")
Back to Journals » Therapeutics and Clinical Risk Management » Volume 13
New developments in the treatment of primary biliary cholangitis – role of obeticholic acid
Authors Jhaveri MA, Kowdley KV
Received 26 April 2017
Accepted for publication 31 May 2017
Published 21 August 2017 Volume 2017:13 Pages 1053—1060
DOI https://doi.org/10.2147/TCRM.S113052
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Gerry Lake-Bakaar
Manan A Jhaveri, Kris V Kowdley
Liver Care Network, Swedish Medical Center, Seattle, WA, USA
Abstract: Primary biliary cholangitis (PBC) is a chronic autoimmune cholestatic liver disease that predominantly affects women in early to middle age. It is typically associated with autoantibodies to mitochondrial antigens and results in immune-mediated destruction of small and medium-sized intrahepatic bile ducts leading to cholestasis, hepatic fibrosis and may progress to cirrhosis or hepatic failure and, in some cases, hepatocellular carcinoma. The clinical presentation and the natural history of PBC have improved over the years due to recognition of earlier widespread use of ursodeoxycholic acid (UDCA); about one-third of patients show suboptimal biochemical response to UDCA with poor prognosis. Until recently, UDCA was the only US Food and Drug Administration approved agent for this disease for more than two decades; obeticholic acid was approved in 2016 for treatment of patients with PBC with a suboptimal response or intolerance to UDCA. Currently, liver transplantation is the most effective treatment modality for PBC patients with end-stage liver disease. This review will focus on the recent advances in therapy of primary biliary cholangitis, with emphasis on obeticholic acid.
Keywords: primary biliary cholangitis, obeticholic acid, ursodeoxycholic acid
Background
Primary biliary cholangitis (PBC) is a chronic, autoimmune, slowly progressive cholestatic liver disease that predominantly affects middle-aged women at a ratio of ~10:1 of women to men.1 Although the reason for this gender difference is not fully understood, X chromosome monosomy is observed in women with PBC, and it has been found that genes related to X-linked immunodeficiencies can lead to granuloma formation and elevated immunoglobulin (Ig) M levels, a frequent observation in PBC.2 The worldwide prevalence rate of PBC is estimated to be between 67 and 940 cases per million population and the incidence is between 0.7 and 49 cases per million population per year.3 Human Development Index estimated that there is a positive association between the incidence of PBC and socioeconomic status; moreover, the incidence rate is low in developing countries.4 The prevalence rates of PBC are high in UK, Scandinavia, Canada and USA.4
The exact pathogenesis of PBC remains unclear. However, there is compelling evidence that the mechanism is likely related to a combination of genetic predisposition and environmental factors,3,5 explaining the geographic variation as well as familial occurrence of this disease. The immune-mediated destruction of bile duct epithelial cells drives the pathogenesis of PBC.6,7 Persistent destruction of the bile ducts leads to intrahepatic accumulation of cytotoxic bile acid, resulting in hepatocellular injury, fibrosis and cirrhosis.8
The diagnosis of PBC is confirmed by the presence of at least two of the following three objective criteria: 1) biochemical evidence of intrahepatic cholestasis based on elevated levels of alkaline phosphatase (ALKP) of ≥1.5 times the upper limit of normal (ULN) for >24 weeks; 2) the presence of serum titers of antimitochondrial antibodies (AMAs) of more than or equal to 1:40; and 3) liver histology compatible with features of PBC, characterized by nonsuppurative cholangitis and granulomatous destruction of interlobular bile ducts with exclusion of drug-induced liver injury.8,9 The disease usually presents with a cholestatic pattern of liver test abnormalities (predominant elevation of serum ALKP, out of proportion to elevation of serum aminotransferases [alanine aminotransferase {ALT}, aspartate aminotransferase {AST}]), and the vast majority of patients also have a positive serum AMA. AMAs are highly sensitive and specific, usually present in 95% of patients and given 98% specificity, liver biopsy is rarely required for the diagnosis of PBC.10 Liver biopsy is helpful in staging and if there is suspicion of overlap syndromes (ie, with autoimmune hepatitis [AIH]) or coexisting liver conditions (ie, nonalcoholic steatohepatitis, drug-induced liver injury).11
Autoimmune liver diseases consist of PBC, primary sclerosing cholangitis, AIH, and IgG4 sclerosing cholangitis. In addition, some patients present with features of other autoimmune liver diseases and will be categorized into a separate class called “overlap syndromes”.12–15 According to Paris criteria,12,16,17 an AIH–PBC overlap syndrome is accepted when two or three criteria of PBC as well as AIH are fulfilled. For AIH, the criteria are:12 1) serum ALT level of ≥5 times the ULN; 2) portal or periportal lymphocytic inflammation; and 3) moderate to severe periportal lymphocytic piecemeal necrosis. For PBC, the criteria are: 1) ALKP >2 times the ULN or gamma-glutamyl transferase (GGT) >5 times the ULN; 2) AMA positivity; and 3) florid duct lesions or destructive cholangitis on histologic examinations. Kuiper et al18 conducted a study on 134 patients of PBC–AIH overlap syndrome. They found that the sensitivity and specificity of Paris criteria for diagnosing overlap syndrome are 92% and 97%, respectively.
The associations between PBC and extrahepatic autoimmune diseases have been well established.19,20 More than 50% of patients with PBC have other autoimmune disease. The extrahepatic autoimmune diseases that have been associated with PBC are Sjogren’s syndrome, systemic lupus erythematosus, rheumatoid arthritis, Raynaud’s disease, CREST syndrome, inflammatory bowel diseases and insulin-dependent diabetes mellitus. Among these conditions, Sjogren’s syndrome and Raynaud’s disease are strongly associated with PBC.20,21
The main aim in the management of PBC is to reverse injury from bile duct inflammation, improve the symptoms, slow down the disease progression, improve liver biochemistries, and prevent the long-term consequences of chronic cholestasis, such as pruritus, fatigue, osteoporosis and fat-soluble vitamin deficiencies.22 Since the introduction of hydrophilic bile acids such as ursodeoxycholic acid (UDCA) and their widespread use, the clinical presentation and progression of natural disease history of PBC patients have improved significantly over the past two decades.23–25 However, about one-third of PBC patients show suboptimal biochemical responses to UDCA and remain at risk for continued progression of disease to more advanced diseases, including cirrhosis.26–28
Ursodeoxycholic acid
UDCA was the only drug approved by the US Food and Drug Administration for the treatment of PBC until the approval of obeticholic acid (OCA) in 2016.29 Chronic cholestasis will result in intrahepatic and systemic accumulation of cytotoxic bile acids that will initially promote hepatocyte proliferation but eventually lead to hepatocellular injury, apoptosis, fibrosis and cirrhosis. UDCA has multiple mechanisms of action in cholestatic conditions including protection of cholangiocytes and periportal hepatocytes from the cytotoxic effects of hydrophobic bile acids, stimulation of hepatocellular and ductular secretion of hydrophobic bile acids and hepatocyte protection against bile acid–induced apoptosis. UDCA also has additional anti-inflammatory and immunomodulatory effects.30
Administration of UDCA increases the bile acid saturation in bile, resulting in increased bile acid clearance from blood and reduced cholestatic symptoms, specifically pruritus. These effects of UDCA occur with the optimal dose of 13–15 mg/kg/day, and it has been proposed to have choleretic and anti-inflammatory effects. It shows improvement in liver biochemistries, slows the histologic progression of the disease and also improves overall survival, at least in those treated in the early stages of the disease.31,32 ter Borg et al33 conducted a prospective cohort study of 297 PBC patients and showed that administration of UDCA in patients with early histologic changes significantly improved transplant-free survival (1 year =99.7%, 5 years =87% and 10 years =71%) than that predicted by the Mayo model. Poupon et al34 conducted a meta-analysis of French, Canadian and Mayo Clinical trials and demonstrated that time to liver transplantation was significantly improved in patients with moderate to severe disease (serum bilirubin level of ≥1.4 mg/dL, stage 3 and 4 histologic abnormalities).
PBC is a rare and slowly progressive disease, and therefore, in most cases, individual clinical trials lacked power to demonstrate clinically significant differences in outcomes. So, the rates of clinical outcomes in patients treated with UDCA are compared to predicted rates of outcomes based upon the natural history models applied to a similar population, assuming that they had not been treated with UDCA.35 A Markov model36 has been used to compare survival of UDCA-treated patients to historical controls using the Mayo natural history model for PBC. This model predicted a significantly better liver transplant-free survival with UDCA as compared to the spontaneous survival rate predicted by Mayo model.36 The overall survival rates without liver transplantation were 84% and 66% at 10 and 20 years, respectively. In early-stage patients, 6% and 22% were predicted to progress to liver transplantation or death after 10 and 20 years, respectively.37 On the other hand, the probability of death or liver transplantation was significantly increased in patients in late stages of the disease.
However, ~30%–40% of PBC patients show suboptimal biochemical responses to UDCA or are intolerant to UDCA and remain at risk for continued progression to advanced diseases including cirrhosis.26,27 The biochemical response to UDCA after 1 year of treatment is a strong predictor of long-term clinical outcomes and, thus, is very helpful in identifying the patient population in need for additional therapies.38 Multiple studies have proposed various criteria for improvement in liver biochemistries as predictors of treatment success with UDCA.39,40 Large cohorts from France and UK showed that reduction of ALKP to <1.5 times the ULN and a normal total bilirubin after 1 year of treatment with UDCA (Paris II criteria) are associated with excellent long-term survival and identified patients at low risk for disease progression.40 The collaboration of multiple international cohorts of >4,500 PBC patients established the PBC GLOBE score which examined the risk of liver transplantation or death as a function of serum ALKP and total bilirubin after 1 year of UDCA treatment.39 The PBC GLOBE score incorporates age at the time of initiating UDCA therapy, serum ALKP, total bilirubin, albumin and platelet counts. A recent meta-analysis evaluated the impact of ALKP and total bilirubin levels on the long-term outcomes, such as liver transplantation and death.41 The authors concluded that the 10-year survival rate without a liver transplant after 1 year of treatment with UDCA was 84% in patients with ALKP level of ≤2 times the ULN and 86% in patients with bilirubin levels ≤1 times the ULN, as compared to 62% in patients with ALKP level of >2 times the ULN (P<0.0001) and 41% in patients with bilirubin levels >1 times the ULN (P<0.0001).41
Obeticholic acid
OCA (6α-ethyl-chenodeoxycholic acid) is a selective farnesoid-X-receptor (FXR) agonist derived from the primary human bile acid chenodeoxycholic acid that has been modified chemically to make it 100 times more potent than chenodeoxycholic acid.42,43 The FXR nuclear receptor is expressed in the liver, intestine, adrenal glands and kidneys; this nuclear receptor has a significant role in the synthesis and enterohepatic circulation of the bile acids. FXR activation in the liver reduces the conversion of cholesterol to bile acids by downregulating cytochrome P450 7A1 (CYP7A1) and CYP8B1, the primary enzymes involved in the synthesis of bile acids, and increases the expression of bilirubin exporter pumps. In the ileum, activation of FXR receptor inhibits the uptake of bile acids by down regulating the sodium-dependent bile acid transporter and also decreased the production of bile acids by increasing the expression of fibroblast growth factor in the liver through inhibition of CYP7A1.42,44,45 Therefore, OCA increases bile flow in cholestatic conditions, and thereby protects the hepatocytes from accumulation of cytotoxic bile acids. In addition, various preclinical studies have found that OCA also has antifibrotic and anti-inflammatory properties.45 OCA has shown improvement in the biochemical markers of liver function in multiple clinical trials.46–48
Clinical trials
Two major Phase II studies46,47 and a Phase III study48 have evaluated the safety and efficacy of OCA in patients with PBC. The summary of OCA clinical trials is presented in Table 1.
 | Table 1 Summary of major clinical trials of obeticholic acid in primary biliary cholangitis |
The efficacy of OCA as monotherapy, comparing 10 and 50 mg OCA, was evaluated in a Phase II, international, double-blind, placebo-controlled trial in patients with PBC with persistent elevation of ALKP (>1.5–2 times the ULN) and who had not been taking UDCA for at least 6 months.47 The authors reported significant improvement in ALKP levels with both doses of OCA (the 10 mg OCA group showed the greatest decrease in ALKP, from 3.9× ULN to 1.9× ULN) as compared to placebo. The OCA group also showed significant reduction in the values of GGT, conjugated bilirubin, C-reactive protein, IgM and tumor necrosis factor-α. Pruritus was the only main clinical adverse event in the OCA group as compared to placebo, and was more severe among patients with higher doses.47
Hirschfield et al46 published a randomized, double-blind, placebo-controlled trial evaluating the safety and efficacy of three doses of OCA (10, 25 and 50 mg/day) compared against placebo and UDCA. The study included patients with persistent elevation of ALKP of >1.5 times the ULN on a stable dose of UDCA for at least 6 months. The primary endpoint of significant reduction in serum ALKP from baseline was met in all three OCA dose groups vs placebo. The mean relative change in ALKP from baseline was a decrease of 24%, 25% and 21% for the 10, 25, and 50 mg OCA groups, respectively, compared with a 3% decrease in the placebo group. The secondary endpoints including significant reductions in the values of GGT, ALT and AST were also met across all OCA treatment groups as compared to placebo. Moreover, OCA groups showed significant improvements in the inflammatory markers, such as C-reactive protein and IgM values.46
The authors also performed an open-label extension of the double-blind trial to evaluate the long-term safety and efficacy of OCA in patients with PBC.49 A total of 78 patients were enrolled and 61 patients completed the first year. The subjects were started on a 10 mg daily dose of OCA and titrated every 8 weeks to a maximum dose of 50 mg. The mean final daily dose of OCA at 12 months was 20 mg. The mean ALKP levels were decreased by 71 U/L (19%) at the 10 mg OCA dose, with a further decrease of 23 U/L (9%) after titrating the OCA dose from 10 to 25 mg. However, the mean ALKP increased by 8 U/L with titration from the 25 to 50 mg dose. The major side effect was dose-related pruritus, which was reported in 87% of patients.49
The results from the only Phase III clinical trial of OCA,48 the PBC OCA International Study of Efficacy (POISE), were the basis for the US Food and Drug Administration’s approval of OCA in the treatment of PBC patients with incomplete response to UDCA. POISE is an international, multicenter, randomized, double-blind, placebo-controlled, Phase III clinical trial that studied the safety and efficacy of OCA in PBC patients with an incomplete response to, or who are unable to, tolerate UDCA.48 An inadequate response to UDCA was defined as ALKP of ≥1.67 times the ULN and/or total bilirubin of more than the ULN but <2 times the ULN. Most of the participants had been on UDCA for at least 12 months and were on a stable dose for at least 3 months prior to enrollment. The primary endpoint was a composite of ALKP level of <1.67 times the ULN, with a reduction of at least 15% from baseline, and a normal total bilirubin level after 12 months of therapy. Two hundred and seventeen subjects were randomized to receive either placebo, 10 or 5 mg of OCA titrated to 10 mg of OCA on the basis of the side effects and biochemical response at 6 months. If patients had side effects such as severe pruritus or had already met the composite primary endpoint, their dose was not increased. The median UDCA dose was 15.5 mg/kg/day and 7% of patients were UDCA intolerant. The composite primary endpoint was met in an intention-to-treat analysis with rates of 10% in the placebo group compared to 47% in the 10 mg OCA group and 46% in the dose-titrated 5–10 mg OCA group (P<0.0001 for both intervention groups vs placebo). The mean decrease in ALKP from baseline was 39% in 10 mg OCA dose group, 33% in 5–10 mg titrated dose OCA group vs 5% in the placebo group (P<0.0001 for both intervention groups vs placebo). In addition to the improvement in the serum level of ALKP, patients treated with 5 and 5–10 mg titrated dose had a greater decrease in total bilirubin compared to placebo (−0.02 and −0.05, respectively, vs 0.12; P-value for both OCA dose groups was <0.001 vs placebo). Moreover, both the OCA intervention groups met predefined secondary endpoints including significant improvement of serum AST, serum ALT, serum GGT and the markers of inflammation (P-value for both OCA dose groups was <0.0005 vs placebo).48
Ninety-seven percent of the patients who completed the double-blind phase of the POISE trial entered an open-label extension phase, which is ongoing and will continue for a total of 5 years. The subjects who received OCA in the double-blind trial showed sustained improvements in ALKP levels; the placebo patients initiating treatment with OCA showed similar improvements in ALKP and bilirubin level as compared to the OCA-treated groups in the double-blind phase.48
Safety and tolerability
Side effects related to OCA were usually mild to moderate and mostly related to pruritus. Treatment with OCA has been associated with increase in low-density lipoprotein-cholesterol and decrease in high-density lipoprotein-cholesterol (HDL-C) and triglycerides. The clinical significance of HDL-C reduction in PBC patients is unclear, as the patients had relatively high levels of HDL-C at baseline. Also, the absolute differences are relatively small. Other commonly reported adverse outcomes were fatigue, headache, and gastrointestinal side effects.46–48
Pruritus
Pruritus is the most common adverse effect in the intervention group requiring change in dose and/or discontinuation of the treatment. The current evidence suggests that there is increased frequency and severity of pruritus, especially in OCA treatment groups at higher doses. Assessment of pruritus as a side effect in PBC is confounded by the fact that it is a common symptom in PBC patients. In the randomized clinical trial conducted by Hirschfield et al,46 the incidence of pruritus was 50% in the placebo group as compared to 85% in the 25 mg OCA group (P<0.0003), 80% in the 50 mg OCA group (P<0.006) and 47% in the 10 mg OCA group (P=0.7949). Sixteen percent of patients in the 10 mg, 24% in the 25 mg and 37% in the 50 mg groups discontinued the trial due to severe pruritus, as compared to 0% in the placebo group. Overall, 10% of the OCA-treated patients discontinued the trial due to pruritus. The similar effects of OCA related to pruritus were also observed in the POISE trial.48 The incidence rates of pruritus were 38%, 56% and 68% in the placebo, 5–10 and 10 mg groups, respectively. The severity of pruritus was less in the titration group (<1% discontinued the trial due to pruritus) as compared to the 10 mg OCA group (10% discontinued the trial due to pruritus). Overall, fewer than 6% of OCA-treated patients discontinued the trial due to pruritus. In the open-label extension phase with up to 2 years of follow-up, new incidence of pruritus is 15% in the 5–10 mg group and 21% in the 10 mg group, which is lower than the double-blind phase.50,51 In summary, pruritus is the major side effect of OCA in PBC, although this symptom can be managed in most patients by the use of bile acid sequestrants, antihistamines, dose reduction or symptomatic treatment.52
Dyslipidemia
PBC patients with early disease have elevated total cholesterol likely due to high HDL-C levels. Changes in serum cholesterol levels were observed in clinical trials of OCA.46–48 Hirschfield et al46 reported changes in the serum lipid level, especially decrease in HDL and total cholesterol was observed in the OCA group. A dose-related decrease in total cholesterol of 3%, 5% and 13% was observed in the OCA 10, 25 and 50 mg groups, respectively, which was due to a decrease in HDL levels. There was a decrease in HDL level in all intervention groups; the mean reduction in HDL was 8 mg/dL (12%) in the 10 mg OCA group and 9 mg/dL (13%) in both the 25 and 50 mg OCA groups, as compared to the placebo group that had relatively no change in HDL. The HDL levels were increased in all intervention groups during the off-treatment phase, which is suggestive of the OCA-induced effect on HDL. The POISE trial48 also observed similar effects of OCA on HDL level. It found reduction of 16% in the titrated group, 26% in the 10 mg group and 3% in the placebo group. Additionally, a modest decrease in triglycerides with no change in low-density lipoprotein-cholesterol was observed in the OCA-treated groups. This could be explained by the fact that the elimination of cholesterol is reduced by attenuating hepatic bile acid synthesis.53 The changes in cholesterol levels are summarized in Table 2.
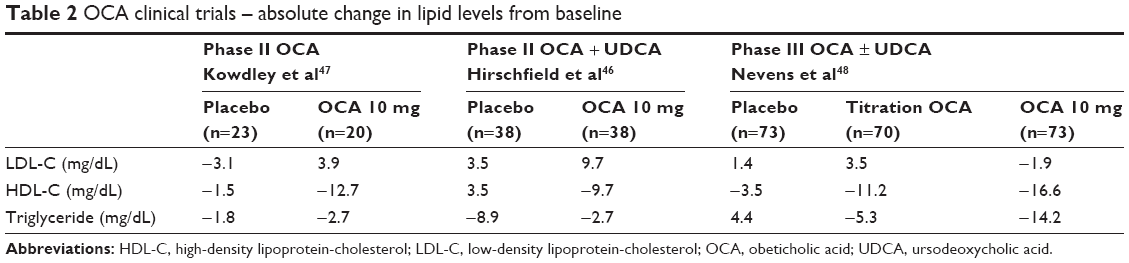 | Table 2 OCA clinical trials – absolute change in lipid levels from baseline |
Other adverse events
Other common side effects that were observed in the treatment groups were headache, rash, and gastrointestinal symptoms such as abdominal discomfort, nausea and vomiting.
Emerging treatments
The requirement for additional treatment for PBC is clearly recognized. Multiple noncontrolled studies found significant improvement in serum ALKP following administration of fibrates in PBC patients. However, these studies are limited due to lack of appropriate controls, smaller follow-up and ill-defined patient population. A Phase III randomized trial is currently underway. Novel immunologic treatments such as anti-interleukin-12, anti-CD80, anti-CD20 (rituximab), mesenchymal stem cells and cytotoxic T-lymphocyte antigen 4 (abatacept) are currently under investigation for the treatment of PBC.54–57
Summary
The recent approval of OCA in the treatment of PBC is an important advance for patients who are at an increased risk of liver-related complications despite UDCA therapy or are unable to tolerate UDCA. However, additional data are awaited to examine the effects of OCA in long-term clinical outcomes such as quality of life measures, decompensation of liver disease or liver-related mortality. The major adverse events related to OCA in PBC are dose-related side effects and possible negative effects on lipid profiles, although the clinical significance of this problem in cholestatic liver disease is unknown. A long-term study is underway to determine whether liver-related outcomes are improved with prolonged OCA treatment. Meanwhile, it is appropriate for clinicians to screen their patients with PBC and evaluate whether their ALKP is 1.67 times the ULN despite UDCA therapy, or in patients intolerant of UDCA to determine whether they are appropriate candidates for treatment with OCA.
Disclosure
KVK: Grants/Research: Evidera, Gilead, Immuron, Intercept, Tobira. Advisory Board: Abbvie, Achillion, BMS, Evidera, Gilead, Merck, Novartis. MAJ reports no conflicts of interest in this work.
References
Selmi C, Bowlus CL, Gershwin ME, Coppel RL. Primary biliary cirrhosis. Lancet. 2011;377(9777):1600–1609. | ||
Invernizzi P, Miozzo M, Battezzati PM, et al. Frequency of monosomy X in women with primary biliary cirrhosis. Lancet. 2004;363(9408):533–535. | ||
Selmi C, Mayo MJ, Bach N, et al. Primary biliary cirrhosis in monozygotic and dizygotic twins: genetics, epigenetics, and environment. Gastroenterology. 2004;127(2):485–492. | ||
Pan HY, Dai YN, Zheng JN, et al. National incidence of autoimmune liver diseases and its relationship with the human development index. Oncotarget. 2016;7(29):46273–46282. | ||
Hirschfield GM, Gershwin ME. The immunobiology and pathophysiology of primary biliary cirrhosis. Annu Rev Pathol. 2013;8:303–330. | ||
Boonstra K, Beuers U, Ponsioen CY. Epidemiology of primary sclerosing cholangitis and primary biliary cirrhosis: a systematic review. J Hepatol. 2012;56(5):1181–1188. | ||
Lazaridis KN, Talwalkar JA. Clinical epidemiology of primary biliary cirrhosis: incidence, prevalence, and impact of therapy. J Clin Gastroenterol. 2007;41(5):494–500. | ||
Kim KA, Jeong SH. The diagnosis and treatment of primary biliary cirrhosis. Korean J Hepatol. 2011;17(3):173–179. | ||
Silveira MG, Brunt EM, Heathcote J, Gores GJ, Lindor KD, Mayo MJ. American Association for the Study of Liver Diseases endpoints conference: design and endpoints for clinical trials in primary biliary cirrhosis. Hepatology. 2010;52(1):349–359. | ||
Cancado EL, Harriz M. The importance of autoantibody detection in primary biliary cirrhosis. Front Immunol. 2015;6:309. | ||
Zein CO, Angulo P, Lindor KD. When is liver biopsy needed in the diagnosis of primary biliary cirrhosis? Clin Gastroenterol Hepatol. 2003;1(2):89–95. | ||
Chazouilleres O, Wendum D, Serfaty L, Montembault S, Rosmorduc O, Poupon R. Primary biliary cirrhosis-autoimmune hepatitis overlap syndrome: clinical features and response to therapy. Hepatology. 1998;28(2):296–301. | ||
Czaja AJ. Frequency and nature of the variant syndromes of autoimmune liver disease. Hepatology. 1998;28(2):360–365. | ||
Floreani A, Baragiotta A, Guido M. Primary biliary cirrhosis-autoimmune hepatitis overlap syndrome: a cause of resistance to ursodeoxycholic treatment. Dig Liver Dis. 2003;35(2):128–129. | ||
Neuhauser M, Bjornsson E, Treeprasertsuk S, et al. Autoimmune hepatitis-PBC overlap syndrome: a simplified scoring system may assist in the diagnosis. Am J Gastroenterol. 2010;105(2):345–353. | ||
Boberg KM, Chapman RW, Hirschfield GM, et al. Overlap syndromes: the International Autoimmune Hepatitis Group (IAIHG) position statement on a controversial issue. J Hepatol. 2011;54(2):374–385. | ||
European Association for the Study of the Liver. EASL Clinical Practice Guidelines: management of cholestatic liver diseases. J Hepatol. 2009;51(2):237–267. | ||
Kuiper EM, Zondervan PE, van Buuren HR. Paris criteria are effective in diagnosis of primary biliary cirrhosis and autoimmune hepatitis overlap syndrome. Clin Gastroenterol Hepatol. 2010;8(6):530–534. | ||
Floreani A, Franceschet I, Cazzagon N, et al. Extrahepatic autoimmune conditions associated with primary biliary cirrhosis. Clin Rev Allergy Immunol. 2015;48(2–3):192–197. | ||
Fox RI. Sjogren’s syndrome. Lancet. 2005;366(9482):321–331. | ||
Nakamura T, Higashi S, Tomoda K, Tsukano M, Sugi K. Primary biliary cirrhosis (PBC)-CREST overlap syndrome with coexistence of Sjogren’s syndrome and thyroid dysfunction. Clin Rheumatol. 2007;26(4):596–600. | ||
Czul F, Peyton A, Levy C. Primary biliary cirrhosis: therapeutic advances. Clin Liver Dis. 2013;17(2):229–242. | ||
Carey EJ, Ali AH, Lindor KD. Primary biliary cirrhosis. Lancet. 2015;386(10003):1565–1575. | ||
Pares A, Caballeria L, Rodes J. Excellent long-term survival in patients with primary biliary cirrhosis and biochemical response to ursodeoxycholic Acid. Gastroenterology. 2006;130(3):715–720. | ||
Poupon R, Chretien Y, Poupon RE, Ballet F, Calmus Y, Darnis F. Is ursodeoxycholic acid an effective treatment for primary biliary cirrhosis? Lancet. 1987;1(8537):834–836. | ||
Carbone M, Mells GF, Pells G, et al. Sex and age are determinants of the clinical phenotype of primary biliary cirrhosis and response to ursodeoxycholic acid. Gastroenterology. 2013;144(3):560–569. e567; quiz e513–e564. | ||
Corpechot C, Abenavoli L, Rabahi N, et al. Biochemical response to ursodeoxycholic acid and long-term prognosis in primary biliary cirrhosis. Hepatology. 2008;48(3):871–877. | ||
Kuiper EM, Hansen BE, de Vries RA, et al; Dutch PBC Study Group. Improved prognosis of patients with primary biliary cirrhosis that have a biochemical response to ursodeoxycholic acid. Gastroenterology. 2009;136(4):1281–1287. | ||
Bowlus CL. Obeticholic acid for the treatment of primary biliary cholangitis in adult patients: clinical utility and patient selection. Hepat Med. 2016;8:89–95. | ||
Poupon R. Ursodeoxycholic acid and bile-acid mimetics as therapeutic agents for cholestatic liver diseases: an overview of their mechanisms of action. Clin Res Hepatol Gastroenterol. 2012;36(Suppl 1):S3–S12. | ||
Angulo P, Dickson ER, Therneau TM, et al. Comparison of three doses of ursodeoxycholic acid in the treatment of primary biliary cirrhosis: a randomized trial. J Hepatol. 1999;30(5):830–835. | ||
Angulo P, Batts KP, Therneau TM, Jorgensen RA, Dickson ER, Lindor KD. Long-term ursodeoxycholic acid delays histological progression in primary biliary cirrhosis. Hepatology. 1999;29(3):644–647. | ||
ter Borg PC, Schalm SW, Hansen BE, van Buuren HR; Dutch PBCSG. Prognosis of ursodeoxycholic acid-treated patients with primary biliary cirrhosis. Results of a 10-year cohort study involving 297 patients. Am J Gastroenterol. 2006;101(9):2044–2050. | ||
Poupon RE, Lindor KD, Cauch-Dudek K, Dickson ER, Poupon R, Heathcote EJ. Combined analysis of randomized controlled trials of ursodeoxycholic acid in primary biliary cirrhosis. Gastroenterology. 1997;113(3):884–890. | ||
Kim WR, Wiesner RH, Poterucha JJ, et al. Adaptation of the Mayo primary biliary cirrhosis natural history model for application in liver transplant candidates. Liver Transpl. 2000;6(4):489–494. | ||
Talwalkar JA. Markov models in primary biliary cirrhosis. Gastroenterology. 2002;123(5):1750–1751. | ||
Corpechot C, Carrat F, Bahr A, Chretien Y, Poupon RE, Poupon R. The effect of ursodeoxycholic acid therapy on the natural course of primary biliary cirrhosis. Gastroenterology. 2005;128(2):297–303. | ||
Weinmann A, Sattler T, Unold HP, et al. Predictive scores in primary biliary cirrhosis: a retrospective single center analysis of 204 patients. J Clin Gastroenterol. 2015;49(5):438–447. | ||
Lammers WJ, Hirschfield GM, Corpechot C, et al. Development and validation of a scoring system to predict outcomes of patients with primary biliary cirrhosis receiving ursodeoxycholic acid therapy. Gastroenterology. 2015;149(7):1804–1812. e1804. | ||
Papastergiou V, Tsochatzis EA, Rodriguez-Peralvarez M, et al. Biochemical criteria at 1 year are not robust indicators of response to ursodeoxycholic acid in early primary biliary cirrhosis: results from a 29-year cohort study. Aliment Pharmacol Ther. 2013;38(11–12):1354–1364. | ||
Lammers WJ, van Buuren HR, Hirschfield GM, et al. Levels of alkaline phosphatase and bilirubin are surrogate end points of outcomes of patients with primary biliary cirrhosis: an international follow-up study. Gastroenterology. 2014;147(6):1338–1349. | ||
Lindor KD. Farnesoid X receptor agonists for primary biliary cirrhosis. Curr Opin Gastroenterol. 2011;27(3):285–288. | ||
Pellicciari R, Fiorucci S, Camaioni E, et al. 6alpha-ethyl-chenodeoxycholic acid (6-ECDCA), a potent and selective FXR agonist endowed with anticholestatic activity. J Med Chem. 2002;45(17):3569–3572. | ||
Flores A, Mayo MJ. Primary biliary cirrhosis in 2014. Curr Opin Gastroenterol. 2014;30(3):245–252. | ||
Verbeke L, Farre R, Trebicka J, et al. Obeticholic acid, a farnesoid X receptor agonist, improves portal hypertension by two distinct pathways in cirrhotic rats. Hepatology. 2014;59(6):2286–2298. | ||
Hirschfield GM, Mason A, Luketic V, et al. Efficacy of obeticholic acid in patients with primary biliary cirrhosis and inadequate response to ursodeoxycholic acid. Gastroenterology. 2015;148(4):751–761. e758. | ||
Kowdley K, Jones D, Luketic V; The OCA PBC Study Group, An international study evaluating the farnesoid X receptor agonist obeticholic acid as monotherapy in PBC. J Hepatol. 2012;54:S13. | ||
Nevens F, Andreone P, Mazzella G, et al. A placebo-controlled trial of obeticholic acid in primary biliary cholangitis. N Engl J Med. 2016;375(7):631–643. | ||
Hirschfield GM, Mason A, Gordon SC, Luketic V, Lindor K. A long term safety extension trial of the farnesoid X receptor (FXR) agonist obeticholic acid (OCA) and UDCA in primary biliary cirrhosis (PBC). Hepatology. 2011;54:429A. | ||
Peters Y, Hooshmand-Rad R, Pencek R. Long-term safety of oeticholic acid in patients wiht primary biliary cirrhosis. Dig Liver Dis. 2016;(48):e117–e118. | ||
Beuers U, Jones DE, Mayo M. FXR agonist obeticholic acid. Pruritus, a common side effect ameliorated by dose titration. Hepatology. 2014;60(8). | ||
Neuschwander-Tetri BA, Loomba R, Sanyal AJ, et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo-controlled trial. Lancet. 2015;385(9972):956–965. | ||
Hambruch E, Miyazaki-Anzai S, Hahn U, et al. Synthetic farnesoid X receptor agonists induce high-density lipoprotein-mediated transhepatic cholesterol efflux in mice and monkeys and prevent atherosclerosis in cholesteryl ester transfer protein transgenic low-density lipoprotein receptor (-/-) mice. J Pharmacol Exp Ther. 2012;343(3):556–567. | ||
Floreani A, Franceschet I, Perini L, Cazzagon N, Gershwin ME, Bowlus CL. New therapies for primary biliary cirrhosis. Clin Rev Allergy Immunol. 2015;48(2–3):263–272. | ||
Beuers U, Gershwin ME. Unmet challenges in immune-mediated hepatobiliary diseases. Clin Rev Allergy Immunol. 2015;48(2–3):127–131. | ||
Invernizzi P, Gershwin ME. New therapeutics in primary biliary cirrhosis: will there ever be light? Liver Int. 2014;34(2):167–170. | ||
Baghdasaryan A, Claudel T, Gumhold J, et al. Dual farnesoid X receptor/TGR5 agonist INT-767 reduces liver injury in the Mdr2-/- (Abcb4-/-) mouse cholangiopathy model by promoting biliary HCO(-) (3) output. Hepatology. 2011;54(4):1303–1312. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.