")
Back to Journals » Journal of Experimental Pharmacology » Volume 12
New Insights into the Mechanism of Action of Viloxazine: Serotonin and Norepinephrine Modulating Properties
Authors Yu C, Garcia-Olivares J, Candler S, Schwabe S, Maletic V
Received 4 April 2020
Accepted for publication 25 July 2020
Published 25 August 2020 Volume 2020:12 Pages 285—300
DOI https://doi.org/10.2147/JEP.S256586
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Bal Lokeshwar
Chungping Yu,1 Jennie Garcia-Olivares,1 Shawn Candler,1 Stefan Schwabe,1 Vladimir Maletic2
1Supernus Pharmaceuticals, Inc., Rockville, MD, USA; 2Department of Psychiatry/Behavioral Science, University of South Carolina School of Medicine, Greenville, SC, USA
Correspondence: Chungping Yu
Supernus Pharmaceuticals, Inc., 9715 Key West Avenue, Rockville, MD 20850, USA
Tel +1 301 838 2679
Email [email protected]
Background: Viloxazine was historically described as a norepinephrine reuptake inhibitor (NRI). Since NRIs have previously demonstrated efficacy in attention deficit/hyperactivity disorder (ADHD), viloxazine underwent contemporary investigation in the treatment of ADHD. Its clinical and safety profile, however, was found to be distinct from other ADHD medications targeting norepinephrine reuptake. Considering the complexity of neuropsychiatric disorders, understanding the mechanism of action (MoA) is an important differentiating point between viloxazine and other ADHD medications and provides pharmacology-based rationale for physicians prescribing appropriate therapy.
Methods: Viloxazine was evaluated in a series of in vitro binding and functional assays. Its effect on neurotransmitter levels in the brain was evaluated using microdialysis in freely moving rats.
Results: We report the effects of viloxazine on serotoninergic (5-HT) system. In vitro, viloxazine demonstrated antagonistic activity at 5-HT2B and agonistic activity at 5-HT2C receptors, along with predicted high receptor occupancy at clinical doses. In vivo, viloxazine increased extracellular 5-HT levels in the prefrontal cortex (PFC), a brain area implicated in ADHD. Viloxazine also exhibited moderate inhibitory effects on the norepinephrine transporter (NET) in vitro and in vivo, and elicited moderate activity at noradrenergic and dopaminergic systems.
Conclusion: Viloxazine’s ability to increase 5-HT levels in the PFC and its agonistic and antagonistic effects on certain 5-HT receptor subtypes, which were previously shown to suppress hyperlocomotion in animals, indicate that 5-HT modulating activity of viloxazine is an important (if not the predominant) component of its MoA, complemented by moderate NET inhibition. Supported by clinical data, these findings suggest the updated psychopharmacological profile of viloxazine can be best explained by its action as a serotonin norepinephrine modulating agent (SNMA).
Keywords: viloxazine, ADHD, serotonin norepinephrine modulating agent, norepinephrine transporter, mechanism of action, SPN-812
Introduction
Viloxazine was first approved in the 1970s in the United Kingdom and in several other European countries for the treatment of depression in adults, spurring a series of mechanistic studies into the compound during the 1970s and 1980s.1,2 During these decades, viloxazine was characterized as a norepinephrine (NE) reuptake inhibitor (NRI), which was supported by its similarity to the tricyclic antidepressants in its ability to potentiate noradrenergic effects across multiple animal models.3–5
Based on the original mechanism of action (MoA) and the established therapeutic benefit of targeting NE neurotransmission in the treatment of attention-deficit/hyperactivity disorder (ADHD), an extended-release formulation of this molecule, SPN-812 (viloxazine extended-release), is currently being developed for the treatment of ADHD in children, adolescents, and adults.6–9 In a proof-of-concept study and recently completed Phase 3 trials,6–8 SPN-812 was effective in reducing symptoms of hyperactivity, impulsivity, and inattention in children and adolescents with ADHD.10 Treatment with SPN-812 exhibited a safety profile somewhat different from that of commonly prescribed NRI antidepressants and ADHD pharmacotherapies with potent NRI activity. Specifically, in contrast to potent noradrenergic agents, SPN-812 demonstrated a low incidence of cardiac-related adverse events.10−14 The low incidence of cardiovascular events (such as increased blood pressure or heart rate) associated with viloxazine—both historically and in these contemporary investigations—suggests that viloxazine possesses only moderate noradrenergic activity.2,10,12
In addition to its clinical profile, historical in vitro pharmacology data indicate that the inhibitory potency of viloxazine for NE and 5-HT uptake is different from that of other known NRIs, such as atomoxetine and reboxetine. Previous data have demonstrated that viloxazine exhibits a more moderate inhibitory effect on NE uptake, with an inhibitory constant (Ki) of 2300 nM, compared to the Ki of 3.4 and 8.2 nM for atomoxetine and reboxetine, respectively (Table 1).15–18 Viloxazine is almost devoid of any 5-HT uptake inhibitory activity (Ki > 10,000 nM), while atomoxetine and reboxetine have a moderate potency for 5-HT uptake inhibition (Ki = 390 and 1070 nM, respectively). Likewise, viloxazine has consistently demonstrated a relatively low affinity towards the NE transporter (NET) in in vitro binding competition assays. Still, viloxazine exhibits higher selectivity towards NET over the 5-HT transporter (SERT) (dissociation constant KD ratio = 110) and almost negligible affinity for the dopamine (DA) transporter (DAT) (KD > 100,000 nM; Table 1).
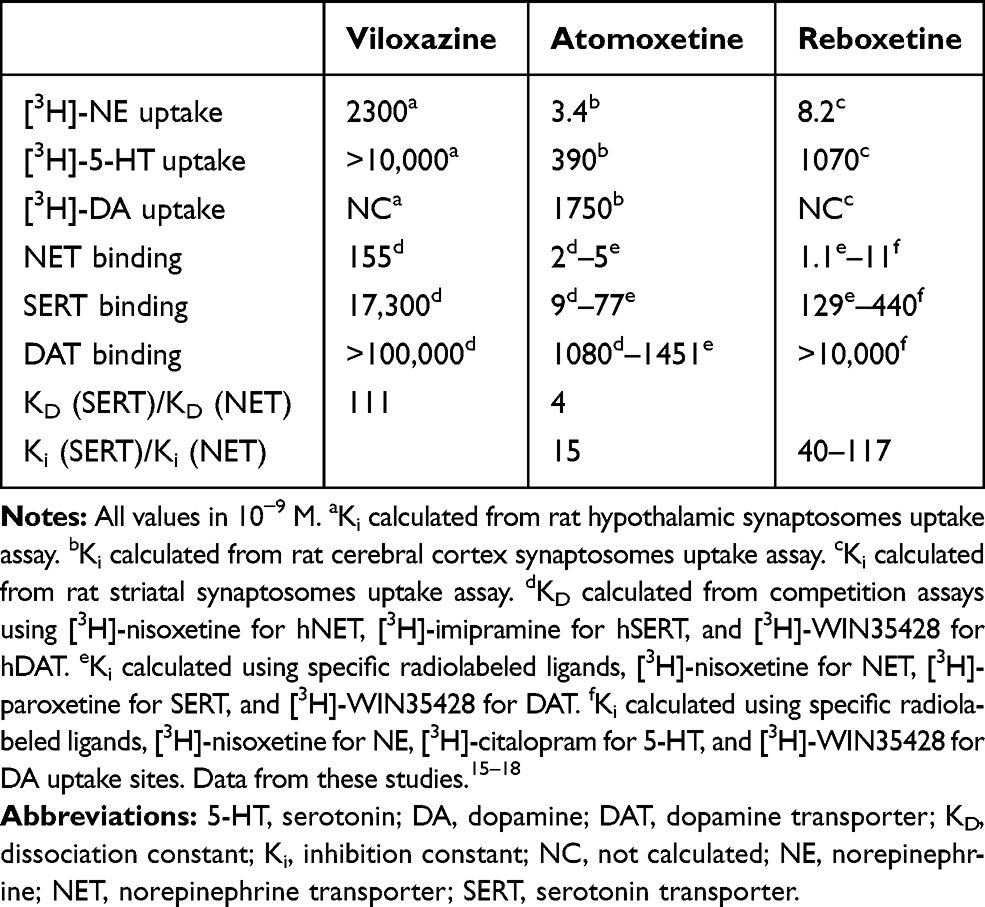 |
Table 1 Monoamine Uptake Inhibition, Transporter Binding Affinity, and Selectivity for Viloxazine, Atomoxetine, and Reboxetine. |
Furthermore, several in vitro and in vivo observations from previous studies have indicated that viloxazine might enhance 5-HT neurotransmission.19 Evidence also suggests that viloxazine lacks the characteristic neurochemical properties of the amphetamines or monoamine oxidase inhibitors, and is absent of inhibitory activity towards peripheral acetylcholine (ACh) or histamine (His).2,3
Distinct clinical profile of viloxazine compared to NRIs observed in recent ADHD clinical trials, along with limited in vitro and animal data emerging from previous studies, led us to hypothesize a more complex MoA of viloxazine than was previously suggested. A series of pharmacological studies using current techniques and methodologies were conducted in order to further elucidate the activity of viloxazine in neurotransmitter systems, including comprehensive radioligand binding affinity assays and in vitro cellular functional activity assays at key monoamine transporters and receptors, and a brain microdialysis study. The microdialysis study focused on the prefrontal cortex (PFC), nucleus accumbens (Acb), and amygdala (Amg). The PFC is an established area associated with ADHD pathophysiology, involving the regulation of attention, behavior, and emotions through extensive projections to multiple regions including Acb and Amg, which also play a key role in substance use disorders.20,21 To better understand the role of viloxazine in modulating neurotransmitter systems involved in ADHD and its potential risk of misuse, the neurotransmitter levels in all three brain regions were investigated.
These studies will help improve current understanding of the MoA of viloxazine providing a better pharmacology-based rationale to treat patients with ADHD and possibly other neuropsychiatric disorders.
Materials and Methods
Compound Tested
Viloxazine hydrochloride was manufactured by Supernus Pharmaceuticals, Inc., Rockville, MD, USA; compound ID 100047426–1.
Radioligand Binding Assays
Viloxazine was assessed by using the Cerep BioprintTM panel (Eurofins, France) equipped with 132 cell-based proprietary assays, including a variety of receptors (82 assays), ion channels (14 assays), monoamine oxidase-A (MAO-A), and neurotransmitter transporters such as the NE, DA, 5-HT, gamma-aminobutyric acid (GABA), and ACh transporters. Binding affinity of viloxazine (10 µM) was represented as a percent inhibition of the binding of a radiolabeled ligand specific for each target. Inhibition higher than 50% was considered significant.
Cellular Uptake and Receptor Functional Assays
Evaluation of the functional activity of viloxazine was performed using Eurofins proprietary cellular uptake and receptor functional activity assays (Eurofins, France). Viloxazine was tested at eight concentrations in duplicates or triplicates for the determination of IC50 (concentration causing a half-maximal inhibition of control response) or EC50 (concentration producing a half-maximal response). The agonistic response of viloxazine was calculated as a percentage of control response to a known reference agonist for each target. The cellular antagonistic response was calculated as a percentage inhibition of control reference agonistic response for each target. Results showing an inhibition or stimulation higher than 50% are considered to represent significant effects. IC50 and EC50 values were determined by nonlinear regression analysis of the concentration-response curves. The apparent dissociation constants (KB) were determined using the modified Cheng-Prusoff equation. Analyses were performed using commercial software SigmaPlot® 4.0 for Windows®.
Briefly, cellular uptake assay measured the effect of viloxazine on [3H]-NE uptake in rat hypothalamic synaptosomes using 0.2 μCi/mL [3H]-NE incubated for 20 min at 37ºC.22 The amount of [3H]-NE incorporated into synaptosomes was measured by scintillation counting. In addition, the effect of viloxazine on [3H]-5-HT uptake was performed in human embryonic kidney (HEK)-293 cells expressing the human SERT gene (hSERT). The accumulation of [3H]-5-HT after a 15 min incubation at room temperature was measured by scintillation counting.
Evaluation of agonistic and antagonistic activity of viloxazine was conducted by Eurofins (formerly Cerep), Poitiers, France, using receptor and transporter functional assays (commercially available from Eurofins) performed in 26 recombinant cell lines. All the recombinant cell lines are created and selected at Eurofins sites or MDS-Millipore, STC, MO, USA. For agonist assays, 5-HT was used as the control reference compound. For antagonist assays, 5-HT was used as a stimulus, and the inhibitory effect of different concentrations of viloxazine on the induced response was expressed as percent inhibition of response. Effects were evaluated via fluorimetry or homogeneous time-resolved fluorescence (HTRF) for a 20 to 30 min period: at room temperature for measuring intracellular calcium (for 5-HT1A), at 28°C for measuring impedance (for 5-HT1D), or at 37ºC for measuring cyclic adenosine monophosphate (for 5-HT1B, 5-HT6, and 5-HT7) or inositol phosphate-1 (IP1, for 5-HT2A, 5-HT2B, and 5-HT2C). EC50 and IC50 values were determined as described above, and apparent dissociation constants (KB) were determined using the modified Cheng-Prusoff equation.
Prediction of Receptor Occupancy
An estimate receptor occupancy (RO) for NET, 5-HT2B, and 5-HT2C was calculated with the following equation:23,24
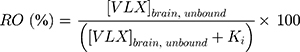
where the RO represents the percentage of receptors occupied in relation to the unbound brain concentration of viloxazine at the target receptor (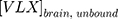 ) and the constant of binding (Ki) was experimentally measured for the target receptor. The human unbound brain concentration was calculated based on the human unbound plasma concentration at clinical dose and the unbound plasma/brain concentration ratio in rats assuming the unbound plasma/brain concentration ratio in rats is similar to that of humans. The human unbound plasma concentration was derived from the human plasma concentration at clinical dose and the unbound fraction in human plasma.
) and the constant of binding (Ki) was experimentally measured for the target receptor. The human unbound brain concentration was calculated based on the human unbound plasma concentration at clinical dose and the unbound plasma/brain concentration ratio in rats assuming the unbound plasma/brain concentration ratio in rats is similar to that of humans. The human unbound plasma concentration was derived from the human plasma concentration at clinical dose and the unbound fraction in human plasma.
Microdialysis in Rat Prefrontal Cortex, Nucleus Accumbens, and Amygdala
Adult male Sprague–Dawley rats (age 7–8 weeks, body weight 300–400 g, Charles River Laboratories, San Francisco, CA) were used for all studies. Animals were maintained on a 12/12-hour light/dark cycle in a temperature- (22 ± 2°C) and humidity- (~50%) controlled room.
On day 0, rats (n = 24) were anesthetized using isoflurane (2%, 800 mL/min O2). Each rat from Groups 1 and 3 (n = 6 per group) received a single I-shaped microdialysis probe (polyacrylonitrile membrane, 2 mm membrane, BrainLink, Netherlands) inserted into the PFC, and each animal from the Groups 2 and 4 (n = 6 per group) received two microdialysis probes inserted into the Acb and Amg.25 Coordinates for the tips of the probes in the PFC included: anteroposterior (AP) = +3.4 mm from bregma; lateral (L) = -0.8 mm to midline; and ventral (V) = -4.0 mm to dura, with the toothbar set at -3.3 mm. Coordinates for the tips of the probes in the Acb were: AP = +2.0 mm from bregma; L = -1.2 mm to midline; and V = -7.9 mm to dura, with the toothbar set at -3.3 mm. Coordinates for the tips of the probes in the Amg were: AP = -3.3 mm from bregma; L = -4.5 mm to midline; and V = -9.0 mm to dura, with the toothbar set at -3.3 mm. On day 1, animals were connected to the fraction collection system and a microperfusion pump (Harvard PHD 2000 Syringe pump), and perfused with artificial cerebrospinal fluid (aCSF) containing 150 nM NaCl, 3 mM KCl, 1.7 mM CaCl2, and 0.9 mM MgCl2, at a flow rate of 1.5 μL/min. After a 2-hour stabilization period, four basal samples were collected (Baseline), followed by the intraperitoneal (IP) administration of vehicle (0.9% NaCl) to each animal in Groups 1 and 2 or viloxazine (50 mg/kg) to each animal in Groups 3 and 4 in a volume of 5 mL/kg. Notably, 50 mg/kg is approximately the animal equivalent dose derived from the human therapeutic dose (400 mg/day)10 on the basis of body surface area. After dosing, interstitial fluid (ISF) samples were collected for 240 min at intervals of 30 min.
At the end of the microdialysis experiment, rats were euthanized with CO2 and a gross histological confirmation of probe location in the PFC, Acb, and Amg was performed. The study was performed as a non-GLP discovery stage study in accordance with the study plan and CRL standard operating procedures. The animal research was performed at Charles River Laboratories (South San Francisco, CA, USA) and approved by the Institutional Animal Care and Use Committee of Charles River Laboratories, SSF. Animals were cared for in accordance with the NIH Guide for the Care and Use of Laboratory Animals, 8th edition.
Samples were analyzed for the levels of NE, DA, 5-HT, GABA, glutamate (Glu), His, and ACh using fit-for-purpose liquid chromatography tandem mass spectrometry (LC-MS/MS) methods. The detection limits were set to 0.03 nM for NE, 0.04 nM for DA and 5-HT, 2 nM for GABA, 10 nM for Glu, 0.1 nM for His, and 0.05 nM for ACh.
Neurotransmitter levels in the ISF were expressed as the percentage of baseline (mean value of the four basal samples, T = 120 to T = 0 min). Samples were excluded from further analysis if levels were below the detection limit, or if prior to two-way analysis of variance (ANOVA) analysis or Grubbs’ outlier test revealed it was a significant outlier. Statistical evaluation was performed using SigmaStat for Windows, Version 4 (Systat Software, Inc., 2011–2012). If significant, the two-way ANOVA analysis was followed by a Dunnett’s Multiple Comparison Method post hoc analysis of vehicle treated vs viloxazine treated at each time point (T = 0 to T = 240 min). The level of statistical significance was set to p < 0.05.
Results
Radioligand Binding Assays
Viloxazine at 10 µM was evaluated in binding competition assays including a series of receptors, ion channels, and transporters in the Eurofins/Cerep BioprintTM panel. Only seven targets from the 132 assessed targets showed >50% inhibition of binding of a specific radiolabeled ligand for each target (Figure 1). These seven targets included NET, 5-HT receptors 5-HT2B, 5-HT2C, 5-HT1B, and 5-HT7, as well as adrenergic alpha1A (ADRα1A) and alpha1B (ADRα1B) receptors. The binding assays showed a weak competitive inhibition (<25%) at DA receptors D1, D2S, and D3, His receptors H1 and H2, and muscarinic cholinergic receptors M1, M2, M3, and M4. Moreover, viloxazine showed a selective binding profile at NET, with notable low activity (~10%) towards DAT and monoamine oxidase A (MAO-A), and negligible binding to SERT, the GABA transporter (GAT), and the choline transporter (CHT-1).
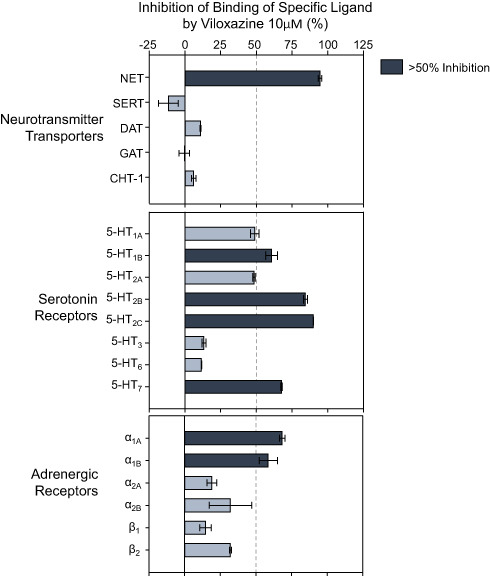 |
Figure 1 Neurotransmitter transporters and receptors binding profile of viloxazine. Representative results of binding competition assays of viloxazine (10 µM). Viloxazine binding was calculated as a percent inhibition of the binding of a radiolabeled ligand specific for each target (mean ± SEM, n = 2). Targets that presented an inhibition higher than 50% were considered a significant effect for viloxazine binding. |
Cellular Uptake and Receptor Functional Assays
Results from the representative functional activity assays are shown in Figure 2. The function of viloxazine as a NET inhibitor was assessed based on its ability to inhibit the uptake of [3H]-NE in rat hypothalamic synaptosomes. Consistent with previous reports, viloxazine showed potency towards NET at submicromolar levels, with an IC50 value of 0.26 µM. Its inhibitory effect on 5-HT uptake in HEK293 cells expressing hSERT was also evaluated. Results showed that viloxazine had inhibited SERT with an IC50 of 257 µM.
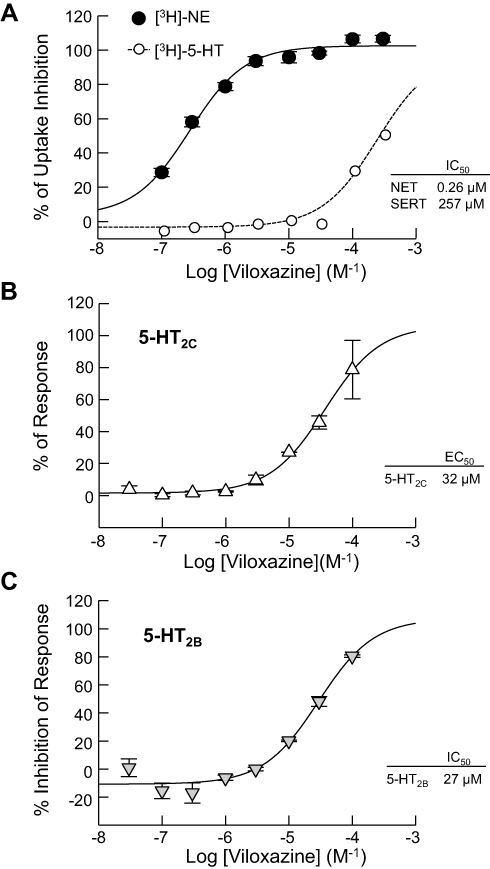 |
Figure 2 Effects of viloxazine on uptake and cellular functional activity in vitro. (A) Dose-dependent inhibition of NET-mediated [3H]-NE uptake (n = 3) measured in rat hypothalamic synaptosomes and [3H]-5-HT uptake (n = 3) measured in HEK293 cells expressing hSERT. (B) Viloxazine activated the response of 5-HT2C (agonist) in a CHO cell-based IP1 HTRF® assay (n = 2). (C) Viloxazine antagonized the activity of 5-HT2B after stimulation with 5-HT in a CHO cell-based IP1 HTRF® assay (n = 2). Data presented as mean ± SEM. |
Across the evaluated receptors, viloxazine was found to exhibit significant agonistic activity towards 5-HT2C (EC50 = 32.0 µM, Emax=78.6%) and antagonistic activity towards 5-HT2B (IC50 = 27.0 µM, KB = 4.2 µM).
In functional assays of 5-HT2A, viloxazine showed no agonistic effect at concentrations up to 300 µM and a very low potent antagonism with an IC50>300 µM. The evaluation of the functional activity of viloxazine at the 5-HT7 receptor showed no agonistic activity at concentrations up to 100 µM and low potency antagonistic activity (52% inhibition) at 100 µM. Viloxazine did not show any significant agonistic or antagonistic activity towards 5-HT1A, 5-HT1B, 5-HT1D, or 5-HT6 receptors. Among the evaluated adrenergic receptors, viloxazine exhibited weak antagonistic activity at ADRβ2 (IC50 = 68 µM, KB = 6.2 µM) and ADRα1B (IC50 = 93.0 µM, KB = 10 µM) receptors, and either very weak (IC50 > 150 µM) or no significant agonistic or antagonistic activity towards ADRα1A, ADRα2A, ADRα2B, ADRα2C and ADRβ1. Of note, viloxazine showed no significant agonistic activity at concentrations up to 300 µM and either very weak (IC50 > 150 µM) or no antagonistic activity towards dopaminergic receptors D1, D2S, and D3; the histaminic receptor H1; and cholinergic muscarinic receptors M1, M3, and M4.
Effects of Viloxazine on Neurotransmitter Levels in Rat Prefrontal Cortex, Nucleus Accumbens, and Amygdala
To further understand the functional effects of viloxazine on neurotransmitter systems, extracellular levels of the neurotransmitters NE, DA, 5-HT, GABA, Glu, His, and ACh were measured in the PFC, Acb, and Amg of freely moving Sprague–Dawley rats from the vehicle control groups (Groups 1 and 2) and viloxazine treatment groups (Groups 3 and 4).
As shown in Figure 3A, viloxazine (50 mg/kg) caused an increase in extracellular levels of 5-HT in the PFC with the peak value of 506±133% from baseline. Two-way ANOVA analysis found a statistically significant increase in the 5-HT level in the viloxazine-treated Group 3 compared to the vehicle-treated Group 1 (F1,10=38.28, p<0.001, Table 2). The increase vs vehicle was observed at 30 to 120 min (p< 0.05, Dunnett’s post hoc test) after viloxazine treatment (Figure 3A). The NE and DA levels in the PFC, also increased with peak levels 649±57% and 670±105% from baseline, respectively. The observed increase was statistically significant in viloxazine-treated group compared to vehicle (F1,10=448.561, p<0.001 and F1,10=4.496, p<0.001, respectively), which was maintained throughout the 4-hour measurement period (p< 0.001, Dunnett’s post hoc test).
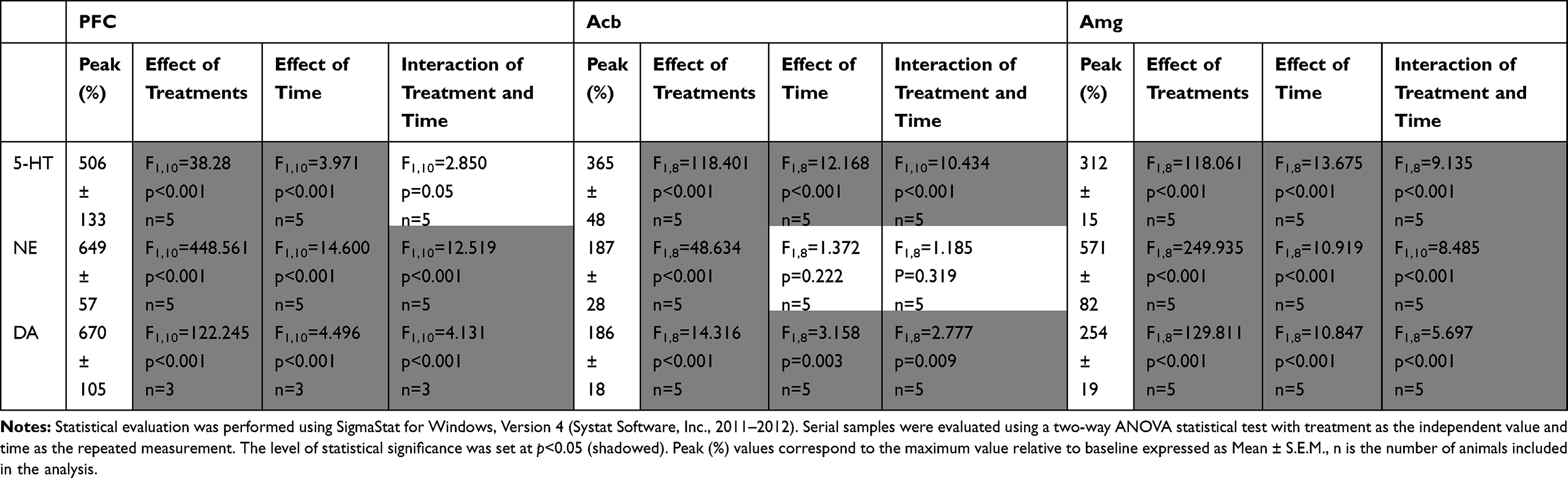 |
Table 2 Statistical Analysis of the Effects of Viloxazine Administration on Monoamine Levels in the Three Brain Regions as Measured by Microdialysis in Freely Moving Rats |
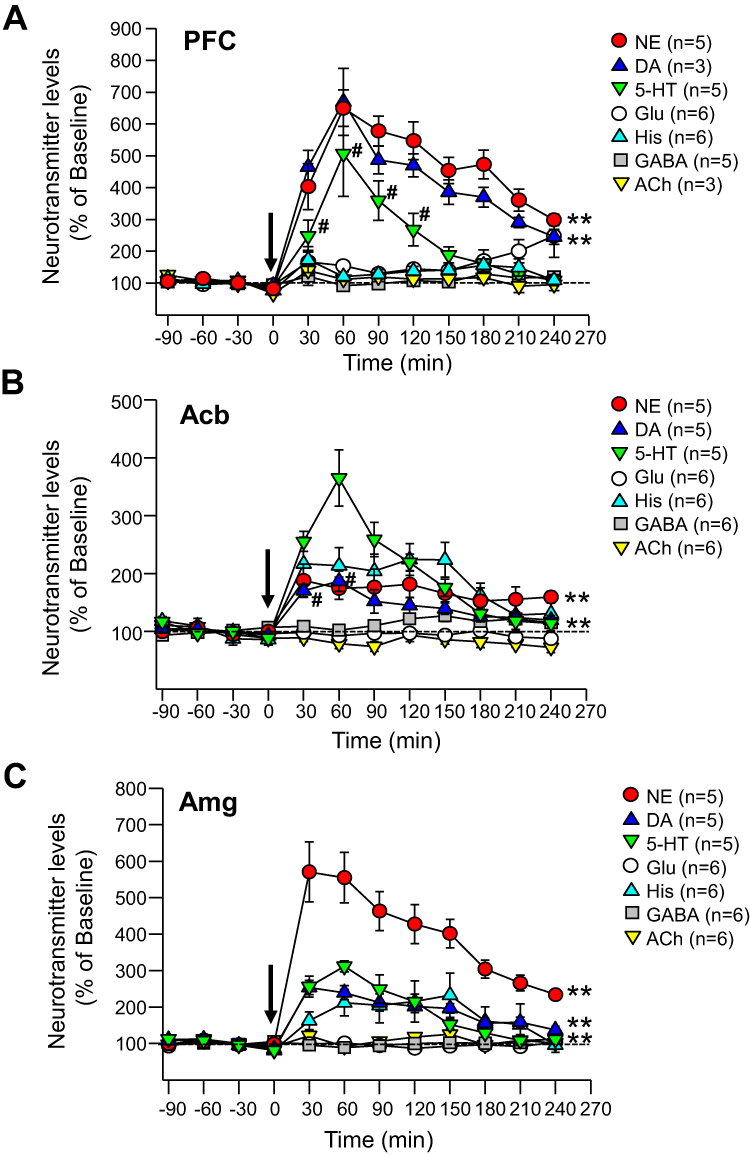 |
Figure 3 Effects of viloxazine on neurotransmitter levels in the PFC, Acb, and Amg of freely moving rats. Administration of viloxazine (50 mg/kg, IP) at time 0 is indicated by the arrow. (A) In the PFC, viloxazine significantly increased extracellular levels of NE and DA throughout the 4 h period (**p < 0.001, Dunnett’s post hoc test) and 5-HT from 30 to 120 minutes (#p < 0.05 Dunnett’s post hoc test) in comparison to vehicle treated groups. (B) In the Acb, extracellular levels of 5-HT and NE increased throughout the 4 h period (**p < 0.001, Dunnett’s post hoc test) and DA from 30 to 60 min (#p < 0.05, Dunnett’s post hoc test). (C) In the Amg, extracellular levels of 5-HT, NE, and DA increased throughout the 4 h period (**p < 0.001, Dunnett’s post hoc test). Measured neurotransmitter levels (mean ± SEM) are reported as the percent of pre-treatment baseline. The statistical post-hoc analyses of vehicle vs viloxazine at each time point (T = 0 to T = 240) and significant interactions (two-way ANOVA with p<0.05) are presented in the Table 2. |
The evaluation of neurotransmitter levels in the Acb and Amg was performed in viloxazine-treated group (Group 4) and vehicle-treated group (Group 2) using double microdialysis probes. The extracellular levels of 5-HT, NE, and DA in the Acb increased (Figure 3B), with the peak values of 365±48%, 187±28%, and 186±18%, respectively. Statistically significant increase was found in viloxazine-treated rats compared to vehicle-treated rats for all three monoamines (F1,8=118.401, p<0.001; F1,8=48.634, p<0.001; F1,8=14.316, p<0.001, respectively; Table 2). For 5-HT and NE, the effect was maintained through 4 h measurement period (p< 0.001, Dunnett’s post hoc test; Figure 3B); for DA, the increase was observed at 30 to 60 min following viloxazine administration (p<0.05, Dunnett’s post hoc test; Figure 3B). Elevated levels of all three neurotransmitters were observed in Amg (Figure 3C) with the peak values of 312±15% (5-HT), 571±82% (NE), and 254±19% (DA) from baseline. The increase in 5-HT, NE, and DA was statistically significant compared to vehicle (F1,8=118.061, p<0.001; F1,8=249.935, p<0.001; F1,8=129.811, p<0.001, respectively; Table 2). The effect was maintained throughout the 4 h period (p<0.001, Dunnett’s post hoc test; Figure 3C).
In the three evaluated brain regions, no significant effects within the vehicle-treated groups were observed. No significant changes in the extracellular levels of GABA, Glu, His, and ACh were observed in the viloxazine-treated groups compared to baseline or vehicle-treated groups.
Prediction of Receptor Occupancy
The receptor occupancies at clinical doses of 100, 200, 400, and 600 mg/day were calculated based on the Ki values of 630, 3900, and 6400 nM for NET, 5-HT2C, and 5-HT2B, respectively (Table 3). The dose and calculated receptor occupancy relationship is depicted in Figure 4.
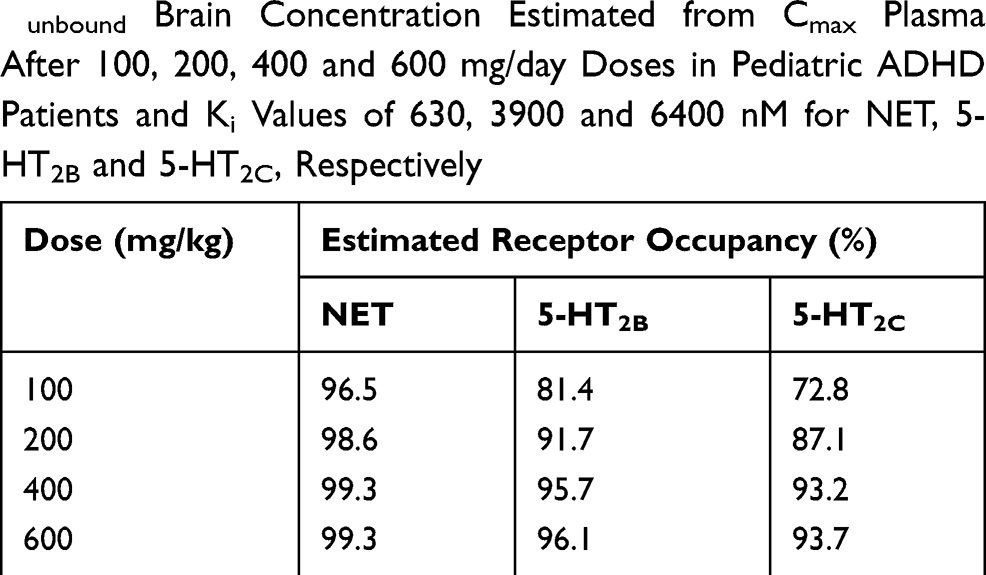 |
Table 3 Receptor Occupancy Calculated Based on Obtained Cunbound Brain Concentration Estimated from Cmax Plasma After 100, 200, 400 and 600 mg/day Doses in Pediatric ADHD Patients and Ki Values of 630, 3900 and 6400 nM for NET, 5-HT2B and 5-HT2C, Respectively |
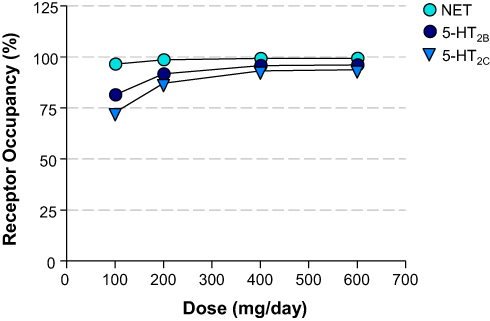 |
Figure 4 Dose – Calculated Receptor Occupancy Curves. Relationship between the dose of administered viloxazine to pediatric ADHD patients (31.5 kg mean body weight) and the estimated receptor occupancy for NET, 5-HT2B, and 5-HT2C based on the estimated unbound brain concentration of viloxazine at target receptor and its binding affinity (Ki) at clinical doses of 100, 200, 400, and 600 mg/day. |
Discussion
Mechanism of Action of Viloxazine
Collectively, the data presented here have demonstrated that the MoA of viloxazine predominantly involves serotonergic and noradrenergic pathways. Historically, viloxazine was found to inhibit reuptake of [3H]-labeled NE in the mouse heart2,3 and potentiate L-adrenaline-induced mydriasis in rabbits, supporting the ability of viloxazine to block NE uptake.5 However, the data presented here show moderate inhibitory activity of viloxazine towards the NET (IC50 in the 0.3 µM range), consistent with the observed low rate of cardiac-related effects in the clinical setting, which are commonly seen in drugs whose primary mechanism is via NRI activity. For example, CNS stimulant treatments and the NRI atomoxetine have been associated with serious cardiovascular events in children, adolescents, and adults.26–28
The involvement of serotonergic system in the drug action of viloxazine is supported by previous data demonstrating that viloxazine: 1) increased the basal release of 5-HT in rat striatal slices,29 2) enhanced the electrophysiological responses of rat cortical neurons to locally applied 5-HT,30–32 and 3) increased the induced hind-limb reflex in spinalized rats in vivo, indicative of a potentiation of the 5-hydroxytryptophan (5-HTP) behavioral syndrome.3
To further evaluate the role of viloxazine in 5-HT modulation, its selectivity and mode of action were tested at eight subtypes of 5-HT receptors using cellular functional assays. As agonist, viloxazine stimulated response in 5-HT2C expressing cells, with an EC50=32 µM. As antagonist, it inhibited the response to 5-HT in cells expressing 5-HT2B with an IC50=27 µM and showed a marginal 52% inhibition at 100 µM in cells expressing 5-HT7. These results together with no effect found on 5-HT reuptake via SERT (IC50=257 µM) suggest that viloxazine action as a serotonin modulator might involve the direct activation of 5-HT2C and/or inactivation of 5-HT2B, with IC50 and EC50 values within clinically relevant concentration range (data not shown). The in vitro pharmacology studies also demonstrated that viloxazine is practically devoid of activity towards dopamine receptors, DAT and shows very weak antagonistic activity towards ADRβ2 and ADRα1B.
According to the calculated receptor occupancy at clinical doses (Table 3 and Figure 4), viloxazine has shown higher than 80% 5-HT2B and 5-HT2C occupancy at all concentrations (except for 5-HT2C at 100 mg/day dose). Therefore, 5-HT2B and 5-HT2C are likely to be involved in the MoA of viloxazine at clinical doses of 100, 200, 400, and 600 mg/day.
Findings from a previous study in rats have demonstrated inhibitory effect of 5-HT2B antagonists (RS 127445 and LY 266097) on cocaine-induced hyperlocomotion possibly involving an action on striatal and/or Acb DA transmission.33 Furthermore, 5-HT2C agonist (Ro 60–0175) has shown ability to modulate the activity of mesoaccumbens pathway at postsynaptic level and, therefore, suppress cocaine-induced hyperlocomotion.34 Based on these data and considering antagonistic activity of viloxazine at 5-HT2B and agonistic activity at 5-HT2C observed in this study, it can be hypothesized that therapeutic action of viloxazine to improve hyperactivity in ADHD involves inhibition of 5-HT2B and activation of 5-HT2C receptors.
The effect of viloxazine on the neurotransmitter systems within the PFC, an established target area associated with ADHD pathophysiology and attention control,20,35-37 was further evaluated using brain microdialysis. The observed upregulation of extracellular 5-HT in the PFC of freely moving rats has demonstrated the modulatory effect of viloxazine on serotonergic neurotransmission (Figure 3). It is worth noting the increase in 5-HT by viloxazine (~500%) is higher than that of the selective 5-HT reuptake inhibitors (200%-400%), as well as that of atomoxetine.17,22,38 Viloxazine also significantly increased the levels of NE and DA in the PFC via NET inhibition as expected,18 and minimally increased DA in the Acb (a brain region with a key role in substance use disorders). Further, even though the level of NE could increase up to approximately 570% in the Amg of rats, (an effect previously associated with increased anxiety in animal models39), in the clinical setting, viloxazine has been shown to have anxiolytic effect.2
Given the lack of affinity to SERT in vitro, the underlying mechanism of the 5-HT upregulation in the PFC observed here remains undefined. However, a recent study in rats has confirmed that blocking of 5-HT2B receptor located in GABAergic interneurons in dorsal raphe nucleus with a selective 5-HT2B antagonist (RS 127445) can activate 5-HT neuron and enhance 5-HT outflow in medial PFC.40 A similar mechanism is, therefore, postulated for viloxazine – its antagonistic effect on 5-HT2B receptors could lead to reducing GABA inhibitory tone to 5-HT neurons and increasing 5-HT outflow in medial PFC (Figure 5).
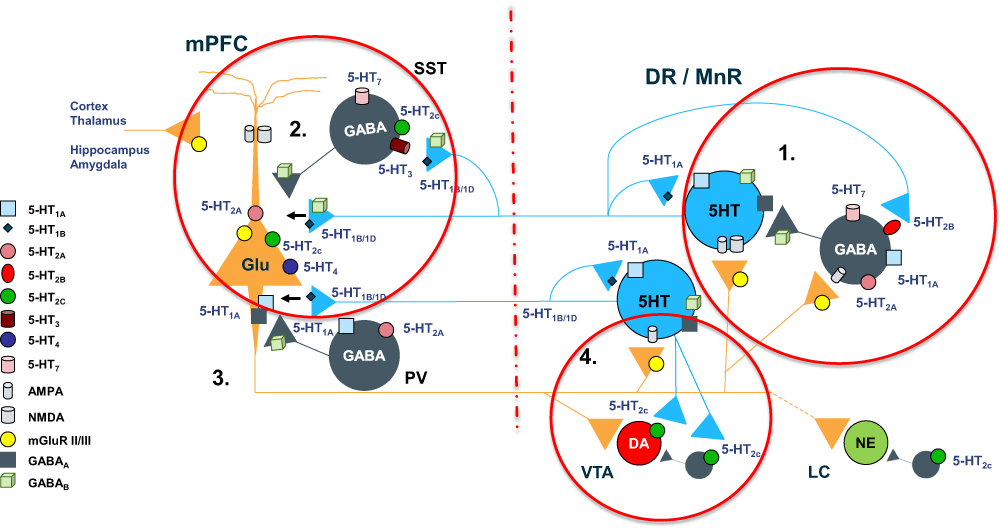 |
Figure 5 Complex regulation of 5-HT projections to prefrontal cortex. 1. “Proximal” regulatory loop includes feedback collateral branches and 5-HT1A autoreceptors, whose activation by excessive neurotransmitter results in reduced 5-HT synthesis and neuronal firing rate. Feedback collaterals also synapse with 5-HT2B receptors on DNR GABA interneurons. Inhibition of these receptors may be one of the principal regulators of 5-HT tonic neurotransmission. Viloxazine, for instance, may inhibit 5-HT2B receptors leading to inhibition of GABAergic transmission and consequently to upregulation of 5-HT tonic neurotransmission in mPFC, which may explain the increase of 5-HT in PFC observed in current microdialysis study. 2. At the terminal end 5-HT1B/1D autoreceptors regulate the 5-HT release. Serotoninergic projections in mPFC interface with SST-expressing and PV-expressing GABA interneurons as well as directly modulate glutamatergic pyramidal neurons via postsynaptic inhibitory 5-HT1A receptors and excitatory 5-HT2A receptors. SST GABA interneurons have a primary role in “signal-to-noise” gating of pyramidal neurons, while PV interneurons via axonal receptors regulate “volume” of glutamate transmission. 3. Serotoninergic input, indirectly, via mPFC GABA interneurons, and directly, via pyramidal neurons, regulates initiation of the “distal” regulatory loop. Additionally, the balance between activation of 5-HT2C receptors on GABA interneurons vs direct stimulation of 5-HT2C receptors located on pyramidal neurons, ultimately tunes glutamatergic output from mPFC. 4. Glutamatergic projections from mPFC to LC noradrenergic neurons, VTA dopaminergic neurons, and DNR/MNR serotoninergic neurons, as well as their corresponding GABA interneurons, directly and indirectly regulate function of the brainstem monoaminergic nuclei. Local balance between stimulation of 5-HT2C receptors on GABA interneurons vs ones located directly on VTA DA neurons, modulates DA output from the nucleus. At the termination point of the “distal” loop, glutamate fibers via excitatory AMPA and NMDA receptors located on 5-HT neurons and adjacent GABA interneurons, provide balanced regulatory input to serotoninergic nuclei. |
Providing a mechanistic explanation for the relationship between 5-HT2C agonism and ADHD is a more complex task, although early literature, relying on rodent preclinical models, has postulated a relationship between 5-HT2C agonism and ameliorative effect on stress-induced anhedonia, antidepressant effect and potential benefit in treatment of obesity and addictive disorders.41–43 The 5-HT2C receptors are located both on GABA interneurons and the VTA dopamine neurons, exerting opposing effects (Figure 5). Similar scenario is encountered in PFC, where 5-HT2C receptors populate both GABA interneurons and pyramidal neurons.44,45 Hence, the final outcome of 5-HT2C stimulation will be influenced by local biochemical environment and relative density and activation of opposing sub-populations of 5-HT2C receptors.46,47
Subcortical 5-HT2C receptors may have a pivotal role in modulating ascending nigrostriatal and mesocorticolimbic dopamine projections. Here are a few notable findings: stimulation of 5-HT2C receptors facilitates DA release in the striatum, while intra-PFC administration of 5HT2C agonists enhances dopaminergic transmission in Acb, induced by cocaine.45 More contemporary preclinical studies, using tissue sampling method, described increased 5-HT concentration in the orbitofrontal cortex, motor cortex-2, as well as increased DA turnover in the medial orbitofrontal cortex, in direct response to administration of 5-HT2C selective agonist.47,48
In aggregate, literature instantiates a plausible explanation for impact of 5-HT2C agonism on change in 5-HT and DA transmission (as well as their relative balance) in brain pathways and regions relevant for impulse control, regulation of motoric activity, reward processes, and emotional regulation.
Collectively, the results of the current study demonstrate that viloxazine not only modulates noradrenergic but also serotoninergic pathways and, therefore, exhibits psychopharmacological action as a serotonin norepinephrine modulating agent (SNMA).
A Different Mechanism of Action from Known ADHD and Depression Pharmacotherapies
This proposed MoA for viloxazine is similar in taking a multimodal approach; however, it differs from that of other known psychopharmacotherapies used to treat ADHD and/or depression, such as the serotonin norepinephrine dopamine reuptake inhibitors (SNDRIs) centanafadine, and dasotraline,9 the serotonin norepinephrine reuptake inhibitors (SNRIs) duloxetine and venlafaxine, as well as the norepinephrine reuptake inhibitors (NRIs) atomoxetine15,49 and reboxetine.16,50 It is also different from the psychostimulant methylphenidate (a norepinephrine dopamine reuptake inhibitor; NDRI).51 Moreover, the proposed MoA of viloxazine differs from that of other agents, such as the alpha-2-adrenergic agonists clonidine and guanfacine, which mechanistically target NE dysregulation directly at the level of the receptor.
In contrast to viloxazine, the NRIs atomoxetine and reboxetine are highly potent NET inhibitors, with inhibition of NE reuptake in the nanomolar range (Table 1). Additionally, unlike the observed viloxazine effects on serotonergic receptors as shown in this study, atomoxetine does not possess appreciable affinity for neuronal receptors investigated in a binding affinity study.17
Regarding atomoxetine, it is noteworthy that despite exhibiting a high level of both NET and SERT occupancy (>90% and >85%, respectively),49 it was not found to functionally increase 5-HT in rat and mouse PFC by microdialysis.17,38 This finding is possibly due to the low expression levels of SERT in the PFC.52,53
The classification of both atomoxetine and reboxetine as NRIs can be attributed to the direct NET inhibitory activity; ie, NET inhibition is the primary driver for the therapeutic efficacy of these compounds.15,16,49,50 In contrast, as reported here, viloxazine exhibits serotonergic modulating activity with moderate noradrenergic effects, implying the inhibitory activity on NET is not solely responsible for its major therapeutic mechanism. Therefore, the classification of viloxazine should be reflected as such—namely an SNMA.
Evaluation of the Risk for Substance Abuse
Drugs of abuse exert their initial reinforcing effects by triggering supraphysiologic surges of dopamine in the Acb, which activates the direct striatal pathway via D1 receptors and inhibits the indirect striato-cortical pathway via D2 receptors.21 Microdialysis studies in animals have shown that addictive drugs preferentially increase extracellular DA in the Acb of freely moving rats.54 For example, psychostimulants such as amphetamine can significantly increase DA (d- and l-amphetamine increase DA to 700 and 1,200%, respectively) from the baseline level in the striatum of freely moving spontaneously hypertensive rats, whereas the saline-treated increases DA to approximately 20%.55
As mentioned above, administration of viloxazine demonstrated a minimal increase of DA in the Acb to approximately 180% in freely moving rats. Vehicle-treated rats demonstrated an increase in DA of approximately 120%. These results are likely due to the lack of binding affinity of viloxazine at the DAT (KD >100 µM).18 In addition, viloxazine does not directly interact with the dopaminergic receptors D1 and D2S. Based on the minimal effect on DA in the Acb, viloxazine is expected to have low substance abuse liability.
This is consistent with previous data obtained in animal models, such as self-administration studies in monkeys, which demonstrated that viloxazine is physical dependence-free and exhibits very low potential for dependence development.56 In the absence of human studies specifically probing the abuse potential of viloxazine, contemporary clinical studies investigating viloxazine extended-release in the treatment of ADHD suggest that it is well tolerated and at tested doses has not been indicative of side effects pertaining to abuse potential.10 Finally, human behavioral studies and real-world evidence of approximately 30 years of its use as an antidepressant in Europe do not show any signals of drug-seeking behavior with viloxazine, any anticipated recreational value, or risk for prescription misuse or abuse.2,19,57,58
Assessment of the Cardiac Valvulopathy Risk
Sustained overstimulation of 5-HT2B, either by 5-HT2B agonists or high plasma 5-HT levels, can contribute to the development of valvular heart disease by stimulating myofibroblast mitogenesis and extracellular matrix deposition.59 This process eventually leads to thickened valve leaflets, pulmonary hypertension, regurgitation caused by improper closing during diastole, and ultimately can lead to heart failure.59 In contrast, blockade of 5-HT2B receptors has been shown in animal models to inhibit fibrosis and cardiac hypertrophy, thereby protecting against ventricular heart failure.60,61 Therefore, antagonists of 5-HT2B like viloxazine would be unlikely to contribute to cardiac valvulopathy and would be predicted to have a low risk of cardiac-related side effects. This is consistent with the long-term cardiac safety profile of viloxazine during its use as an antidepressant in Europe.2,12,62-64 A recently completed randomized clinical trial evaluating the efficacy and safety of extended release viloxazine in the treatment of ADHD, also demonstrated very low rates of cardiovascular adverse events.10
CNS Inflammatory Signaling
5-HT2B antagonism may reduce inflammatory signaling originating both from microglia in the brain and peripheral immune cells.65,66 Dysregulation of inflammatory signaling may be a relevant pathophysiological mechanism of ADHD. For example, Oades et al have described correlations between increases of interleukins IL-16 and IL-13 and total ADHD symptom ratings, elevation of IL-16 and hyperactivity, and association between increased IL-13 and inattention.67 Furthermore, increased CSF concentration of inflammatory cytokines has been associated with increased 5-HT and DA turnover, most likely reflecting inefficient signaling.68 Viloxazine via 5-HT2B inhibition may ameliorate aberrant inflammatory signaling in the brain and help restore homeostasis in DA and 5-HT signaling.
Therapeutic Implications in ADHD
Recognizing that the underlying mechanism for the enhancement of 5-HT is not yet fully understood, the ability of viloxazine to prominently modulate both 5-HT and NE supports the therapeutic relevance of viloxazine to neuropsychiatric disorders impacted by monoaminergic transmission, such as ADHD and depression (Figure 6).51,69,70 In particular, neuropharmacological studies have provided a wealth of evidence that dysregulation of NE and DA contributes to symptoms of inattention and deficits in executive decision-making in ADHD.51 Recently, 5-HT has also emerged as a potential therapeutic target in ADHD.9,71 In rodent models, hyperlocomotor activity, for instance, has been attenuated by SERT inhibitors, suggesting a role for serotonergic signaling in regulating locomotor activity.72 Serotonergic antagonists directly targeting 5-HT2B and agonists of 5-HT2C receptors have also been specifically implicated in the attenuation of locomotor activity in rats in vivo.33,34 The role of viloxazine as a 5-HT2C agonist and 5-HT enhancer – along with its inhibitory activity towards NET – identified in the current study demonstrates the relevance of this compound to target core symptomatology in ADHD.9,13,36
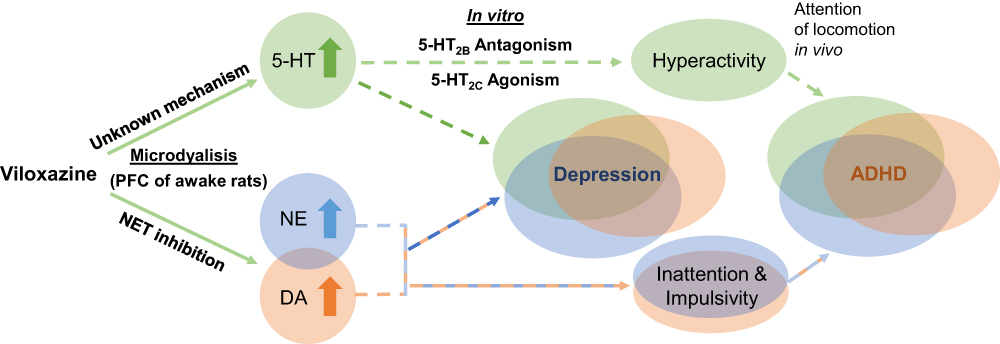 |
Figure 6 Proposed dual mechanism of action of viloxazine. The schematic shows purported clinical implications (dashed lines) of the observed neurotransmitter modulating effects of viloxazine. Microdialysis and in vitro studies demonstrate that viloxazine increases 5-HT, NE, and DA levels in the PFC of awake rats, as well as exhibits antagonistic activity towards 5-HT2B receptors and agonistic activity towards 5-HT2C receptors. These effects of viloxazine on modulating both the 5-HT and NE systems help explain why viloxazine is therapeutically relevant to neuropsychiatric disorders, such as the core symptoms of ADHD and depression. |
Two recent imaging studies, comparing clinically identified ADHD patients with typically developing participants identified an altered pattern of functional network anatomy and connectivity as an essential feature of ADHD pathophysiology.73,74 Structural differences implicated components of the sensorimotor network, default-mode network (DMN), and salience network.73,74 Furthermore, functional imaging studies have identified aberrant functional connectivity between sensorimotor, DMN, and cognitive and attentional networks.75,76 Proper functional interaction between sensorimotor, DMN, and salience networks appears to be regulated by input from the thalamus and striatum, relying on optimal homeostatic balance between 5-HT and DA brainstem projections into these regions.77 To our knowledge, the only study evaluating monoamine metabolite levels in an ADHD cohort has found an association between homovanillic acid (HVA)/5-hydroxyindolacetic acid (5-HIAA) ratio in ADHD subjects with clinical measures of hyperactivity, and 5-HIAA levels with measures of aggression and hyperactivity.78 Another study comparing monoamine metabolites in a 24-hour urine collection of children with ADHD and typically developing children, noted that HVA/5-HIAA levels were significantly lower in ADHD subjects compared to typically developing children. The same HVA/5-HIAA ratio was negatively correlated with measures of impulsivity.79
In aggregate, this evidence suggests that balance between monoamines, and especially the role of 5-HT may have been grievously neglected, as it likely has bearing on adaptive cortico-striatal-thalamic-cortical pathway regulation and by extension, harmonious interaction between, sensory-motor, DMN, salience and cognitive/attentional networks in ADHD. Novel ADHD treatments regulating 5-HT as well as catecholamine transmission may provide additional therapeutic benefits, hitherto unavailable with existing stimulant and noradrenalin-modulating ADHD medications. Viloxazine may correct monoamine imbalance by robustly elevating 5-HT, NE, and DA signaling in the PFC. It may, therefore, reestablish more harmonious interactions among major resting-state signaling networks. Future studies including contemporary methods of electrophysiology and imaging may help further improve our understanding of the role of viloxazine in these processes.
Therapeutic Implications in Depression
5-HT is broadly considered to play a key role in regulating anxiety, stress levels, patience,62,63 coping capability, and mood in depression.80–82 A wide range of evidence has previously been reported supporting the role of 5-HT2C receptors in depression, and correspondingly, the mechanistic basis for the antidepressant properties and behavioral effects of 5-HT2C receptor agonists.41,83 Indeed, pharmacological restoration of 5-HT synthesis with 5-HTP or treatment with a 5-HT2C receptor agonist (CP809.101) reduced cognitive deficits in Tph2-KI mice and contributed to an antidepressant effect.84 The enhancing action of viloxazine on 5-HT levels within the rat PFC and its antagonistic/agonist activity at selected 5-HT receptors suggests this MoA contributes to the therapeutic profile of viloxazine as an antidepressant.
Conclusions
Evaluating the collective data, we conclude that viloxazine currently being developed as an extended-release medication for ADHD is mechanistically distinct from NRIs, other known antidepressants, and other known pharmacotherapies that have been used to treat ADHD. Viloxazine exhibits selective antagonistic activity towards 5-HT2B receptors and agonistic activity towards 5-HT2C receptors, complemented by moderate NET inhibition. Therefore, we propose that, due to its multimodal MoA that synergistically contribute to its psychopharmacological activity, viloxazine is best described as a serotonin norepinephrine modulating agent (SNMA).
Abbreviations
5-HT, serotonin; 5-HTP, 5-hydroxytryptophan; Acb, nucleus accumbens; ACh, acetylcholine; aCSF, artificial cerebrospinal fluid; ADHD, attention-deficit/hyperactivity disorder; ADR, adrenergic receptor; Amg, amygdala; ANOVA, analysis of variance; AP, anteroposterior; CHT-1, choline transporter; CHO, Chinese hamster ovary; CNS, central nervous system; DA, dopamine; DAT, dopamine transporter; EC50, concentration producing a half-maximal response; GABA, gamma-aminobutyric acid; GAT, GABA transporter; Glu, glutamate; HEK, human embryonic kidney; His, histamine; hSERT, human serotonin transporter gene; HTRF, homogeneous time-resolved fluorescence; IC50, concentration causing a half-maximal inhibition of control response; IP, intraperitoneal; IP1, inositol phosphate-1; ISF, interstitial fluid; KB, apparent dissociation constant; KD, dissociation constant; Ki, inhibitory constant; L, lateral; LC-MS/MS, liquid chromatography tandem mass spectrometry; MAO-A, monoamine oxidase-A; MoA, mechanism of action; NE, norepinephrine; NET, norepinephrine transporter; NDRI, norepinephrine-dopamine reuptake inhibitor; NRI, norepinephrine reuptake inhibitor; PFC, prefrontal cortex; SERT, serotonin transporter; SNDRI, serotonin-norepinephrine-dopamine reuptake inhibitor; SNMA, serotonin norepinephrine modulating agent; SNRI, serotonin-norepinephrine reuptake inhibitor; V, ventral.
Data Sharing Statement
Not available.
Acknowledgments
The authors would like to thank Eurofins (formerly Cerep), Poitiers, France, for their assistance in the assay techniques; and IMPRINT Science, New York, NY, USA, for providing editorial support for this manuscript.
Author Contributions
All authors contributed toward data analysis, drafting and revising the paper, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
CY, JGO, SC, and SS are employees of Supernus Pharmaceuticals, Inc. VM is an employee of the University of South Carolina School of Medicine. He is a consultant for ACADIA Pharmaceuticals Inc.; Alfasigma USA, Inc.; Alkermes, Inc.; Allergan; Eisai-Purdue; Intra-Cellular Therapies; Janssen; H. Lundbeck A/S; Otsuka America Pharmaceutical, Inc.; Sage Pharmaceuticals; Sunovion Pharmaceuticals Inc.; Supernus Pharmaceuticals, Inc.; and Takeda Pharmaceutical Company Limited. He serves on the speakers bureau of ACADIA Pharmaceuticals Inc.; Alkermes, Inc.; Allergan; Ironshore; Intra-Cellular; Janssen; H. Lundbeck A/S; Otsuka America Pharmaceutical, Inc.; Sunovion Pharmaceuticals Inc.; and Takeda Pharmaceutical Company Limited. The authors report no other conflicts of interest in this work.
References
1. Foye WO. Pharmacodynamic agents. In: Foye’s Principles of Medicinal Chemistry. Lemke TL, Williams DA, Roche VF, and Zito SW, eds. UK: Wolters Kluwer Health/Lippincott Williams & Wilkins; 2013:610.
2. Pinder RM, Brogden RN, Speight TM, Avery GS. Viloxazine: a review of its pharmacological properties and therapeutic efficacy in depressive illness. Drugs. 1977;13(6):401–421. doi:10.2165/00003495-197713060-00001.
3. Greenwood DT. International vivalan symposium; Animal pharmacology of viloxazine (Vivalan). J Int Med Res. 1975;3.
4. Carlier PR, Lo MM, Lo PC, et al. Synthesis of a potent wide-spectrum serotonin-, norepinephrine-, dopamine-reuptake inhibitor (SNDRI) and a species-selective dopamine-reuptake inhibitor based on the gamma-amino alcohol functional group. Bioorg Med Chem Lett. 1998;8(5):487–492. doi:10.1016/S0960-894X(98)00062-6.
5. Lippman W, Pugsley TA. Effects of viloxazine, an antidepressant agent, on biogenic amine uptake mechanisms and related activities. Can J Physiol Pharmacol. 1976;54(4):494–509. doi:10.1139/y76-069.
6. ClinicalTrials.gov. Evaluation of SPN-812 ER low dose in children with ADHD. Available from: https://clinicaltrials.gov/ct2/show/NCT03247530?term=NCT03247530&rank=1. 2018.
7. Clinicaltrials.gov. Evaluation of SPN-812 ER low dose in adolescents with ADHD. Available from: https://clinicaltrials.gov/ct2/show/NCT03247517?term=NCT03247517&rank=1.
8. Clinicaltrials.gov. Evaluation of SPN-812 ER high dose in children with ADHD. Available from: https://clinicaltrials.gov/ct2/show/NCT03247543?term=NCT03247543&rank=1.
9. Nageye F, Cortese S. Beyond stimulants: a systematic review of randomised controlled trials assessing novel compounds for ADHD. Expert Rev Neurother. 2019;19(7):707–717. doi:10.1080/14737175.2019.1628640.
10. Johnson JK, Liranso T, Saylor K, et al. A Phase II double-blind, placebo-controlled, efficacy and safety study of SPN-812 (Extended-Release Viloxazine) in children with ADHD. J Atten Disord. 2020;24(2):348–358. doi:10.1177/1087054719836159.
11. Nasser A, Faison SL, Liranso T et al. An assessment of QTc effects with SPN-812 (Extended-Release Viloxazine) in healthy adults. Neuroscience Education Institute 15th Annual Congress; November 7–10, 2019; Colorado Springs, CO.
12. Corona GL, Frattini P, Cucchi ML, et al. Viloxazine in depressed women: clinical response and cardiovascular effects. Int J Clin Pharmacol Ther Toxicol. 1987;25(6):322–327.
13. Faraone SV. The pharmacology of amphetamine and methylphenidate: relevance to the neurobiology of attention-deficit/hyperactivity disorder and other psychiatric comorbidities. Neurosci Biobehav Rev. 2018;87:255–270. doi:10.1016/j.neubiorev.2018.02.001.
14. Green EA, Raj V, Shibao CA, et al. Effects of norepinephrine reuptake inhibition on postural tachycardia syndrome. J Am Heart Assoc. 2013;2(5):e000395. doi:10.1161/JAHA.113.000395.
15. Wong DT, Threlkeld PG, Best KL, Bymaster FP. A new inhibitor of norepinephrine uptake devoid of affinity for receptors in rat brain. J Pharmacol Exp Ther. 1982;222(1):61–65.
16. Wong EH, Sonders MS, Amara SG, et al. Reboxetine: a pharmacologically potent, selective, and specific norepinephrine reuptake inhibitor. Biol Psychiatry. 2000;47(9):818–829. doi:10.1016/S0006-3223(99)00291-7.
17. Bymaster FP, Katner JS, Nelson DL, et al. Atomoxetine increases extracellular levels of norepinephrine and dopamine in prefrontal cortex of rat: a potential mechanism for efficacy in attention deficit/hyperactivity disorder. Neuropsychopharmacology. 2002;27(5):699–711. doi:10.1016/S0893-133X(02)00346-9.
18. Tatsumi M, Groshan K, Blakely RD, Richelson E. Pharmacological profile of antidepressants and related compounds at human monoamine transporters. Eur J Pharmacol. 1997;340(2–3):249–258. doi:10.1016/S0014-2999(97)01393-9.
19. Greenwood DT. Viloxazine and neurotransmitter function. Adv Biochem Psychopharmacol. 1982;31:287–300.
20. Arnsten AF. The emerging neurobiology of attention deficit hyperactivity disorder: the key role of the prefrontal association cortex. J Pediatr. 2009;154(5):I–S43. doi:10.1016/j.jpeds.2009.01.018.
21. Volkow ND, Morales M. The brain on drugs: from reward to addiction. Cell. 2015;162(4):712–725. doi:10.1016/j.cell.2015.07.046.
22. Bymaster FP, Zhang W, Carter PA, et al. Fluoxetine, but not other selective serotonin uptake inhibitors, increases norepinephrine and dopamine extracellular levels in prefrontal cortex. Psychopharmacology (Berl). 2002;160(4):353–361. doi:10.1007/s00213-001-0986-x.
23. Li D, Kerns EH, edited by. Blood-Brain Barrier in Drug Discovery: Optimizing Brain Exposure of CNS Drugs and Minimizing Brain Side Effects for Peripheral Drugs;2015. ISBN 978-1-118-78835-6 (hardback).
24. Settimo L, Taylor D. Evaluating the dose-dependent mechanism of action of trazodone by estimation of occupancies for different brain neurotransmitter targets. J Psychopharmacol. 2018;32(1):96–104. doi:10.1177/0269881117742101.
25. Paxinos G. The Rat Brain in Stereotaxic Coordinates.
26. STRATTERA®. Prescribing Information. Indianapolis, IN: Lilly USA, LLC; 2017.
27. VYVANSE®. Prescribing Information. Lexington, MA: Shire US Inc; 2018.
28. CONCERTA®. Prescribing Information. Titusville, NJ: Janssen Pharmaceuticals, Inc; 2017.
29. Martin IL, Baker GB, Mitchell PR. The effect of viloxazine hydrochloride on the transport of noradrenaline, dopamine, 5-hydroxytryptamine and gamma-amino-butyric acid in rat brain tissue. Neuropharmacology. 1978;17(6):421–423. doi:10.1016/0028-3908(78)90018-7.
30. Jones RS, Roberts MH Effects of viloxazine on cortical neurone responses to monoamines and acetylcholine
31. Jones RS, Roberts MH Potentiation of monoamine responses of denervated cells by a noradrenaline uptake inhibitor (viloxazine)
32. Jones RS, Roberts MH Potentiation of responses to monoamines by antidepressants after destruction of monoamine afferents. Paper presented at: Proceedings of the British Association for Psychopharmacology; March, 1979.
33. Devroye C, Cathala A, Di Marco B, et al. Central serotonin(2B) receptor blockade inhibits cocaine-induced hyperlocomotion independently of changes of subcortical dopamine outflow. Neuropharmacology. 2015;97:329–337. doi:10.1016/j.neuropharm.2015.06.012.
34. Cathala A, Devroye C, Maitre M, et al. Serotonin2C receptors modulate dopamine transmission in the nucleus accumbens independently of dopamine release: behavioral, neurochemical and molecular studies with cocaine. Addict Biol. 2015;20(3):445–457. doi:10.1111/adb.12137.
35. Shaw P, Stringaris A, Nigg J, Leibenluft E. Emotion dysregulation in attention deficit hyperactivity disorder. Am J Psychiatry. 2014;171(3):276–293. doi:10.1176/appi.ajp.2013.13070966.
36. Arnsten AF. Fundamentals of attention-deficit/hyperactivity disorder: circuits and pathways. J Clin Psychiatry. 2006;67(Suppl 8):7–12.
37. Maletic V, Eramo A, Gwin K, Offord SJ, Duffy RA. The role of norepinephrine and its alpha-adrenergic receptors in the pathophysiology and treatment of major depressive disorder and schizophrenia: a systematic review. Front Psychiatry. 2017;8:42. doi:10.3389/fpsyt.2017.00042.
38. Koda K, Ago Y, Cong Y, Kita Y, Takuma K, Matsuda T. Effects of acute and chronic administration of atomoxetine and methylphenidate on extracellular levels of noradrenaline, dopamine and serotonin in the prefrontal cortex and striatum of mice. J Neurochem. 2010;114(1):259–270. doi:10.1111/j.1471-4159.2010.06750.x.
39. McCall JG, Siuda ER, Bhatti DL, et al. Locus coeruleus to basolateral amygdala noradrenergic projections promote anxiety-like behavior. Elife. 2017;6.
40. Cathala A, Devroye C, Drutel G, Revest JM, Artigas F, Spampinato U. Serotonin2B receptors in the rat dorsal raphe nucleus exert a GABA-mediated tonic inhibitory control on serotonin neurons. Exp Neurol. 2019;311:57–66. doi:10.1016/j.expneurol.2018.09.015.
41. Moreau JL, Bos M, Jenck F, Martin JR, Mortas P, Wichmann J. 5HT2C receptor agonists exhibit antidepressant-like properties in the anhedonia model of depression in rats. Eur Neuropsychopharmacol. 1996;6(3):169–175. doi:10.1016/0924-977X(96)00015-6.
42. Jenck F, Moreau JL, Berendsen HH, et al. Antiaversive effects of 5HT2C receptor agonists and fluoxetine in a model of panic-like anxiety in rats. Eur Neuropsychopharmacol. 1998;8(3):161–168. doi:10.1016/S0924-977X(97)00055-2.
43. Palacios JM, Pazos A, Hoyer D. A short history of the 5-HT2C receptor: from the choroid plexus to depression, obesity and addiction treatment. Psychopharmacology (Berl). 2017;234(9–10):1395–1418.
44. Leggio GM, Cathala A, Moison D, Cunningham KA, Piazza PV, Spampinato U. Serotonin2C receptors in the medial prefrontal cortex facilitate cocaine-induced dopamine release in the rat nucleus accumbens. Neuropharmacology. 2009;56(2):507–513. doi:10.1016/j.neuropharm.2008.10.005.
45. Berg KA, Clarke WP, Cunningham KA, Spampinato U. Fine-tuning serotonin2c receptor function in the brain: molecular and functional implications. Neuropharmacology. 2008;55(6):969–976. doi:10.1016/j.neuropharm.2008.06.014.
46. Navailles S, Moison D, Cunningham KA, Spampinato U. Differential regulation of the mesoaccumbens dopamine circuit by serotonin2C receptors in the ventral tegmental area and the nucleus accumbens: an in vivo microdialysis study with cocaine. Neuropsychopharmacology. 2008;33(2):237–246. doi:10.1038/sj.npp.1301414.
47. Chagraoui A, Whitestone S, Baassiri L, Manem J, Di Giovanni G, De Deurwaerdere P. Neurochemical impact of the 5-HT2C receptor agonist WAY-163909 on monoamine tissue content in the rat brain. Neurochem Int. 2019;124:245–255. doi:10.1016/j.neuint.2019.01.019.
48. Whitestone S, Deurwaerdere P, Baassiri L, et al. Effect of the 5-HT2C receptor agonist WAY-163909 on serotonin and dopamine metabolism across the rat brain: a quantitative and qualitative neurochemical study. Int J Mol Sci. 2019;20:12. doi:10.3390/ijms20122925.
49. Ding YS, Naganawa M, Gallezot JD, et al. Clinical doses of atomoxetine significantly occupy both norepinephrine and serotonin transports: implications on treatment of depression and ADHD. Neuroimage. 2014;86:164–171. doi:10.1016/j.neuroimage.2013.08.001.
50. Hajos M, Fleishaker JC, Filipiak-Reisner JK, Brown MT, Wong EH. The selective norepinephrine reuptake inhibitor antidepressant reboxetine: pharmacological and clinical profile. CNS Drug Rev. 2004;10(1):23–44. doi:10.1111/j.1527-3458.2004.tb00002.x.
51. Curatolo P, Paloscia C, D’Agati E, Moavero R, Pasini A. The neurobiology of attention deficit/hyperactivity disorder. Eur J Paediatr Neurol. 2009;13(4):299–304. doi:10.1016/j.ejpn.2008.06.003.
52. Zhou FC, Sari Y, Zhang JK. Expression of serotonin transporter protein in developing rat brain. Brain Res Dev Brain Res. 2000;119(1):33–45. doi:10.1016/S0165-3806(99)00152-2.
53. Chen X, Petit EI, Dobrenis K, Sze JY. Spatiotemporal SERT expression in cortical map development. Neurochem Int. 2016;98:129–137. doi:10.1016/j.neuint.2016.05.010.
54. Di Chiara G, Bassareo V, Fenu S, et al. Dopamine and drug addiction: the nucleus accumbens shell connection. Neuropharmacology. 2004;47(Suppl 1):227–241. doi:10.1016/j.neuropharm.2004.06.032.
55. Heal DJ, Smith SL, Gosden J, Nutt DJ. Amphetamine, past and present–a pharmacological and clinical perspective. J Psychopharmacol. 2013;27(6):479–496. doi:10.1177/0269881113482532.
56. Yanagita T, Wakasa Y, Kiyohara H. Drug dependence potential of viloxazine hydrochloride tested in rhesus monkeys. Pharmacol Biochem Behav. 1980;12(1):155–161. doi:10.1016/0091-3057(80)90430-X.
57. Cryan JF, Valentino RJ, Lucki I. Assessing substrates underlying the behavioral effects of antidepressants using the modified rat forced swimming test. Neurosci Biobehav Rev. 2005;29(4–5):547–569. doi:10.1016/j.neubiorev.2005.03.008.
58. European Monitoring Centre for Drugs and Drug Addiction (EMCDDA), Drug Misuse Research Division, Health Research Board. Literature Review on the Relation Between Drug Use, Impaired Driving and Traffic Accidents (CT.97.EP.14). Lisbon: Portugal; 1999.
59. Papoian T, Jagadeesh G, Saulnier M, et al. Regulatory forum review*: utility of in vitro secondary pharmacology data to assess risk of drug-induced valvular heart disease in humans: regulatory considerations. Toxicol Pathol. 2017;45(3):381–388. doi:10.1177/0192623317690609.
60. Bharti S, Rani N, Bhatia J, Arya DS. 5-HT2B receptor blockade attenuates beta-adrenergic receptor-stimulated myocardial remodeling in rats via inhibiting apoptosis: role of MAPKs and HSPs. Apoptosis. 2015;20(4):455–465. doi:10.1007/s10495-014-1083-z.
61. Janssen W, Schymura Y, Novoyatleva T, et al. 5-HT2B receptor antagonists inhibit fibrosis and protect from RV heart failure. Biomed Res Int. 2015;2015:438403. doi:10.1155/2015/438403.
62. Canal CE, Morgan D, Felsing D, et al. A novel aminotetralin-type serotonin (5-HT) 2C receptor-specific agonist and 5-HT2A competitive antagonist/5-HT2B inverse agonist with preclinical efficacy for psychoses. J Pharmacol Exp Ther. 2014;349(2):310–318. doi:10.1124/jpet.113.212373.
63. Brody T. FDA’s Drug Review Process and the Package Label: Strategies for Writing Successful FDA Submissions. Academic Press; 2018:441–511.
64. Maroteaux L, Ayme-Dietrich E, Aubertin-Kirch G, et al. New therapeutic opportunities for 5-HT2 receptor ligands. Pharmacol Ther. 2017;170:14–36. doi:10.1016/j.pharmthera.2016.10.008.
65. Herr N, Bode C, Duerschmied D. The effects of serotonin in immune cells. Front Cardiovasc Med. 2017;4:48. doi:10.3389/fcvm.2017.00048.
66. Krabbe G, Matyash V, Pannasch U, Mamer L, Boddeke HW, Kettenmann H. Activation of serotonin receptors promotes microglial injury-induced motility but attenuates phagocytic activity. Brain Behav Immun. 2012;26(3):419–428. doi:10.1016/j.bbi.2011.12.002.
67. Oades RD, Myint AM, Dauvermann MR, Schimmelmann BG, Schwarz MJ. Attention-deficit hyperactivity disorder (ADHD) and glial integrity: an exploration of associations of cytokines and kynurenine metabolites with symptoms and attention. Behav Brain Funct. 2010;6(1):32. doi:10.1186/1744-9081-6-32.
68. Felger JC, Lotrich FE. Inflammatory cytokines in depression: neurobiological mechanisms and therapeutic implications. Neuroscience. 2013;246:199–229. doi:10.1016/j.neuroscience.2013.04.060.
69. Brus R, Nowak P, Szkilnik R, Mikolajun U, Kostrzewa RM. Serotoninergics attenuate hyperlocomotor activity in rats. Potential new therapeutic strategy for hyperactivity. Neurotox Res. 2004;6(4):317–325. doi:10.1007/BF03033442.
70. Kostrzewa RM, Brus R, Kalbfleisch JH, Perry KW, Fuller RW. Proposed animal model of attention deficit hyperactivity disorder. Brain Res Bull. 1994;34(2):161–167. doi:10.1016/0361-9230(94)90013-2.
71. Whitney MS, Shemery AM, Yaw AM, Donovan LJ, Glass JD, Deneris ES. Adult brain serotonin deficiency causes hyperactivity, circadian disruption, and elimination of siestas. J Neurosci. 2016;36(38):9828–9842. doi:10.1523/JNEUROSCI.1469-16.2016.
72. Davids E, Zhang K, Kula NS, Tarazi FI, Baldessarini RJ. Effects of norepinephrine and serotonin transporter inhibitors on hyperactivity induced by neonatal 6-hydroxydopamine lesioning in rats. J Pharmacol Exp Ther. 2002;301(3):1097–1102. doi:10.1124/jpet.301.3.1097.
73. Yoo JH, Kim JI, Kim BN, Jeong B. Exploring characteristic features of attention-deficit/hyperactivity disorder: findings from multi-modal MRI and candidate genetic data. Brain Imaging Behav. 2019. doi:10.1007/s11682-019-00164-x.
74. Qureshi MNI, Oh J, Min B, Jo HJ, Multi-modal LB. Multi-measure, and multi-class discrimination of ADHD with hierarchical feature extraction and extreme learning machine using structural and functional brain MRI. Front Hum Neurosci. 2017;11:157.
75. Sudre G, Szekely E, Sharp W, Kasparek S, Shaw P. Multimodal mapping of the brain’s functional connectivity and the adult outcome of attention deficit hyperactivity disorder. Proc Natl Acad Sci U S A. 2017;114(44):11787–11792. doi:10.1073/pnas.1705229114.
76. Cao M, Shu N, Cao Q, Wang Y, He Y. Imaging functional and structural brain connectomics in attention-deficit/hyperactivity disorder. Mol Neurobiol. 2014;50(3):1111–1123. doi:10.1007/s12035-014-8685-x.
77. Conio B, Martino M, Magioncalda P, et al. Opposite effects of dopamine and serotonin on resting-state networks: review and implications for psychiatric disorders. Mol Psychiatry. 2020;25(1):82–93. doi:10.1038/s41380-019-0406-4.
78. Castellanos FX, Elia J, Kruesi MJ, et al. Cerebrospinal fluid monoamine metabolites in boys with attention-deficit hyperactivity disorder. Psychiatry Res. 1994;52(3):305–316. doi:10.1016/0165-1781(94)90076-0.
79. Oades RD. Dopamine may be ‘hyper’ with respect to noradrenaline metabolism, but ‘hypo’ with respect to serotonin metabolism in children with attention-deficit hyperactivity disorder. Behav Brain Res. 2002;130(1–2):97–102. doi:10.1016/S0166-4328(01)00440-5.
80. Carhart-Harris RL, Nutt DJ. Serotonin and brain function: a tale of two receptors. J Psychopharmacol. 2017;31(9):1091–1120. doi:10.1177/0269881117725915.
81. Miyazaki KW, Miyazaki K, Doya K. Activation of dorsal raphe serotonin neurons is necessary for waiting for delayed rewards. J Neurosci. 2012;32(31):10451–10457. doi:10.1523/JNEUROSCI.0915-12.2012.
82. Miyazaki K, Miyazaki KW, Doya K. The role of serotonin in the regulation of patience and impulsivity. Mol Neurobiol. 2012;45(2):213–224. doi:10.1007/s12035-012-8232-6.
83. Cryan JF, Lucki I. Antidepressant-like behavioral effects mediated by 5-Hydroxytryptamine(2C) receptors. J Pharmacol Exp Ther. 2000;295(3):1120–1126.
84. Del’Guidice T, Lemay F, Lemasson M, et al. Stimulation of 5-HT2C receptors improves cognitive deficits induced by human tryptophan hydroxylase 2 loss of function mutation. Neuropsychopharmacology. 2014;39(5):1125–1134. doi:10.1038/npp.2013.313.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.