")
Back to Journals » Drug, Healthcare and Patient Safety » Volume 6
New developments in the treatment of metastatic melanoma – role of dabrafenib–trametinib combination therapy
Received 4 April 2014
Accepted for publication 24 April 2014
Published 24 June 2014 Volume 2014:6 Pages 77—88
DOI https://doi.org/10.2147/DHPS.S39568
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Jason J Luke, Patrick A Ott
Melanoma Disease Center, Dana-Farber Cancer Institute and Harvard Medical School, Boston, MA, USA
Abstract: Development of selective inhibitors of BRAF has improved the survival of patients with BRAF-mutant melanoma. The progression-free survival after treatment with a BRAF inhibitor is modest, however, and BRAF inhibitors induce cutaneous toxicity, likely due to paradoxical activation of the mitogen-activated protein kinase pathway. Combining selective BRAF and MEK inhibition, such as the BRAF inhibitor dabrafenib and the MEK inhibitor trametinib, has been shown to improve the response rate and progression-free survival in patients with advanced melanoma while significantly alleviating the paradoxical activation of mitogen-activated protein kinase. This combination treatment results in a reduction in skin toxicity relative to that seen with a BRAF inhibitor alone; however, addition of the MEK inhibitor adds other toxicities, such as pyrexia and gastrointestinal or ocular toxicity. While combined BRAF–MEK inhibition appears primed to become a standard molecular approach for BRAF-mutant melanoma, the utility of the combination has to be considered in the rapidly changing landscape of immunotherapeutics, such as immune checkpoint blockade using anti-cytotoxic T lymphocyte antigen-4 and anti-programmed death-1/programmed death-L1 antibodies. Here we review the development of the dabrafenib plus trametinib combination, the characteristics of each drug and the combination, and the role of this combination in the management of patients with BRAF-mutant melanoma.
Keywords: BRAF, dabrafenib, trametinib, melanoma
Introduction
Malignant melanoma is among the most lethal of the cutaneous neoplasms. In the USA, the annual incidence of melanoma in 2010 was approximately 70,230 cases according to Surveillance, Epidemiology, and End Results data.1 The incidence of melanoma has been rising over the past few decades.2 Multiple risk factors for the development of melanoma have been proposed; however, the only parameter that has been consistently associated with development of melanoma is ultraviolet light exposure.3,4 The outcomes are good for patients diagnosed with early-stage disease, with 5-year recurrence rates of <10% for stage I patients. However, as stages progress, disease-free survival rates begin to decrease significantly, such that patients rendered disease-free but who are stage IIIC have 5-year recurrence rates of 70%–80%.5
Outcomes continue to be poor for most patients with metastatic disease. Prior to 2011, the diagnosis of unresectable, advanced malignant melanoma carried a prognosis that included a 5-year survival of 6% and a median survival of 7.5 months.6,7 Since 2011, two major advances have come into clinical practice, ie, immune-checkpoint blockade and targeting of the BRAFV600 protein in patients harboring this mutation. Advances in immunotherapy were initially spurred with the development of ipilimumab (Bristol-Myers Squibb, Princeton, NJ, USA), the human anti-cytotoxic T lymphocyte antigen-4 (CTLA-4) monoclonal antibody. This drug was demonstrated to improve survival in two Phase III clinical trials.8,9 In studies of ipilimumab, objective radiographic responses at preselected time points occurred in 8%–15% of patients, with an overall response rate of 10%–20%.10–12 Further, improvements in overall survival were observed in patients with advanced melanoma when ipilimumab was compared with a gp100 vaccine8 and when the combination of ipilimumab plus dacarbazine was compared with dacarbazine alone.9 Moving forward, two anti-programmed death-1 (PD-1) receptor antibodies, nivolumab (Bristol-Myers Squibb) and MK-3475 (Merck, Whitehouse Station, NJ, USA) are also likely to be approved for the treatment of advanced melanoma in the next few years. These immunotherapies demonstrate higher response rates and an impressive safety profile, with preliminary data suggesting an improvement in overall survival, while Phase III data from the clinical trials are not yet mature.13
Regarding BRAF-directed therapy, the BRAF inhibitor vemurafenib (F Hoffmann-La Roche Ltd, Basel, Switzerland) was also shown to significantly improve overall survival in patients with advanced melanoma in a Phase III trial,14 with a 30% improvement as compared with dacarbazine.15 Notably, the response rate of vemurafenib was much higher (approximately 50%) when compared with chemotherapy (dacarbazine); however, nearly all patients eventually had progression of melanoma on BRAF inhibitor therapy. These clinical gains have significantly advanced the care of patients with melanoma; however, they are not applicable to all patients. As such, the median overall survival of this population continues to be likely less than 2 years.16
While BRAF targeting has a high response rate, the benefit in terms of progression-free survival appears to be finite in nearly all patients with melanoma. To address the eventual progression of melanoma while on a BRAF inhibitor, rational approaches have been studied to improve the therapeutic potential and durability of BRAF inhibition. An important step in this regard has been the addition of concurrent downstream inhibition of the mitogen-activated protein kinase (MAPK) pathway at MEK in conjunction with BRAF. Dual BRAF–MEK inhibition by dabrafenib and trametinib (both GlaxoSmithKline, Philadelphia, PA, USA) in BRAFV600E/K-mutant melanoma has been observed to be more durable as compared with monotherapy using a BRAF inhibitor.17 This drug combination received fast-track approval from the US Food and Drug Administration (FDA) in 2014 and is now available in standard clinical practice. Here we review the biology of BRAF and the rationale for this combination therapy, the pharmacology and mechanisms of action of these drugs, the clinical efficacy and toxicity associated with these agents, the potential implications regarding quality of life and patient satisfaction, and finally the role of this drug combination in the management of advanced BRAF-mutant melanoma.
BRAF biology and rationale for combination
With the increase in molecular profiling of human tumor specimens, mutations that are common across multiple malignancies are increasingly being recognized. Some of these are potentially targetable with molecular therapeutics, and may necessitate the advent of a new paradigm in which the molecular phenotype of a tumor is treated as opposed to its site of origin. Mutations in the BRAF oncogene may be an example of this. BRAF mutations have been characterized to constitute up to approximately 60% of the driver lesions in cutaneous melanoma,18,19 and they have also been observed in several other tumor types. More specifically, BRAF mutations have been characterized in 10%–15% of colorectal carcinomas,18 3% of lung adenocarcinomas20 and breast cancers,21 20%–50% of serous ovarian cancers,22–24 and 29%–69% of papillary thyroid cancers.25,26 In addition, BRAF mutations may confer a worse clinical prognosis in several of these tumor types compared with cancers without the BRAF mutation.27,28 In melanoma, the presence of BRAF mutation in the primary lesion has not been shown to impact on disease-free interval (time to metastasis) or overall survival.29,30 However, after the development of metastatic melanoma, the median survival of patients with BRAF mutations has been described as shorter relative to patients with wild-type BRAF tumors, although this appears no longer to be the case given the development of selective BRAF inhibitors.31
Multiple laboratory and clinical reports have started to detail mechanisms of both primary and acquired resistance to BRAFV600E inhibition in malignant melanoma.32,33 These mechanisms include acquired NRAS mutations, activation of non-MAPK growth pathways such as phosphatidylinositol 3-kinase/AKT34 via receptor tyrosine kinases (ie, platelet-derived growth factor receptor-β), overexpression of COT kinase,35 mutation of MEK,36 development of RAS-independent BRAFV600E isoform splice variants,37 BRAF amplification,38 and overexpression of hepatocyte growth factor/activation of MET.39 Adaptive upregulation of the AKT pathway has also been shown to modulate the utility of BRAF inhibitors in patients.40 These mechanisms of resistance are shown in Figure 1.
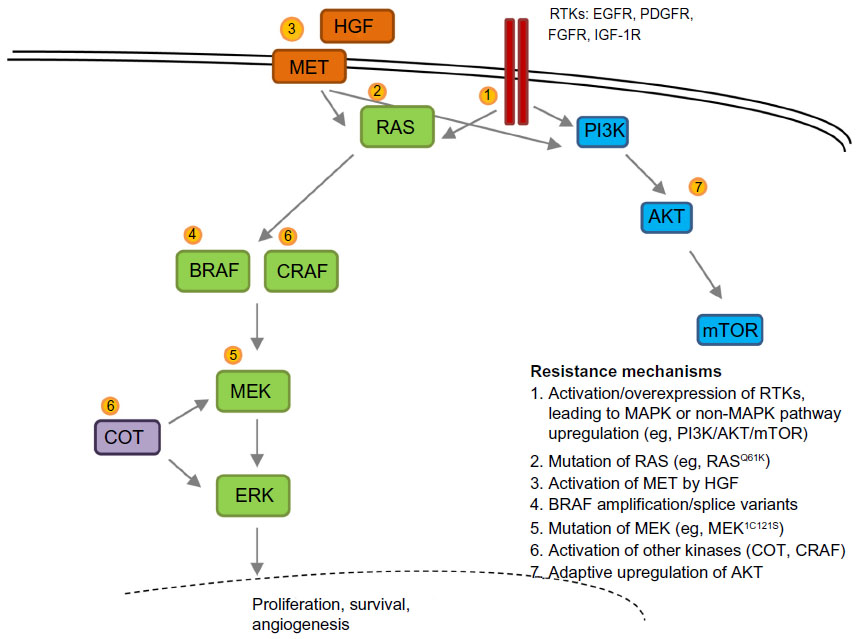 | Figure 1 Mechanisms of resistance to BRAF inhibitor treatment. |
Mechanisms of resistance to treatment with BRAF inhibitors have recently been published in larger cohorts demonstrating approximately 50%–70% as harboring MAPK reactivating changes, with RAS mutations, amplification of BRAF, and BRAF splice variants being the most common.41,42 Mutations not previously described but thought likely to reactivate MAPK were also discovered, and include MAP2K2, MITF, and NF1, as well as mutations with less clear relevance to MAPK, such as HOXD8 and RAC1.42 Alterations in the phosphatidylinositol 3-kinase/AKT pathway were also observed in approximately 20% of patients.41,42
The outcomes for patients with BRAF-mutant melanoma after development of resistance to BRAF targeting are poor, and there are preliminary data suggesting that these patients deteriorate faster than patients not treated with vemurafenib. In a post hoc analysis of the Phase I study of vemurafenib presented at the 2011 American Society of Clinical Oncology meeting, 18 of 48 patients were allowed to continue the study drug beyond the development of initial resistance after receiving local therapy to control isolated disease progression.43 In this group, the median overall survival was not reached during a follow-up period of 15.5 months from initiation of vemurafenib and median overall survival from the time of initial progressive disease was also not reached (median follow-up of 6.0 months). For the 28 patients who did not continue treatment with vemurafenib after progression, the median overall survival after development of resistance was 1.4 months.43 This analysis also detailed the sites of progression after development of resistance, with a notable finding that 25% of patients had disease progression in the brain and central nervous system (19% of patients had metastasis to the brain as the only site). Similarly, an analysis of patient outcomes in advanced BRAF-mutant melanoma showed that patients receiving BRAF inhibitor therapy prior to immunotherapy had worse outcomes when compared with the opposite approach of immunotherapy followed by BRAF inhibitor therapy.44 These data are consistent with models of molecular resistance which suggest that upon removal of inhibition of the primary oncogenic kinase pathway (such as BRAF in melanoma), patients experience a tumor flare as the kinase is reactivated.45–47
The molecular biology of mutant RAF signaling is complex, and significant efforts have been made to elucidate it. Briefly, RAS is activated upon upstream activation of receptor tyrosine kinase, leading to signal transduction through wild-type RAF isoforms (including BRAF, CRAF, and ARAF). These RAF isoforms produce homodimers and heterodimers in different combinations (eg, BRAF: BRAF, BRAF: CRAF, etc). These dimer complexes then lead to activation of MEK1 or MEK2, with subsequent signaling through activation of the ERK and MAPK pathways. In contrast, mutated BRAF exists as a monomer and signals in constitutive fashion independent of upstream activation by receptor tyrosine kinase and RAS. Vemurafenib blocks this activation of mutant BRAF but “paradoxically” can also lead to downstream MEK activation in physiologic MAPK (non-BRAF-mutated cells) cells through ARAF or CRAF homodimerization and heterodimerization.48 This is caused by transactivation of the non-drug-bound partner in BRAF to CRAF heterodimers or CRAF to CRAF homodimers by BRAF inhibitors.49 This process is described in Figure 2.
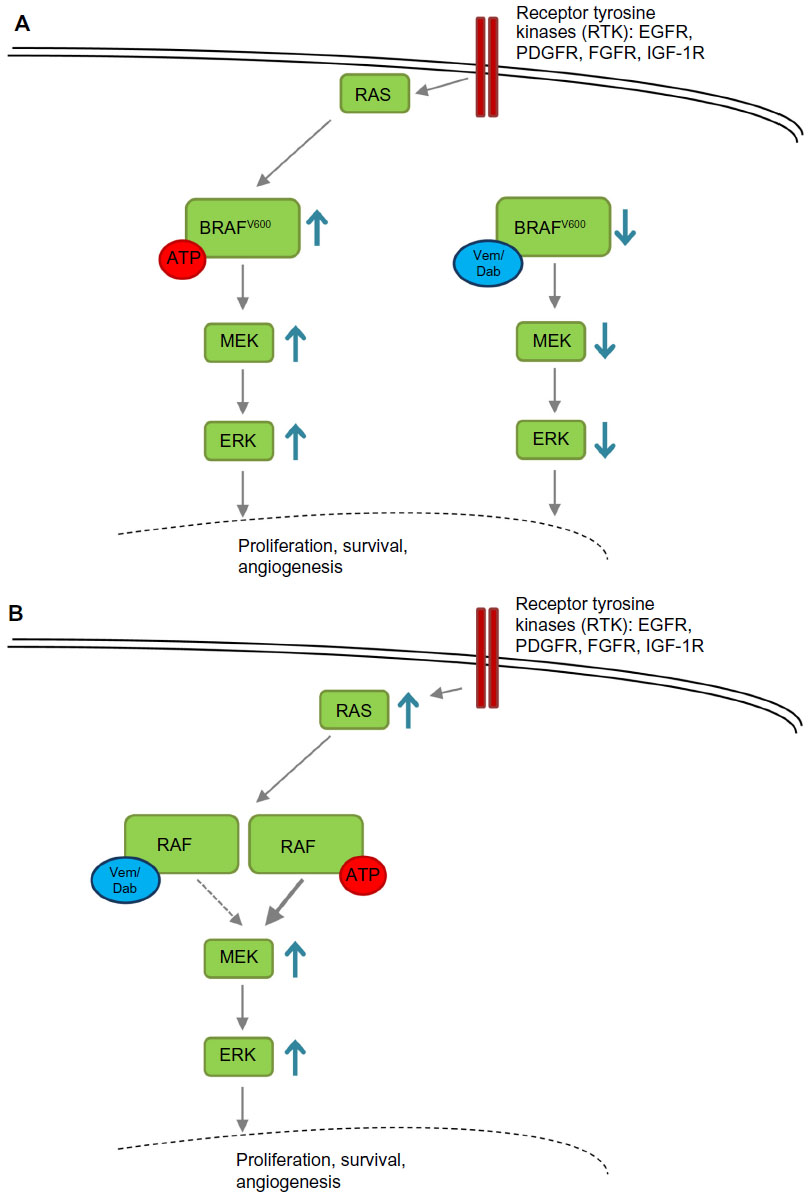 | Figure 2 Paradoxical activation of the MAPK pathway. |
These various RAF dimers, and especially the paradoxical downstream activation of MEK by nonmutant RAF dimers, help to explain some of the mechanisms of vemurafenib resistance and also shed light on the initially unexpected side effects of keratoacanthoma and cutaneous squamous cell carcinoma. Activation of MEK after inhibition of BRAF by vemurafenib is mediated by dimerization of BRAF to CRAF, leading to subsequent CRAF signaling.50,51 There is some suggestion that this may occur in a dose-dependent fashion.49 Further, it is now apparent that upstream activation of RAS, predominantly by HRAS mutation, significantly promotes this effect in non-BRAF-mutant tissues.52 Activation of RAS is not unexpected in the skin, given the prevalence of such mutations due to ultraviolet light exposure from the sun.
Given that the mechanisms of BRAF inhibitor resistance overwhelmingly reactivate MAPK as well as the observation of RAF-induced skin effects, there has been interest in combining oncogenic BRAF inhibition with downstream MAPK inhibition, such as a MEK inhibitor. Preclinical evaluations of BRAF plus MEK inhibition in cell lines and xenografts that have acquired resistance to BRAFV600E targeting have demonstrated that the combination is active.53 Further, combined BRAF–MEK inhibition has been shown to induce greater cell killing and to allow a longer time to development of resistance than treatment with a BRAF or MEK inhibitor alone in naïve BRAFV600E-mutant melanoma models. Also of note is that preclinical rat models of the combination therapy have demonstrated that development of keratoacanthoma and squamous cell carcinoma appears to be abrogated to a significant degree by the combination54 and clinical toxicity data bear this out.55 Most importantly, the Phase I–II clinical trial of dabrafenib and trametinib showed impressive efficacy in terms of response rate and progression-free survival for the combination.17 While pending further validation in Phase III trials, these preliminary data demonstrated a median progression-free survival at the highest dose for the BRAF–MEK combination arm of 9.4 months (dabrafenib 150 mg twice per day, trametinib 2 mg daily), as compared with 5.8 months in the BRAF monotherapy group. Further, the rate of complete or partial response with BRAF–MEK combination therapy was 76%, as compared with 54% with BRAF monotherapy. As such, the combination of BRAF plus MEK inhibition by dabrafenib plus trametinib appears to be an improvement on single-agent BRAF inhibition. Early efforts have been made to examine the mechanisms of resistance to BRAF–MEK combination therapy, with the first report revealing that reactivation of MAPK continues to be the major driver of resistance. In three of five samples from patients who had become resistant to the combination, a novel mutation in MEK2 (Q60P) was observed, while two previously described mechanisms to single-agent BRAF inhibitor, BRAF splice variant and BRAF amplification, were observed.56 Why treatment with a MEK inhibitor did not overcome these mechanisms is not clear; however, this suggests that further investigations regarding the optimal approach to inhibit the MAPK pathway in BRAF-mutant melanoma are in order.
Pharmacology and mechanism of action of dabrafenib and trametinib
Pharmacokinetics of dabrafenib
Dabrafenib has a median terminal half-life of approximately 8 hours after a single dose, with plasma concentrations peaking at a median of 2 hours and declining thereafter.57 In single-dose studies, the peak concentration and area under the curve were dose-proportional through 300 mg of dose escalation. At the approved dose of dabrafenib (150 mg twice per day), the peak concentration and area under the curve were approximately 40% lower by day 15 as compared with day 8. Despite this, the area under the curve appears to remain stable thereafter with subsequent dosing. The mechanism for this decrease is not clear, but may relate to autoinduction pharmacokinetics by dabrafenib itself.
The metabolism of dabrafenib is predominantly hepatic, with preclinical studies suggesting that the cytochrome P450 (CYP) superfamily of mono-oxygenases plays a major role. More specifically, CYP2C8 and CYP3A4 appear to be the predominant enzymes; however, CYP2C19 may also contribute. Dabrafenib is also a substrate for ATP-binding cassette subfamily B member 1 and G member 2, also known as multidrug resistance protein 1 and breast cancer resistance protein, respectively. As such, drugs that are strong inducers of these CYPs or transport proteins are contraindicated during treatment with dabrafenib, and care must be taken when administering other drugs metabolized by these pathways.
The major route of excretion of dabrafenib in humans is fecal (>70%), while renal excretion accounts for less than 20%. The effects of fatty foods on the absorption of dabrafenib have been somewhat equivocal; however, it is currently recommended that dabrafenib be taken on an empty stomach.
Pharmacokinetics of trametinib
Trametinib has a median terminal half-life of approximately 4.5 days after a single dose, with plasma concentrations peaking at a median of one and a half days.58 In single-dose studies, the peak concentration and area under the curve were dose-proportional through dose escalation of up to 6 mg. At the approved dose of trametinib (2 mg daily), the peak concentration and area under the curve were significantly influenced by fatty meals, with an approximately 70% decrease in peak concentration and a 10% decrease in area under the curve when compared with fasting. Trametinib demonstrates a small peak to trough ratio of approximately 2 which, in the context of the extended half-life, allows more constant MEK inhibition within a narrow range of exposure.59
As opposed to dabrafenib, the metabolism of trametinib is predominantly nonhepatic, involving deacetylation as well as secondary modifications including oxidation and glucuronidation. Trametinib does not appear to have significant inhibitory activity toward CYP isozymes or transport proteins, thus limiting potential drug–drug interactions.
Pharmacokinetics of dabrafenib and trametinib
Dabrafenib has a somewhat higher plasma exposure in combination with trametinib, although this finding does not appear to be clinically significant and no clear rationale has yet been advanced to explain it. The pharmacokinetics of trametinib does not appear to be influenced when given in combination with dabrafenib.
Mechanism of action of dabrafenib and trametinib
Dabrafenib is an ATP-competitive, selective inhibitor of RAF kinases, with the strongest effect being on mutant BRAFV600 relative to wild-type BRAF or CRAF. Dabrafenib has minor activity (50% inhibitory concentration <100 nM) against a small number of other kinases, including LIMK1, ALK5, NEK11, SIK, SIK2, PKD2, and BRK. Trametinib is an allosteric, ATP-noncompetitive inhibitor of MEK1 and MEK2 at subnanomolar concentrations. Trametinib did not have significant effects on other kinases when compared in a 183 kinase panel.60
Safety and efficacy of dabrafenib and trametinib as single agents and in combination
Dabrafenib is the second BRAF inhibitor approved by the FDA less than 2 years after vemurafenib. An initial Phase I study of patients with solid tumors harboring BRAFV600E/K mutations examined escalating doses of dabrafenib in 184 patients. The recommended Phase II dose of 150 mg twice daily was established despite no observed maximum tolerated dose.61 Common toxicities included photosensitivity, rash, and fatigue, similar to vemurafenib. Serious adverse events were reported in 39% of patients, specifically noting rash (13%), squamous cell carcinoma (11%), and pyrexia (6%). Pyrexia was the most common adverse event leading to dose interruption. Tumor shrinkage was observed in nine of ten patients with previously untreated brain metastases.61 A Phase II study (known as BREAK-MB, ClinicalTrials.gov identifier NCT01266967) evaluated the use of dabrafenib in patients with BRAFV600E/K-mutant melanoma who were previously untreated or had been locally treated only for brain metastases.62 Significant clinical activity was observed in a cohort of 172 patients, where response rates of 39.2% and 30.8% were observed for untreated versus locally treated patients, respectively. The toxicity profile was manageable, noting the occurrence of pyrexia (6%) and squamous cell carcinoma (6%). BREAK-3 (ClinicalTrials.gov identifier NCT01227889) was a Phase III clinical trial evaluating the effects of dabrafenib as compared with dacarbazine chemotherapy. This study demonstrated an improvement in progression-free survival as the primary endpoint and overall survival as a secondary endpoint.63 The study included 250 patients randomized 3:1 to receive dabrafenib, with crossover at progression. The median progression-free survival for the dabrafenib group was 5.1 months compared with 2.7 months for dacarbazine. These data have been updated to show a median progression-free survival of 6.9 months for dabrafenib and 2.7 months for dacarbazine after extended follow-up. Overall survival data were initially immature, but reported a hazard ratio for survival of 0.61 (confidence interval 0.25–1.48); however, with subsequent follow-up and adjustment for crossover to dabrafenib from the dacarbazine arm, an overall survival of 18.2 months was observed for dabrafenib as compared with 15.6 months for dacarbazine.64 Toxicities were consistent with those observed in previous dabrafenib trials.
Trametinib was initially evaluated in a Phase I study of patients with advanced solid tumors. In a group of 206 patients, a recommended Phase II dose of 2 mg per day was determined with a response rate of 10% in nonmolecularly selected patients.59 Common adverse events included rash and other cutaneous events as well as diarrhea. Dose-limiting toxicities were cutaneous and gastrointestinal, with some ocular events including central serous retinopathy. In a substudy within this clinical trial, a cohort of patients with melanoma was evaluated more specifically.65 Ninety-seven patients with melanoma were included and differentiated by molecular status including BRAFV600E/K (n=36, of whom 30 were not previously treated with a BRAF inhibitor), BRAF wild-type (n=39), BRAF status unknown (n=6), and uveal melanoma (n=16). Among the population of patients with tumors harboring BRAF mutations, the response rate and median progression-free survival were found to be 33% and 5.7 months, respectively. Within the group of patients whose tumors were wild-type for BRAF, the response rate was 10%, but no responses were observed in seven patients with NRAS mutation. Clinical activity of trametinib has also been observed in patients non-BRAFV600E/K-mutant melanoma, with long-term stable disease observed in two patients with BRAFK601E-mutant and BRAFV600R-mutant disease.66 As was seen in the total Phase I population, the toxicity profile in these trials was predominantly cutaneous and gastrointestinal events. The activity observed in patients with BRAF-mutant tumors (BRAFV600E/K) was significant enough to recommend the pursuit of a Phase III study (METRIC, ClinicalTrials.gov identifier NCT01245062) comparing trametinib with chemotherapy (dacarbazine or paclitaxel).67 This clinical trial included 322 patients randomized 2:1 to trametinib and the possibility of crossover from chemotherapy to trametinib at the time of progression. When considering the intention-to-treat population, progression-free survival on trametinib was 4.8 months as compared with 1.5 months on chemotherapy. Although the data were not fully mature at the time of reporting, the hazard ratio for death significantly favored trametinib at 0.54 (95% confidence interval 0.32–0.92), even with the consideration that nearly half of patients received trametinib in crossover after progression on dacarbazine or paclitaxel. As in the Phase I study, common toxicities included cutaneous events, diarrhea, and fatigue. Toxicities or events of special interest included cardiac-related toxicity, such as a decrease in ejection fraction or ventricular dysfunction, which was observed in 7% of patients, and ocular toxicity (blurred vision or reversible chorioretinopathy) in 9%. Notably, no cases of retinal vein occlusion or squamous cell carcinoma were observed.
Given that most molecular mechanisms of resistance to selective BRAF inhibitors described from patient biopsy specimens appear to reactivate signaling through MAPK/ERK, an interest arose regarding the potential sequencing or combining of BRAF and MEK inhibitors. Studies have now been reported describing both sequences (BRAF then MEK inhibition or MEK then BRAF inhibition); however, neither appears to be as efficacious as concurrent BRAF–MEK. Regarding the sequence of a BRAF inhibitor followed by a MEK inhibitor, patients with BRAF-mutant melanoma who had previously been treated with a BRAF inhibitor (either vemurafenib or dabrafenib) or with no prior BRAF inhibitor exposure were treated in a Phase II study using trametinib. Progression-free survival was 4.0 months in the BRAF inhibitor-naïve patients as compared with 1.8 months in those who had previously received a BRAF inhibitor.66 The response rate was also markedly different in naïve versus BRAF inhibitor-pretreated patients (25% and 0%, respectively). Inhibition of MEK by a single agent thus has limited value in patients with melanoma after progression on a BRAF inhibitor.
The reverse sequence of agents has also been investigated, whereby patients with BRAF-mutant melanoma were first treated with a MEK inhibitor and upon progression of disease transitioned to a BRAF inhibitor.68 In this retrospective study of 23 patients, total treatment time was similar to that observed using the BRAF followed by MEK inhibitor sequence; however, time to progression was similar in both groups (4.8 months for trametinib and 4.5 months for dabrafenib). This sequence of agents was observed to achieve a higher rate of response to MEK inhibition when given first (39%).
As compared with the sequencing approach of BRAF and MEK inhibitors, upfront combination of BRAF and MEK inhibitors appears to be more promising. A Phase I/II study explored the combination of dabrafenib and trametinib, eventually describing respective combination doses of 150 mg twice per day and 2 mg daily as tolerable. All patients in this study had advanced melanoma harboring mutations in BRAFV600E/K. Combination treatment at 150 mg and 2 mg as well as 150 mg and 1 mg were compared with dabrafenib 150 mg twice per day. In the 150/2 arm, a significant improvement in both response rate (76% versus 54%, P=0.03) and progression-free survival (9.4 versus 5.8 months, hazard ratio 0.39; 95% confidence interval 0.25–0.62; P<0.001) was observed when compared with dabrafenib monotherapy.17 The combination treatment arm demonstrated fewer adverse events as compared with the dabrafenib monotherapy arm, eg, for squamous cell carcinoma (7% versus 19%, respectively), although the incidence of pyrexia (71% versus 26%) was increased. These data formed the basis for the FDA approval of this combination therapy.
Mature data from the Phase III clinical trial evaluating dabrafenib plus trametinib as compared with dabrafenib plus placebo are eagerly awaited. A preliminary communication in January 2014 reported that the primary endpoint of progression-free survival had been met. An initial description included a median progression-free survival of 9.3 months for combination therapy as compared with 8.8 months with dabrafenib (hazard ratio 0.75, P=0.035). The response rate for the combination treatment was reported to be 67% as compared with 51% for dabrafenib. Toxicity was described as similar to the Phase II results, although a decrease in total pyrexia events was described (51% in Phase III versus 70% in Phase II). Overall survival data are not yet mature.69
Patient-focused perspectives: quality of life and patient satisfaction
The development of selective BRAF inhibitors marked a shift away from standard chemotherapeutic approaches for the management of BRAF-mutant melanoma. This clearly led to a reduction in the toxicities associated with chemotherapy; however, a new spectrum of cutaneous adverse events, including rash, photosensitivity, and development of keratoacanthoma and squamous cell carcinoma, was observed. Overall these toxicities seem to be more tolerable for patients. Additionally, a quality of care analysis suggested more than a doubling of quality-adjusted life years by vemurafenib treatment relative to the prior standard of care.70
Whether the combination of dabrafenib and trametinib will demonstrate an improvement in quality of life as compared with vemurafenib (or dabrafenib alone) is still an open question. Although BRAF plus MEK inhibition decreases the cutaneous toxicity substantially, treatment with dabrafenib plus trametinib has demonstrated a different set of toxicities. The primary problematic toxicity with dabrafenib is pyrexia, which in some patients can be intolerable. Although many patients experience an initial episode of pyrexia that resolves with a drug holiday and dabrafenib rechallenge without any issues,71 other patients have to discontinue the drug altogether. Class effect MEK inhibitor toxicities must be taken into consideration for trametinib. These predominantly include an acneiform rash, edema, and gastrointestinal events (diarrhea and nausea), but can also include more serious toxicities, such as ocular (central serous retinopathy) and cardiac (decreased ejection fraction) toxicities. Algorithms for the management of these adverse events have been developed in parallel with the clinical trials; however, their applicability in the general oncology practice setting has yet to be tested.
No studies to date have assessed quality of life parameters or patient satisfaction of dabrafenib plus trametinib as compared with chemotherapy or BRAF inhibition alone. This is likely to be an important issue, especially if overall survival on the two-drug combination is not improved in a manner that is clinically impressive as compared with current standards of care. The available data to date suggest that dabrafenib plus trametinib improves progression-free survival, but overall survival has not yet been clearly demonstrated, and long-term tolerability of the combination may be a contributing factor. This issue will likely play out in the next several years as longer-term, real-world experience provides insight into patient perspectives and other treatments for melanoma become standard practice.
Conclusion
Given the impressive tumor responses, and progression-free and overall survival benefit associated with treatment with a selective BRAF inhibitor, it is clear that this approach is now a cornerstone in the management of patients with BRAF-mutant melanoma. The development of dabrafenib plus trametinib has now improved upon that in terms of both response rate and progression-free survival and seems likely to also improve overall survival. Nevertheless, whether combined BRAF–MEK inhibition with dabrafenib plus trametinib will displace single-agent BRAF inhibition entirely is yet to be determined and may depend on the clinical circumstance in which this treatment is being utilized. Although the study of BRAF and MAPK inhibition with selective BRAF and MEK inhibitors has been an exciting development in the field of melanoma therapeutics, on a parallel time course, major advancements have been made in immunotherapeutics with immune-checkpoint blockade. These advances have included FDA approval of the anti-CTLA4 antibody ipilimumab, and the likely approval of anti-PD1 antibodies, MK-3475, and nivolumab in the near future. Whereas targeted therapies such as BRAF and MEK inhibitors have a high response rate but modest progression-free survival, immunotherapies have the potential for durable disease control in a higher number of patients. Ipilimumab notably has a much lower response rate and longer time to onset of efficacy as compared with BRAF inhibition.8 The response rate of anti-PD1 antibodies is higher, however, and demonstrate a much more rapid onset of activity compared with ipilimumab.72,73 Further, combination approaches using existing immunotherapies (anti-CTLA4 plus anti-PD1)74 and potentially other novel approaches (eg, anti-KIR, anti-LAG3, anti-OX40) have the potential to increase the response rate and durable disease control rates even further.75
An important consideration for the future role of BRAF and MAPK inhibition must then include the most effective combination or sequence of both targeted and immunotherapies. A robust body of data suggests that combining a BRAF inhibitor with immunotherapy in some fashion is likely to be efficacious.76 However, the initial attempt to combine vemurafenib with ipilimumab had to be aborted due to high rates of immune-mediated hepatitis.77 Beyond this, laboratory experiments suggest that MEK inhibition is likely to have immune-dampening effects and thus may not be an appropriate targeted therapy to combine with immunotherapy.78 Thus, at this time, it is unclear whether targeted therapies may be synergistic or antagonistic with immunotherapy.79 Table 1 describes ongoing clinical trials examining the combination of BRAF and/or MEK inhibitors with immunotherapy.
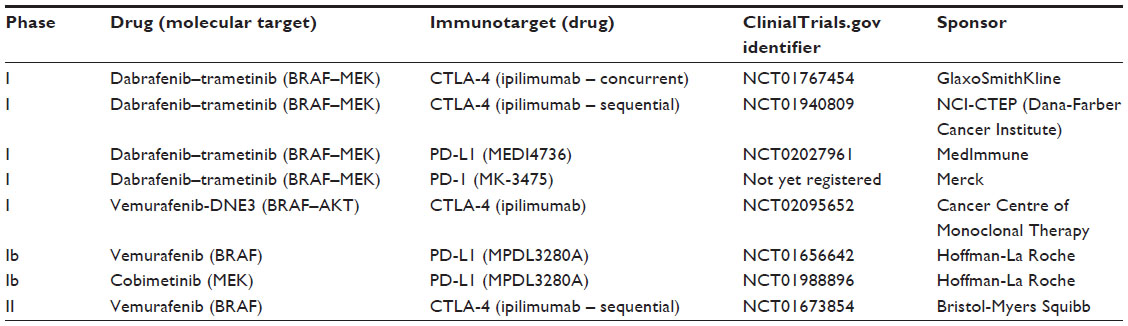 | Table 1 Combination BRAF or BRAF–MEK with immunotherapy studies relevant to patients with melanoma |
Another important issue when considering BRAF inhibitor monotherapy or treatment with dabrafenib plus trametinib will be the potential toxicity and quality of life for the patient. In a robust patient, it is likely that the absolute improvement in response rate and progression-free survival would be enough to drive a treatment choice with dabrafenib and trametinib. However, in a patient of lesser performance status who would be less able to tolerate pyrexia or in a patient who has pre-existing gastrointestinal, ocular, or cardiac toxicity, treatment with a single-agent BRAF inhibitor may be preferable. It is also worth noting that other BRAF–MEK inhibitor combinations, as well as “paradox-breaker” RAF inhibitors that both block mutant BRAF and mitigate physiologic signaling through other RAF isoforms, are in clinical development. The side effect profiles of these other approaches and their clinical efficacy as compared with BRAF inhibitor monotherapy as well as dabrafenib plus trametinib will also significantly influence appropriate sequencing or combinations of available drugs. Table 2 describes ongoing clinical trials examining other BRAF inhibitors and BRAF–MEK inhibitor combinations.
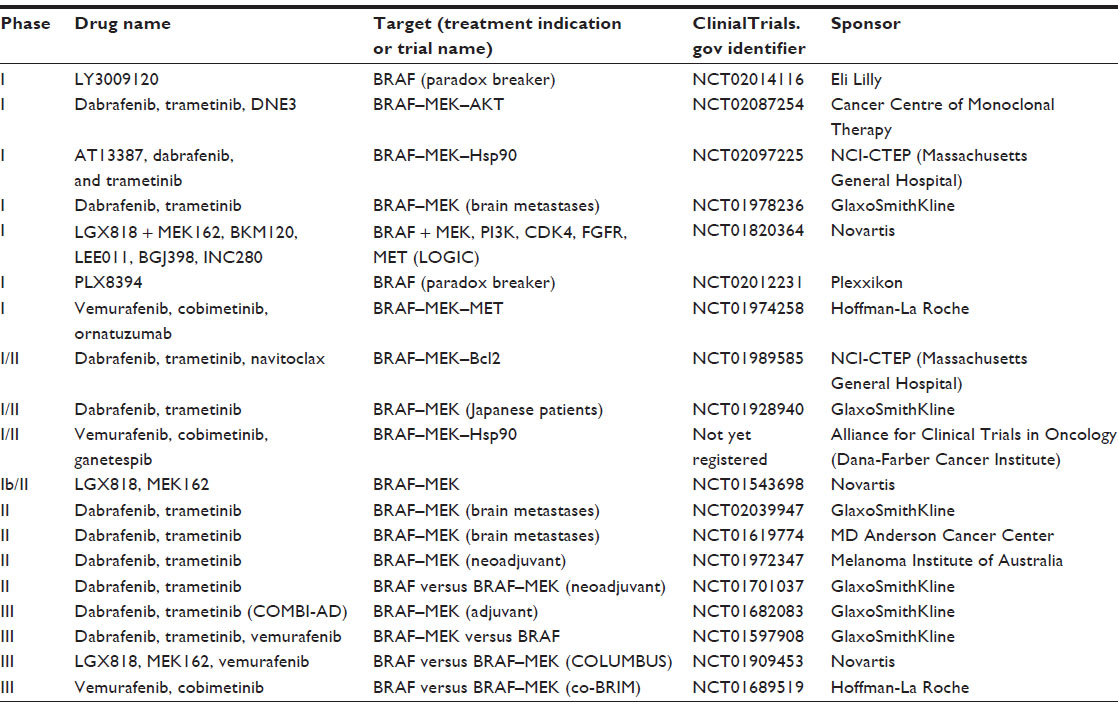 | Table 2 Clinical trials evaluating BRAF and MEK inhibitors in combination |
It is clearly an exciting time in the field of melanoma therapeutics. Nevertheless, many questions remain regarding combining and sequencing agents, in order to obtain the greatest long-term benefit for patients, in addition to the investigation of novel agents. As such, participation in a clinical trial should be offered to all patients when possible, as while many options exist, there is not yet a defined standard of care. For a patient who is not a candidate for a clinical trial, the front-line standard of care is debatable; however, we would advocate that all patients who have the opportunity be treated first with immunotherapy, given the potential for long-term disease control with this approach. In those patients who progress through immunotherapy, are not candidates for such treatment, or have rapidly progressive disease that necessitates immediate palliation, treatment with dabrafenib plus trametinib is emerging as an efficacious option in the management of BRAF-mutant advanced melanoma.
Disclosure
The authors report no conflicts of interest in this work.
References
Howlader N, Noone AM, Krapcho M, et al. Surveillance, Epidemiology, and End Results Program (SEER) Cancer Statistics Review, 1975–2008. Available from: http://seer.cancer.gov/csr/1975_2008/. Accessed May 17, 2014. | |
Geller AC, Miller DR, Annas GD, Demierre MF, Gilchrest BA, Koh HK. Melanoma incidence and mortality among US whites, 1969–1999. JAMA. 2002;288:1719–1720. | |
Veierod MB, Weiderpass E, Thorn M, et al. A prospective study of pigmentation, sun exposure, and risk of cutaneous malignant melanoma in women. J Natl Cancer Inst. 2003;95:1530–1538. | |
Rhodes AR, Weinstock MA, Fitzpatrick TB, Mihm MC Jr, Sober AJ. Risk factors for cutaneous melanoma. A practical method of recognizing predisposed individuals. JAMA. 1987;258:3146–3154. | |
Xing Y, Chang GJ, Hu CY, et al. Conditional survival estimates improve over time for patients with advanced melanoma: results from a population-based analysis. Cancer. 2010;116:2234–2241. | |
Barth A, Wanek LA, Morton DL. Prognostic factors in 1,521 melanoma patients with distant metastases. J Am Coll Surg. 1995;181: 193–201. | |
Balch CM, Soong SJ, Gershenwald JE, et al. Prognostic factors analysis of 17,600 melanoma patients: validation of the American Joint Committee on Cancer melanoma staging system. J Clin Oncol. 2001;19:3622–3334. | |
Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–723. | |
Robert C, Thomas L, Bondarenko I, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med. 2011;364: 2517–2526. | |
Weber J, Thompson JA, Hamid O, et al. A randomized, double-blind, placebo-controlled, phase II study comparing the tolerability and efficacy of ipilimumab administered with or without prophylactic budesonide in patients with unresectable stage III or IV melanoma. Clin Cancer Res. 2009;15:5591–5598. | |
Wolchok JD, Neyns B, Linette G, et al. Ipilimumab monotherapy in patients with pretreated advanced melanoma: a randomised, double-blind, multicentre, phase 2, dose-ranging study. Lancet Oncol. 2010;11: 155–164. | |
O’Day SJ, Maio M, Chiarion-Sileni V, et al. Efficacy and safety of ipilimumab monotherapy in patients with pretreated advanced melanoma: a multicenter single-arm phase II study. Ann Oncol. 2010;21: 1712–1717. | |
Topalian SL, Sznol M, McDermott DF, et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol. 2014;32:1020–1030. | |
Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507–2516. | |
McArthur GA, Chapman PB, Robert C, et al. Safety and efficacy of vemurafenib in BRAF(V600E) and BRAF(V600K) mutation-positive melanoma (BRIM-3): extended follow-up of a phase 3, randomised, open-label study. Lancet Oncol. 2014;15:323–332. | |
Seetharamu N, Tu TJ, Christos P, et al. Enrollment in clinical trials correlates with improved survival in metastatic melanoma. Oncology. 2012;81:403–409. | |
Flaherty KT, Infante JR, Daud A, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med. 2012;367:1694–1703. | |
Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. | |
Curtin JA, Fridlyand J, Kageshita T, et al. Distinct sets of genetic alterations in melanoma. N Engl J Med. 2005;353:2135–2147. | |
Paik PK, Arcila ME, Fara M, et al. Clinical characteristics of patients with lung adenocarcinomas harboring BRAF mutations. J Clin Oncol. 2011;29:2046–2051. | |
Santarpia L, Qi Y, Stemke-Hale K, et al. Mutation profiling identifies numerous rare drug targets and distinct mutation patterns in different clinical subtypes of breast cancers. Breast Cancer Res Treat. 2012;134: 333–343. | |
Singer G, Oldt R 3rd, Cohen Y, et al. Mutations in BRAF and KRAS characterize the development of low-grade ovarian serous carcinoma. J Natl Cancer Inst. 2003;95:484–486. | |
Singer G, Kurman RJ, Chang HW, Cho SK, Shih IeM. Diverse tumorigenic pathways in ovarian serous carcinoma. Am J Pathol. 2002;160: 1223–1228. | |
Sieben NL, Macropoulos P, Roemen GM, et al. In ovarian neoplasms, BRAF, but not KRAS, mutations are restricted to low-grade serous tumours. J Pathol. 2004;202:336–340. | |
Kimura ET, Nikiforova MN, Zhu Z, Knauf JA, Nikiforov YE, Fagin JA. High prevalence of BRAF mutations in thyroid cancer: genetic evidence for constitutive activation of the RET/PTC-RAS-BRAF signaling pathway in papillary thyroid carcinoma. Cancer Res. 2003;63: 1454–1457. | |
Lupi C, Giannini R, Ugolini C, et al. Association of BRAF V600E mutation with poor clinicopathological outcomes in 500 consecutive cases of papillary thyroid carcinoma. J Clin Endocrinol Metab. 2007;92: 4085–4090. | |
Caronia LM, Phay JE, Shah MH. Role of BRAF in thyroid oncogenesis. Clin Cancer Res. 2011;17:7511–7517. | |
Hutchins G, Southward K, Handley K, et al. Value of mismatch repair, KRAS, and BRAF mutations in predicting recurrence and benefits from chemotherapy in colorectal cancer. J Clin Oncol. 2011;29:1261–1270. | |
Shinozaki M, Fujimoto A, Morton DL, Hoon DS. Incidence of BRAF oncogene mutation and clinical relevance for primary cutaneous melanomas. Clin Cancer Res. 2004;10:1753–1757. | |
Maldonado JL, Fridlyand J, Patel H, et al. Determinants of BRAF mutations in primary melanomas. J Natl Cancer Inst. 2003;95:1878–1890. | |
Long GV, Menzies AM, Nagrial AM, et al. Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. J Clin Oncol. 2011;29:1239–1246. | |
Luke JJ, Hodi FS. Vemurafenib and BRAF inhibition: a new class of treatment for metastatic melanoma. Clin Cancer Res. 2012;18:9–14. | |
Luke JJ, Hodi FS. Ipilimumab, vemurafenib, dabrafenib, and trametinib: synergistic competitors in the clinical management of BRAF mutant malignant melanoma. Oncologist. 2013;18:717–125. | |
Nazarian R, Shi H, Wang Q, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010;468:973–977. | |
Johannessen CM, Boehm JS, Kim SY, et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature. 2010;468:968–972. | |
Wagle N, Emery C, Berger MF, et al. Dissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profiling. J Clin Oncol. 2011;29:3085–3096. | |
Poulikakos PI, Persaud Y, Janakiraman M, et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature. 2011;480:387–390. | |
Shi H, Moriceau G, Kong X, et al. Melanoma whole-exome sequencing identifies (V600E)B-RAF amplification-mediated acquired B-RAF inhibitor resistance. Nat Commun. 2012;3:724. | |
Straussman R, Morikawa T, Shee K, et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature. 2012;487:500–504. | |
Shi H, Hong A, Kong X, et al. A novel AKT1 mutant amplifies an adaptive melanoma response to BRAF inhibition. Cancer Discov. 2014;4:69–79. | |
Shi H, Hugo W, Kong X, et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov. 2014;4: 80–93. | |
Van Allen EM, Wagle N, Sucker A, et al. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov. 2014;4:94–109. | |
Kim KB, Flaherty KT, Chapman PB, et al. Pattern and outcome of disease progression in phase I study of vemurafenib in patients with metastatic melanoma (MM). J Clin Oncol. 2011;29 Suppl:8519. | |
Ackerman A, Klein O, McDermott DF, et al. Outcomes of patients with metastatic melanoma treated with immunotherapy prior to or after BRAF inhibitors. Cancer. February 27, 2014. [Epub ahead of print.] | |
Katayama R, Khan TM, Benes C, et al. Therapeutic strategies to overcome crizotinib resistance in non-small cell lung cancers harboring the fusion oncogene EML4-ALK. Proc Natl Acad Sci U S A. 2011;108: 7535–7340. | |
Chaft JE, Oxnard GR, Sima CS, Kris MG, Miller VA, Riely GJ. Disease flare after tyrosine kinase inhibitor discontinuation in patients with EGFR-mutant lung cancer and acquired resistance to erlotinib or gefitinib: implications for clinical trial design. Clin Cancer Res. 2011;17:6298–6303. | |
Le Cesne A, Ray-Coquard I, Bui BN, et al. Discontinuation of imatinib in patients with advanced gastrointestinal stromal tumours after 3 years of treatment: an open-label multicentre randomised phase 3 trial. Lancet Oncol. 2010;11:942–949. | |
Joseph EW, Pratilas CA, Poulikakos PI, et al. The RAF inhibitor PLX4032 inhibits ERK signaling and tumor cell proliferation in a V600E BRAF-selective manner. Proc Natl Acad Sci U S A. 2010;107: 14903–14908. | |
Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature. 2010;464:427–430. | |
Hatzivassiliou G, Song K, Yen I, et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature. 2010;464:431–435. | |
Heidorn SJ, Milagre C, Whittaker S, et al. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell. 2010;140:209–221. | |
Oberholzer PA, Kee D, Dziunycz P, et al. RAS mutations are associated with the development of cutaneous squamous cell tumors in patients treated with RAF inhibitors. J Clin Oncol. 2012;30:316–321. | |
Greger J, Eastman S, Zhang V, et al. Combinations of BRAF, MEK, and PI3K/mTOR inhibitors overcome acquired resistance to the BRAF inhibitor GSK2118436 dabrafenib, mediated by NRAS or MEK mutations. Mol Cancer Ther. 2012;11:909–920. | |
Su F, Viros A, Milagre C, et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med. 2012;366:207–215. | |
Infante JR, Falchook GS, Lawrence DP, et al. Phase I/II study to assess safety, pharmacokinetics, and efficacy of the oral MEK 1/2 inhibitor GSK1120212 (GSK212) dosed in combination with the oral BRAF inhibitor GSK2118436 (GSK436). J Clin Oncol. 2011; 29 Suppl;CRA8503. | |
Wagle N, Van Allen EM, Treacy DJ, et al. MAP kinase pathway alterations in BRAF-mutant melanoma patients with acquired resistance to combined RAF/MEK inhibition. Cancer Discov. 2014;4: 61–68. | |
Tafinlar [full prescribing information]. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/label/2013/202806s000lbl.pdf. Accessed May 17, 2014. | |
Mekinist [full prescribing information]. Available from: http://www.accessdata.fda.gov/drugsatfda_docs/label/2013/204114s000lbl.pdf. Accessed May 17, 2014. | |
Infante JR, Fecher LA, Falchook GS, et al. Safety, pharmacokinetic, pharmacodynamic, and efficacy data for the oral MEK inhibitor trametinib: a phase 1 dose-escalation trial. Lancet Oncol. 2012;13: 773–781. | |
Yamaguchi T, Kakefuda R, Tajima N, Sowa Y, Sakai T. Antitumor activities of JTP-74057 (GSK1120212), a novel MEK1/2 inhibitor, on colorectal cancer cell lines in vitro and in vivo. Int J Oncol. 2011;39: 23–31. | |
Falchook GS, Long GV, Kurzrock R, et al. Dabrafenib in patients with melanoma, untreated brain metastases, and other solid tumours: a phase 1 dose-escalation trial. Lancet. 2012;379:1893–1901. | |
Long GV, Trefzer U, Davies MA, et al. Dabrafenib in patients with Val600Glu or Val600Lys BRAF-mutant melanoma metastatic to the brain (BREAK-MB): a multicentre, open-label, phase 2 trial. Lancet Oncol. 2012;13:1087–1095. | |
Hauschild A, Grob JJ, Demidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380:358–365. | |
Hauschild A, Grob J, Demidov L, et al. An update on BREAK-3, a phase III, randomized trial: Dabrafenib (DAB) versus dacarbazine (DTIC) in patients with BRAF V600E-positive mutation metastatic melanoma (MM). J Clin Oncol. 2013;Suppl 31:9013. | |
Falchook GS, Lewis KD, Infante JR, et al. Activity of the oral MEK inhibitor trametinib in patients with advanced melanoma: a phase 1 dose-escalation trial. Lancet Oncol. 2012;13:782–789. | |
Kim KB, Kefford R, Pavlick AC, et al. Phase II study of the MEK1/MEK2 inhibitor Trametinib in patients with metastatic BRAF-mutant cutaneous melanoma previously treated with or without a BRAF inhibitor. J Clin Oncol. 2013;31:482–489. | |
Flaherty KT, Robert C, Hersey P, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med. 2012;367: 107–114. | |
Goldinger SM, Zimmer L, Schulz C, et al. Upstream mitogen-activated protein kinase (MAPK) pathway inhibition: MEK inhibitor followed by a BRAF inhibitor in advanced melanoma patients. Eur J Cancer. 2014;50:406–410. | |
GlaxoSmithKline. GSK announces headline results for Phase III study of the combination of Tafinlar® (dabrafenib) and Mekinist® (trametinib) in metastatic melanoma. Available from: http://us.gsk.com/html/media-news/pressreleases/2014/gsk-announces-headline-results-for-phase-iii-study-of-the-combin.html. Accessed May 17, 2014. | |
Beale S, Dickson R, Bagust A, et al. Vemurafenib for the treatment of locally advanced or metastatic BRAF V600 mutation-positive malignant melanoma: a NICE single technology appraisal. Pharmacoeconomics. 2013;31:1121–1129. | |
Daud A, Ashworth M, Kefford RF, et al. Pyrexia in patients treated with dabrafenib and trametinib for BRAFV600 mutation-positive metastatic melanoma. Pigment Cell Melanoma Res. 2013;26:945. | |
Hamid O, Robert C, Daud A, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med. 2013;369: 134–144. | |
Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366: 2443–2454. | |
Wolchok JD, Kluger H, Callahan MK, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369:122–133. | |
Ott PA, Hodi FS, Robert C. CTLA-4 and PD-1/PD-L1 blockade: new immunotherapeutic modalities with durable clinical benefit in melanoma patients. Clin Cancer Res. 2013;19:5300–5309. | |
Boni A, Cogdill AP, Dang P, et al. Selective BRAFV600E inhibition enhances T-cell recognition of melanoma without affecting lymphocyte function. Cancer Res. 2010;70:5213–5219. | |
Ribas A, Hodi FS, Callahan M, Konto C, Wolchok J. Hepatotoxicity with combination of vemurafenib and ipilimumab. N Engl J Med. 2013;368:1365–1346. | |
Ott PA, Henry T, Baranda SJ, et al. Inhibition of both BRAF and MEK in BRAF(V600E) mutant melanoma restores compromised dendritic cell (DC) function while having differential direct effects on DC properties. Cancer Immunol Immunother. 2013;62:811–822. | |
Luke JJ, Ott PA. Kinase inhibitors and immune check-point blockade for the treatment of metastatic melanoma and advanced cancer: synergistic or antagonistic? Expert Opin Pharmacother. 2013;14:2457–2462. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.