")
Back to Archived Journals » Advances in Genomics and Genetics » Volume 5
New insight into the gut microbiome through metagenomics
Received 15 September 2014
Accepted for publication 20 October 2014
Published 29 January 2015 Volume 2015:5 Pages 77—91
DOI https://doi.org/10.2147/AGG.S57215
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr John Martignetti
Boyang Ji, Jens Nielsen
Department of Chemical and Biological Engineering, Chalmers University of Technology, Göteborg, Sweden
Abstract: The human gut is colonized by different types of microorganisms, which are known to play important roles in the human host by maintaining physiological homeostasis. The human host provides a nutrient-rich environment, and the microbiota provides some necessary functions that humans cannot perform. A comprehensive analysis of the human gut microbiome is thus important for revealing the mechanisms of these host–microbe interactions. The development of high-throughput sequencing technology and related computational frameworks enables exploration of the metabolic interactions and their roles in human health and diseases. Herein, we describe the metagenomic methods used in human gut microbiome studies and review the roles of gut microbiota as well as the integrative analyses of metagenomic data with other omics data. Finally, we discuss the application of constraint-based modeling to elucidate the microbe–microbe interaction and host–microbe interaction in the human gut microbiota.
Keywords: dysbiosis, host–microbe interaction, metabolic modeling
Introduction
Trillions of microorganisms have been colonized in the human gastrointestinal tract.1 Anaerobic bacteria dominate the gut microbial community, but it also includes aerobic or facultative anaerobic bacteria, archaea, viruses, and eukaryotes. These microbial communities outnumber the total number of human cells by a factor of 10 and contain 150 times as many genes as those encoded by the human genome.2 The human gut microbiota can therefore be considered as an organ or super-organism that coevolved with the human host to achieve a beneficial symbiotic relationship.3,4 The human host provides a nutrient-rich environment, and the microbiota contributes to broad ranges of biochemical and metabolic capacities that the human body does not possess, such as the digestion of resistant starch or polysaccharides, production of some vitamins, and bile acid biotransformation. Unlike the human genome, the gut microbiome is dynamic and can be altered by environmental factors (diet,4–6 drugs,4,7,8 probiotics,4,9 etc).
Development of cultivation-independent approaches based on the 16S ribosomal RNA (rRNA) sequences expands our understanding of the composition and diversity of the gut microbiota. Bacteria from the Bacteroidetes and Firmicutes phyla dominate the gut microbiome, with members from Proteobacteria, Actinobacteria, and Fusobacteria also being abundant.2 With the emergence of next-generation sequencing (NGS) technology, the role of the gut microbiome in human health and diseases is becoming clearer, and the dysbiosis of gut microbiota can be associated with numerous diseases (type 2 diabetes [T2D], inflammatory bowel disease [IBD], nonalcoholic fatty liver disease [NAFLD], etc).4,10,11 Herein, we aim to summarize the metagenomic methods used in the human gut microbiome studies, review the role of the human gut microbiota, and describe integrative analyses of metagenomic data and other omics data. Finally, we will discuss the application of genome-scale metabolic models in the metabolic interactions of microbial communities.
Metagenomics: toward a better understanding of the gut microbiome
Traditional microbiology was dependent on growing pure cultures of species in specific media. However, numerous uncultivated microbial species have been identified in diverse environments and their cultures are still limited.12 Metagenomics initially started with the cloning of DNA fragments from environmental samples, followed by functional genomic screening,13 and were quickly extended by the application of whole-genome shotgun sequencing in microbial populations.14 In these initial projects, large DNA insert metagenomic libraries were constructed and sequenced, hence giving insights into the functional, structural, and phylogenetic composition of microbial communities in environmental samples in a culture-independent way. The revolution of NGS technologies,15,16 such as 454 pyrosequencing,16,17 SOLiD™ (sequencing by ligation),16 and Illumina (sequencing by synthesis),16 have vividly accelerated the development of sequencing-based metagenomic projects. The rapid and substantial reduced cost of NGS technologies make it possible to sequence numerous environmental samples in parallel and in a cost-effective manner, and the development of analytical tools for NGS also enables researchers to understand the microbial communities of interest from a phylogenetic and functional perspective.
The human gut microbiome is vast, and consists of about 1014 bacterial cells, which is ten times the number of cells in the human body. A detailed inventory of this microbial “factory” is nearly unreachable by conventional microbial techniques.18,19 Culture-based studies have characterized hundreds of microbial species in the gut microbiota, including archaea (eg, Methanobrevibacter genus),20 Gram-positive bacteria (eg, Bifidobacterium, Clostridium, Eubacterium, Lactobacillus, and Ruminococcus genera),21–23 and Gram-negative bacteria (eg, Bacteroides and Fusobacteria genera).21,22 However, these approaches generally work on the “fast-growth” or “easy-to-grow” species, which are just 10% to 50% of the gut bacteria.24 With the application of culture-independent metagenomics approaches to study the human gut microbiota, more than 1,000 species-level microbial phylotypes have been identified, most of which belong to just a few phyla (Bacteroidetes and Firmicutes).2,19,25 In addition to the taxonomic diversity of the microbiota, metagenomic analysis also provides a more comprehensive view of functional association related to the human body by identifying microbial genes involved in specific metabolic pathways or functions. Such functional profiles of the gut microbiome have been subjected to intensive study in recent years, driven by large-scale sequencing projects such as the Human Microbiome Project26 and the Metagenomics of the Human Intestinal Tract (MetaHIT) project.2 The Human Microbiome Project characterized the diversity of human-associated microbial communities at different body sites, including the gastrointestinal tract, in the healthy human population,26 whereas MetaHIT focused on the correlation between the gut microbiome and intestinal diseases, such as obesity and IBD.2 These studies have demonstrated the interpersonal microbiome composition variability and shared core functionalities,2,27 which provide a useful knowledge base for understanding gut microbiome functions in human health. For example, the shotgun metagenome analysis of two healthy adults revealed a significant metabolic enrichment of glycans, amino acids, and xenobiotics; methanogenesis; and 2-methyl-D-erythritol 4-phosphate pathway-mediated biosynthesis of vitamins and isoprenoids in the human gut microbiome.28 Moreover, the gut microbiota is not constant, and can be altered by diet,5,6,29 antibiotic usage,7,8 aging,30,31 and many other factors. As well as aiding investigation of the relationship between gut microbiota and human health, metagenomic studies have also necessitated a better understanding of the dynamics of gut microbiome. For instance, Davenport et al29 found that seasonal differences in diet shape the gut microbiome compositions of Hutterite individuals differently between the winter and summer. Rampelli et al30 observed an age-related reduction of the abundance of genes involved in short-chain fatty acid (SCFA) production, and identified 116 microbial genes that significantly correlated with aging. Altogether, a metagenomic-based approach is not only a powerful tool for exploring the microbiome community dynamics, but also for characterizing the functional profiling of communities.
Bioinformatics challenges: the current state of gut metagenomic data analysis
Although DNA sequencing has been used since the 1970s,32 it was formerly too expensive and time-consuming for application in high-throughput studies. With the development of NGS technologies in 2004,16 massively parallel DNA sequencing became economically affordable for most scientists and enabled sequencing of environmental DNA samples, and metagenomic studies have since then become gradually more common.33,34 As shown in Figure 1, two different types of NGS-based approaches have been used for human gut microbiome studies: 1) amplicon/target sequencing using specific primers (ie, primers for 16S rRNA genes or other marker genes), and 2) whole-genome shotgun sequencing with genome amplification using random primers.
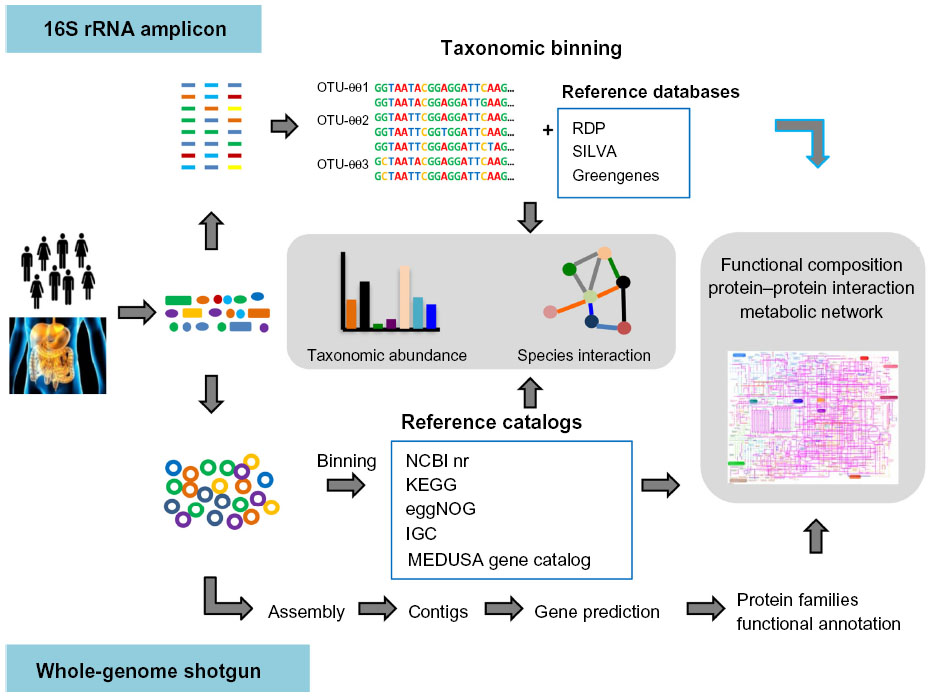 | Figure 1 Overview of bioinformatics methods for gut microbiota metagenomic analysis. |
In typical 16S rRNA marker gene-based metagenomic studies, DNA is extracted from samples without the need to culture microbes in the laboratory. The amplification of 16S rRNA genes is performed with specific primers, and is followed by parallel sequencing with a selected NGS platform (Figure 1). A bioinformatics challenge that arises immediately from 16S rRNA sequencing analysis is the precise definition of a “unique” sequence.33 Considering that errors can be induced during polymerase chain reaction amplification and sequencing processes, the utilization of 100% identity would be unrealistic, and treat the same clonal genomes as different organisms. Thus, a user-defined similarity cutoff of 95%, 97%, or 99% is usually used in binning 16S rRNA gene sequences into operational taxonomic units (OTUs) or sometimes phylotypes in practice.33 In general, there are two main approaches for OTU clustering. The most commonly used is de novo OTU clustering, which compares all the sequences to each other for clustering into OTUs without any external references. Tools such as CD-HIT35 or UCLUST,36 which execute the alignment-free clustering method, are broadly used to cluster OTUs independently of taxonomy, as it has been suggested that alignment quality has a significant impact on OTU clustering.37 In contrast, the taxonomy-dependent approach searches sequences against a 16S rRNA reference database (usually Greengenes,38 the Ribosomal Database Project [RDP],39 or SILVA40), and sequences failing to match the reference database are discarded. This generates better results and includes less erroneous sequences in the OTU, because all OTUs have been predefined and well checked in the reference databases. However, it also discards numerous sequences due to the failure to match to reference databases. Alternatively, the third approach is a two-step process consisting of reference-based OTU clustering followed by de novo clustering of sequences that fail to match the reference database. Once OTUs are determined, representative sequences from each OTU can be chosen and used to assign taxonomy to the OTUs. As described previously, SINA,41 RDP Classifier,42 and the NAST algorithm43 can be used to assign taxonomic affiliations to sequences using corresponding 16S rRNA databases. Following taxonomic binning, further bioinformatics analysis of taxonomic abundance and microbiome diversity can be performed. Diversity within a sample is usually described as alpha-diversity, which captures both the organismal richness of a sample and the evenness of the organism’s abundance,33 whereas the beta-diversity is often referred to as the diversity between multiple microbial samples.33 Moreover, multivariate analysis methods, such as principal coordinates analysis44 or principal component analysis,44 are frequently used in human gut microbiome studies to test multivariate hypotheses concerning the effects of metadata (eg, diet, age, genotype, and diseases) to the microbial diversity. With the taxonomic composition and abundance data in different samples, it is possible to predict the ecological interaction among those gut microbes by inferring co-occurrence or correlation network.45 In conclusion, 16S rRNA-based metagenomic sequencing provides insights into the population characteristics of the human gut microbiota. However, 16S rRNA genes do not provide direct evidence of the community’s functional capabilities. Recently, computational approaches (ie, PICRUSt46 and Genometraits47) were successfully employed to predict the functional composition of metagenomes using marker genes and a reference genome database. Therefore, combination of 16S rRNA metagenomics and single species genomics could be an efficient way to provide insight into both organismal and functional diversity in the gut microbiome.
As an alternative to 16S rRNA sequencing, the total DNA is extracted and amplified using random primers and then sequenced by shotgun metagenomic analysis. Shotgun metagenomic analysis provides the opportunity to simultaneously explore the taxonomic composition and bimolecular functions in microbial communities at the gene level. Usually, there are two general strategies for whole-genome shotgun metagenomic data analysis. The simplest method is assembly-free, while, with the other, the reads are subjected to assembly. If assembly is not performed, the produced reads are treated as 16S rRNAs or gene fragments and are analyzed directly. After preprocessing to reduce sequencing errors, taxonomic binning is performed to determine the taxonomic affiliation for each read (Figure 1). Similarity-based taxonomic assignment utilizes the similarity between query sequences and sequences from reference databases (ie, NCBI RefSeq,48 KEGG,49 and eggNOG50). Recently, several catalogs of reference genes were constructed, such as the human gut microbial gene catalog from MetaHIT,2 the global human microbial gene catalog from MEDUSA,51 and the integrated gene catalog (IGC).52 These gene catalogs facilitate the quantitative characterization of human gut microbiome data to understand the community composition and functional variations. Sequence composition-based approaches are also widely used for taxonomic binning, which utilizes the genome-wide taxonomic signals contained in the fragment-wide GC content, codon usage, or the frequency of short oligomers (K-mers).53 As compositional signature is a global character of sequence, sufficient numbers of sequences instead of the whole genome are required for interference of the composition. Consequently, these compositional binning methods generally do not need the alignments of reads to the reference sequences and are able to process large datasets relatively rapidly. With these two approaches, several computational frameworks (ie, MEGAN,54 PhymmBL,55 MetaPhlAn,56 and mOTU57) were developed to assign the microbial taxonomy efficiently. As described previously, further bioinformatics analysis of taxonomic abundance and microbiome diversity can be performed following taxonomic binning. With an assembly-based analytical strategy, the collinear metagenomic reads from the same genome are merged into contigs and generate much longer sequences,34 which can simplify bioinformatics analysis relative to short reads. After assembly, the functional diversity of the microbial communities can be quantified by inferring the functional annotations of these metagenomic sequences. Generally, two steps make up the annotation process: gene prediction and functional annotation. The first step identifies encoding genes in the sequence fragments. This step can be performed in different ways. For example, some tools (ie, MetaGeneMark,58 Glimmer-MG59) identify genes de novo. While another approach involves translating each sequence into all six possible encoding frames and searching the resulting protein sequences against the annotated reference protein database to identify genes that encode proteins similar to the reference proteins. Once the encoding sequences are predicted, they can be subjected to functional annotation. Numerous databases and online tools, such as KEGG,49 the Clusters Of Orthologous Groups database (COG),60 Pfam,61 SEED,62 eggNOG,50 MG-RAST,63 CAMERA,64 and IMG/M,65 can be used to infer the functional annotation and classification. With functional classifications and abundance data, the functional enrichment of pathways in a sample or between multiple samples can be inferred by a statistical test (ie, Fisher’s exact test, gene set enrichment analysis). An increasing number of strategies and toolboxes (ie, ShotgunFunctionalizeR,66 STAMP,67 MetaPath,68 metagenomeSeq69) have been developed for identification of the substantial functional differences between microbial samples at the function level and association of the abundance differences with the corresponding metadata. By comparing the metabolic gene abundance in different samples, one can determine the differences in the metabolic capacity of microbial community and identify possible interactions between environment and microbiome. In addition to inferring the differential abundance in metabolic pathways, Abubucker et al70 recently described a promising methodology, named HUMAnN, for reconstructing the functional potential of human-associated microbial communities from shotgun data. Such metabolic reconstruction methods for the whole microbial community provide a way to characterize the metabolic pathways in the community level, and enable the comparison of community roles in different communities.
From the perspective of both taxonomic and functional composition from metagenomes, the gut microbiota might be linked to and contribute to many complex diseases in humans. For example, a number of studies have revealed that obesity is associated with an increase in Firmicutes and a relatively lower abundance of Bacteroidetes.27,71 A T2D study also showed that the proportion of Firmicutes and Clostridia was significantly reduced in patients compared with the normal group.72 However, more detailed information is required to associate the gut microbial compositional alternations with its impact on human health and diseases. Integrating metagenomic abundance data with a systems-level metabolic network analysis73 shows that both gene-level and network-level topological differences are associated with obesity and IBD. The enzymes enriched in obese or IBD microbiomes exhibit unique topological features, which suggest that diseases may be associated with high-order modes of deviation in the microbiome.73 Metagenome-wide association study based on shotgun metagenomic sequencing of the stool samples from T2D patients and nondiabetic controls identified nearly 60,000 T2D-associated markers potentially useful for classifying T2D.74 Recently, another association study identified 26 metagenomic gene clusters to be differentially abundant between a T2D group and the normal glucose tolerance group.11 A mathematical model was developed based on the species and metagenomic gene cluster profiles, and application of this model showed better prediction in classification of T2D individuals than traditional markers, such as body mass index and waist–hip ratio.11 Moreover, comparative analysis of gut microbiome from 98 patients with liver cirrhosis and 85 healthy controls found that 38 and 28 metagenomic species are enriched in healthy individuals and patients, respectively. With 15 gene markers, a support vector machine classifier was constructed and confirmed that these gene markers can be used for identifying patients with liver cirrhosis.75
Functional metagenomics of gut microbiome
Although metagenomic studies have provided deep understanding about the relationships between gut microbiome and human health, it is still worth noting that other omics approaches with functional metagenomics can be used as complementary ways to achieve comprehensive insights into the host–microbiome interaction. Functional metagenomic screens, originally proposed as one method by which to characterize the uncultured microbes, have been adapted to characterize the functions of human microbial communities, representing an interesting interdisciplinarity between ecological microbiology and biomedicine.76 As previously noted, the gut microbiota is involved in the host metabolism regulation.77 Functional metagenomic screening is thus a powerful tool for detecting the capacities of microbial metabolism. One example of such application is the discovery of new carbohydrate active enzyme (CAZyme) genes in the human gastrointestinal microbiota. The gut bacteria produce a vast amount of CAZymes in order to digest dietary fibers into metabolizable monosaccharides and disaccharides;78,79 15,882 different CAZymes have been detected in a mini-microbiome composed of 177 sequenced members of the human gut microbiota, representing its major phylogenetic lineages.78 Tasse et al79 applied high-throughput functional screens and identified 310 clones showing beta-glucanase, hemicellulase, galactanase, amylase, or pectinase activities. In addition, the functional genomic screens have also been employed to identify potential enzymes from the gut microbiome for biotechnological applications, such as biofuel production.80 For example, new xylanases81 and butyryl-CoA:acetate CoA-transferase82 have been obtained by screening the human gut microbial communities. Another area of application for functional metagenomic screens is in the discovery of antibiotic resistance genes. Antibiotic resistance, which reduces the clinical ability to fight infectious diseases and increases treatment costs, is a worldwide public health threat. Recent metagenomic resistome sequencing analysis detected resistance genes for 50 of the 68 known antibiotic classes and subclasses from 252 fecal samples of 207 individuals from three countries.83 However, the sequencing-based method is limited to resistance genes that are already known, as sequences with low similarity to known reference antibiotic sequences cannot be identified. Thus, functional genomic screens, which are involved in cloning and expression of gene fragments in heterologous hosts (such as E. coli), and screens for resistance genes by growing transformants on antibiotic containing media at concentrations that the original host is susceptible, will provide the chance to discover new resistance genes. Sommer et al84,85 characterized 95 unique functional antibiotic resistance genes in saliva and fecal samples from two unrelated healthy humans and 115 unique antibiotic genes from cultured aerobic gut microbiome isolates. Although most of these resistance genes were highly related to known resistance genes in pathogenic bacteria, ten previously unknown beta-lactamase genes were identified, which showed from 35% to 61% identity to proteins from GenBank. Moreover, another functional metagenomic screen for human microbiome also identified eight new antibiotic resistance genes against amoxicillin, D-cycloserine, and kanamycin.86
The role of the human gut microbiome
At the functional level, numerous gut metagenomic studies have shown that the gut microbiota has coevolved with the human host to perform important biochemical functions that impact human metabolism.87–89 The gut microbiota has been suggested as an auxiliary metabolic organ due to the microbes’ metabolic capacity.88,90 The metabolic functions of the gut microbiota include energy harvest and utilization, macromolecule production (eg, SCFA, amino acids), vitamin biosynthesis, bile acid deconjugation and dehydroxylation, and xenobiotic metabolism, among others (Table 1). The human host is able to adsorb monosaccharides such as glucose and galactose in the small intestine, and it can also hydrolyze certain disaccharides such as sucrose and lactose to their constituent monosaccharides.91 However, the human host has limited enzymatic ability to degrade complex polysaccharides from the diet such as resistant starch, non-starch polysaccharides, and plant fibers. Therefore, simple sugars (eg, glucose, galactose) are absorbed in the proximal small intestine by active transport, and undigested dietary polysaccharides enter into the distal small intestine and colon, where they are degraded by microbes. The anaerobic degradation of these undigested dietary polysaccharides is a fermentation process involving numerous gut microbes, including Bacteroides, Bifidobacterium, Ruminococcus, and Roseburia spp., as well as some microbes from Clostridium, Eubacterium, and Enterococcus genera92 (Table 1). The major end products of polysaccharide fermentation are SCFA acetate, propionate, and butyrate, which provide energy for the host and are involved in a number of physiological functions (Figure 2). These SCFAs can be used as the energy source of epithelial cells or peripheral tissues.4 Previous study have also suggested that SCFAs stimulate the colonic sodium and fluid absorption, modify the microbial composition, and regulate the glucose and energy homeostasis.93 Acetate and propionate can be taken up by the liver and be used as substrates for liver cholesterol and fatty acid biosynthesis; propionate can also act as a substrate for gluconeogenesis.94 Butyrate has been shown to improve insulin sensitivity, increase energy expenditure, and modulate immune responses by macrophages.95,96 After carbohydrate fermentation in proximal colon, the carbohydrate availability decreases, and protein becomes the main energy source for the gut microbiota in the distal colon.97,98 The predominant proteolytic species include strains from Bacteroides, Propionibacterium, Streptococcus, Clostridium, Bacillus, and Staphylococcus.97 In addition to SCFA, hydrogen, CO2, ammonia, and branched chain volatile fatty acids are produced during protein degradation.97,98 The intestinal gut microbiota can also act as an important supplier of vitamin K as well as water-soluble vitamin B (ie, biotin, cobalamin, riboflavin, thiamine, and folate) (Figure 2).87 With numerous gut microbioal genomic data, the vatamin biosynthesis capacities are extensively explored. For example, the Bifidobacterium and Lactobacillus spp. have been verified to have folate biosynthetic properties;99,100 Lactobacillus reuteri also has the ability to produce cobalamin (vitamin B12).101 Besides production of SCFA and vitamins, the gut microbiome is also involved in bile acid metabolism and the metabolism of choline. Bile acids, which are important for the metabolism of dietary fat and the absorption of cholesterol and fat-soluble vitamins, are steroid acids that are synthesized from cholesterol by the liver and secreted in the bile. During the enterohepatic circulation between the gut and liver, 90%–95% of bile acids are reabsorbed by the intestine and returned to the liver. Bile acid metabolism performed by the gut microbiome consists of conjugation and hydroxyl group oxidation, which transform the primary bile acids into secondary bile acids (Figure 2).102 Choline is one of the essential dietary nutrients that is metabolized in the liver, and is important for lipid metabolism and the synthesis of very-low-density lipoprotein.4,77 Gut microbial enzymes (eg, glycyl radical enzymes in Desulfovibrio)103 catalyze the conversion of choline to trimethylamine, which can be transferred into trimethylamine N-oxide in the liver (Figure 2). The microbial conversion of choline to trimethylamine is a metabolic hallmark associated with NAFLD104 and cardiovascular disease.105
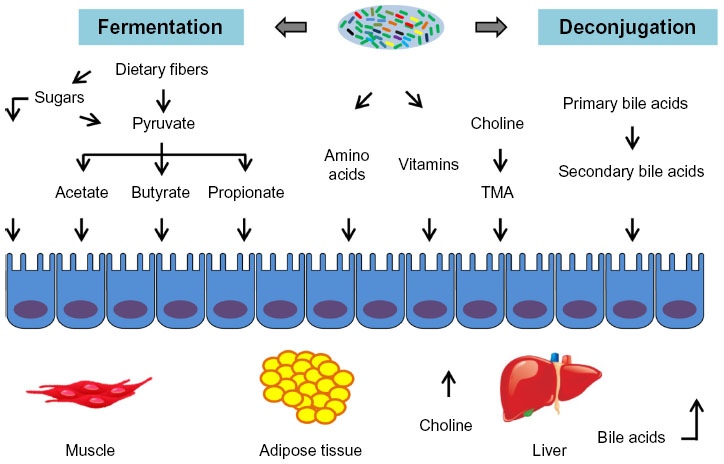 | Figure 2 Metabolic interaction between gut microbiota and host. |
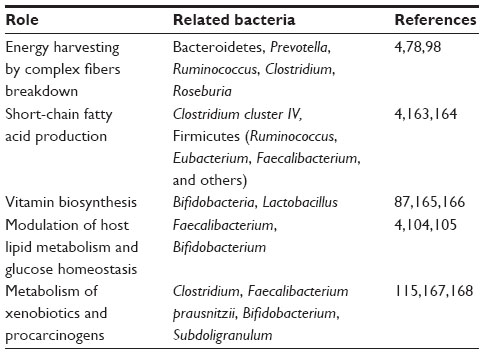 | Table 1 Beneficial metabolic contributions of intestinal microbes to human health |
As described above, the gut microbiota establish a mutualistic relationship with the human host and provide beneficial metabolic functions. There is increasing evidence to link the gut microbiome with human disease. IBD is a group of inflammation disorders of the small intestine and colon, which can be classified into two forms: Crohn’s disease (CD) and ulcerative colitis (UC). Evidence has suggested that IBD is a complex disease arising from the interaction of genetic and environmental factors.106 From a genetics perspective, genome-wide association studies and subsequent meta-analyses have identified a total of 163 IBD loci.10 From the metagenomic perspective, it has been shown that IBD patients have an altered gut microbiome composition with decreased diversity, in particular, reduced diversity in the Firmicutes phylum (eg, Clostridium leptum).106,107 Along with reduced Clostridium spp, a decrease in Bifidobacterium spp. was also observed in fecal samples from CD patients.108 As previously described, Clostridium and Bifidobacterium spp are important producers of butyrate. In agreement with the decrease of Clostridium and Bifidobacterium, metabolomic analysis of fecal samples from patients with CD and UC showed decreased levels of butyrate and acetate compared with the control group.106,109 Moreover, the increased abundances of bacteria, including Enterobacteriaceae, Pasteurellaceae, Veillonellaceae, and Fusobacteriaceae, and decreased abundances of Erysipelotrichales, Bacteroidales, and Clostridiales, correlate strongly with IBD status. Microbiome comparison between CD patients with and without antibiotic exposure also indicates that antibiotic use amplifies the microbial dysbiosis associated with CD.110 Comparing the microbial signatures between the ileum, the rectum, and fecal samples indicates that, at the early stage of disease, assessing the rectal mucosal-associated microbiome offers potential for convenient and early diagnosis of CD,110 The alternation in the gut microbial composition as well as the reduced bacterial diversity have been associated with obesity. Significant enrichment of Firmicutes and decrease of Bacteroidetes has been observed in obese individuals compared with lean indiviuals.71,111,112 Metagenomic analyses demonstrated that these changes affect the metabolic potential of the gut microbiota, and the obese microbiome has an increased capacity for energy harvest.27,71 The transplantation of lean and obese fecal microbiota into germ-free mice confirmed that the trait of obesity is transmissible through fecal transplants from obese mice.71,113 T2D is one type of metabolic disorder whose primary cause is obesity-linked insulin resistance. Recent studies showed that the proportions of Firmicutes and Clostridia were significantly reduced in the T2D group compared to the control group, and the ratio of Bacteroidetes to Firmicutes as well as the ratio of Bacteroides-Prevotella group to Clostridium coccoides-Eubacterium rectale group correlated positively and significantly with plasma glucose concentration but not with body mass index.72 With the application of metagenome-wide association studies in gut microbiome samples from T2D patients, a moderate degree of gut microbial dysbiosis was observed in the T2D patients,11,71 and potential gut microbial markers useful for T2D classification and early diagnosis were thereby identified.
Integrative analysis: beyond metagenomics
Metagenomics is a useful tool for describing the genetic potential and compositional changes of the gut microbiota, but it cannot provide any information about the gene expression in the microbial community. One way to overcome this limitation is environmental meta-transcriptomics to assess which genes are expressed in the gut habitat. Gosalbes et al114 applied meta-transcriptomics and 16S rRNA gene sequencing to analyze the human gastrointestinal tract microbiota in ten healthy individuals. Carbohydrate metabolism, energy production, and cellular components synthesis were the main functions of the expressed genes. In contrast, housekeeping activities such as amino acid and lipid metabolism were expressed at a lower level in the meta-transcriptome. Moreover, a study combining flow cytometry, 16S rRNA gene sequencing, and meta-transcriptomics demonstrated that the gut contains a distinctive set of active microorganisms, primarily Firmicutes.115 Short-term exposure to a panel of xenobiotics significantly alters the gene expression of this active gut microbiome, and the changes are involved in antibiotic resistance, drug metabolism, and stress response pathways.115 Another approach to evaluating functions of the gut microbiome is to perform human gut microbial metaproteomics. The first human fecal microbiota metaproteomic analysis from two infants obtained proteins by two-dimensional gel electrophoresis, followed by excision and de novo sequencing of targeted spots on the gel.116 High-throughput, non-targeted mass spectrometry approaches, which can determine the identity of thousands of microbial proteins, have also been successfully applied to human fecal samples from two Swedish inviduals.117 The resulting metaproteomes had more proteins for translation, energy production, and carbohydrate metabolism when compared to what was earlier predicted from metagenomics. In a recent study, the composition and temporal stability of the intestinal metaproteome was determined by using fecal samples from three healthy subjects over a period of 6 to 12 months.118 Combined with metagenome and single bacterial genome sequence data, it showed that the fecal metaproteome is subject-specific and stable during a 1-year period. Meta-metabolomics is another widely used omics approach to studying the human gut microbiome, and has been applied to fecal water extracts, plasma, and urine to study numerous disorders (eg, colorectal cancer, obesity, and IBD).119,120 Dumas et al104 tested the effects of dietary change from a 5% low-fat diet to a 40% high-fat diet on the plasma and urine metabolic profiles in a mouse strain susceptible to NAFLD. The results showed that NAFLD is associated with dysbiosis in choline metabolism, and the conversion of choline into methylamines by the microbiota on a high-fat diet decreases the bioavailability of choline and mimics the effect of choline-deficient diets, causing NAFLD. Moreover, metabolic profiles from germ-free and conventional mice show that the gut microbiome strongly impacts bile acid metabolism.121 Furthermore, Wikoff et al122 directly measured the metabolomic profiles of plasma extracts from germ-free and conventional mice. More than 10% of all detectable serum metabolites showed at least 50% variation in concentration between the mice with and without gut microbes. Metabolomic approaches combined with metagenomic analysis have also proved useful for assessing the metabolic changes associated with disease. For example, Sellitto et al123 characterized the differences between the developing microbiota of infants with genetic predisposition for celiac disease and with a nonselected genetic background and found a lack of Bacteroidetes in the gut microbiota of infants genetically susceptible to celiac disease. Simultaneous metabolomic analysis also revealed several metabolites (eg, amino acids, SCFA, sugars) that can be used as potential biomarkers for the prediction of celiac disease.123,124
Predictive gut microbial metagenomics: from genomes to models
As discussed above, metagenomic data can help us understand how the gut microbiota and its corresponding metabolic capacity affect human health. The biological functions of gut microbiome can be inferred by using metagenomics annotation information. From shotgun metagenomic data, a network-based method, called PRMT, was employed to explore the metabolic capacities of microbial communities.125 This approach first generates an environmental metabolome matrix (EMM) containing metabolites and reaction information according to the enzyme reactions in the metagenomic sample. Next, the normalized sequence abundances associated with metabolic reactions are compared across multiple metagenomes to identify the relative change in the metabolites to be produced or consumed. Therefore, PRMT predicts the potential for relative changes in the production or consumption of the metabolites inferred to be present in the environment. For more detailed predictions of metabolic phenotypes for microbial communities, two metabolic modeling frameworks, topology-based network models and constraints-based models, can be applied.126 In the topology-based network approach, the metabolic network is usually constructed according to the automatic homology-based enzyme destination using databases such as KEGG and following enzyme-reaction destination.49 Thus, the network can be represented as a simple directed graph, wherein nodes denote metabolites and edges represent enzymes that convert substrates and products in the different reactions, and the corresponding network properties can be inferred using different tools.127,128 Borenstein et al129 define the concept of a metabolic network’s “seed set” according to the network topology. The seed set is a set of metabolites that are exogenously acquired and which provide a computational framework to infer the environmental interface of a given metabolic network.129 With the seed sets, the metabolic competition index and the metabolic complementary index are defined to describe the metabolic interaction between two pairwise species. Applying this computational approach to the human gut microbiome showed that co-occurring species usually strongly compete, suggesting that microbiome assembly is dominated by habitat filtering.130 This approach has been applied to metabolic exchanges between two coresident intracellular symbionts, Baumannia cicadellinicola and Sulcia muelleri, and their insect host, Homalodisca coagulata, and indicated that the seeds that appear to be obligatory for metabolite synthesis are involved in the symbiotic function.131 Constraint-based modeling is another way of mathematically representing metabolic networks known as the stoichiometric matrix, and aims to define the set of metabolic flux constraints in the given species (Figure 3).132 The reconstruction of genome-scale metabolic models includes four main phases: 1) generating the draft model based on the genome annotation; 2) manual curations according to the literature; 3) functional validation with experimental data; and 4) simulation and analysis.133 The manual reconstruction of genome-scale metabolic models is complex and time-consuming, but numerous tools (eg, Model SEED,134 Pathway Tools,135 RAVEN136) that utilize the biological knowledge from different databases (such as KEGG,49 Reactome,137 BioCyc,138 BRENDA,139 and PubMed140) have been developed to perform automatic or semiautomatic reconstruction. The RAVEN toolbox136 was developed recently and has been used to reconstruct metabolic models for Pichia stipitis,141 Pichia pastoris,141 Saccharomyces cerevisiae,142 and Penicillium chrysogenum.136 This toolbox reconstructs draft models based on the homology to the reference organism and performs automatic gap-filling to ensure that the the different parts of metabolic network are connected. With these available tools, genome-scale metabolic models have been assembled for a growing number of microbes, including numerous gut microbes, such as Bifidobacterium adolescentis143 and Faecalibacterium prausnitzii.143 The availability of multiple microbial metabolic models provides the chance to model the microbial communities to elucidate interactions between different species (Figure 3). Stolyar et al144 constructed a two-species model to predict the syntrophic growth of Desulfovibrio vulgaris and Methanococcus maripaludis, while Zhuang et al145 predicted relative proportions of Geobacter sulfurreducens and Rhodoferax ferrireducens in geochemically distinct zones via a dynamic multispecies metabolic modeling framework. Moreover, using 118 bacterial metabolic models, Freilich et al146 predicted possible cooperation, competition, or neutral interactions in pairs of species and identified a high level of competition between species with mutually exclusive distribution patterns. However, constraint-based modeling of the microbial community mainly focuses on simple systems, ie, one or two species. Recently, the genome-scale metabolic models for three gut microbial species (Bacteroides thetaiotaomicron, E. rectale, and Methanobrevibacter smithii) as representatives of three important phyla in the human gut (Bacteroidetes, Firmicutes, and Euryarchaeota) were reconstructed.147 The integration of these three models with transcriptomics data demonstrated that the models can be used as a scaffold for understanding bacterial interactions in the gut. Moreover, genome-scale, manually curated human metabolic reconstructions (EHMN,148 HMR,149 Recon1,150 Recon2,151 and HMR 2.0152) have also been published. The newest version of HMR includes 7,707 reactions and 5,997 metabolites. With both human and microbial metabolic models, the interaction between host and microbe can be inferred (Figure 3).153 For example, Bordbar et al154 built an integrated host-pathogen metabolic model to decipher the intracellular infection of Mycobacterium tuberculosis in human alveolar macrophage phagosomes. Interestingly, the host-pathogen model is much more accurate in predicting M. tuberculosis gene essentiality than the single pathogen model, and integration of the host-pathogen model with transcriptional data shows wide heterogeneity of macrophages in three different infection stages. An integrated metabolic model of B. thetaiotaomicron (a prominent representative of human gut microbiota) and mouse with a joint compartment allowing metabolite exchanges was reconstructed.155 This integrated model successfully identified metabolite exchanges and stimulated the interaction (cross-feeding and competition) between these two organisms.
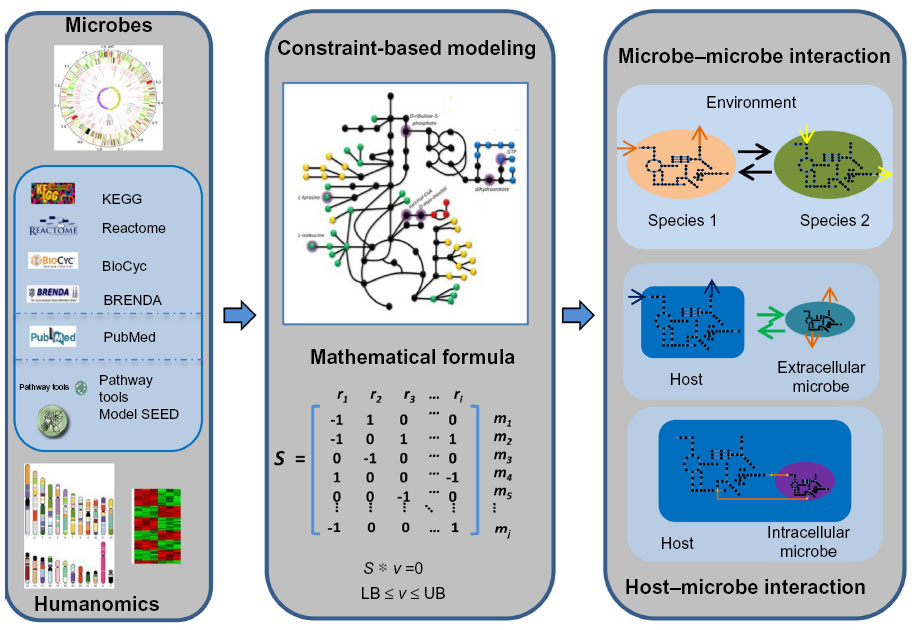 | Figure 3 Reconstruction of microbe–microbe and host–microbe metabolic models. |
Conclusion and perspective
Metagenomics is useful for human gut microbiome studies, and standardized experimental procedures and bioinformatics analysis tools have been developed. The application of association studies or other machine-learning methods to metagenomics data also provides the potential for development of new microbial markers for early disease diagnostics. However, how the functions and mechanisms of the gut microbiota contribute to human health is still not clear, and our understanding of the gut microbiome will require more detailed functional analysis of the gut microbiota. Thus, integrative analysis of multiple datasets from metagenomics, meta-transcriptomics, metaproteomics, and meta-metabolomics will facilitate the discovery of impaired metabolic pathways linked to disease status. Such integrative analyses will also be important for investigation of transcriptional regulation, signal transduction, or dynamic response of the gut microbial community. In addition, an interesting next step will be the opportunity to investigate the dynamic behavior of the gut microbiome. Numerous studies, which have included temporal variability in healthy adults,156 temporal response to antibiotic perturbations,157 and dietary changes,6 have provided insights into the dynamic changes of microbiome, but most of these studies only utilized the composition data of microbial species and lacked simple analytical tools to infer the time series of microbiome data. Consequently, the development of new computational frameworks for integrative dynamic analysis of different microbiome data (transcriptomics, proteomics, metabolomics) will be necessary to explore the microbial behaviors in the gut ecosystem.
Genome-scale metabolic models have been successfully used for predicting the metabolic capacity of single organisms in metabolic engineering. Recently, constraint-based modeling has also been applied to the human gut microbiome, providing a systematic view of metabolic interaction between host and microbes.158 Although the gut microbiome has been simulated by two-species models143,155 or three-species models.147 Considering the numerous species within the gut microbiome, integration of multiple single species and reconstruction of the gut microbial community model remain too complex and need to be scaled up. Although some modeling frameworks (ie, DMMM,145 OptCom,159 d-OptCom,160 cFBA,161 COMETS,162) for community metabolic modeling have been developed, several conceptual challenges still need to be addressed.126 For example, the composition of the gut microbial community is highly flexible and environmentally dependent, and traditional objective functions (eg, maximizing biomass) are not appropriate and need to be replaced by putative energy, biosynthesis principles, or the tradeoff between different species. In addition, integrating metabolic models with composition data from metagenomic sequencing will also be valuable for evaluating the functional role of single species in the gut microbial community.
Acknowledgments
We gratefully thank Leif Väremo, Manuel Garcia-Albornoz, and Saeed Shoaie for critical review of the manuscript. This work was financially supported by the Knut och Alice Wallenberg Stiftelse, Torsten Söderberska Stiftelse, and the EU Framework VII project MetaCardis (grant agreements HEALTH-F4-2012-305312/METACARDIS).
Disclosure
Jens Nielsen is a shareholder in MetaboGen AB. The authors report no other conflicts of interest in this work.
References
Turnbaugh PJ, Gordon JI. The core gut microbiome, energy balance and obesity. J Physiol. 2009;587(Pt 17):4153–4158. | |
Qin J, Li R, Raes J, et al; MetaHIT Consortium, Bork P, Ehrlich SD, Wang J. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464(7285):59–65. | |
Glendinning L, Free A. Supra-organismal interactions in the human intestine. Front Cell Infect Microbiol. 2014;4:47. | |
Tremaroli V, Bäckhed F. Functional interactions between the gut microbiota and host metabolism. Nature. 2012;489(7415):242–249. | |
David LA, Maurice CF, Carmody RN, et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature. 2014;505(7484):559–563. | |
Wu GD, Chen J, Hoffmann C, et al. Linking long-term dietary patterns with gut microbial enterotypes. Science. 2011;334(6052):105–108. | |
Dethlefsen L, Huse S, Sogin ML, Relman DA. The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS Biol. 2008;6(11):e280. | |
Dethlefsen L, Relman DA. Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proc Natl Acad Sci U S A. 2011;108 Suppl 1:4554–4561. | |
Martin FP, Wang Y, Sprenger N, et al. Probiotic modulation of symbiotic gut microbial-host metabolic interactions in a humanized microbiome mouse model. Mol Syst Biol. 2008;4:157. | |
Jostins L, Ripke S, Weersma RK, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491(7422):119–124. | |
Karlsson FH, Tremaroli V, Nookaew I, et al. Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature. 2013;498(7452):99–103. | |
Rappé MS, Giovannoni SJ. The uncultured microbial majority. Annu Rev Microbiol. 2003;57(1):369–394. | |
Handelsman J, Rondon MR, Brady SF, Clardy J, Goodman RM. Molecular biological access to the chemistry of unknown soil microbes: a new frontier for natural products. Chem Biol. 1998;5(10):R245–R249. | |
Venter JC, Remington K, Heidelberg JF, et al. Environmental genome shotgun sequencing of the Sargasso Sea. Science. 2004;304(5667):66–74. | |
Metzker ML. Sequencing technologies – the next generation. Nat Rev Genet. 2010;11(1):31–46. | |
Mardis ER. Next-generation DNA sequencing methods. Annu Rev Genomics Hum Genet. 2008;9:387–402. | |
Ronaghi M. Pyrosequencing sheds light on DNA sequencing. Genome Res. 2001;11(1):3–11. | |
Neish AS. Microbes in gastrointestinal health and disease. Gastroenterology. 2009;136(1):65–80. | |
Maccaferri S, Biagi E, Brigidi P. Metagenomics: key to human gut microbiota. Dig Dis. 2011;29(6):525–530. | |
Samuel BS, Hansen EE, Manchester JK, et al. Genomic and metabolic adaptations of Methanobrevibacter smithii to the human gut. Proc Natl Acad Sci U S A. 2007;104(25):10643–10648. | |
Lagier JC, Million M, Hugon P, Armougom F, Raoult D. Human gut microbiota: repertoire and variations. Front Cell Infect Microbiol. 2012;2:136. | |
Moore WE, Holdeman LV. Human fecal flora: the normal flora of 20 Japanese-Hawaiians. Appl Microbiol. 1974;27(5):961–979. | |
Goodman AL, Kallstrom G, Faith JJ, et al. Extensive personal human gut microbiota culture collections characterized and manipulated in gnotobiotic mice. Proc Natl Acad Sci U S A. 2011;108(15):6252–6257. | |
Eckburg PB, Bik EM, Bernstein CN, et al. Diversity of the human intestinal microbial flora. Science. 2005;308(5728):1635–1638. | |
Lozupone CA, Stombaugh JI, Gordon JI, Jansson JK, Knight R. Diversity, stability and resilience of the human gut microbiota. Nature. 2012;489(7415):220–230. | |
Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature. 2012;486(7402):207–214. | |
Turnbaugh PJ, Hamady M, Yatsunenko T, et al. A core gut microbiome in obese and lean twins. Nature. 2009;457(7228):480–484. | |
Gill SR, Pop M, DeBoy RT, et al. Metagenomic analysis of the human distal gut microbiome. Science. 2006;312(5778):1355–1359. | |
Davenport ER, Mizrahi-Man O, Michelini K, Barreiro LB, Ober C, Gilad Y. Seasonal variation in human gut microbiome composition. PLoS One. 2014;9(3):e90731. | |
Rampelli S, Candela M, Turroni S, et al. Functional metagenomic profiling of intestinal microbiome in extreme ageing. Aging (Albany N Y). 2013;5(12):902–912. | |
Yatsunenko T, Rey FE, Manary MJ, et al. Human gut microbiome viewed across age and geography. Nature. 2012;486(7402):222–227. | |
Sanger F, Coulson AR. A rapid method for determining sequences in DNA by primed synthesis with DNA polymerase. J Mol Biol. 1975;94(3):441–448. | |
Morgan XC, Huttenhower C. Chapter 12: human microbiome analysis. PLoS Comput Biol. 2012;8(12):e1002808. | |
Teeling H, Glöckner FO. Current opportunities and challenges in microbial metagenome analysis – a bioinformatic perspective. Brief Bioinform. 2012;3(6):728–742. | |
Fu L, Niu B, Zhu Z, Wu S, Li W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics. 2012;28(23):3150–3152. | |
Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26(19):2460–2461. | |
Schloss PD. The effects of alignment quality, distance calculation method, sequence filtering, and region on the analysis of 16S rRNA gene-based studies. PLoS Comput Biol. 2010;6(7):e1000844. | |
DeSantis TZ, Hugenholtz P, Larsen N, et al. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol. 2006;72(7):5069–5072. | |
Cole JR, Wang Q, Fish JA, et al. Ribosomal Database Project: data and tools for high throughput rRNA analysis. Nucleic Acids Res. 2014;42(D1):D633–D642. | |
Quast C, Pruesse E, Yilmaz P, et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2013;41(D1):D590–D596. | |
Pruesse E, Peplies J, Glöckner FO. SINA: accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics. 2012;28(14):1823–1829. | |
Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. 2007;73(16):5261–5267. | |
DeSantis TZ Jr, Hugenholtz P, Keller K, et al. NAST: a multiple sequence alignment server for comparative analysis of 16S rRNA genes. Nucleic Acids Res. 2006;34(Web Server issue):W394–W399. | |
Ramette A. Multivariate analyses in microbial ecology. FEMS Microbiol Ecol. 2007;62(2):142–160. | |
Faust K, Raes J. Microbial interactions: from networks to models. Nat Rev Microbiol. 2012;10(8):538–550. | |
Langille MG, Zaneveld J, Caporaso JG, et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol. 2013;31(9):814–821. | |
Keller A, Horn H, Förster F, Schultz J. Computational integration of genomic traits into 16S rDNA microbiota sequencing studies. Gene. 2014;549(1):186–191. | |
Pruitt KD, Tatusova T, Brown GR, Maglott DR. NCBI Reference Sequences (RefSeq):current status, new features and genome annotation policy. Nucleic Acids Res.2012;40(Database issue):D130–D135. | |
Kanehisa M, Goto S, Sato Y, Kawashima M, Furumichi M, Tanabe M. Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res. 2014;42(Database issue):D199–D205. | |
Powell S, Forslund K, Szklarczyk D, et al. eggNOG v4.0: nested orthology inference across 3686 organisms. Nucleic Acids Res. 2014;42(D1):D231–D239. | |
Karlsson FH, Nookaew I, Nielsen J. Metagenomic data utilization and analysis (MEDUSA) and construction of a global gut microbial gene catalogue. PLoS Comput Biol. 2014;10(7):e1003706. | |
Li J, Jia H, Cai X, et al; MetaHIT Consortium. An integrated catalog of reference genes in the human gut microbiome. Nat Biotechnol. 2014;32(8):834–841. | |
Dröge J, McHardy AC. Taxonomic binning of metagenome samples generated by next-generation sequencing technologies. Brief Bioinform. 2012;13(6):646–655. | |
Huson DH, Mitra S, Ruscheweyh HJ, Weber N, Schuster SC. Integrative analysis of environmental sequences using MEGAN4. Genome Res. 2011;21(9):1552–1560. | |
Brady A, Salzberg SL. Phymm and PhymmBL: metagenomic phylogenetic classification with interpolated Markov models. Nat Methods. 2009;6(9):673–676. | |
Segata N, Waldron L, Ballarini A, Narasimhan V, Jousson O, Huttenhower C. Metagenomic microbial community profiling using unique clade-specific marker genes. Nat Methods. 2012;9(8):811–814. | |
Sunagawa S, Mende DR, Zeller G, et al. Metagenomic species profiling using universal phylogenetic marker genes. Nat Methods. 2013;10(12):1196–1199. | |
Zhu W, Lomsadze A, Borodovsky M. Ab initio gene identification in metagenomic sequences. Nucleic Acids Res. 2010;38(12):e132. | |
Kelley DR, Liu B, Delcher AL, Pop M, Salzberg SL. Gene prediction with Glimmer for metagenomic sequences augmented by classification and clustering. Nucleic Acids Res. 2012;40(1):e9. | |
Tatusov RL, Galperin MY, Natale DA, Koonin EV. The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000;28(1):33–36. | |
Finn RD, Bateman A, Clements J, et al. Pfam: the protein families database. Nucleic Acids Res. 2014;42(D1):D222–D230. | |
Overbeek R, Begley T, Butler RM, et al. The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucleic Acids Res. 2005;33(17):5691–5702. | |
Meyer F, Paarmann D, D’Souza M, et al. The metagenomics RAST server – a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics. 2008;9:386. | |
Sun S, Chen J, Li W, et al. Community cyberinfrastructure for Advanced Microbial Ecology Research and Analysis: the CAMERA resource. Nucleic Acids Res. 2011;39(suppl 1):D546–551. | |
Markowitz VM, Ivanova NN, Szeto E, et al. IMG/M: a data management and analysis system for metagenomes. Nucleic Acids Res. 2008;36(Database issue):D534–D538. | |
Kristiansson E, Hugenholtz P, Dalevi D. ShotgunFunctionalizeR: an R-package for functional comparison of metagenomes. Bioinformatics. 2009;25(20):2737–2738. | |
Parks DH, Beiko RG. Identifying biologically relevant differences between metagenomic communities. Bioinformatics. 2010;26(6):715–721. | |
Liu B, Pop M. MetaPath: identifying differentially abundant metabolic pathways in metagenomic datasets. BMC Proc. 2011;5 Suppl 2:S9. | |
Paulson JN, Stine OC, Bravo HC, Pop M. Differential abundance analysis for microbial marker-gene surveys. Nature Methods. 2013; 10(12):1200–1202. | |
Abubucker S, Segata N, Goll J, et al. Metabolic reconstruction for metagenomic data and its application to the human microbiome. PLoS Comput Biol. 2012;8(6):e1002358. | |
Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444(7122):1027–1131. | |
Larsen N, Vogensen FK, van den Berg FW, et al. Gut microbiota in human adults with type 2 diabetes differs from non-diabetic adults. PLoS One. 2010;5(2):e9085. | |
Greenblum S, Turnbaugh PJ, Borenstein E. Metagenomic systems biology of the human gut microbiome reveals topological shifts associated with obesity and inflammatory bowel disease. Proc Natl Acad Sci U S A. 2012;109(2):594–599. | |
Qin J, Li Y, Cai Z, et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature. 2012;490(7418):55–60. | |
Qin N, Yang F, Li A, et al. Alterations of the human gut microbiome in liver cirrhosis. Nature. 2014;513(7516):59–64. | |
Moore AM, Munck C, Sommer MO, Dantas G. Functional metagenomic investigations of the human intestinal microbiota. Front Microbiol. 2011;2:188. | |
Nicholson JK, Holmes E, Kinross J, et al. Host-gut microbiota metabolic interactions. Science. 2012;336(6086):1262–1267. | |
El Kaoutari A, Armougom F, Gordon JI, Raoult D, Henrissat B. The abundance and variety of carbohydrate-active enzymes in the human gut microbiota. Nat Rev Microbiol. 2013;11(7):497–504. | |
Tasse L, Bercovici J, Pizzut-Serin S, et al. Functional metagenomics to mine the human gut microbiome for dietary fiber catabolic enzymes. Genome Res. 2010;20(11):1605–1612. | |
Xing MN, Zhang XZ, Huang H. Application of metagenomic techniques in mining enzymes from microbial communities for biofuel synthesis. Biotechnol Adv. 2012;30(4):920–929. | |
Hong PY, Iakiviak M, Dodd D, Zhang M, Mackie RI, Cann I. Two new xylanases with different substrate specificities from the human gut bacterium Bacteroides intestinalis DSM 17393. Appl Environ Microbiol. 2014;80(7):2084–2093. | |
Louis P, Young P, Holtrop G, Flint HJ. Diversity of human colonic butyrate-producing bacteria revealed by analysis of the butyryl-CoA:acetate CoA-transferase gene. Environ Microbiol. 2010;12(2):304–314. | |
Forslund K, Sunagawa S, Kultima JR, et al. Country-specific antibiotic use practices impact the human gut resistome. Genome Res. 2013;23(7):1163–1169. | |
Sommer MO, Dantas G, Church GM. Functional characterization of the antibiotic resistance reservoir in the human microflora. Science. 2009;325(5944):1128–1131. | |
Sommer MO, Church GM, Dantas G. The human microbiome harbors a diverse reservoir of antibiotic resistance genes. Virulence. 2010; 1(4):299–303. | |
Cheng G, Hu Y, Yin Y, et al. Functional screening of antibiotic resistance genes from human gut microbiota reveals a novel gene fusion. FEMS Microbiol Lett. 2012;336(1):11–16. | |
LeBlanc JG, Milani C, de Giori GS, Sesma F, van Sinderen D, Ventura M. Bacteria as vitamin suppliers to their host: a gut microbiota perspective. Curr Opin Biotechnol. 2013;24(2):160–168. | |
Bäckhed F, Ley RE, Sonnenburg JL, Peterson DA, Gordon JI. Host-bacterial mutualism in the human intestine. Science. 2005; 307(5717):1915–1920. | |
Cénit MC, Matzaraki V, Tigchelaar EF, Zhernakova A. Rapidly expanding knowledge on the role of the gut microbiome in health and disease. Biochim Biophys Acta. 2014;1842(10):1981–1992. | |
O’Hara AM, Shanahan F. The gut flora as a forgotten organ. EMBO Rep. 2006;7(7):688–693. | |
Hooper LV, Midtvedt T, Gordon JI. How host-microbial interactions shape the nutrient environment of the mammalian intestine. Annu Rev Nutr. 2002;22:283–307. | |
Bernalier-Donadille A. [Fermentative metabolism by the human gut microbiota]. Gastroenterol Clin Biol. 2010;34 Suppl 1:S16–S22. French. | |
Puertollano E, Kolida S, Yaqoob P. Biological significance of short-chain fatty acid metabolism by the intestinal microbiome. Curr Opin Clin Nutr Metabo Care. 2014;17(2):139–144. | |
Scheppach W. Effects of short chain fatty acids on gut morphology and function. Gut. 1994;35(Suppl 1):S35–S38. | |
Gao Z, Yin J, Zhang J, et al. Butyrate improves insulin sensitivity and increases energy expenditure in mice. Diabetes. 2009;58(7):1509–1517. | |
Chang PV, Hao L, Offermanns S, Medzhitov R. The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proc Natl Acad Sci U S A. 2014;111(6):2247–2252. | |
Macfarlane G, Cummings J, Allison C. Protein degradation by human intestinal bacteria. J Gen Microbiol. 1986;132(6):1647–1656. | |
Kovatcheva-Datchary P, Tremaroli V, Bäckhed F. The gut microbiota. In: The Prokaryotes: Springer; 2013:3–24. | |
Pompei A, Cordisco L, Amaretti A, Zanoni S, Matteuzzi D, Rossi M. Folate production by bifidobacteria as a potential probiotic property. Appl Environ Microbiol. 2007;73(1):179–185. | |
Santos F, Wegkamp A, de Vos WM, Smid EJ, Hugenholtz J. High-level folate production in fermented foods by the B12 producer Lactobacillus reuteri JCM1112. Appl Environ Microbiol. 2008;74(10):3291–3294. | |
Taranto MP, Vera JL, Hugenholtz J, De Valdez GF, Sesma F. Lactobacillus reuteri CRL1098 produces cobalamin. J Bacteriol. 2003;185(18):5643–5647. | |
Ridlon JM, Kang DJ, Hylemon PB. Bile salt biotransformations by human intestinal bacteria. J Lipid Res. 2006;47(2):241–259. | |
Craciun S, Balskus EP. Microbial conversion of choline to trimethylamine requires a glycyl radical enzyme. Proc Natl Acad Sci U S A. 2012;109(52):21307–21312. | |
Dumas ME, Barton RH, Toye A, et al. Metabolic profiling reveals a contribution of gut microbiota to fatty liver phenotype in insulin-resistant mice. Proc Natl Acad Sci U S A. 2006;103(33):12511–12516. | |
Wang Z, Klipfell E, Bennett BJ, et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature. 2011;472(7341):57–63. | |
Kostic AD, Xavier RJ, Gevers D. The microbiome in inflammatory bowel disease: current status and the future ahead. Gastroenterology. 2014;146(6):1489–1499. | |
Manichanh C, Rigottier-Gois L, Bonnaud E, et al. Reduced diversity of faecal microbiota in Crohn’s disease revealed by a metagenomic approach. Gut. 2006;55(2):205–211. | |
Joossens M, Huys G, Cnockaert M, et al. Dysbiosis of the faecal microbiota in patients with Crohn’s disease and their unaffected relatives. Gut. 2011;60(5):631–637. | |
Marchesi JR, Holmes E, Khan F, et al. Rapid and noninvasive metabonomic characterization of inflammatory bowel disease. J Proteome Res. 2007;6(2):546–551. | |
Gevers D, Kugathasan S, Denson LA, et al. The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe. 2014;15(3):382–392. | |
Ley RE, Bäckhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI. Obesity alters gut microbial ecology. Proc Natl Acad Sci U S A. 2005;102(31):11070–11075. | |
Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature. 2006;444(7122):1022–1023. | |
Turnbaugh PJ, Bäckhed F, Fulton L, Gordon JI. Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell Host Microbe. 2008;3(4):213–223. | |
Gosalbes MJ, Durbán A, Pignatelli M, et al. Metatranscriptomic approach to analyze the functional human gut microbiota. PLoS One. 2011;6(3):e17447. | |
Maurice CF, Haiser HJ, Turnbaugh PJ. Xenobiotics shape the physiology and gene expression of the active human gut microbiome. Cell. 2013;152(1–2):39–50. | |
Klaassens ES, de Vos WM, Vaughan EE. Metaproteomics approach to study the functionality of the microbiota in the human infant gastrointestinal tract. Appl Environ Microbiol. 2007;73(4):1388–1392. | |
Verberkmoes NC, Russell AL, Shah M, et al. Shotgun metaproteomics of the human distal gut microbiota. ISME J. 2009;3(2):179–189. | |
Kolmeder CA, de Been M, Nikkilä J, et al. Comparative metaproteomics and diversity analysis of human intestinal microbiota testifies for its temporal stability and expression of core functions. PLoS One. 2012;7(1):e29913. | |
Lepage P, Leclerc MC, Joossens M, et al. A metagenomic insight into our gut’s microbiome. Gut. 2013;62(1):146–158. | |
Ursell LK, Haiser HJ, Van Treuren W, et al. The intestinal metabolome: an intersection between microbiota and host. Gastroenterology. 2014;146(6):1470–1476. | |
Claus SP, Tsang TM, Wang Y, et al. Systemic multicompartmental effects of the gut microbiome on mouse metabolic phenotypes. Mol Syst Biol. 2008;4:219. | |
Wikoff WR, Anfora AT, Liu J, et al. Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc Natl Acad Sci U S A. 2009;106(10):3698–3703. | |
Sellitto M, Bai G, Serena G, et al. Proof of concept of microbiome-metabolome analysis and delayed gluten exposure on celiac disease autoimmunity in genetically at-risk infants. PLoS One. 2012;7(3):e33387. | |
Bertini I, Calabrò A, De Carli V, et al. The metabonomic signature of celiac disease. J Proteome Res. 2008;8(1):170–177. | |
Larsen PE, Collart F, Field D, et al. Predicted Relative Metabolomic Turnover (PRMT):determining metabolic turnover from a coastal marine metagenomic dataset. Microb Inform Exp. 2011;1(1):4. | |
Borenstein E. Computational systems biology and in silico modeling of the human microbiome. Brief Bioinform. 2012;13(6):769–780. | |
Ma H, Zeng AP. Reconstruction of metabolic networks from genome data and analysis of their global structure for various organisms. Bioinformatics. 2003;19(2):270–277. | |
Barabási AL, Oltvai ZN. Network biology: understanding the cell’s functional organization. Nat Rev Genet. 2004;5(2):101–113. | |
Borenstein E, Kupiec M, Feldman MW, Ruppin E. Large-scale reconstruction and phylogenetic analysis of metabolic environments. Proc Natl Acad Sci U S A. 2008;105(38):14482–14487. | |
Levy R, Borenstein E. Metabolic modeling of species interaction in the human microbiome elucidates community-level assembly rules. Proc Natl Acad Sci U S A. 2013;110(31):12804–12809. | |
Cottret L, Milreu PV, Acuña V, et al. Graph-based analysis of the metabolic exchanges between two co-resident intracellular symbionts, Baumannia cicadellinicola and Sulcia muelleri, with their insect host, Homalodisca coagulata. PLoS Comput Biol. 2010;6(9):e1000904. | |
Bordbar A, Monk JM, King ZA, Palsson BO. Constraint-based models predict metabolic and associated cellular functions. Nat Rev Genet. 2014;15(2):107–120. | |
Thiele I, Palsson BØ. A protocol for generating a high-quality genome-scale metabolic reconstruction. Nat Protoc. 2010;5(1):93–121. | |
Henry CS, DeJongh M, Best AA, Frybarger PM, Linsay B, Stevens RL. High-throughput generation, optimization and analysis of genome-scale metabolic models. Nat Biotechnol. 2010;28(9):977–982. | |
Karp PD, Paley SM, Krummenacker M, et al. Pathway Tools version 13.0: integrated software for pathway/genome informatics and systems biology. Brief Bioinform. 2010;11(1):40–79. | |
Agren R, Liu L, Shoaie S, Vongsangnak W, Nookaew I, Nielsen J. The RAVEN toolbox and its use for generating a genome-scale metabolic model for Penicillium chrysogenum. PLoS Comput Biol. 2013;9(3):e1002980. | |
Croft D, Mundo AF, Haw R, et al. The Reactome pathway knowledgebase. Nucleic Acids Res. 2014;42(Database issue):D472–D477. | |
Caspi R, Altman T, Dreher K, et al. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res. 2012;40(Database issue):D742–D753. | |
Chang A, Scheer M, Grote A, Schomburg I, Schomburg D. BRENDA, AMENDA and FRENDA the enzyme information system: new content and tools in 2009. Nucleic Acids Res. 2009;37(Database issue):D588–D592. | |
Roberts RJ. PubMed Central: The GenBank of the published literature. Proc Natl Acad Sci U S A. 2001;98(2):381–382. | |
Caspeta L, Shoaie S, Agren R, Nookaew I, Nielsen J. Genome-scale metabolic reconstructions of Pichia stipitis and Pichia pastoris and in silico evaluation of their potentials. BMC Syst Biol. 2012;6:24. | |
Österlund T, Nookaew I, Bordel S, Nielsen J. Mapping condition-dependent regulation of metabolism in yeast through genome-scale modeling. BMC Syst Biol. 2013;7:36. | |
El-Semman IE, Karlsson FH, Shoaie S, Nookaew I, Soliman TH, Nielsen J. Genome-scale metabolic reconstructions of Bifidobacterium adolescentis L2-32 and Faecalibacterium prausnitzii A2-165 and their interaction. BMC Syst Biol. 2014;8:41. | |
Stolyar S, Van Dien S, Hillesland KL, et al. Metabolic modeling of a mutualistic microbial community. Mol Syst Biol. 2007;3:92. | |
Zhuang K, Izallalen M, Mouser P, et al. Genome-scale dynamic modeling of the competition between Rhodoferax and Geobacter in anoxic subsurface environments. ISME J. 2011;5(2):305–316. | |
Freilich S, Zarecki R, Eilam O, et al. Competitive and cooperative metabolic interactions in bacterial communities. Nat Commun. 2011;2:589. | |
Shoaie S, Karlsson F, Mardinoglu A, Nookaew I, Bordel S, Nielsen J. Understanding the interactions between bacteria in the human gut through metabolic modeling. Sci Rep. 2013;3:2532. | |
Ma H, Sorokin A, Mazein A, et al. The Edinburgh human metabolic network reconstruction and its functional analysis. Mol Syst Biol. 2007;3:135. | |
Agren R, Bordel S, Mardinoglu A, Pornputtapong N, Nookaew I, Nielsen J. Reconstruction of genome-scale active metabolic networks for 69 human cell types and 16 cancer types using INIT. PLoS Comput Biol. 2012;8(5):e1002518. | |
Duarte NC, Becker SA, Jamshidi N, et al. Global reconstruction of the human metabolic network based on genomic and bibliomic data. Proc Natl Acad Sci U S A. 2007;104(6):1777–1782. | |
Thiele I, Swainston N, Fleming RM, et al. A community-driven global reconstruction of human metabolism. Nat Biotechnol. 2013;31(5):419–425. | |
Mardinoglu A, Agren R, Kampf C, Asplund A, Uhlen M, Nielsen J. Genome-scale metabolic modelling of hepatocytes reveals serine deficiency in patients with non-alcoholic fatty liver disease. Nat Commun. 2014;5:3083. | |
Thiele I, Heinken A, Fleming RM. A systems biology approach to studying the role of microbes in human health. Curr Opin Biotechnol. 2013;24(1):4–12. | |
Bordbar A, Lewis NE, Schellenberger J, Palsson BØ, Jamshidi N. Insight into human alveolar macrophage and M. tuberculosis interactions via metabolic reconstructions. Mol Syst Biol. 2010;6:422. | |
Heinken A, Sahoo S, Fleming RM, Thiele I. Systems-level characterization of a host-microbe metabolic symbiosis in the mammalian gut. Gut Microbes. 2013;4(1):28–40. | |
Caporaso JG, Lauber CL, Costello EK, et al. Moving pictures of the human microbiome. Genome Biol. 2011;12(5):R50. | |
Pérez-Cobas AE, Gosalbes MJ, Friedrichs A, et al. Gut microbiota disturbance during antibiotic therapy: a multi-omic approach. Gut. 2013;62(11):1591–1601. | |
Shoaie S, Nielsen J. Elucidating the interactions between the human gut microbiota and its host through metabolic modeling. Front Genet. 2014;5:86. | |
Zomorrodi AR, Maranas CD. OptCom: a multi-level optimization framework for the metabolic modeling and analysis of microbial communities. PLoS Comput Biol. 2012;8(2):e1002363. | |
Zomorrodi AR, Islam MM, Maranas CD. d-OptCom: dynamic multi-level and multi-objective metabolic modeling of microbial communities. ACS Synth Biol. 2014;3(4):247–257. | |
Khandelwal RA, Olivier BG, Röling WF, Teusink B, Bruggeman FJ. Community flux balance analysis for microbial consortia at balanced growth. PLoS One. 2013;8(5):e64567. | |
Harcombe WR, Riehl WJ, Dukovski I, et al. Metabolic resource allocation in individual microbes determines ecosystem interactions and spatial dynamics. Cell Rep. 2014;7(4):1104–1115. | |
Topping DL, Clifton PM. Short-chain fatty acids and human colonic function: roles of resistant starch and nonstarch polysaccharides. Physiol Rev. 2001;81(3):1031–1064. | |
Hijova E, Chmelarova A. Short chain fatty acids and colonic health. Bratisl Lek Listy. 2007;108(8):354–358. | |
Turroni F, Ventura M, Buttó LF, et al. Molecular dialogue between the human gut microbiota and the host: a Lactobacillus and Bifidobacterium perspective. Cell Mol Life Sci. 2014;71(2):183–203. | |
Said HM. Intestinal absorption of water-soluble vitamins in health and disease. Biochem J. 2011;437(3):357–372. | |
Zheng X, Xie G, Zhao A, et al. The footprints of gut microbial-mammalian co-metabolism. J Proteome Res. 2011;10(12):5512–5522. | |
Björkholm B, Bok CM, Lundin A, Rafter J, Hibberd ML, Pettersson S. Intestinal microbiota regulate xenobiotic metabolism in the liver. PLoS One. 2009;4(9):e6958. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.