC13).14,51 The QLQ-C30 includes five functional domains, eight symptoms, global health, and financial impact.52,53 Patient reported outcomes are based on their responses in accordance with a four-point scale from “not at all” to “very much”. The raw scores are then converted to a scale from 0 to 100. Higher scores reflect a greater symptom burden, but also reflect a higher level of function. The QLQ-LC13 includes 13 symptom scales common to lung cancer patients undergoing treatment with chemotherapy or radiation. The Phase II study reported a ≥10 point improvement in pain, cough, dyspnea, and fatigue; the majority of these improvements were noted by the second cycle of therapy.51 Likewise, the Phase III trial demonstrated a significant reduction in alopecia, cough, dyspnea, fatigue, and pain from baseline in the group treated with crizotinib.14 Progression of symptoms was also significantly delayed in the crizotinib group (median 5.6 months crizotinib vs median 1.4 months chemotherapy). Finally, there was a meaningful improvement in global quality of life by cycle 4 in the crizotinib group that was not observed in the chemotherapy treated patients.
Conclusion and future perspective
Although the frequency of the ALK gene rearrangement is relatively low (approximately 4% of NSCLC patients), this translates into 70,000 cases annually worldwide. Accurately identifying these patients is critical given the degree of benefit that has been observed with ALK inhibitors. Both IHC and FISH have been identified as potential diagnostic resources; however, differences in accuracy, availability, timeliness, and cost need to be completely resolved. Additionally, the question of whether to test all advanced non-squamous cell carcinoma patients or to stratify testing based on clinicopathologic and molecular features needs to be answered. One potential pitfall of using an algorithmic approach to answer these important genetic testing questions is that ALK has been identified in patients who do not fit the defined “standard” phenotype and coexisting mutations have been described. Given the potential therapeutic impact of ALK inhibitor therapy, the authors favor erring on the side of testing while remaining within the bounds of guidelines and good clinical practice.
Activation of ALK appears to be a “driver” genetic alteration. Crizotinib is an oral drug that is the first ALK inhibitor to show promising clinical activity in tumors with ALK activation. Crizotinib is generally well tolerated, with nausea, emesis, and visual changes reported as the main adverse events. Grade 3/4 toxicities are infrequent, with liver transaminase elevations being the most common.
As anticipated, resistance to crizotinib ultimately develops. The molecular alterations that result in resistance appear to be highly diverse, including but not limited to secondary mutations within the ALK-TKI domain, gene amplification, alternate signaling pathway activation, and coexisting driver mutation. Other ALK inhibitors currently in development may have activity in some of these crizotinib-resistant tumors. Certain second-generation ALK inhibitors have shown promising activity in tumors resistant to crizotinib, and several clinical trials are ongoing in this arena. Since ALK is a chaperone protein of HSP90, inhibitors of HSP90 may prove to be beneficial in patients with ALK gene-rearranged NSCLC.
The discovery of the ALK gene rearrangement and effective targeted therapy represents a new model for translational cancer research (Figure 2).54,55 Only 2 years elapsed from the time of the discovery of ALK to the objective tumor responses seen in patients on ALK inhibitors. Further, it only took 3 years to advance from Phase I to Phase III clinical trials, roughly half the time required to move EGFR inhibitors forward. Finally, mechanisms of resistance are already being elucidated and clinical trials are currently underway in this drug-resistant patient population. Thus, in the ALK-positive NSCLC patient population, the gap between the “bench” and the “bedside” has been bridged in record time.
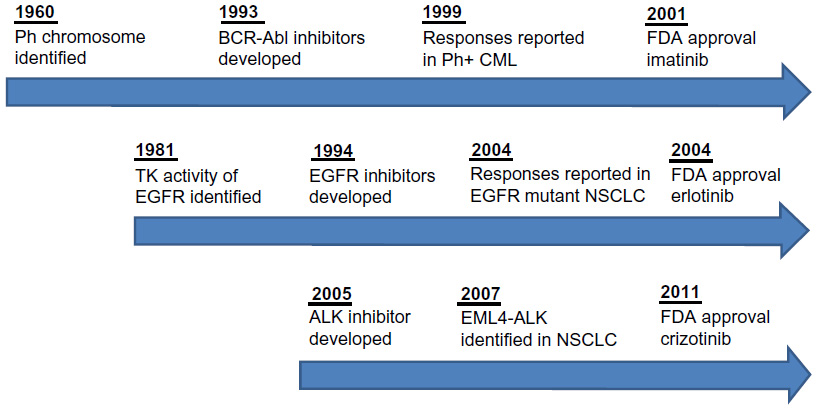 | Figure 2 Timeline of targeted therapy development. |
Disclosure
Dr Gadgeel is a member of the Advisory Board of Novartis International AG, the Advisory Board of Genentech, Inc., and the Speaker’s Bureau Genentech, Inc. Dr Weise reports no conflicts of interest.
References
Shibuya K, Mathers CD, Boschi-Pinto C, Lopez AD, Murray CJ. Global and regional estimates of cancer mortality and incidence by site: II. Results for the global burden of disease 2000. BMC Cancer. 2002;2:37. | |
Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350(21):2129–2139. | |
Paez JG, Janne PA, Lee JC, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304(5676):1497–1500. | |
Soda M, Choi YL, Enomoto M, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448(7153):561–566. | |
Kwak EL, Camidge DR, Clark J, et al. Clinical activity observed in a phase I dose escalation trial of an oral c-met and ALK inhibitor, PF-02341066. J Clin Oncol 2009;27(Suppl): Abstract 3509. | |
Ardini E, Magnaghi P, Orsini P, Galvani A, Menichincheri M. Anaplastic Lymphoma Kinase: role in specific tumours, and development of small molecule inhibitors for cancer therapy. Cancer Lett. 2010; 299(2):81–94. | |
Grande E, Bolós MV, Arriola E. Targeting oncogenic ALK: a promising strategy for cancer treatment. Mol Cancer Ther. 2011;10(4):569–579. | |
Deuel TF. Anaplastic lymphoma kinase: “Ligand Independent Activation” mediated by the PTN/RPTPβ/ζ signaling pathway. Biochim Biophys Acta. 2013;1834(10):2219–2223. | |
Moran T, Quiroga V, de los Llanos Gil M, et al. Targeting EML4-ALK driven non-small cell lung cancer (NSCLC). Transl Lung Cancer Res. 2013;2(2):128–141. | |
Solomon B, Varella-Garcia M, Camidge DR. ALK gene rearrangements: a new therapeutic target in a molecularly defined subset of non-small cell lung cancer. J Thorac Oncol. 2009;4(12):1450–1454. | |
Takeuchi K, Choi YL, Togashi Y, et al. KIF5B-ALK, a novel fusion oncokinase identified by an immunohistochemistry-based diagnostic system for ALK-positive lung cancer. Clin Cancer Res. 2009;15(9):3143–3149. | |
Rikova K, Guo A, Zeng Q, et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell. 2007;131(6):1190–1203. | |
Takeuchi K, Choi YL, Soda M, et al. Multiplex reverse transcription-PCR screening for EML4-ALK fusion transcripts. Clin Cancer Res. 2008;14(20):6618–6624. | |
Shaw AT, Kim DW, Nakagawa K, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med. 2013;368(25):2385–2394. | |
Murakami Y, Mitsudomi T, Yatabe Y. A screening method for the ALK fusion gene in NSCLC. Front Oncol. 2012;2:24. | |
Conklin CM, Craddock KJ, Have C, Laskin J, Couture C, Ionescu DN. Immunohistochemistry is a reliable screening tool for identification of ALK rearrangement in non-small-cell lung carcinoma and is antibody dependent. J Thorac Oncol. 2013;8(1):45–51. | |
Minca EC, Portier BP, Wang Z, et al. ALK status testing in non-small cell lung carcinoma: correlation between ultrasensitive IHC and FISH. J Mol Diagn. 2013;15(3):341–346. | |
Martinez P, Hernández-Losa J, Montero MÁ, et al. Fluorescence in situ hybridization and immunohistochemistry as diagnostic methods for ALK positive non-small cell lung cancer patients. PLoS One. 2013;8(1): e52261. | |
Park HS, Lee JK, Kim DW, et al. Immunohistochemical screening for anaplastic lymphoma kinase (ALK) rearrangement in advanced non-small cell lung cancer patients. Lung Cancer. 2012;77(2):288–292. | |
Sholl LM, Weremowicz S, Gray SW, et al. Combined use of ALK immunohistochemistry and FISH for optimal detection of ALK-rearranged lung adenocarcinomas. J Thorac Oncol. 2013;8(3):322–328. | |
Shaw AT, Yeap BY, Mino-Kenudson M, et al. Clinical features and outcome of patients with non-small-cell lung cancer who harbor EML4-ALK. J Clin Oncol. 2009;27(26):4247–4253. | |
Paik JH, Choi CM, Kim H, et al. Clinicopathologic implication of ALK rearrangement in surgically resected lung cancer: a proposal of diagnostic algorithm for ALK-rearranged adenocarcinoma. Lung Cancer. 2012;76(3):403–409. | |
Kwak EL, Bang YJ, Camidge DR, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med. 2010;363(18):1693–1703. | |
Kris MG, Johnson BE, Kwiatkowski DJ, et al. Identification of driver mutations in tumor specimens from 1,000 patients with lung adenocarcinoma: The NCI’s Lung Cancer Mutation Consortium (LCMC). J Clin Oncol. 2011;29(Suppl): Abstract CRA7506. | |
Horn L, Pao W. EML4-ALK: honing in on a new target in non-small-cell lung cancer. J Clin Oncol. 2009;27(26):4232–4235. | |
Lindeman NI, Cagle PT, Beasley MB, et al. Molecular testing guideline for selection of lung cancer patients for egfr and alk tyrosine kinase inhibitors: summary of recommendations [webpage on the Internet]. International Association for the Study of Lung Cancer (IASLC). 2013. Available at https://www.iaslc.org/sites/default/files/wysiwyg-assets/cap_iaslc_amp_summary_of_recommendations.pdf. Accessed May 22, 2014. | |
Gridelli C, Peters S, Sgambato A, Casaluce F, Adjei AA, Ciardiello F. ALK inhibitors in the treatment of advanced NSCLC. Cancer Treat Rev. 2014;40(2):300–306. | |
Cui JJ, Tran-Dubé M, Shen H, et al. Structure based drug design of crizotinib (PF-02341066), a potent and selective dual inhibitor of mesenchymal-epithelial transition factor (c-MET) kinase and anaplastic lymphoma kinase (ALK). J Med Chem. 2011;54(18):6342–6363. | |
Zou HY, Li Q, Lee JH, et al. An orally available small-molecule inhibitor of c-Met, PF-2341066, exhibits cytoreductive antitumor efficacy through antiproliferative and antiangiogenic mechanisms. Cancer Res. 2007;67(9):4408–4417. | |
Camidge DR, Bang YJ, Kwak EL, et al. Activity and safety of crizotinib in patients with ALK-positive non-small-cell lung cancer: updated results from a phase 1 study. Lancet Oncol. 2012;13(10):1011–1019. | |
Riely GJ, Evans TL, Salgia R, et al. Results of a global phase II study of crizotinib in advanced ALK-positive non-small cell lung cancer. Presented at the IASLC Chicago Multidisciplinary Symposium in Thoracic Oncology 2012, September 6–8, 2012. Abstract 166. | |
Giaccone G, Wang Y. Strategies for overcoming resistance to EGFR family tyrosine kinase inhibitors. Cancer Treat Rev. 2011;37(6):456–464. | |
Deininger M, Buchdunger E, Druker BJ. The development of imatinib as a therapeutic agent for chronic myeloid leukemia. Blood. 2005;105(7):2640–2653. | |
Sequist LV, Waltman BA, Dias-Santagata D, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med. 2011;3(75):75ra26. | |
Choi YL, Soda M, Yamashita Y, et al; ALK Lung Cancer Study Group. EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors. N Engl J Med. 2010;363(18):1734–1739. | |
Katayama R, Khan TM, Benes C, et al. Therapeutic strategies to overcome crizotinib resistance in non-small cell lung cancers harboring the fusion oncogene EML4-ALK. Proc Natl Acad Sci U S A. 2011;108(18):7535–7540. | |
Sakamoto H, Tsukaguchi T, Hiroshima S, et al. CH5424802, a selective ALK inhibitor capable of blocking the resistant gatekeeper mutant. Cancer Cell. 2011;19(5):679–690. | |
Doebele RC, Pilling AB, Aisner DL, et al. Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell lung cancer. Clin Cancer Res. 2012;18(5):1472–1482. | |
Mehra R, Camidge DR, Sharma S, et al. First-in-human phase I study of the ALK inhibitor LDK378 in advanced solid tumors. J Clin Oncol. 2012;30(Suppl): Abstract 3007. | |
Shaw AT, Mehra M, Kim D-W, et al. Clinical activity of the ALK inhibitor LDK378 in advanced, ALK-positive NSCLC. J Clin Oncol. 2013;31(Suppl): Abstract 8010. | |
Shaw AT, Kim DW, Mehra R, et al. Ceritinib in ALK-rearranged non-small-cell lung cancer. N Engl J Med. 2014;370(13):1189–1197. | |
Shaw AT, Mok T, Spigel DR, et al. A phase II single-arm study of LDK378 in patients with ALK-activated (ALK+) non-small cell lung cancer (NSCLC) previously treated with chemotherapy and crizotinib (CRZ). J Clin Oncol. 2013;31(Suppl): Abstract TPS8119. | |
Kiura K, Seto T, Yamamoto N, et al. A first-in-human phase I/II study of ALK inhibitor CH5424802 in patients with ALK-positive NSCLC. J Clin Oncol. 2012;30(Suppl): Abstract 7602. | |
Nakagawa K, Kiura K, Nishio M, et al. A phase I/II study with a highly selective ALK inhibitor CH5424802 in ALK-positive non-small cell lung cancer (NSCLC) patients: Updated safety and efficacy results from AF-001JP. J Clin Oncol. 2013;31(Suppl): Abstract 8033. | |
Gadgeel S, Ou SH, Chiappori A, et al. A phase I dose escalation study of a new ALK inhibitor, SH542480202, in ALK + non-small cell lung cancer patients who have failed crizotinib. Presented at the 15th World Conference on Lung Cancer; October 27–30, 2013; Sydney, Australia. Abstract O16.06. | |
Ou SH, Gadgeel S, Chiappori AA, et al. Consistent therapeutic efficacy of CH5424802/RO5424802 in brain metastases among crizotinib-refractory ALK-positive non-small cell lung cancer (NSCLC) patients in an ongoing phase I/II study. Presented at the 15th World Conference on Lung Cancer; October 27–30, 2013; Sydney, Australia, October 29, 2013. Abstract O16.07. | |
Camidge DR, Bazhenova L, Salgia R, et al. First-in-human dose-finding study of the ALK/EGFR inhibitor AP26113 in patients with advanced malignancies: Updated results. J Clin Oncol. 2013;31(Suppl): Abstract 8031. | |
Trepel J, Mollapour M, Giaccone G, Neckers L. Targeting the dynamic HSP90 complex in cancer. Nat Rev Cancer. 2010;10(8):537–549. | |
Sang J, Acquaviva J, Friedland JC, et al. Targeted inhibition of the molecular chaperone Hsp90 overcomes ALK inhibitor resistance in non-small cell lung cancer. Cancer Discov. 2013;3(4):430–443. | |
Sequist LV, Gettinger S, Senzer NN, et al. Activity of IPI-504, a novel heat-shock protein 90 inhibitor, in patients with molecularly defined non-small-cell lung cancer. J Clin Oncol. 2010;28(33):4953–4960. | |
Crino L, Kim D, Riely GJ, et al. Initial phase II results with crizotinib in advanced ALK-positive non-small cell lung cancer (NSCLC): Profile 1005. J Clin Oncol. 2011;29(Suppl): Abstract 7514. | |
Snyder CF, Blackford AL, Okuyama T, et al. Using the EORTC-QLQ-C30 in clinical practice for patient management: identifying scores requiring a clinician’s attention. Qual Life Res. 2013;22(10):2685–2691. | |
Bergman B, Aaronson NK, Ahmedzai S, Kaasa S, Sullivan M. The EORTC QLQ-LC13: a modular supplement to the EORTC Core Quality of Life Questionnaire (QLQ-C30) for use in lung cancer clinical trials. EORTC Study Group on Quality of Life. Eur J Cancer. 1994;30A(5):635–642. | |
Chin L, Andersen JN, Futreal PA. Cancer genomics: from discovery science to personalized medicine. Nat Med. 2011;17(3):297–303. | |
Gandhi L, Jänne PA. Crizotinib for ALK-rearranged non-small cell lung cancer: a new targeted therapy for a new target. Clin Cancer Res. 2012;18(14):3737–3742. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.