")
Back to Journals » Journal of Hepatocellular Carcinoma » Volume 1
New insights on the role of epigenetic alterations in hepatocellular carcinoma
Authors Frau M, Feo CF, Feo F, Pascale RM
Received 21 January 2014
Accepted for publication 9 April 2014
Published 12 June 2014 Volume 2014:1 Pages 65—83
DOI https://doi.org/10.2147/JHC.S44506
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Video abstract presented by Francesco Feo.
Views: 851
Maddalena Frau,1 Claudio F Feo,2 Francesco Feo,1 Rosa M Pascale1
1Department of Clinical and Experimental Medicine, Division of Experimental Pathology and Oncology, 2Department of Clinical and Experimental Medicine, Division of Surgery. University of Sassari, Sassari, Italy
Abstract: Emerging evidence assigns to epigenetic mechanisms heritable differences in gene function that come into being during cell development or via the effect of environmental factors. Epigenetic deregulation is strongly involved in the development of hepatocellular carcinoma (HCC). It includes changes in methionine metabolism, promoter hypermethylation, or increased proteasomal degradation of oncosuppressors, as well as posttranscriptional deregulation by microRNA or messenger RNA (mRNA) binding proteins. Alterations in the methylation of the promoter of methyl adenosyltransferase MAT1A and MAT2A genes in HCC result in decreased S-adenosylmethionine level, global DNA hypomethylation, and deregulation of signal transduction pathways linked to methionine metabolism and methyl adenosyltransferases activity. Changes in S-adenosylmethionine levels may also depend on MAT1A mRNA destabilization associated with MAT2A mRNA stabilization by specific proteins. Decrease in MAT1A expression has also been attributed to miRNA upregulation in HCC. A complex deregulation of miRNAs is also strongly involved in hepatocarcinogenesis, with up-regulation of different miRNAs targeting oncosuppressor genes and down-regulation of miRNAs targeting genes involved in cell-cycle and signal transduction control. Oncosuppressor gene down-regulation in HCC is also induced by promoter hypermethylation or posttranslational deregulation, leading to proteasomal degradation. The role of epigenetic changes in hepatocarcinogenesis has recently suggested new promising therapeutic approaches for HCC on the basis of the administration of methylating agents, inhibition of methyl adenosyltransferases, and restoration of the expression of tumor-suppressor miRNAs.
Keywords: hepatocarcinogenesis, DNA methylation, microRNA, Piwi-interacting RNAs, stem cells, therapeutic targets
Introduction
Hepatocellular carcinoma (HCC) is a frequent and deadly human cancer, with 0.25–1 million new cases per year and a life expectancy of about 6 months from the time of diagnosis.1,2 HCC incidence changes with age, sex, ethnic group, and geographic region3 and is rising in Western countries.4,5 Major risk factors associated with HCC development are chronic hepatitis B virus (HBV) and hepatitis C virus (HCV) infections, alcoholic steatohepatitis, Aflatoxin B1,1–3 and some metabolic diseases.6 HCC incidence shows differences within human population in response to risk factors,7 suggesting a pathogenetic role of genetic predisposition. Studies on genetic predisposition of chemically induced rodent hepatocarcinogenesis and of human families at risk suggest the implication of a polygenic control of HCC incidence.8,9 The interaction of genetic and environmental risk factors creates a wide genotypic and phenotypic heterogeneity within human HCC.10
A body of evidence indicates that the interaction of DNA with carcinogens and reactive radicals, generated during carcinogen metabolism and/or inflammation in early phases of liver carcinogenesis, induces genomic instability (GI) with consequent genomic alterations and signaling pathway deregulation.2,8,11,12 This allows initiated cells to evolve to dysplastic nodules and malignant lesions.8,11,12
This review provides an analysis of the epigenetic mechanisms implicated in signaling pathway deregulation during hepatocarcinogenesis. We explore the contribution of epigenetic changes to identify new putative prognostic markers and opportunities for targeted therapies.
Role of epigenetics
Epigenetics refers to a gene activity stable over long time that may be inherited and reprogrammed, affecting gene function, during cell development or as a consequence of environmental factors, without any change of DNA sequence. Epigenetic mechanisms include DNA methylation, nucleosome remodeling, histone modification (acetylation, methylation of arginine or lysine residues, and ubiquitination), mRNA and protein turnover, and small and microRNA (miRNA) noncoding sequences. These processes contribute to control gene function by affecting DNA structure, DNA-binding protein architecture, DNA access to transcription factors, and RNA and protein degradation.
DNA methylation
One of the best-understood molecular mechanisms of epigenetic gene function control involves methylation of specific cytosine lining upstream of a promoter region. The methylation of cytosine residues of CpG dinucleotides changes the major trench of the DNA chain, thus altering the proteins and transcription factors binding to DNA.
Around 4%–8% of DNA cytosines are methylated, mostly in CpG sequences.13 DNA-methyltransferases (DNMTs) catalyze DNA methylation, using S-adenosylmethionine (SAM) as methyl donor. DNMT-1 acts as maintenance methylase during cell replication and, using the parental methylated DNA strand as template, provides the epigenetic information between cell generations.14 DNMT3a and DNMT3b are responsible for de novo methylation during development.15 The presence of the methyl group directly interferes with the binding of a protein to its cognate DNA sequence. Many transcription factors fail their binding ability when the CpGs are methylated, leading to transcription repression.16 Significant correlations between loss of E-cadherin, resulting from promoter methylation, and tumor stage and location have been observed in a subset of gastric cancer patients, supporting a correlation between promoter hypermethylation and tumor aggressiveness and metastasis.17
The mechanisms limiting genomic methylation to a minority of CpG islands are poorly understood. A simple mechanism creating an unmethylated DNA domain is the presence into a DNA stretch of proteins causing DNMT exclusion.18 According to recent observations,19 RNA arising from the C/EBPA (CCAAT/enhancer binding protein α) transcription factor gene locus binds to DNMT-1 and prevents C/EBPA gene locus methylation. These interesting findings suggest that the interaction between DNMT-1 and RNA plays a role in selective DNA hypomethylation.
Chromatin and histone modification
An additional mechanism of epigenetic control is the association of histone proteins with DNA. Chromatin formation and the chemical modification of chromatin by covalent changes, as acetylation, arginine and/or lysine methylation, phosphorylation, SUMOylation and ubiquitination, can change DNA accessibility. Hypo-acetylation of lysine residues on core histone H4 is coupled with chromatin compaction and gene repression, while euchromatin displays higher levels of histone acetylation.20 The methylation of arginine or lysine residues does not alter the charge of histones but, depending on which arginine or lysine residues are modified, it can positively or negatively affect gene expression.21 Chromatin heavily acetylated, forming the so called “open configuration”, allows transcription factors to interact with gene promoters.22 Chemical marks in the N/C terminal tails of histone proteins are effectors of gene modulations, they influence also gene splicing, and DNA replication, repair, and recombination.
mRNA decay and protein turnover
An important molecular mechanism involved in the modification of gene expression is the mRNA decay process. Several signal transduction pathways contribute to controlling mRNA stability. The lability of mRNAs codifying signal transduction factors, cytokines, and oncoproteins contributes to determine their required levels in accordance with endogenous or exogenous stimuli. Most mRNAs start their decay by shortening their poly(A)-tail. Deadenylated mRNA that loses its 5′-cap (decapping) becomes suitable for digestion by exonucleases.23
mRNA instability is determined by AREs (elements rich of Au dinucleotides in the untranslated regions [UTRs]). AREs generally promote rapid mRNA degradation by deadenylation. They are grouped into class I, containing one or more AUUUA sequences in the U-rich region; class II, characterized by AUUUA tandem repeats; and U-rich class III, devoid of AUUUA. mRNAs encoding oncoproteins contain class I and class II sequences. Cytokines and chemokines encoding mRNAs often contain class II ARE sequences. AREs should interact with ARE-interacting proteins (ARE-BP) to regulate mRNA stability. ARE-BPs include AUF1/hnRNP, Hsc70, Hsp70 HuH/Hur, and several other proteins.23 RNA interaction with specific elements can confer transcript stability. Stable transcripts encode housekeeping proteins. Phosphorylation (ensured by activated protein kinases, such as p38MAPK, ERK, and Wnt/β-catenin), isomerization, methylation, and relocalization are ways to modulate RNA binding to proteins and, consequently, contribute to coordinating gene expression.
Noncoding RNA
In recent years, much attention focused on noncoding RNA (ncRNA), a family of untranslated transcripts monoallelicly expressed, deriving from the classically denominated “junk” DNA. ncRNA may be distinguished into long ncRNA (>200 nucleotides), transcribed by RNA polymerase II, and short ncRNA (20–40 nucleotides). Other species less well-characterized also may exist. Long ncRNA positioned in cis within large chromatin domains is involved in silencing multiple genes.24 Short ncRNAs are represented by miRNAs and Piwi-interacting RNAs.
miRNAs are noncoding minute RNAs implicated in posttranscriptional regulation of gene expression (Figure 1).
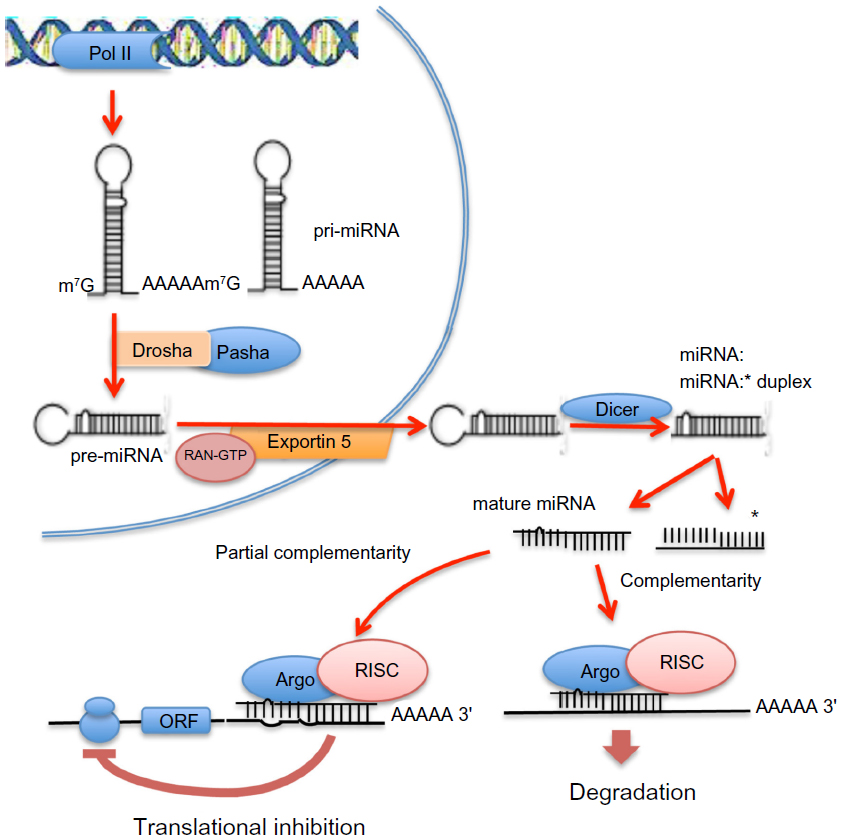 | Figure 1 Intranuclear and extranuclear phases of the process of maturation of microRNAs (miRNAs) and mechanisms of inhibition of messenger RNA transcription. In the nucleus, long primary transcripts (primary miRNAs) are cleaved by RNase III Drosha, with its cofactor Pasha, to release the precursor miRNAs, which are exported to the cytoplasm by exportin 5. In the cytoplasm, precursor miRNAs are processed by RNase III Dicer, with the release double-stranded duplexes of 20–23 RNA that contain the mature miRNA and the passenger miRNA strand (asterisked). The mature miRNAs interact with the 3′ untranslated region of its targeted mRNAs to form, with Argonaute proteins, the RNA-induced silencing complex (RISC). RISC binds to the 3′ untranslated region of its target or targets, inhibiting translation of single or multiple proteins; complete miRNA complementarity induces mRNA degradation, whereas partial complementarity represses translation. |
In the nucleus, RNA polymerase II transcribes primary miRNAs (pri-miRNAs) constituted by 5′-7-methyl guanylate cap, stem–loop secondary structures, and a 32-poly(A) tail.25,26 The endoribonuclease III, Drosha, with its cofactor Pasha, cleaves pri-miRNAs into 50–150 nucleotide precursor miRNAs, which are exported to cytoplasm by Exportin 5. In the cytoplasm, RNAse III Dicer cleaves precursor miRNAs into 20–23 nucleotide double-stranded duplexes. Each duplex splits into single-stranded mature miRNA, which associates with Argonaute proteins constituting the RNA-induced silencing complex (RISC). RISC binds to the 3′-UTR of its mRNA target or targets, inhibiting their translation.27 Totally complementary miRNAs induce mRNA degradation, whereas partial complementarity inhibits translation. A body of evidence assigns to miRNAs a key role in the regulation of gene expression. miRNA deregulation is strongly involved in cancer development.28
Piwi-interacting RNAs silence gene functions through base-pairing with mRNA 3′-UTRs. They can either repress translation or determine mRNA degradation by a RNAi (interference)-like mechanism. Piwi-interacting RNAs constitute a novel class of small RNAs that specifically protect mammalian DNA germline from transposon activity.29
The disruption of epigenetic regulatory mechanisms in oncogenesis may lead to inappropriate expression of genes regulating key cellular processes.30 Cancer cell genome is often globally hypomethylated, with a loss of 20%–60% of 5′-methylcytosine leading to activation of previously repressed protooncogenes. Several oncosuppressor genes are frequently hypermethylated in cancer. DNA repair genes may be subjected to promoter methylation. Cancers with hypermethylated methylguanosintransferase genes are susceptible to p53 and K-ras mutations.31 Hypermethylation of MLH1 (MutL, Escherichia coli, homolog of, 1), a mismatch repair gene, can cause microsatellite instability in colorectal and endometrial cancers.32 A clear example of epigenetic–genetic cooperation occurs in the colon cancer cell line HCT116, in which one allele of MLH1 and CDKN2A (cyclin-dependent kinase inhibitor 2a) genes is mutated and the other allele is silenced by DNA methylation, with consequent functional loss of DNA mismatch repair and cell cycle regulation.33
Epigenetic deregulation in hepatocellular carcinoma
Different observations showed global DNA hypomethylation and steatohepatitis, followed by HCC development even without carcinogens administration, in rats fed methyl-deficient diets (MDDs).34–36 MDDs induce a fall in SAM, the major methyl donor, and in SAM/S-adenosylhomocysteine (SAH) ratio. Remarkably, persistent decrease in SAM content and SAM/SAH ratio was also found in the liver, dysplastic nodules, and HCC of rats fed an adequate diet and subjected to the administration of different carcinogens in various experimental models.37–41 Global DNA hypomethylation and SAM decrease, with no change in SAH, was also found in human HCC and, to a lesser extent, in cirrhotic liver.42 These observations associate HCC development with global DNA hypomethylation and decrease in SAM.
Deregulation of methionine metabolism and global DNA hypomethylation in HCC
Liver is the main source of SAM, which is synthesized from methionine and adenosine triphosphate (ATP) in a reaction catalyzed by methionine adenosyltransferases (MATs)42,43 (Figure 2). SAM may undergo decarboxylation, followed by polyamine synthesis, or conversion to SAH throughout transmethylation reactions. SAH hydroxylase (SAHH) catalyzes the conversion of SAH to homocysteine and adenosine. Homocysteine is a precursor of GSH (reduced glutathione) via the transsulfuration pathway. Alternatively, betaine-homocysteine methyltransferase (BHMT) catalyzes the synthesis of methionine and dimethylglicine from homocysteine and betaine, whereas 5-methyltetrahydrofolate homocysteine methyltransferase (MTHF-HMT) catalyzes the synthesis of methionine and tetrahydrofolate from homocysteine plus 5-methyltetrahydrofolate. SAH and 5′-methylthioadenosine, produced during polyamine synthesis, may inhibit transmethylation reactions.
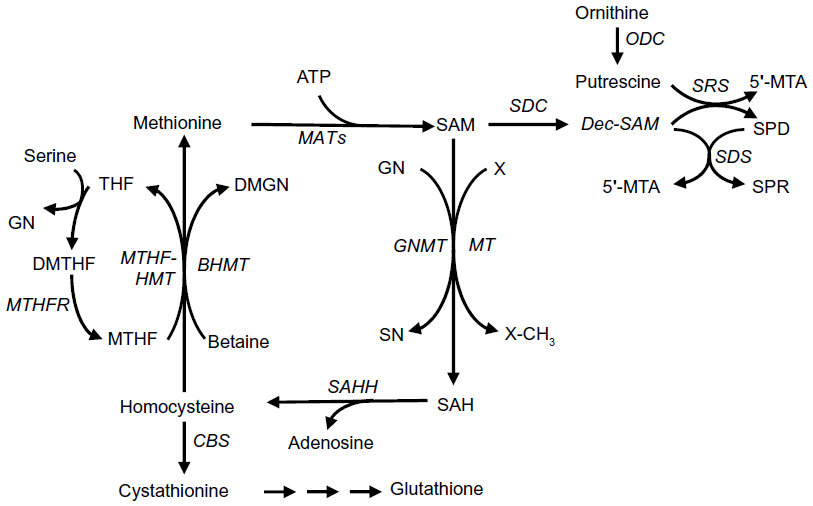 | Figure 2 Methionine metabolism. |
Liver-specific MAT1A codifies the isozymes MATI and MATIII, which are a tetramer and dimer of the subunit α1, respectively.44 MAT2A codifies an α2-subunit, the enzyme MATII isoform, which is widely distributed. MAT2A expression prevails in fetal liver and is progressively substituted by MAT1A in adult liver.43,45 MATI and MATIII isozymes are characterized by intermediate (23 μM–1 mM) and high (215 μM–7 mM) Michaelis constant (Km) for methionine. As a consequence, the physiological level of liver SAM (~60 μM) poorly inhibits MATI, whereas it stimulates MATIII activity.43 The low Km (~4–10 μM) for methionine of MATII causes its inhibition by the reaction product.43 A β-subunit of MATII, devoted of catalytic action, is encoded by MAT2B gene. This subunit regulates MATII by lowering its Km for methionine and Ki for SAM.44 As a consequence, the association of β and α subunits potentiates MATII inhibition by SAM. Recent observations show a correlation of glycine N-methyltransferase (GNMT) involved in hepatic SAM catabolism (Figure 2), with MAT1A and betaine-homocysteine methyltransferase hepatic enzymes, indicating a coordinate regulation of methionine cycle enzymes that determine SAM level.45
Various observations indicate that the reconstitution of a normal SAM level and the SAM/SAH ratio results in HCC chemoprevention. Huh7 cell transfectants, stably overexpressing MAT1A, exhibit higher SAM content; lower DNA synthesis, proliferative activity, microvessel density, and CD31 and Ki-67 expression; and higher apoptosis than controls.46,47 Further, the administration to rats of highly purified SAM during hepatocarcinogenesis significantly inhibits the development of early preneoplastic liver lesions, dysplastic nodules, HCC, and induces a decrease in labeling index and an increase in apoptosis of preneoplastic cells.37–40 It must be noted, however, that SAM infusion for 24 days did not affect the size of already-established tumors in mice transplanted with Huh7 cell transfectants.47 It has been hypothesized that a compensatory induction of hepatic GNMT (Figure 2) prevents SAM accumulation, thus hampering a therapeutic effect.
Fall in MAT1A expression associated with MAT2A up-regulation occurs in liver cirrhosis and rodent and human HCC, leading to a decrease in the MAT1A:MAT2A ratio (called MAT1A/MAT2A switch).41,48,49 In cirrhotic liver, MATI/III down-regulation, resulting from the oxidation of cysteine residue in the ATP binding site, is associated with GSH fall.50,51 Protection of MATI/III and reconstitution of the GSH pool by SAM administration strongly inhibits liver fibrosis both in rats and humans.50,52 Limited MATII up-regulation in HCC, because of its inhibition by the reaction product,43 cannot compensate for MATI/III decrease. Therefore, a sharp decrease in MATI/III:MATII activity ratio and an increase in polyamine synthesis (Figure 2) contribute to SAM fall.37 These observations strongly suggest a role in hepatocarcinogenesis of MAT1A/MAT2A switch and fall in SAM level. Accordingly, MAT1A-knockout mice, characterized by chronic SAM deficiency, undergo steatosis and mononuclear cell infiltration in periportal areas.53 HCC develops in a high percentage of these mice at 18 months of age.53
MAT2A up-regulation may also contribute to HCC cell proliferation. In H35 hepatocellular carcinoma cells, mitogen-activated protein kinase (MAPK) and phpsphatidylinositol 3 kinase/v-AKT murine thymoma viral oncogene homolog (PI3K/AKT) pathways are involved in HGF-induced cell proliferation and MAT2A up-regulation.54 Their inhibition results in proliferation restraint and a decrease in MAT1A:MAT2A switch.54 Moreover, the transfection of MAT2B in Huh7 cells that do not express this subunit, followed by the interaction of the β-subunit with the α2-subunit, increases DNA synthesis and inhibits SAM production, whereas the down-regulation of β-subunit in HepG2 cells, overexpressing MAT2B, inhibits DNA synthesis.55
The alterations of methionine metabolism have been comparatively evaluated in c-Myc and c-Myc/transforming growth factor α (TGF-α) transgenic mice to assess their relationships with tumor progression: c-Myc transgenics show lower progression of dysplastic liver to HCC and development of less-aggressive HCC with respect to the double transgenics.41 Dysplastic nodules and HCC of both transgenic models exhibit a reduction in MatI/III:MatII activity ratio and Cbs (cystathionine beta-synthase) expression, and a rise in Sahh expression. This suggests that in these lesions, a relatively high homocysteine production is presumably not associated with an increase in GSH level (Figure 2).41 In the lesions of both transgenics, Bhmt expression decreases, whereas Mthf-hmt expression abruptly increases in HCC of Myc/TGFα transgenics and does not change in the lesions of c-Myc transgenics.41 The expression of genes involved in polyamine synthesis, Sdc (SAM decarboxylase), Odc (ornithine decarboxylase), Srs (spermine synthase), and Sds (spermidine synthase), increases in both transgenic mice, with the highest values in the lesions of c-Myc/Tgf-a transgenics (Figure 2). Finally, the expression of the Mtap1 (5′-methylthioadenosine phosphorylase) gene, encoding a key enzyme of the methionine salvage pathway,56 exhibits a significant increase in dysplastic nodules and HCC of Myc/TGFα transgenics, suggesting a rise in methionine resynthesis.41
Experiments with human HCC showed a similar situation when HCCs with better prognosis (based on survival >3 years after partial liver resection; HCCB) and poorer prognosis (survival <3 years; HCCP), and their corresponding surrounding liver, were examined.41 In these lesions, MAT1A:MAT2A expression ratio and MATI:MATII activity ratio and BHMT, CBS, and MTAP1 expression decrease and SDC, ODC, SMR, and SMS expression increases with no variation of SAHH and MTHF-HMT expression in surrounding liver and HCC. These changes are higher in HCC than the surrounding liver and in HCCP with respect to HCCB.41
The comparative evaluation of the deregulation of methionine metabolism in dysplastic nodules and HCC induced in the Fisher-344 (F344) strain, which is genetically susceptible to hepatocarcinogenesis, and the Brown Norway (BN) genetically resistant strain8 showed a lower MATI/III:MATII activity ratio and Mat1A:Mat2A expression ratio in fast-growing and progressing F344 rat lesions.57 Notably, these ratios were negatively correlated with DNA synthesis and DNA methylation and positively correlated with apoptosis in rat liver lesions.57 A positive correlation was found between cell proliferation and global DNA hypomethylation and GI of transgenic mice and human HCC, whereas an inverse correlation of MAT1A:MAT2A and MATI/III:MATII ratios with proliferation rate and GI, and a direct correlation with apoptosis and DNA methylation, occurred in human HCC.41 Finally, prediction of patient’s survival length by MATI/III:MATII ratio was shown by Cox analysis.41
Altogether, these findings indicate a close association of reduced methionine resynthesis, increased in SAM use for polyamine synthesis, and a MAT1A/MAT2A switch with HCC progression and prognosis.
Mechanisms of MATs deregulation
Numerous observations associate methylthioadenosine’s deregulation with alterations in promoter methylation of MAT genes. MAT1A down-regulation in rat cirrhotic liver, induced by chronic CCl4 treatment, and in the human HepG2 hepatoblastoma cell line has been ascribed to the methylation of CCGG sequences of the MAT1A promoter.58 CCGG methylation at +10 and +80 of the coding region in Huh7, HEK293, and HepG2 cell lines has been associated with underexpression of MAT1A.59 Conversely, MAT2A overexpression of human HCC was associated with CCGG hypomethylation of gene promoter.60 In accordance with these results, recent observations showed the association between Mat1A/Mat2A switch and reduced SAM levels with CpG hypermethylation and histone H4 deacetylation of Mat1A promoter of fast-growing HCC induced in F344 rats.57 In the Mat2A promoter, CpG hypomethylation and histone H4 acetylation occurred.57 These changes were low or absent in slowly growing HCC of BN rats.57 Accordingly, MAT1A promoter hypermethylation and MAT2A promoter hypomethylation were highest in human HCCP compared with HCCB.57 These findings suggest that transcriptional regulation of MAT1A and MAT2A by promoter methylation may be strongly involved in HCC aggressivity.
Posttranscriptional mechanisms involving mRNA binding proteins may also be implicated in the deregulation of methyl adenosyltransferases. It is known that AUF1 may promote mRNA decay, whereas HuR enhances mRNA stabilization by selective binding to AUrich elements.61,62 Increased HuR and AUF1 protein levels have been found in human livers with HCC.63 Recent work documented a sharp increase of AUF1 and HuR, as well as of MAT1A-AUF1 and MAT2A-HuR ribonucleoproteins, in F344 HCC and human HCCP.57 Notably, these changes were very low or absent in slowly progressing HCC of BN rats.57
MAT1A under-regulation has also been attributed to up-regulation in HCC of miR-664, miR-485-3p, and miR-495.64 Individual knockdown of these miRNAs in Hep3B and HepG2 cells enhanced MAT1A expression. The knockdown of miRNAs-664/485-3p/495 increased Hep3B cell tumorigenesis in nude mice, whereas the opposite occurred when these miRNAs were stably overexpressed.64
Deregulation of HCC oncosuppressors
The inactivation of oncosuppressor genes plays a crucial role in hepatocarcinogenesis. Numerous studies on oncosuppressor gene deregulation in HCC have shown that aberrant promoter methylation is the mechanism most frequently responsible for down-regulation of these genes.65–68 Variable percentages of HCC show hypermethylation of APC, E-CAD, P14ARF, P15, P16INK4, P21WAF1, P27KIP1, P57KIP2, P73, P130, RAR-B (retinoic acid receptor, beta), SOCS-1 (suppressor of cytokine signaling 1), PRM2 (PR domain-containing protein 2), GSTP (glutathione S-transferase), O6-MDM (O6-methylguanine-DNA methyltransferase), RASSF1A (Ras association domain family 1A), and FOXO1 (Forkhead box O1). The degree of promoter methylation in HCC is significantly higher than in dysplastic nodules and cirrhotic liver.66–68
P53 gene has been intensively studied in HCC. The P53 pathway has crucial roles in cell cycle control, transcriptional regulation, and apoptosis.69 P53 deregulation in HCC mostly depends on mutational events.69 However, it has been observed that HBx protein binds to the p53 protein in the cytoplasm, resulting in the blockage of p53 entry into the nucleus.70 Furthermore, the 14-3-3σ gene, implicated in G2/M cell cycle arrest by p53, is frequently hypermethylated in HCC.71
Promoter hypermethylation of oncosuppressors is never present in all HCC cases. Recent studies have analyzed some alternative epigenetic mechanisms that could induce oncosuppressor down-regulation. Fast proliferation of dysplastic nodules and HCC requires the inactivation of cell cycle inhibitors. The inhibition of CDK4 and CDK6 by p16INK4 blocks G1 phase by preventing pRb phosphorylation. p16INK4 inactivation in 60%–85% of human HCC depends on promoter GpG methylation.72 p16INK4 inactivation may also be achieved by the linkage of cyclin D1 kinases with HSP90 (heat shock protein 90) and CDC37 (cell division cycle 37) proteins,72 as well as through the transfer of E2F4, a p16INK4 effector, from nucleus to cytoplasm by CRM1 (chromosome region maintenance 1).73,74 These mechanisms are particularly active in fast-growing dysplastic nodules and HCC of F344 rats.75 Moreover, the highest formation of p16INK4/HSP90/CDC37 complexes was found in human HCCP.75
P21WAF1, P27KIP1, P57KIP2, P130, RASSF1A, and FOXO1, inhibitors of CYCLIN E- and CYCLIN A-dependent CDK2, are down-regulated by promoter methylation in variable percentages of human HCC.76 In unmethylated tumors, these inhibitors of G1-S transition are ubiquitinated by the ubiquitin ligase complex SKP2-CSK1 (S-phase kinase-associated protein 2–Cdc28 protein kinase 1) ligase, followed by proteasomal degradation.76 CDK2 inhibitors are more frequently down-regulated in HCCP than in HCCB, either by promoter hypermethylation or proteasomal degradation.76 These observations and the existence of a positive correlation between SKP2 expression and HCC cell proliferation and microvascularization, and a negative correlation with apoptosis,76 suggest a connection of the degradation of CDK2 inhibitors with HCC aggressivity. Researches on rat liver carcinogenesis showed higher ubiquitination of CDK2 inhibitors by Skp2–Cks1 in fast-growing dysplastic nodules and HCC of genetically susceptible F344 rats than in slow-growing lesions of resistant BN rats,77 thus suggesting a genetic control of cell cycle inhibitor inactivation by proteasomal degradation.
SKP2 activity is regulated by the SKP2 suppressor HINT1 (histidine triad nucleotide binding protein 1) and CDC14B (cell division cycle 14, Saccharomyces cerevisiae homolog B) phosphatase, which induces SKP2 dephosphorylation, followed by degradation mediated by the ubiquitin ligase (APC/C)CDH1 (anaphase-promoting complex/cyclosome and its activator CDH1).78 The investigation of the interstrain differences in the degradation of CDK2 inhibitors indicated the involvement of the up-regulation of HINT1 and CDC14B.79 Interestingly, the mechanisms blocking SKP2 degradation, such as down-regulation of CDC14B, associated with CDK2-dependent serine phosphorylation (which impedes the interaction between CDH1 and SKP2), and HINT1 inactivation, are much higher in HCCP than in HCCB.77
Among the genes involved in methionine and SAM metabolism, GNMT is underexpressed both in rat and human HCC, with the lowest expression in fast-growing F344 HCC and human HCCP with respect to slowly growing BN HCC and HCCB.80 Remarkably, loss of GNMT was found to induce liver steatosis and HCC.81 Recent work indicates that DNA hypermethylation plays an important role in GNMT suppression in HCC.82 Loss of GNMT causes SAM accumulation.74 Histone and genome-wide methylation analyses suggest that GNMT deregulation in HCC could be involved in promoter-specific DNA methylation of oncosuppressor genes.82
DNA methylation status of liver cancer stem cells
An emerging aspect of the studies on the role of epigenetic alterations in carcinogenesis is the possibility that these alterations are involved in the regulation of cancer stem/progenitor cells.83 Side population cells, possessing typical cancer stem/progenitor cell-like properties, isolated from HCC cell lines Huh7 and PLC/PRF/5, induce HCC in NOD/SCID mice.83 Genome-wide DNA methylation microarray analysis showed the presence, in the two cell lines compared with their corresponding non-side population cells, of both hypermethylated and hypomethylated genes, which were related to four and eight pathways, respectively.84
In keeping with these observations, recent studies revealed altered methylation status of the promoter of OCT4, SOX2, KLF4, and c-MYC genes in HCC.85 The overexpression of these genes induces pluripotency in somatic cells, leading to the generation of embryonic stem cell-like pluripotent cells.86,87
Compared with normal liver cells L02, which displayed a modest methylation level, DNA hypomethylation of NANOG was found in the HCC cell lines PLC (primary HCC) and MHCC97L and MHCC97H, derived from metastatic HCC, with the lowest decrease in DNA methylation in the low metastatic MHCC97L cells.85 A reduced methylation pattern of OCT4 promoter and c-MYC exon 3 was also observed in PLC and MHCC97L cells. These changes were associated with NANOG, OCT4, and c-MYC up-regulation. NANOG promoter hypomethylation and gene up-regulation were also observed in human HCC primary tumor tissue.85 Importantly, a cross-regulation between OCT4 and NANOG in cancer cells by reprogramming of promoter methylation was demonstrated, suggesting a role of epigenetic reprogramming of gene expression in the acquisition of stem cell-like properties.85 This conclusion is enforced by recent observations indicating a role of DNMT1 depletion in the enhancement of cancer stem cell properties in WRL68 HCC and HepG2 hepatoblastoma cell lines.88
These observations suggest that DNA methylation status is a key epigenetic mechanism that determines the pool of cancer stem cells in liver cancer by maintaining the pluripotency and regulating the differentiation of stem cells during cancer progression.
Signaling deregulation
Role of altered methionine metabolism
The role of MAT1A/MAT2A switch and SAM fall in HCC has been evaluated by studying the effect of reconstituting MAT1A expression and SAM level on signal transduction. Forced expression of MAT1A in the Huh7 and HepG2 cell lines down-regulates cyclin D1, E2F1, IKK, NF-kB, and antiapoptotic BCL2 and XIAP genes and up-regulates proapoptotic BAK and BAX genes.57,89 Nuclear factor kB activation in rat preneoplastic foci is inhibited by SAM.90 The latter also restrains AMP activated protein kinase (AMPK), phosphorylated AKT (pAKT), and phosphorylated extracellular signal-regulated protein kinase (pERK) activities by up-regulating the oncosuppressor PP2A.91,92 SAM levels and PP2A expression are inversely correlated with pAKT and pERK expression both in rat and human HCC.91,92 Recent research aimed at investigating the mechanism of a SAM inhibitory effect on ERK1/2 activity showed a possible interference of SAM with the activity of DUSP1, a specific ERK inhibitor. HCC of F344 rats and human HCC exhibit low DUSP1 expression associated with ERK1/2 up-regulation.93,94 DUSP1 phosphorylation at Ser296 by active ERK1/2 leads to its ubiquitination by SKP2-CKS1 ubiquitin ligase, followed by proteasomal degradation.93,94 In addition, ERK1/2 supports SKP2-CKS1 activity through the activation of FOXM1.95 Recent work discovered a reduction of DUSP1 mRNA and, at a higher extent, of DUSP1 protein levels in the livers of MAT1A-KO mice and in in vitro cultures of mouse and human hepatocytes.96 SAM treatment increased DUSP1 mRNA and protein levels, suggesting a regulation of DUSP1 expression at transcriptional and posttranslational levels.96 Interestingly, hypoxia could also be involved in SAM deregulation of ERK1/2 expression. It is known that the tumor necrosis factor (TNF)-a/HIF-1a axis sustains the expression of FOXM1,97 which is a mediator of ERK1/2 effects on cell proliferation.95 The observation that HIF-1a can bind MAT2A promoter suggests the possibility that ERK1/2 up-regulation in BEL-7404 and Hep3B HCC cells and HepG2 cells could be at least partially attributed to a reduction of SAM level during hypoxia.98
Recent work99 showed that Apex1 mRNA and protein levels are significantly reduced in the liver of MAT1A-KO mice. Apex1 expression was correlated to the increase of AP sites and the reduction of the expression of Apex1 targets Bax, Fas, and p21.99 An association of MAT1A mRNA decrease with APEX1 and c-MYC mRNAs increase was found in cell cultures of human and mouse hepatocytes in which, however, APEX1 protein level decreased to 60% of baseline.99 The prevention of these changes by SAM was attributed to possible proteasome inhibition.100
Signaling regulation by miRNA
Different studies on experimental models have ascertained the deregulation of miRNAs even in early stages of liver carcinogenesis. Up-regulation of miR-17-92 cluster, miR-106a, and miR-34 was observed during tamoxifen-induced hepatocarcinogenesis in female rats.101 Thirty differential expressed miRNAs have been identified by microarray analysis of mice fed a MDD.102 Down-regulation of miR-15/16, miR-26a, miR-34a, miR-150, and miR-195 occurs in c-Myc-transgenic mice.103
Numerous studies showed the enhancement of cell cycle and signal transduction in HCC induced by up-regulation of miRNAs targeting oncosuppressor or apoptogenic genes (Table 1). MiR-423104 and miR-221105 target wildtype p53-activated fragment (WAF) and cyclin-dependent kinase inhibitor (KIP) family inhibitors of cell cycle, respectively. MiR-602 up-regulation may enhance RAS/ERK and MSP/YAP pathways through RASSF1A inhibition.106 PP2A and/or PTEN inhibition results from up-regulation of miR-221, miR-222, and miR-21.107,108 Furthermore, miR-221 overexpression inhibits DDIT4 (DNA damage-inducible transcript 4), a regulator of the mammalian target of rapamycin (mTOR) kinase, by enhancing the activity of the putative oncosuppressor TSC1/2 (tuberous sclerosis 1/2) complex.109 Liver cell migration and metastasis may be favored by MiR-221, miR-222, and miR-181b/d overexpression that enhances MMP2 and MMP9 (matrix metalloproteinase 2 and 9) activity through TIMP3 (tissue inhibitor of metalloproteinase 3) inhibition110 (Table 1). Finally, a miR-221 and miR-106b-25 cluster, containing miR-106b, miR-93, and miR-25, may modulate apoptosis by targeting the proapoptotic genes BMF and BIM genes111,112 (Table 1).
Surprisingly, up-regulation of miR-93 and miR-106b, targeting E2F1, a key gene of G1-S transition, was observed in HCC.112 It has been hypothesized113 that miR-93 and miR-106b up-regulation hinders the accumulation of high E2F1 levels that could induce apoptosis.114
A contribution of miRNA deregulation to DNA damage accumulation in HCC has been suggested on the basis of the discovery of MiR-192 up-regulation, leading to inhibition nucleotide excision repair by targeting ERCC3 and ERCC4 (excision-repair, complementing defective, in Chinese hamster 3 and 4) in the HBV-related HCC cell line HepG2.2.15115 (Table 1).
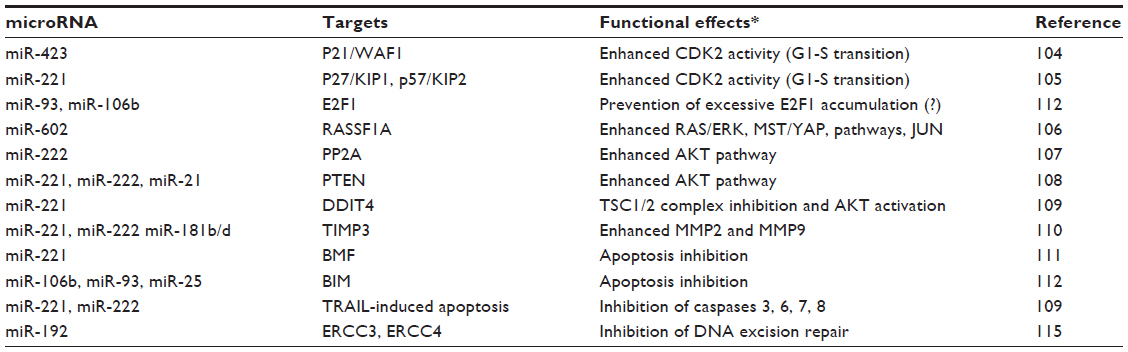 | Table 1 Targets and functional effects of microRNAs up-regulated in hepatocarcinogenesis |
Several miRNAs exhibit an oncosuppressor activity, and their down-regulation in HCC enhances signal transduction and growth (Table 2). Activation of cell cycle may result from the down-regulation of miR-26a,116 targeting Cyclins D2 and E2 genes; of miR-195,117 targeting Cyclin D1, CDK6, and E2F3; and of miR-122a,118 which targets Cyclin G1. miR-122 underexpression is also associated with overexpression of PBF (pituitary tumor-transforming gene 1 binding factor), which promotes tumor growth.119 G2-M transition may be favored by STMN1 (Stathmin) overexpression and microtubules stabilization120 induced by miR-223 down-regulation.
Numerous observations attribute oncosuppressor activity to Let-7 miRNA family (Table 2). The underexpression of hsa-Let-7g, a positive regulator of c-MYC and a negative regulator of p16INK4A,121 is higher in metastatic HCC than in metastasis-free HCC.122 The deregulation of collagen synthesis and the enhancement of epithelial- mesenchymal transition (EMT) are involved in the prometastatic effect of Let-7g down-regulation. Let-7g targets COL1A2 (type I collagen a2), and cell migration is suppressed in Let-7g overexpressing huH1-let-7g cells. This latter effect is eliminated by the addition to the cell culture of type I collagen a2.122
Stimulation of HCC cell migration and EMT transition follows the activation of ZEB1/2 (zinc finger e box-binding homeobox 1/2) by down-regulation of miR-200b123 and of ROCK2 (RHO-associated coiled-coil-containing protein kinase 2) and EZH2 (enhancer of ZESTE, drosophila, homolog 2) by down-regulation of miR-124.124 Furthermore, miR-194 overexpression suppresses N-cadherin expression, thus inhibiting the migration of mesenchymal-like cancer cells.125 Accordingly, miR-148a forced expression in MHCC97-H cells inhibits the expression of SNAIL and N-cadherin, suggesting EMT suppression.126,127 Migration of human HCC cells is also suppressed by miR-23b targeting uPA and c-MET 128 (Table 2).
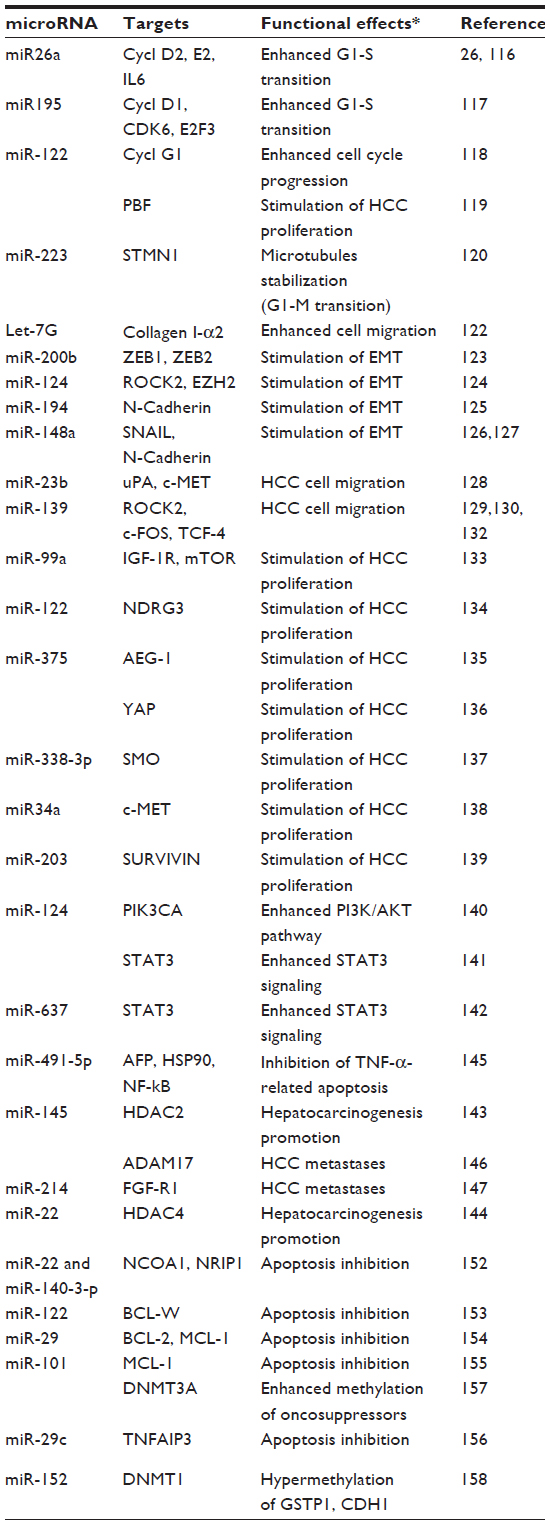 | Table 2 Targets and functional effects of microRNAs down-regulated in hepatocarcinogenesis |
A crucial role in HCC cell migration is also played by miR-139. MiR-139 down-regulation in HCC is significantly correlated to the poor prognosis and metastatic potential of this tumor.129,130 Forced overexpression of miR-139 in human HCC cell lines SMMC-7721 and BEL7402 significantly inhibits cell migration and invasion in vitro, as well as tumor incidence and lung metastasis in mouse orthotropic transplants of HCC. Mechanistic studies demonstrated that miR-139 interacts with the 3′-UTR of ROCK2 and inhibits its expression in HCC cells.129 These observations are consistent with the known capacity of the Rho-kinases to activate the actomyosin contraction, cellular migration, and chemotaxis.131
Additional mechanisms of the prometastatic effect of miR-139 down-regulation in HCC have been recently discovered. Evidence has been presented in favor of a contribution of increased c-FOS expression, associated with miR-139 down-regulation, to the high metastatic potential of human HCC cell sublines.130 Furthermore, it has been observed that the inhibition of HCC cell proliferation and invasion, induced by miR-139 forced overexpression, is associated with suppression of TCF-4 (T-cell factor 4) activity, suggesting a contribution of miR-139 down-regulation to the increase in β-catenin/TCF-4 transcriptional activity in HCC.132
Stimulation of HCC cell proliferation also occurs by down-regulation of miR-99a,133 miR-122,134 miR-375,135,136 miR-338-3p,137 miR-34a,138 and miR-203139 that target IGF-1R/mTOR, NDRG3 (N-myc downstream-regulated gene 3), AEG-1 (astrocyte elevated gene-1)/YAP (Hippo signaling), Hedgehog signaling, c-MET, and SURVIVIN, respectively (Table 2). Interestingly, targeting of c-MET by miR-34a induces a decrease in c-MET-induced phosphorylation of ERK1/2.138 Inhibition of miR-124 and miR-637, leading to enhanced PI3K/AKT140 and/or STAT3141,142 signaling, may result in increased hepatocarcinogenesis.143 Promotion of liver cancer may also result from up-regulation of histone deacetylase 2 (HDAC2) and HDAC4 targeted by miR-145143 and miR-22,144 respectively, HCC development is also facilitated by the under-regulation of miR-491-5p, targeting AFP, HSP90, and NF-kB, through inhibition of TNF-α-induced apoptosis.145 Interestingly, recent observations indicate that miR-145 represses HCC cell invasion by targeting ADAM17 (a disintegrin and metalloprotease 17).146 Notably, ADAM17 is also targeted by miR-122, the down-regulation of which induces HCC metastasis.147 HCC metastasis is also stimulated by miR-214 down-regulation, leading to FGFR-1 (fibroblast growth factor receptor 1) gene overexpression and HCC progression.148
In HBV-related hepatocarcinogenesis, miR-122 expression is inversely correlated to NDRG3 protein level134 (Table 2). The forced expression of miR-122 into the HBV-related HepG2.2.15 cell line suppresses the expression of NDRG3 (n-myc downstream-regulated gene 3), with subsequent reversion of the malignant phenotype. The restoration of miR-122 inhibits HBV replication and HCC cells proliferation.
Numerous other miRNAs exhibit a prometastatic potential. Among these, miR-550a, up-regulated in HCC, inhibits CPRB4 (cytoplasmic polyadenylation element binding protein 4) expression, which impedes HCC cell migration and invasion;149 miR-210, induced by hypoxia, increases the metastatic potential of HCC by targeting VMP1 (vacuole membrane protein 1);150 and targeting of GNAI2 (G protein, alpha-inhibiting 2B) by miR-30d induces HCC cell invasion and metastasis.151
MiRNAs down-regulation may also contribute to hepatocarcinogenesis by increasing cancer cell survival (Table 2). miR-22 and miR-140-3-p down-regulation stimulates antiapoptotic signaling in HCC by targeting NCOA1 (nuclear receptor coactivator 1) and NRIP1 (nuclear receptor-interacting protein 1),152 miR-122 down-regulation inhibits the antiapoptotic gene BCL-w,153 and miR-29 down-regulation favors the expression of the antiapoptotic genes BCL-2 and MCL-1.154 MCL-1 is also targeted by miR-101 that is down-regulated in HBV-related HCC.155 Interestingly, according to recent observations, forced overexpression of miR-29c in HepG2.2.15 cells leads to inhibition of TNFAIP3 (TNF alpha-induced protein 3) expression, HBV DNA replication, and cell proliferation, associated with increase in cell death.156
The down-regulation of miR-101 by the HBx protein also induces aberrant DNA methylation by targeting DNMT3a.157 In HBV-related HCC, aberrant DNA methylation may likewise occur by down-regulation of miR-152, the expression of which is inversely correlated to DNMT1 mRNA.159 Inhibition of miR-152 expression in liver cell lines causes global DNA hypermethylation and increased methylation of oncosuppressors GSTP1 (glutathione S-transferase pi 1) and CDH1 (E-cadherin 1)158 (Table 2).
The induction of miR-23a-27a-24 cluster expression by TGF-β in Huh-7 cells attenuates the antiproliferative and proapoptotic effects of TGF-β.159 miR-23a-27a-24 cluster is overexpressed in HCC cells and promotes their proliferation and survival.160 Furthermore, in mice fed a MDD, TGF-β increases miR-181b expression, resulting in TIMP3 inhibition and stimulation of MMP2 and MMP9 (Table 1), with consequent enhancement of HCC cell proliferation and tumorigenicity.110 These findings, taken altogether, suggest that induction of specific miRNAs by TGF-β may abolish its tumor-suppressive effect for HCC.
Recent observations indicate a possible role of miRNAs as prognostic markers. miR-15b, miR-21, miR-130b, and miR-183 are highly expressed in HCC. A recent cross-center validation study demonstrated that combined miR-15b and miR-130b serum levels are a classifier useful to identify early-stage HCCs not detected by serum α-fetoprotein.161 Furthermore, a cluster of 23 miRNAs arising from imprinted genomic loci and associated with poor clinical outcomes was identified.162 This cluster is encoded within the Dlk1-Gtl2 imprinted region on mouse chromosome 12qF1 and the human DLK1-DIO3 region on chromosome 14q32.2. DLK1-miRNA cluster is overexpressed in HCC of c-MET transgenic mice and human HCC. Interestingly DLK1-miRNA cluster up-regulation in human HCC is positively correlated with HCC stem cell markers, such as CD133, CD90, EpCAM, and Nestin.64
These observations indicate that a complex miRNAs deregulation is strongly involved in hepatocarcinogenesis. The analysis of the mechanisms leading to miRNA deregulation has shown frequent amplification of the 8q24.3 chromosomal region encoding miR-151163 and frequent loss of 18q21.31 region encoding miR-122 in HCC.164 Noticeably, various polymorphisms of Dicer1 and RAN (Ras-related nuclear protein) that, in association with GTP acts as a cofactor of exportin 5 (Figure 1), occur in HCC.165 The association of these polymorphisms with cancer susceptibility and survival has been reported. DICER1 rs1057035 CT/CC variant genotypes are associated with decreased HCC risk compared with wild-type TT. This behavior seems to depend on a rise in affinity binding to hsa-miR-574-3p, which results in a positive feedback of hsa-miR-574-3p.166
Increased susceptibility to HCC has been related to pri-miR-146a,167 miRNA-196a2,168,169 pri-miR-218,170 and miR-34b/c.171 miR-122 binds to the IL1A (interleukin 1 alpha) gene and is abolished by the insertion of TTCA sequence in the in IL1A 3′-UTR region.172 As a consequence, IL1A is overexpressed, likely increasing susceptibility to HCC.172 Interestingly, according to a recent report, the overexpression of miR-25-93-106b cluster suppresses the expression of MICA (MHC class I polypeptide-related sequence A) gene.173 The activity of this gene has been associated with the risk for hepatitis-virus-related HCC.173 These findings seem to connect miRNA deregulation to the susceptibility to HCC, a topic that deserves further attention.
Numerous observations indicate an epigenetic deregulation of miRNAs expression in different tumors, including HCC. MiR-1 down-regulation in human HCC is associated with promoter methylation.174 Treatment of primary HCC cultures with the hypomethylating agent 5-aza-2′-deoxycytidine caused miR-1 reexpression, down-regulation of miR-1 targets, including FoxP1, MET, and HDAC4, overexpressed in HCCs, and inhibition of cell cycle progression and cell survival.174
Epigenetic repression by promoter methylation has been demonstrated for miRNAs exhibiting oncosuppressor activity, such as miR-129-2,175 miR-132,176 miR-335,177 miR-24, and miR-203.178 According to recent observations, HBx is able to inhibit miR-205, a potential tumor-suppressor gene, by inducing hypermethylation of its promoter.179
Notably, hypomethylation of the hsa-miR-191 locus causes miR-191 overexpression and promotes EMT in HCC.180 Recent observations showed up-regulation of HDAC1-3 in HCC.181 Their inhibition by trichostatin A in HCC cell lines resulted in increased levels of hsa-miR-449a, leading to reduced c-MET mRNA level. HDAC1-3 overexpression, by causing miR-449a down-regulation, leads to c-MET overexpression that favors HCC cell growth.181 These observations assign to chromatin-modifying drugs, including HDAC inhibitors, a potential therapeutic role for HCC.182
Gene interactions are also involved in miRNA deregulation. RACK1 (receptor for activated protein kinase C), frequently underexpressed in HCC, interacts with KSRP (KH-type splicing regulatory protein), a member of DICER183 (Figure 1). KSRP binds to and promotes the maturation of miRNA precursors184 and is involved in recruiting mature miRNAs to RISC183 (Figure 1). These findings attribute to a decrease in RACK1; in HCC, a decrease in miRNAs function.
Two oncogenes overexpressed in HCC,185 SND1 (Staphylococcal nuclease domain-containing 1) and AEG1 (also called Metadherin), are components of RISC. RISC activity enables the degradation of oncosuppressor mRNAs by the oncogenic miR-221 (Table 1), thus promoting hepatocarcinogenesis.185 One of the mechanisms involved in miR-221-induced oncogenesis is the up-regulation of angiogenin and CXCL16.186 Up-regulation of miR-221 by AEG1 or SND1 also leads to inhibition of PTEN and P57 (CDKN1C). SND1 and AEG1 also up-regulate various miRNAs targeting oncosuppressors, such as miR-21, which targets PTEN and SPRY2 (Sprouty 2); miR-106b, which targets P21 (CDKN1A); and miR-93-17,18, which targets TGFBR2.186
The role of SND1 in tumorigenesis is strengthened by the observation that human SND1 protein binds PIWIL1 (HIWI), a member of the PIWI protein family that is implicated in RNA silencing.187 Up-regulation of PIWIL1 gene occurs in various cancer types, including HCC.188 Recent observations showed up-regulation of HIWI protein and mRNA in the metastatic HCC cell lines HCCLM3, MHCC97H, and MHCC97L, the proliferation and invasiveness of which decreased after HIWI depletion.189 Positive expression of HIWI was associated with larger tumor size and presence of intrahepatic metastases and was an independent risk factor for overall survival and recurrence-free survival.189
Importantly, the presence of PIWI proteins and Piwi-interacting RNAs is thought to result in aberrant DNA methylation and silencing of genomic regions and to be critical for the maintenance of genomic integrity in germline stem cells.190 Work from different groups suggests the presence in cancer cells of epigenetic and signaling pathways common to stem cells.191 The HIWI-mediated methylation results in DNA hypermethylation with subsequent genetic and epigenetic changes that recapitulate the aberrant “stem-like” status, allowing cells to possess the self-renewal capacity of stem cells, which characterizes tumorigenesis. Accordingly, it has been found that HIWI increases DNA methylation levels and consequent silencing of cyclin-dependent kinase inhibitor (CDKI).188
Finally, recent findings indicate that the binding of the PPARγ (peroxisome proliferator activated receptor-gamma) and RXRα (retinoid X receptor alpha) complex to the DR1 and DR2 consensus site of miR-122 gene promoter enhances miR-122 gene transcription.192 HBx protein binding to PPARγ inhibits miR-122 transcription. This epigenetic mechanism is absent in HCV-related HCC, in which other mechanisms, not yet known, cause miR-122 down-regulation.192
Therapeutic perspectives
The existence in HCC of a global hypomethylation of DNA, associated with promoter hypermethylation of tumor suppressor genes, encouraged the evaluation of therapies aimed at correcting the alterations of DNA methylation. The attempt to contrast global DNA hypomethylation with SAM administration did not exhibit any therapeutic effect.47 However, a well-documented chemoprevention of HCC by SAM, administered during experimental hepatocarcinogenesis, was reported.38–40,47 Furthermore, the observation of a SAM curative effect on alcoholic steatohepatitis and hepatitis C193–196 suggests a possible chemoprevention of some types of human HCC. SAM chemoprevention of hepatitis C is under evaluation by a current phase II clinical trial.193 However, silencing of MAT2A or MAT2B in HepG2 cells inhibits the proliferative response to leptin.197 The intracellular transduction of viral vectors in vivo still presents numerous restrictions.198 Recent observations on the curative effect for HCC of a family of fluorinated N,N-dialkylaminostilbenes are particularly important in this context. Binding of these compounds to MATIIα catalytic subunit inhibits SAM synthesis and in vitro and in vivo proliferation of colorectal cancer cells.199 Moreover, the observation that N,N-dialkylaminostilbenes inhibit the WNT/β-catenin pathway suggests they could be particularly effective against β-catenin mutated HCC.199
The promoter hypermethylation of oncosuppressor genes, in variable percentages of HCC, leads to the suppression of their expression.198,200,201 This is associated with HDAC overexpression, an epigenetic alteration that contributes to the development and maintenance of HCC.202 These findings suggest the possibility of new therapeutic approaches based on modulation of the methylation status of oncosuppressors and the acetylation pattern of histones associated with DNA. Promising results in the treatment of HCC have been obtained by hydroxamate-based histone deacetylase Inhibitors, which synergize with other anticancer agents, such as cytostatic compounds.203,204 Moreover, studies on HCC cell lines and xenografted HCC clearly showed a significant increment of the inhibition of HCC cell growth in vivo by the association of HDAC inhibitors with the demethylating agent 5-aza-2′-deoxycytidine.205 Interestingly, according to recent research, 5-aza-2′-deoxycytidine down-regulates the telomerase reverse transcriptase (hTERT) and represses telomerase activity in SMMC-7721 and HepG2 cell lines.204 This is associated with the reversion of the methylation status of hTERT promoter in SMMC-7721, but not in HepG2 cells, suggesting that hTERT gene expression is not exclusively regulated by promoter methylation in HCC cells treated with 5-aza-2′-deoxycytidine.204
The observation that the overexpression of several miRNAs is involved in hepatocarcinogenesis suggests a possible therapeutic effect of specific anti-miRNAs oligonucleotides. However, miRNA-based therapeutic methods achieving real inhibition and safety in vivo are still lacking. Recently, linear DNA sequences expressing anti-HBV micro-RNA shuttles were shown to efficiently silence HBV markers in cell culture and in vivo.206 Inhibition of HBV replication and HCC growth was obtained by a lentiviral miRNA-based system.207 Further, the systemic administration in a mouse HCC of an adeno-associated virus preparation of miR-26A, underexpressed in this tumor, was found to significantly inhibit cancer cell survival, proliferation, and progression in the absence of toxicity. Harmless and efficacious delivery of anti-miRNAs oligonucleotides waits for novel modification, conjugation, or formulation strategies. Different studies tested the antitumor effect of different miRNA inhibitors, and promising results were obtained in recent clinical trials with 15 nucleotide antisense oligonucleotides and 8 nucleotide versions in nonhuman primates.208,209 Finally, miRNAs were shown to enhance the effect of anticancer drugs such as doxorubicin and sorafenib.210–212
Conclusion
Numerous chromosome aberrations and oncogene and oncosuppressor mutations have been discovered in HCC.2,213–215 Polymorphisms of genes involved in miRNA maturation, such as DICER1 and RAN,165 are involved in HCC risk. Some polymorphisms of DNMT3a and DNMT3b have been recently found in HCC,216,217 but they are not correlated with liver cancer risk. A GNMT polymorphism has been described and considered an early event in hepatocarcinogenesis,218 but its association with susceptibility to HCC has not been unequivocally demonstrated.
Several observations indicate that the epigenetic manipulation of transcription and translation mechanisms is strongly involved in hepatocarcinogenesis (Figure 3).
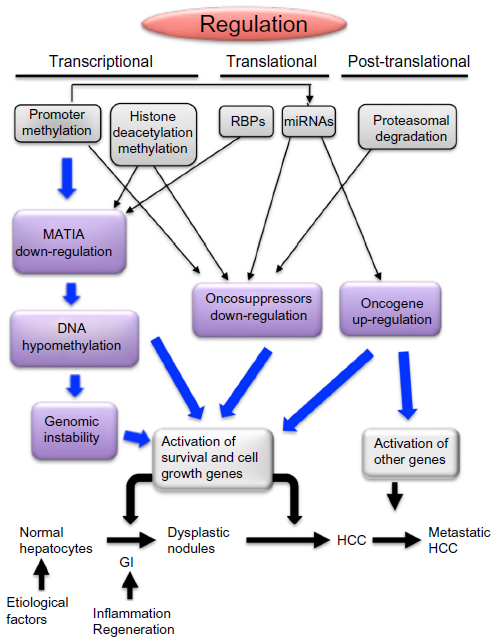 | Figure 3 Transcriptional, translational, and posttranslational mechanisms leading to deregulation of MAT1A, tumor suppressor genes, and oncogenes during hepatocarcinogenesis. |
Transcriptional deregulation implicates down-regulation of oncosuppressors and of MAT1A, leading to global DNA hypomethylation that contributes to signaling deregulation through the increment of GI and possible up-regulation of oncogenes with CpGs-rich promoters. Translational deregulation occurs via the action of RBPs on MATs, as well as by miRNAs affecting MAT1A expression or exhibiting oncosuppressor or oncogene activities. Finally, ubiquitination and proteasomal degradation of oncosuppressors represents an important posttranslational deregulation mechanism strongly involved in hepatocarcinogenesis.
These deregulation mechanisms are correlated with HCC prognosis, and further study on these correlations could allow discovering of new prognostic markers. Furthermore, epigenetic silencing of oncosuppressor genes in HCC, mediated by methylation of CpG islands in gene promoters, histone deacetylation or methylation, and silencing of oncosuppressor miRNAs by epigenetic alterations, offer the possibility of new epigenetic approaches to HCC treatment. Restoring the expression of tumor suppressor miRNAs may be a novel promising therapeutic strategy for HCC.
Acknowledgments
Supported by grants from Associazione Italiana Ricerche sul Cancro (IG8952) and Fondazione Banco di Sardegna.
Disclosure
The authors report no conflicts of interest in this work.
References
Llovet JM, Bruix J. Molecular targeted therapies in hepatocellular carcinoma. Hepatology. 2008;48(4):1312–1327. | |
Feo F, Pascale RM, Simile MM, De Miglio MR, Muroni MR, Calvisi D. Genetic alterations in liver carcinogenesis: implications for new preventive and therapeutic strategies. Crit Rev Oncog. 2000;11(1):19–62. | |
McGlynn KA, London WT. Epidemiology and natural history of hepatocellular carcinoma. Best Pract Res Clin Gastroenterol. 2005;19(1):3–23. | |
Tanaka Y, Hanada K, Mizokami M, et al. A comparison of the molecular clock of hepatitis C virus in the United States and Japan predicts that hepatocellular carcinoma incidence in the United States will increase over the next two decades. Proc Natl Acad Sci U S A. 2002;99(24):15584–15589. | |
Taylor-Robinson SD, Foster GR, Arora S, Hargreaves S, Thomas HC. Increase in primary liver cancer in the UK, 1979–1994. Lancet. 1997;350(9085):1142–1143. | |
Calvisi DF, Frau M, Tomasi ML, Feo F, Pascale RM. Deregulation of signalling pathways in prognostic subtypes of hepatocellular carcinoma: novel insights from interspecies comparison. Biochim Biophys Acta. 2012;1826(1):215–237. | |
Indulski JA, Lutz W. Metabolic genotype in relation to individual susceptibility to environmental carcinogens. Int Arch Occup Environ Health. 2000;73(2):71–85. | |
Feo F, De Miglio MR, Simile MM, et al. Hepatocellular carcinoma as a complex polygenic disease. Interpretive analysis of recent developments on genetic predisposition. Biochim Biophys Acta. 2006;1765(2):126–147. | |
Dragani TA. Risk of HCC: genetic heterogeneity and complex genetics. J Hepatol. 2010;52(2):252–257. | |
Donato F, Gelatti U, Limina RM, Fattovich G. Southern Europe as an example of interaction between various environmental factors: a systematic review of the epidemiologic evidence. Oncogene. 26, 2006;25(27):3756–3770. | |
Frau M, Biasi F, Feo F, Pascale RM. Prognostic markers and putative therapeutic targets for hepatocellular carcinoma. Mol Aspects Med. 2010;31(2):179–193. | |
Villanueva A, Newell P, Chiang DY, Friedman SL, Llovet JM. Genomics and signaling pathways in hepatocellular carcinoma. Semin Liver Dis. 2007;27(1):55–76. | |
Bird AP. The relationship of DNA methylation to cancer. Cancer Surv. 1996;28:87–101. | |
Bestor TH, Ingram VM. Two DNA methyltransferases from murine erythroleukemia cells: purification, sequence specificity, and mode of interaction with DNA. Proc Natl Acad Sci U S A. 1983;80(18):5559–5563. | |
Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99(3):247–257. | |
Bell AC, Felsenfeld G. Methylation of a CTCF-dependent boundary controls imprinted expression of the Igf2 gene. Nature. 2000;405(6785):482–485. | |
Moghbeli M, Moaven O, Memar B, et al. Role of hMLH1 and E-cadherin promoter methylation in gastric cancer progression. J Gastrointest Cancer. 2014;45(1):40–47. | |
Bird AP. CpG-rich islands and the function of DNA methylation. Nature. 1986;321(6067):209–213. | |
Di Ruscio A, Ebralidze AK, Benoukraf T, et al. DNMT1-interacting RNAs block gene-specific DNA methylation. Nature. 2013;503(7476):371–376. | |
Turner BM. Histone acetylation and control of gene expression. J Cell Sci. 1991;99(Pt 1):13–20. | |
Suganuma T, Workman JL. Signals and combinatorial functions of histone modifications. Annu Rev Biochem. 2011;80:473–499. | |
Cross SH, Bird AP. CpG islands and genes. Curr Opin Genet Dev. 1995;5(3):309–314. | |
Brewer G. Messenger RNA decay during aging and development. Ageing Res Rev. 2002;1(4):607–625. | |
Nagano T, Fraser P. Emerging similarities in epigenetic gene silencing by long noncoding RNAs. Mamm Genome. 2009;20(9–10):557–562. | |
Law PT, Wong N. Emerging roles of microRNA in the intracellular signaling networks of hepatocellular carcinoma. J Gastroenterol Hepatol. 2011;26(3):437–449. | |
Braconi C, Henry JC, Kogure T, Schmittgen T, Patel T. The role of microRNAs in human liver cancers. Semin Oncol. 2011;38(6):752–763. | |
Lim LP, Lau NC, Garrett-Engele P, et al. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature. 2005;433(7027):769–773. | |
Kumar A. MicroRNA in HCV infection and liver cancer. Biochim Biophys Acta. 2011;1809(11–12):694–699. | |
Royo H, Cavaillé J. Non-coding RNAs in imprinted gene clusters. Biol Cell. 2008;100(3):149–166. | |
You JS, Jones PA. Cancer genetics and epigenetics: two sides of the same coin? Cancer Cell. 2012;22(1):9–20. | |
Baylin SB, Jones PA. A decade of exploring the cancer epigenome – biological and translational implications. Nat Rev Cancer. 2011;11(10):726–734. | |
Krivtsov AV, Armstrong SA. MLL translocations, histone modifications and leukaemia stem-cell development. Nat Rev Cancer. 2007;7(11):823–833. | |
Baylin SB, Ohm JE. Epigenetic gene silencing in cancer – a mechanism for early oncogenic pathway addiction? Nat Rev Cancer. 2006;6(2):107–116. | |
Mikol YB, Hoover KL, Creasia D, Poirier LA. Hepatocarcinogenesis in rats fed methyl-deficient, amino acid-defined diets. Carcinogenesis. 1983;4(12):1619–1629. | |
Ghoshal AK, Farber E. The induction of liver cancer by dietary deficiency of choline and methionine without added carcinogens. Carcinogenesis. 1984;5(10):1367–1370. | |
Yokoyama S, Sells MA, Reddy TV, Lombardi B. Hepatocarcinogenic and promoting action of a choline-devoid diet in the rat. Cancer Res. 1985;45(6):2834–2842. | |
Garcea R, Pascale R, Daino L, et al. Variations of ornithine decarboxylase activity and S-adenosyl-L-methionine and 5′-methylthioadenosine contents during the development of diethylnitrosamine-induced liver hyperplastic nodules and hepatocellular carcinoma. Carcinogenesis. 1987;8(5):653–658. | |
Pascale RM, Simile MM, Satta G, et al. Comparative effects of L-methionine, S-adenosyl-L-methionine and 5′-methylthioadenosine on the growth of preneoplastic lesions and DNA methylation in rat liver during the early stages of hepatocarcinogenesis. Anticancer Res. 1991;11(4):1617–1624. | |
Garcea R, Daino L, Pascale R, et al. Inhibition of promotion and persistent nodule growth by S-adenosyl-L-methionine in rat liver carcinogenesis: role of remodeling and apoptosis. Cancer Res. 1989;49(7):1850–1856. | |
Pascale RM, Simile MM, De Miglio MR, et al. Chemoprevention by S-adenosyl-L-methionine of rat liver carcinogenesis initiated by 1,2-dimethylhydrazine and promoted by orotic acid. Carcinogenesis. 1995;16(2):427–430. | |
Calvisi DF, Simile MM, Ladu S, et al. Altered methionine metabolism and global DNA methylation in liver cancer: relationship with genomic instability and prognosis. Int J Cancer. 2007;121(11):2410–2420. | |
Finkelstein JD. Methionine metabolism in mammals. J Nutr Biochem. 1990;1(5):228–237. | |
Ramani K, Mato JM, Lu SC. Role of methionine adenosyltransferase genes in hepatocarcinogenesis. Cancers (Basel). 2011;3(2):1480–1497. | |
LeGros L, Halim AB, Chamberlin ME, Geller A, Kotb M. Regulation of the human MAT2B gene encoding the regulatory beta subunit of methionine adenosyltransferase, MAT II. J Biol Chem. 2001;276(27):24918–24924. | |
Ji Y, Nordgren KK, Chai Y, et al. Human liver methionine cycle: MAT1A and GNMT gene resequencing, functional genomics, and hepatic genotype-phenotype correlation. Drug Metab Dispos. 2012;40(10):1984–1992. | |
Li J, Ramani K, Sun Z, et al. Forced expression of methionine adenosyltransferase 1A in human hepatoma cells suppresses in vivo tumorigenicity in mice. Am J Pathol. 2010;176(5):2456–2466. | |
Lu SC, Ramani K, Ou X, et al. S-adenosylmethionine in the chemoprevention and treatment of hepatocellular carcinoma in a rat model. Hepatology. 2009;50(2):462–471. | |
Mato JM, Corrales FJ, Lu SC, Avila MA. S-Adenosylmethionine: a control switch that regulates liver function. FASEB J. 2002;16(1):15–26. | |
Lu SC, Mato JM. Role of methionine adenosyltransferase and S-adenosylmethionine in alcohol-associated liver cancer. Alcohol. 2005;35(3):227–234. | |
Mato JM, Alvarez L, Ortiz P, Mingorance J, Durán C, Pajares MA. S-adenosyl-L-methionine synthetase and methionine metabolism deficiencies in cirrhosis. Adv Exp Med Biol. 1994;368:113–117. | |
Mato JM, Corrales F, Martin-Duce A, Ortiz P, Pajares MA, Cabrero C. Mechanisms and consequences of the impaired trans-sulphuration pathway in liver disease: Part I. Biochemical implications. Drugs. 1990;40 Suppl 3:58–64. | |
Simile MM, Banni S, Angioni E, et al. 5′-Methylthioadenosine administration prevents lipid peroxidation and fibrogenesis induced in rat liver by carbon-tetrachloride intoxication. J Hepatol. 2001;34(3):386–394. | |
Lu SC, Alvarez L, Huang ZZ, et al. Methionine adenosyltransferase 1A knockout mice are predisposed to liver injury and exhibit increased expression of genes involved in proliferation. Proc Natl Acad Sci U S A. 2001;98(10):5560–5565. | |
Pañeda C, Gorospe I, Herrera B, Nakamura T, Fabregat I, Varela-Nieto I. Liver cell proliferation requires methionine adenosyltransferase 2A mRNA up-regulation. Hepatology. 2002;35(6):1381–1391. | |
Martínez-Chantar ML, García-Trevijano ER, Latasa MU, et al. Methionine adenosyltransferase II beta subunit gene expression provides a proliferative advantage in human hepatoma. Gastroenterology. 2003;124(4):940–948. | |
Albers E. Metabolic characteristics and importance of the universal methionine salvage pathway recycling methionine from 5′-methylthioadenosine. IUBMB Life. 2009;61(12):1132–1142. | |
Frau M, Tomasi ML, Simile MM, et al. Role of transcriptional and posttranscriptional regulation of methionine adenosyltransferases in liver cancer progression. Hepatology. 2012;56(1):165–175. | |
Torres L, Avila MA, Carretero MV, et al. Liver-specific methionine adenosyltransferase MAT1A gene expression is associated with a specific pattern of promoter methylation and histone acetylation: implications for MAT1A silencing during transformation. FASEB J. 2000;14(1):95–102. | |
Tomasi ML, Li TW, Li M, Mato JM, Lu SC. Inhibition of human methionine adenosyltransferase 1A transcription by coding region methylation. J Cell Physiol. 2012;227(4):1583–1591. | |
Yang H, Huang ZZ, Zeng Z, Chen C, Selby RR, Lu SC. Role of promoter methylation in increased methionine adenosyltransferase 2A expression in human liver cancer. Am J Physiol Gastrointest Liver Physiol. 2001;280(2):G184–G190. | |
Lal A, Mazan-Mamczarz K, Kawai T, Yang X, Martindale JL, Gorospe M. Concurrent versus individual binding of HuR and AUF1 to common labile target mRNAs. EMBO J. 2004;23(15):3092–3102. | |
Kim MY, Hur J, Jeong S. Emerging roles of RNA and RNA-binding protein network in cancer cells. BMB Rep. 2009;42(3):125–130. | |
Vázquez-Chantada M, Fernández-Ramos D, Embade N, et al. HuR/methyl-HuR and AUF1 regulate the MAT expressed during liver proliferation, differentiation, and carcinogenesis. Gastroenterology. 2010;138(5):1943–1953. | |
Yang H, Cho ME, Li TW, et al. MicroRNAs regulate methionine adenosyltransferase 1A expression in hepatocellular carcinoma. J Clin Invest. 2013;123(1):285–298. | |
Yang B, Guo M, Herman JG, Clark DP. Aberrant promoter methylation profiles of tumor suppressor genes in hepatocellular carcinoma. Am J Pathol. 2003;163(3):1101–1107. | |
Lehmann U, Berg-Ribbe I, Wingen LU, et al. Distinct methylation patterns of benign and malignant liver tumors revealed by quantitative methylation profiling. Clin Cancer Res. 2005;11(10):3654–3660. | |
Herath NI, Leggett BA, MacDonald GA. Review of genetic and epigenetic alterations in hepatocarcinogenesis. J Gastroenterol Hepatol. 2006;21(1 Pt 1):15–21. | |
Zekri Ael-R, Nassar AA, El-Din El-Rouby MN, et al. Disease progression from chronic hepatitis C to cirrhosis and hepatocellular carcinoma is associated with increasing DNA promoter methylation. Asian Pac J Cancer Prev. 2013;14(11):6721–6726. | |
Shiraha H, Yamamoto K, Namba M. Human hepatocyte carcinogenesis (review). Int J Oncol. 2013;42(4):1133–1138. | |
Ueda H, Ullrich SJ, Gangemi JD, et al. Functional inactivation but not structural mutation of p53 causes liver cancer. Nat Genet. 1995;9(1):41–47. | |
Iwata N, Yamamoto H, Sasaki S, et al. Frequent hypermethylation of CpG islands and loss of expression of the 14-3-3 sigma gene in human hepatocellular carcinoma. Oncogene. 2000;19(46):5298–5302. | |
Matsuda Y. Molecular mechanism underlying the functional loss of cyclindependent kinase inhibitors p16 and p27 in hepatocellular carcinoma. World J Gastroenterol. 2008;14(11):1734–1740. | |
Stepanova L, Leng X, Parker SB, Harper JW. Mammalian p50Cdc37 is a protein kinase-targeting subunit of Hsp90 that binds and stabilizes Cdk4. Genes Dev. 1996;10(12):1491–1502. | |
Gaubatz S, Lees JA, Lindeman GJ, Livingston DM. E2F4 is exported from the nucleus in a CRM1-dependent manner. Mol Cell Biol. 2001;21(4):1384–1392. | |
Crosby ME, Almasan A. Opposing roles of E2Fs in cell proliferation and death. Cancer Biol Ther. 2004;3(12):1208–1211. | |
Pascale RM, Simile MM, Calvisi DF, et al. Role of HSP90, CDC37, and CRM1 as modulators of P16(INK4A) activity in rat liver carcinogenesis and human liver cancer. Hepatology. 2005;42(6):1310–1319. | |
Calvisi DF, Ladu S, Pinna F, et al. SKP2 and CKS1 promote degradation of cell cycle regulators and are associated with hepatocellular carcinoma prognosis. Gastroenterology. 2009;137(5):1816–1826. | |
Calvisi DF, Pinna F, Ladu S, et al. The degradation of cell cycle regulators by SKP2/CKS1 ubiquitin ligase is genetically controlled in rodent liver cancer and contributes to determine the susceptibility to the disease. Int J Cancer. 2010;126(5):1275–1281. | |
Rodier G, Coulombe P, Tanguay PL, Boutonnet C, Meloche S. Phosphorylation of Skp2 regulated by CDK2 and Cdc14B protects it from degradation by APC(Cdh1) in G1 phase. EMBO J. 2008;27(4):679–691. | |
Frau M, Simile MM, Tomasi ML, et al. An expression signature of phenotypic resistance to hepatocellular carcinoma identified by cross-species gene expression analysis. Cell Oncol (Dordr). 2012;35(3):163–173. | |
Martínez-Chantar ML, Vázquez-Chantada M, Ariz U, et al. Loss of the glycine N-methyltransferase gene leads to steatosis and hepatocellular carcinoma in mice. Hepatology. 2008;47(4):1191–1199. | |
Huidobro C, Toraño EG, Fernández AF, et al. A DNA methylation signature associated with the epigenetic repression of glycine N-methyltransferase in human hepatocellular carcinoma. J Mol Med (Berl). 2013;91(8):939–950. | |
Feinberg AP, Ohlsson R, Henikoff S. The epigenetic progenitor origin of human cancer. Nat Rev Genet. 2006;7(1):21–33. | |
Zhai JM, Yin XY, Hou X, et al. Analysis of the genome-wide DNA methylation profile of side population cells in hepatocellular carcinoma. Dig Dis Sci. 2013;58(7):1934–1947. | |
Takahashi K, Tanabe K, Ohnuki M, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131(5):861–872. | |
Okita K, Ichisaka T, Yamanaka S. Generation of germline-competent induced pluripotent stem cells. Nature. 2007;448(7151):313–317. | |
Wang XQ, Ng RK, Ming X, et al. Epigenetic regulation of pluripotent genes mediates stem cell features in human hepatocellular carcinoma and cancer cell lines. PLoS One. 2013;8(9):e72435. | |
Raggi C, Factor VM, Seo D, et al. Epigenetic reprogramming modulates malignant properties of human liver cancer. Hepatology. Epub January 21, 2014. | |
Cai J, Mao Z, Hwang JJ, Lu SC. Differential expression of methionine adenosyltransferase genes influences the rate of growth of human hepatocellular carcinoma cells. Cancer Res. 1998;58(7):1444–1450. | |
García-Román R, Salazar-González D, Rosas S, et al. The differential NF-kB modulation by S-adenosyl-L-methionine, N-acetylcysteine and quercetin on the promotion stage of chemical hepatocarcinogenesis. Free Radic Res. 2008;42(4):331–343. | |
Millward TA, Zolnierowicz S, Hemmings BA. Regulation of protein kinase cascades by protein phosphatase 2A. Trends Biochem Sci. 1999;24(5):186–191. | |
Eichhorn PJ, Creyghton MP, Bernards R. Protein phosphatase 2A regulatory subunits and cancer. Biochim Biophys Acta. 2009;1795(1):1–15. | |
Calvisi DF, Pinna F, Pellegrino R, et al. Ras-driven proliferation and apoptosis signaling during rat liver carcinogenesis is under genetic control. Int J Cancer. 2008;123(9):2057–2064. | |
Calvisi DF, Pinna F, Meloni F, et al. Dual-specificity phosphatase 1 ubiquitination in extracellular signal-regulated kinase-mediated control of growth in human hepatocellular carcinoma. Cancer Res. 2008;68(11):4192–4200. | |
Calvisi DF, Pinna F, Ladu S, et al. Forkhead box M1B is a determinant of rat susceptibility to hepatocarcinogenesis and sustains ERK activity in human HCC. Gut. 2009;58(5):679–687. | |
Tomasi ML, Ramani K, Lopitz-Otsoa F, et al. S-adenosylmethionine regulates dual-specificity mitogen-activated protein kinase phosphatase expression in mouse and human hepatocytes. Hepatology. 2010;51(6):2152–2161. | |
Xia L, Mo P, Huang W, et al. The TNF-α/ROS/HIF-1-induced upregulation of FoxMI expression promotes HCC proliferation and resistance to apoptosis. Carcinogenesis. 2012;33(11):2250–2259. | |
Liu Q, Liu L, Zhao Y, et al. Hypoxia induces genomic DNA demethylation through the activation of HIF-1α and transcriptional upregulation of MAT2A in hepatoma cells. Mol Cancer Ther. 2011;10(6):1113–1123. | |
Tomasi ML, Iglesias-Ara A, Yang H, et al. S-adenosylmethionine regulates apurinic/apyrimidinic endonuclease 1 stability: implication in hepatocarcinogenesis. Gastroenterology. 2009;136(3):1025–1036. | |
Tomasi ML, Tomasi I, Ramani K, et al. S-adenosyl methionine regulates ubiquitin-conjugating enzyme 9 protein expression and sumoylation in murine liver and human cancers. Hepatology. 2012;56(3):982–993. | |
Pogribny IP, Tryndyak VP, Boyko A, Rodriguez-Juarez R, Beland FA, Kovalchuk O. Induction of microRNAome deregulation in rat liver by long-term tamoxifen exposure. Mutat Res. 2007;619(1–2):30–37. | |
Wang B, Majumder S, Nuovo G, et al. Role of microRNA-155 at early stages of hepatocarcinogenesis induced by choline-deficient and amino acid-defined diet in C57BL/6 mice. Hepatology. 2009;50(4):1152–1161. | |
Chang TC, Yu D, Lee YS, et al. Widespread microRNA repression by Myc contributes to tumorigenesis. Nat Genet. 2008;40(1):43–50. | |
Lin J, Huang S, Wu S, et al. MicroRNA-423 promotes cell growth and regulates G(1)/S transition by targeting p21Cip1/Waf1 in hepatocellular carcinoma. Carcinogenesis. 2011;32(11):1641–1647. | |
Fornari F, Gramantieri L, Ferracin M, et al. MiR-221 controls CDKN1C/p57 and CDKN1B/p27 expression in human hepatocellular carcinoma. Oncogene. 2008;27(43):5651–5661. | |
Yang L, Ma Z, Wang D, Zhao W, Chen L, Wang G. MicroRNA-602 regulating tumor suppressive gene RASSF1A is overexpressed in hepatitis B virus-infected liver and hepatocellular carcinoma. Cancer Biol Ther. 2010;9(10):803–808. | |
Wong QW, Ching AK, Chan AW, et al. MiR-222 overexpression confers cell migratory advantages in hepatocellular carcinoma through enhancing AKT signaling. Clin Cancer Res. 2010;16(3):867–875. | |
Garofalo M, Di Leva G, Romano G, et al. miR-221&222 regulate TRAIL resistance and enhance tumorigenicity through PTEN and TIMP3 downregulation. Cancer Cell. 2009;16(6):498–509. | |
Pineau P, Volinia S, McJunkin K, et al. miR-221 overexpression contributes to liver tumorigenesis. Proc Natl Acad Sci U S A. 2010;107(1):264–269. | |
Wang B, Hsu SH, Majumder S, et al. TGFbeta-mediated upregulation of hepatic miR-181b promotes hepatocarcinogenesis by targeting TIMP3. Oncogene. 2010;29(12):1787–1797. | |
Gramantieri L, Fornari F, Ferracin M, et al. MicroRNA-221 targets Bmf in hepatocellular carcinoma and correlates with tumor multifocality. Clin Cancer Res. 2009;15(16):5073–5081. | |
Li Y, Tan W, Neo TW, et al. Role of the miR-106b-25 microRNA cluster in hepatocellular carcinoma. Cancer Sci. 2009;100(7):1234–1242. | |
Lim LP, Lau NC, Garrett-Engele P, et al. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature. 2005;433(7027):769–773. | |
Stanelle J, Pützer BM. E2F1-induced apoptosis: turning killers into therapeutics. Trends Mol Med. 2006;12(4):177–185. | |
Xie QH, He XX, Chang Y, et al. MiR-192 inhibits nucleotide excision repair by targeting ERCC3 and ERCC4 in HepG2.2.15 cells. Biochem Biophys Res Commun. 2011;410(3):440–445. | |
Kota J, Chivukula RR, O’Donnell KA, et al. Therapeutic microRNA delivery suppresses tumorigenesis in a murine liver cancer model. Cell. 2009;137(6):1005–1017. | |
Xu T, Zhu Y, Xiong Y, Ge YY, Yun JP, Zhuang SM. MicroRNA-195 suppresses tumorigenicity and regulates G1/S transition of human hepatocellular carcinoma cells. Hepatology. 2009;50(1):113–121. | |
Gramantieri L, Ferracin M, Fornari F, et al. Cyclin G1 is a target of miR-122a, a microRNA frequently down-regulated in human hepatocellular carcinoma. Cancer Res. 2007;67(13):6092–6099. | |
Li C, Wang Y, Wang S, et al. Hepatitis B virus mRNA-mediated miR-122 inhibition upregulates PTTG1-binding protein, which promotes hepatocellular carcinoma tumor growth and cell invasion. J Virol. 2013;87(4):2193–2205. | |
Wong QW, Lung RW, Law PT, et al. MicroRNA-223 is commonly repressed in hepatocellular carcinoma and potentiates expression of Stathmin1. Gastroenterology. 2008;135(1):257–269. | |
Lan FF, Wang H, Chen YC, et al. Hsa-let-7g inhibits proliferation of hepatocellular carcinoma cells by downregulation of c-Myc and upregulation of p16(INK4A). Int J Cancer. 2011;128(2):319–331. | |
Ji J, Zhao L, Budhu A, et al. Let-7g targets collagen type I alpha2 and inhibits cell migration in hepatocellular carcinoma. J Hepatol. 2010;52(5):690–697. | |
Kim T, Veronese A, Pichiorri F, et al. p53 regulates epithelial-mesenchymal transition through microRNAs targeting ZEB1 and ZEB2. J Exp Med. 2011;208(5):875–883. | |
Zheng F, Liao YJ, Cai MY, et al. The putative tumour suppressor microRNA-124 modulates hepatocellular carcinoma cell aggressiveness by repressing ROCK2 and EZH2. Gut. 2012;61(2):278–289. | |
Meng Z, Fu X, Chen X, et al. miR-194 is a marker of hepatic epithelial cells and suppresses metastasis of liver cancer cells in mice. Hepatology. 2010;52(6):2148–2157. | |
Zhang JP, Zeng C, Xu L, Gong J, Fang JH, Zhuang SM. MicroRNA-148a suppresses the epithelial-mesenchymal transition and metastasis of hepatoma cells by targeting Met/Snail signaling. Oncogene. Epub September 9, 2013. | |
Xu X, Fan Z, Kang L, et al. Hepatitis B virus X protein represses miRNA-148a to enhance tumorigenesis. J Clin Invest. 2013;123(2):630–645. | |
Salvi A, Sabelli C, Moncini S, et al. MicroRNA-23b mediates urokinase and c-met downmodulation and a decreased migration of human hepatocellular carcinoma cells. FEBS J. 2009;276(11):2966–2982. | |
Wong CC, Wong CM, Tung EK, et al. The microRNA miR-139 suppresses metastasis and progression of hepatocellular carcinoma by down-regulating Rho-kinase 2. Gastroenterology. 2011;140(1):322–331. | |
Fan Q, He M, Deng X, et al. Derepression of c-Fos caused by microRNA-139 down-regulation contributes to the metastasis of human hepatocellular carcinoma. Cell Biochem Funct. 2013;31(4):319–324. | |
Gu W, Li X, Wang J. miR-139 regulates the proliferation and invasion of hepatocellular carcinoma through the WNT/TCF-4 pathway. Oncol Rep. 2014;31(1):397–404. | |
Liao JK, Seto M, Noma K. Rho kinase (ROCK) inhibitors. J Cardiovasc Pharmacol. 2007;50(1):17–24. | |
Li D, Liu X, Lin L, et al. MicroRNA-99a inhibits hepatocellular carcinoma growth and correlates with prognosis of patients with hepatocellular carcinoma. J Biol Chem. 2011;286(42):36677–36685. | |
Fan CG, Wang CM, Tian C, et al. miR-122 inhibits viral replication and cell proliferation in hepatitis B virus-related hepatocellular carcinoma and targets NDRG3. Oncol Rep. 2011;26(5):1281–1286. | |
He XX, Chang Y, Meng FY, et al. MicroRNA-375 targets AEG-1 in hepatocellular carcinoma and suppresses liver cancer cell growth in vitro and in vivo. Oncogene. 2012;31(28):3357–3369. | |
Liu AM, Poon RT, Luk JM. MicroRNA-375 targets Hippo-signaling effector YAP in liver cancer and inhibits tumor properties. Biochem Biophys Res Commun. 2010;394(3):623–627. | |
Huang XH, Chen JS, Wang Q, et al. miR-338-3p suppresses invasion of liver cancer cell by targeting smoothened. J Pathol. 2011;225(3):463–472. | |
Li N, Fu H, Tie Y, et al. miR-34a inhibits migration and invasion by down-regulation of c-Met expression in human hepatocellular carcinoma cells. Cancer Lett. 2009;275(1):44–53. | |
Wei W, Wanjun L, Hui S, Dongyue C, Xinjun Y, Jisheng Z. miR-203 inhibits proliferation of HCC cells by targeting survivin. Cell Biochem Funct. 2013;31(1):82–85. | |
Lang Q, Ling C. MiR-124 suppresses cell proliferation in hepatocellular carcinoma by targeting PIK3CA. Biochem Biophys Res Commun. 2012;426(2):247–252. | |
Lu Y, Yue X, Cui Y, Zhang J, Wang K. MicroRNA-124 suppresses growth of human hepatocellular carcinoma by targeting STAT3. Biochem Biophys Res Commun. 2013;441(4):873–879. | |
Zhang JF, He ML, Fu WM, et al. Primate-specific microRNA-637 inhibits tumorigenesis in hepatocellular carcinoma by disrupting signal transducer and activator of transcription 3 signaling. Hepatology. 2011;54(6):2137–2148. | |
Noh JH, Chang YG, Kim MG, et al. MiR-145 functions as a tumor suppressor by directly targeting histone deacetylase 2 in liver cancer. Cancer Lett. 2013;335(2):455–462. | |
Zhang J, Yang Y, Yang T, et al. microRNA-22, downregulated in hepatocellular carcinoma and correlated with prognosis, suppresses cell proliferation and tumourigenicity. Br J Cancer. 2010;103(8):1215–1220. | |
Yoon S, Kim TH, Natarajan A, et al. Acute liver injury upregulates microRNA-491-5p in mice, and its overexpression sensitizes Hep G2 cells for tumour necrosis factor-alpha-induced apoptosis. Liver Int. 2010;30(3):376–387. | |
Yang XW, Zhang LJ, Huang XH, et al. miR-145 suppresses cell invasion in hepatocellular carcinoma cells: miR-145 targets ADAM17. Hepatol Res. 2014;44(5):551–559. | |
Tsai WC, Hsu PW, Lai TC, et al. MicroRNA-122, a tumor suppressor microRNA that regulates intrahepatic metastasis of hepatocellular carcinoma. Hepatology. 2009;49(5):1571–1582. | |
Wang J, Li J, Wang X, Zheng C, Ma W. Downregulation of microRNA-214 and overexpression of FGFR-1 contribute to hepatocellular carcinoma metastasis. Biochem Biophys Res Commun. 2013;439(1):47–53. | |
Tian Q, Liang L, Ding J, et al. MicroRNA-550a acts as a pro-metastatic gene and directly targets cytoplasmic polyadenylation element-binding protein 4 in hepatocellular carcinoma. PLoS One. 2012;7(11):e48958. | |
Ying Q, Liang L, Guo W, et al. Hypoxia-inducible microRNA-210 augments the metastatic potential of tumor cells by targeting vacuole membrane protein 1 in hepatocellular carcinoma. Hepatology. 2011;54(6):2064–2075. | |
Yao J, Liang L, Huang S, et al. MicroRNA-30d promotes tumor invasion and metastasis by targeting Galphai2 in hepatocellular carcinoma. Hepatology. 2010;51(3):846–856. | |
Takata A, Otsuka M, Kojima K, et al. MicroRNA-22 and microRNA-140 suppress NF-κB activity by regulating the expression of NF-κB coactivators. Biochem Biophys Res Commun. 2011;411(4):826–831. | |
Lin CJ, Gong HY, Tseng HC, Wang WL, Wu JL. miR-122 targets an anti-apoptotic gene, Bcl-w, in human hepatocellular carcinoma cell lines. Biochem Biophys Res Commun. 2008;375(3):315–320. | |
Xiong Y, Fang JH, Yun JP, et al. Effects of microRNA-29 on apoptosis, tumorigenicity, and prognosis of hepatocellular carcinoma. Hepatology. 2010;51(3):836–845. | |
Su H, Yang JR, Xu T, et al. MicroRNA-101, down-regulated in hepatocellular carcinoma, promotes apoptosis and suppresses tumorigenicity. Cancer Res. 2009;69(3):1135–1142. | |
Wang CM, Wang Y, Fan CG, et al. miR-29c targets TNFAIP3, inhibits cell proliferation and induces apoptosis in hepatitis B virus-related hepatocellular carcinoma. Biochem Biophys Res Commun. 2011;411(3):586–592. | |
Wei X, Xiang T, Ren G, et al. miR-101 is down-regulated by the hepatitis B virus x protein and induces aberrant DNA methylation by targeting DNA methyltransferase 3A. Cell Signal. 2013;25(2):439–446. | |
Huang S, He X, Ding J, et al. Upregulation of miR-23a approximately 27a approximately 24 decreases transforming growth factor-beta- induced tumor-suppressive activities in human hepatocellular carcinoma cells. Int J Cancer. 2008;123(4):972–978. | |
Huang J, Wang Y, Guo Y, Sun S. Down-regulated microRNA-152 induces aberrant DNA methylation in hepatitis B virus-related hepatocellular carcinoma by targeting DNA methyltransferase 1. Hepatology. 2010;52(1):60–70. | |
Petrocca F, Vecchione A, Croce CM. Emerging role of miR-106b-25/miR-17-92 clusters in the control of transforming growth factor beta signaling. Cancer Res. 2008;68(20):8191–8194. | |
Liu AM, Yao TJ, Wang W, et al. Circulating miR-15b and miR-130b in serum as potential markers for detecting hepatocellular carcinoma: a retrospective cohort study. BMJ Open. 2012;2(2):e000825. | |
Luk JM, Burchard J, Zhang C, et al. DLK1-DIO3 genomic imprinted microRNA cluster at 14q32.2 defines a stemlike subtype of hepatocellular carcinoma associated with poor survival. J Biol Chem. 2011;286(35):30706–30713. | |
Ding J, Huang S, Wu S, et al. Gain of miR-151 on chromosome 8q24.3 facilitates tumour cell migration and spreading through downregulating RhoGDIA. Nat Cell Biol. 2010;12(4):390–399. | |
Wu X, Wu S, Tong L, et al. miR-122 affects the viability and apoptosis of hepatocellular carcinoma cells. Scand J Gastroenterol. 2009;44(11):1332–1339. | |
Liu L, An J, Liu J, et al. Potentially functional genetic variants in microRNA processing genes and risk of HBV-related hepatocellular carcinoma. Mol Carcinog. 2013;52 Suppl 1:E148–E154. | |
Gui J, Tian Y, Wen X, et al. Serum microRNA characterization identifies miR-885-5p as a potential marker for detecting liver pathologies. Clin Sci (Lond). 2011;120(5):183–193. | |
Xu T, Zhu Y, Wei QK, et al. A functional polymorphism in the miR-146a gene is associated with the risk for hepatocellular carcinoma. Carcinogenesis. 2008;29(11):2126–2131. | |
Qi P, Dou TH, Geng L, et al. Association of a variant in MIR 196A2 with susceptibility to hepatocellular carcinoma in male Chinese patients with chronic hepatitis B virus infection. Hum Immunol. 2010;71(6):621–626. | |
Li XD, Li ZG, Song XX, Liu CF. A variant in microRNA-196a2 is associated with susceptibility to hepatocellular carcinoma in Chinese patients with cirrhosis. Pathology. 2010;42(7):669–673. | |
Zhang LS, Liang WB, Gao LB, et al. Association between pri-miR-218 polymorphism and risk of hepatocellular carcinoma in a Han Chinese population. DNA Cell Biol. 2012;31(5):761–765. | |
Xu Y, Liu L, Liu J, et al. A potentially functional polymorphism in the promoter region of miR-34b/c is associated with an increased risk for primary hepatocellular carcinoma. Int J Cancer. 2011;128(2):412–417. | |
Gao Y, He Y, Ding J, et al. An insertion/deletion polymorphism at miRNA-122-binding site in the interleukin-1alpha 3′ untranslated region confers risk for hepatocellular carcinoma. Carcinogenesis. 2009;30(12):2064–2069. | |
Kishikawa T, Otsuka M, Yoshikawa T, et al. Regulation of the expression of the liver cancer susceptibility gene MICA by microRNAs. Sci Rep. 2013;3:2739. | |
Datta J, Kutay H, Nasser MW, et al. Methylation mediated silencing of MicroRNA-1 gene and its role in hepatocellular carcinogenesis. Cancer Res. 2008;68(13):5049–5058. | |
Lu CY, Lin KY, Tien MT, Wu CT, Uen YH, Tseng TL. Frequent DNA methylation of MiR-129-2 and its potential clinical implication in hepatocellular carcinoma. Genes Chromosomes Cancer. 2013;52(7):636–643. | |
Wei X, Tan C, Tang C, et al. Epigenetic repression of miR-132 expression by the hepatitis B virus x protein in hepatitis B virus-related hepatocellular carcinoma. Cell Signal. 2013;25(5):1037–1043. | |
Dohi O, Yasui K, Gen Y, et al. Epigenetic silencing of miR-335 and its host gene MEST in hepatocellular carcinoma. Int J Oncol. 2013;42(2):411–418. | |
Furuta M, Kozaki KI, Tanaka S, Arii S, Imoto I, Inazawa J. miR-124 and miR-203 are epigenetically silenced tumor-suppressive microRNAs in hepatocellular carcinoma. Carcinogenesis. 2010;31(5):766–776. | |
Zhang T, Zhang J, Cui M, et al. Hepatitis B virus X protein inhibits tumor suppressor miR-205 through inducing hypermethylation of miR-205 promoter to enhance carcinogenesis. Neoplasia. 2013;15(11):1282–1291. | |
He Y, Cui Y, Wang W, et al. Hypomethylation of the hsa- miR-191 locus causes high expression of hsa-mir-191 and promotes the epithelial-to-mesenchymal transition in hepatocellular carcinoma. Neoplasia. 2011;13(9):841–853. | |
Buurman R, Gürlevik E, Schäffer V, et al. Histone deacetylases activate hepatocyte growth factor signaling by repressing microRNA-449 in hepatocellular carcinoma cells. Gastroenterology. 2012;143(3):811–820. | |
Saito Y, Hibino S, Saito H. Alterations of epigenetics and microRNA in hepatocellular carcinoma. Hepatol Res. 2014;44(1):31–42. | |
Otsuka M, Takata A, Yoshikawa T, et al. Receptor for activated protein kinase C: requirement for efficient microRNA function and reduced expression in hepatocellular carcinoma. PLoS One. 2011;6(9):e24359. | |
Gherzi R, Lee KY, Briata P, et al. A KH domain RNA binding protein, KSRP, promotes ARE-directed mRNA turnover by recruiting the degradation machinery. Mol Cell. 2004;14(5):571–583. | |
Yoo BK, Santhekadur PK, Gredler R, et al. Increased RNA-induced silencing complex (RISC) activity contributes to hepatocellular carcinoma. Hepatology. 2011;53(5):1538–1548. | |
Santhekadur PK, Das SK, Gredler R, et al. Multifunction protein staphylococcal nuclease domain containing 1 (SND1) promotes tumor angiogenesis in human hepatocellular carcinoma through novel pathway that involves nuclear factor κB and miR-221. J Biol Chem. 2012;287(17):13952–13958. | |
Liu K, Chen C, Guo Y, et al. Structural basis for recognition of arginine methylated Piwi proteins by the extended Tudor domain. Proc Natl Acad Sci U S A. 2010;107(43):18398–18403. | |
Siddiqi S, Matushansky I. Piwis and piwi-interacting RNAs in the epigenetics of cancer. J Cell Biochem. 2012;113(2):373–380. | |
Zhao YM, Zhou JM, Wang LR, et al. HIWI is associated with prognosis in patients with hepatocellular carcinoma after curative resection. Cancer. 2012;118(10):2708–2717. | |
Juliano C, Wang J, Lin H. Uniting germline and stem cells: the function of Piwi proteins and the piRNA pathway in diverse organisms. Annu Rev Genet. 2011;45:447–469. | |
Hendrix MJ, Seftor EA, Hess AR, Seftor RE. Vasculogenic mimicry and tumour-cell plasticity: lessons from melanoma. Nat Rev Cancer. 2003;3(6):411–421. | |
Song K, Han C, Zhang J, et al. Epigenetic regulation of MicroRNA-122 by peroxisome proliferator activated receptor-gamma and hepatitis b virus X protein in hepatocellular carcinoma cells. Hepatology. 2013;58(5):1681–1692. | |
Cholongitas E, Papatheodoridis GV. Review article: novel therapeutic options for chronic hepatitis C. Aliment Pharmacol Ther. 2008;27(10):866–884. | |
Halsted CH, Medici V. Aberrant hepatic methionine metabolism and gene methylation in the pathogenesis and treatment of alcoholic steatohepatitis. Int J Hepatol. 2012;2012:959746. | |
Anstee QM, Day CP. S-adenosylmethionine (SAMe) therapy in liver disease: a review of current evidence and clinical utility. J Hepatol. 2012;57(5):1097–1109. | |
Morgan TR. Chemoprevention of hepatocellular carcinoma in chronic hepatitis C. Recent Results Cancer Res. 2011;188:85–99. | |
Ramani K, Yang H, Xia M, Ara AI, Mato JM, Lu SC. Leptin’s mitogenic effect in human liver cancer cells requires induction of both methionine adenosyltransferase 2A and 2beta. Hepatology. 2008;47(2):521–531. | |
Calvisi DF, Pascale RM, Feo F. Dissection of signal transduction pathways as a tool for the development of targeted therapies of hepatocellular carcinoma. Rev Recent Clin Trials. 2007;2(3):217–236. | |
Zhang W, Sviripa V, Chen X, et al. Fluorinated N,N-dialkylaminostilbenes repress colon cancer by targeting methionine S-adenosyltransferase 2A. ACS Chem Biol. 2013;8(4):796–803. | |
Tischoff I, Tannapfe A. DNA methylation in hepatocellular carcinoma. World J Gastroenterol. 2008;14(11):1741–1748. | |
Nishida N, Goel A. Genetic and epigenetic signatures in human hepatocellular carcinoma: a systematic review. Curr Genomics. 2011;12(2):130–137. | |
Wu J, Du C, Lv Z, et al. The up-regulation of histone deacetylase 8 promotes proliferation and inhibits apoptosis in hepatocellular carcinoma. Dig Dis Sci. 2013;58(12):3545–3553. | |
Grassadonia A, Cioffi P, Simiele F, Iezzi L, Zilli M, Natoli C. Role of Hydroxamate-Based Histone Deacetylase Inhibitors (Hb-HDACIs) in the Treatment of Solid Malignancies. Cancers (Basel). 2013;5(3):919–942. | |
Tao SF, Zhang CS, Guo XL, et al. Anti-tumor effect of 5-aza-2′-deoxycytidine by inhibiting telomerase activity in hepatocellular carcinoma cells. World J Gastroenterol. 2012;18(19):2334–2343. | |
Venturelli S, Armeanu S, Pathil A, et al. Epigenetic combination therapy as a tumor-selective treatment approach for hepatocellular carcinoma. Cancer. 2007;109(10):2132–2141. | |
Chattopadhyay S, Ely A, Bloom K, Weinberg MS, Arbuthnot P. Inhibition of hepatitis B virus replication with linear DNA sequences expressing antiviral micro-RNA shuttles. Biochem Biophys Res Commun. 2009;389(3):484–489. | |
Xiangji L, Feng X, Qingbao C, et al. Knockdown of HBV surface antigen gene expression by a lentiviral microRNA-based system inhibits HBV replication and HCC growth. J Viral Hepat. 2011;18(9):653–660. | |
Broderick JA, Zamore PD. MicroRNA therapeutics. Gene Ther. 2011;18(12):1104–1110. | |
Guo J, Friedman SL. The expression patterns and clinical significance of microRNAs in liver diseases and hepatocellular carcinoma. Curr Pharm Des. 2013;19(7):1262–1272. | |
Fornari F, Milazzo M, Chieco P, et al. MiR-199a-3p regulates mTOR and c-Met to influence the doxorubicin sensitivity of human hepatocarcinoma cells. Cancer Res. 2010;70(12):5184–5193. | |
Xu Y, Xia F, Ma L, et al. MicroRNA-122 sensitizes HCC cancer cells to adriamycin and vincristine through modulating expression of MDR and inducing cell cycle arrest. Cancer Lett. 2011;310(2):160–169. | |
Shimizu S, Takehara T, Hikita H, et al. The let-7 family of microRNAs inhibits Bcl-xL expression and potentiates sorafenib-induced apoptosis in human hepatocellular carcinoma. J Hepatol. 2010;52(5):698–704. | |
Bruix J, Boix L, Sala M, Llovet JM. Focus on hepatocellular carcinoma. Cancer Cell. 2004;5(3):215–219. | |
Farazi PA, DePinho RA. Hepatocellular carcinoma pathogenesis: from genes to environment. Nat Rev Cancer. 2006;6(9):674–687. | |
Imbeaud S, Ladeiro Y, Zucman-Rossi J. Identification of novel oncogenes and tumor suppressors in hepatocellular carcinoma. Semin Liver Dis. 2010;30(1):75–86. | |
Zhao C, Yan F, Wu H, Qiao F, Qiu X, Fan H. DNMT3A −448A>G polymorphism and the risk for hepatocellular carcinoma. Biomed Rep. 2013;1(4):664–668. | |
Lao Y, Wu H, Zhao C, Wu Q, Qiao F, Fan H. Promoter polymorphisms of DNA methyltransferase 3B and risk of hepatocellular carcinoma. Biomed Rep. 2013;1(5):771–775. | |
Tseng TL, Shih YP, Huang YC, et al. Genotypic and phenotypic characterization of a putative tumor susceptibility gene, GNMT, in liver cancer. Cancer Res. 2003;63(3):647–654. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.