")
Back to Journals » Journal of Blood Medicine » Volume 7
New developments in the management of moderate-to-severe hemophilia B
Received 6 January 2016
Accepted for publication 9 February 2016
Published 1 April 2016 Volume 2016:7 Pages 27—38
DOI https://doi.org/10.2147/JBM.S81520
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin H Bluth
Moniba Nazeef,1,2 John P Sheehan1,2
1Department of Medicine, Division of Hematology/Oncology, 2UW Carbone Cancer Center, University of Wisconsin School of Medicine and Public Health, Madison, WI, USA
Abstract: Hemophilia B is an X-linked genetic deficiency of coagulation factor IX (FIX) activity associated with recurrent deep tissue and joint bleeding that may lead to long-term disability. FIX replacement therapy using plasma-derived protein or recombinant protein has significantly reduced bleeding and disability from hemophilia B, particularly when used in a prophylactic fashion. Although modern factor replacement has excellent efficacy and safety, barriers to the broader use of prophylaxis remain, including the need for intravenous (IV) access, frequent dosing, variability in individual pharmacokinetics, and cost. To overcome the requirement for frequent factor dosing, novel forms of recombinant FIX have been developed that possess extended terminal half-lives. Two of these products (FIXFc and rIX-FP) represent fusion proteins with the immunoglobulin G1 (IgG1) Fc domain and albumin, respectively, resulting in proteins that are recycled in vivo by the neonatal Fc receptor. The third product has undergone site-specific PEGylation on the activation peptide of FIX, similarly resulting in a long-lived FIX form. Clinical trials in previously treated hemophilia B patients have demonstrated excellent efficacy and confirmed less-frequent dosing requirements for the extended half-life forms. However, gaps in knowledge remain with regard to the risk of inhibitor formation and allergic reactions in previously untreated patient populations, safety in elderly patients with hemophilia, effects on in vivo FIX distribution, and cost-effectiveness. Additional strategies designed to rebalance hemostasis in hemophilia patients include monoclonal-antibody-mediated inhibition of tissue factor pathway inhibitor activity and siRNA-mediated reduction in antithrombin expression by the liver. Both of these approaches are long acting and potentially involve subcutaneous administration of the drug. In this review, we will discuss the biology of FIX, the evolution of FIX replacement therapy, the emerging FIX products possessing extended half-lives, and novel “rebalancing” approaches to hemophilia therapy.
Keywords: hemophilia B, factor IX, bleeding disorders, plasma half-life, therapy
Introduction
Hemophilia B is an X-linked genetic deficiency of coagulation factor IX (FIX) activity, which leads to recurrent and disabling bleeding complications.1 FIX is the zymogen of factor IXa (FIXa), a serine protease critical to amplification of blood coagulation. Numerous mutations in the FIX gene, located on the long arm of chromosome X, are associated with this disorder. In contrast to hemophilia A, FIX deficiency is most commonly caused by single base pair substitutions, resulting in missense, nonsense, or frameshift mutations. Deletions are the second most common gene defect seen in this population.2 The predominance of point mutations, as opposed to the large gene inversions found in hemophilia A, means that a substantial proportion of patients with hemophilia B express some hypofunctioning or nonfunctional protein. The higher prevalence of protein expression in hemophilia B is likely reflected in the lower rates of inhibitor formation (1%–5%) compared to hemophilia A (25%–35%).3,4
Hemophilia B is classified into severe (<1%), moderate (1%–5%), or mild (5%–40%) phenotypes based on the plasma FIX activity of affected individuals.5 The severe phenotype is characterized by spontaneous and recurrent bleeding episodes into joints and muscles, with hemarthroses being the predominant cause of long-term disability.6 The moderate phenotype is characterized by occasional spontaneous bleeds and prolonged bleeding with minor trauma or surgery. Finally, patients with the mild phenotype rarely demonstrate spontaneous bleeding but may have significant bleeding with major trauma or surgery. Aggressive factor replacement is required primarily for patients with moderate and severe hemophilia B phenotypes.
Factor replacement therapy may be provided either “on demand” for symptoms related to bleeding or as “prophylaxis” in which scheduled infusions are undertaken in an attempt to prevent hemorrhage. Primary prophylaxis refers to factor replacement that is started to prevent clinical bleeding episodes in the infant or young child, while secondary prophylaxis refers to replacement therapy that is initiated in response to recurrent bleeding symptoms. Prophylaxis has the potential to change the landscape in hemophilia B by reducing debilitating musculoskeletal complications in patients with severe hemophilia and improving quality of life.7,8 Current clinical research and development efforts are predominantly aimed at manipulating the pharmacokinetic and physiologic properties of FIX to prolong the biological half-life and/or enhance in vivo hemostatic function. Alternative approaches seek to “rebalance” the coagulation response via long-acting agents. Finally, although gene therapy for hemophilia B remains an active area of preclinical and early phase clinical investigation, it is beyond the scope of this review.
Biology of FIX
Biosynthesis, activation, and mechanism of action
FIX is synthesized by hepatocytes as a 461-amino acid precursor polypeptide that undergoes extensive posttranslational modifications including proteolytic removal of the 46-amino acid prepropeptide sequence; vitamin K-dependent γ-carboxylation of selected glutamic acid residues in the N-terminal GLA domain of the mature protein; partial β-hydroxylation of Asp 64; O-linked glycosylation at Ser 61 and 63, Thr 159, 169, 172, and 179; and N-glycosylation (Asn 157 and Asn 167), sulfation (Tyr 155), and phosphorylation (Ser 158) within the activation peptide (Figure 1A–C).9,10 Zymogen FIX (molecular weight =55,000) is secreted into the circulation and may undergo activation to protease (FIXa) by proteolytic cleavage following Arg 145 and Arg 180, with release of the heavily glycosylated activation peptide. FIXa may be generated by the tissue factor–factor VIIa (TF–FVIIa) complex during the initiation phase or by FXIa during the propagation phase of blood coagulation. In the presence of the cofactor factor VIIIa, calcium, and an appropriate phospholipid surface, FIXa is incorporated into the intrinsic “tenase” (FIXa–FVIIIa) complex.11 Ex vivo modeling of blood coagulation demonstrates that factor X activation by the intrinsic tenase complex is the rate-limiting step for thrombin generation.12,13 Reduced or nonexistent activity of the intrinsic tenase complex in hemophilia A and B results in defective thrombin generation and fibrin clot formation that is associated with delayed clinical bleeding.
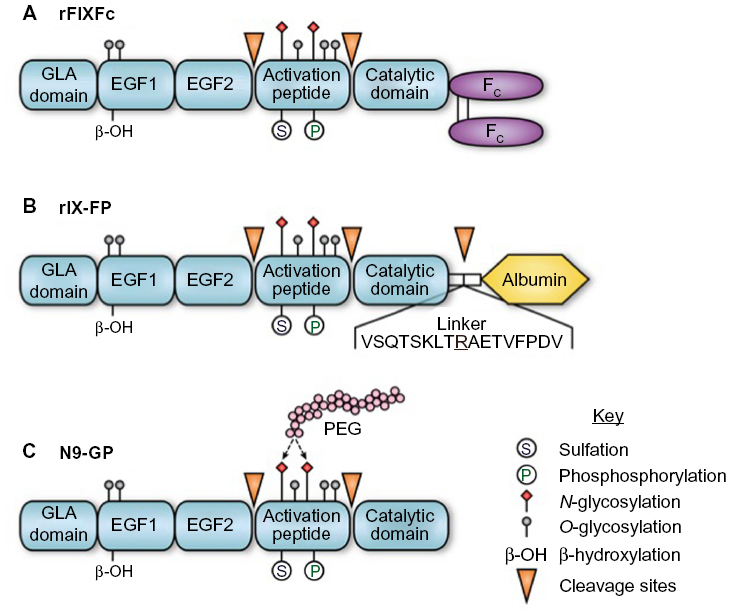 | Figure 1 rFIX forms with extended terminal half-lives. |
Tissue distribution
The procofactor factor VIII (280 kDa) circulates in blood primarily bound to von Willebrand factor (500–2,000 kDa), which largely restricts distribution to the intravascular compartment.14 In contrast, FIX is a smaller protein (55 kDa) with access to both intravascular and extravascular compartments. The apparent volume of distribution for FIX is significantly larger than that for plasma, which supports a two-compartment pharmacokinetic model15 (Figure 2). A number of clinical observations in hemophilia B patients suggest the presence of a significant noncirculating, extravascular “pool” of FIX, including 1) the rapid initial loss of FIX from the circulation following bolus infusion (enhanced in patients who completely lack FIX antigen), 2) a gradual rise in trough levels with repeated bolus dosing, and 3) a reduced dose requirement over time to maintain 100% levels during continuous infusion of plasma-derived FIX (pdFIX) or recombinant FIX (rFIX).16,17 Similarly, infusion of excess bovine FIX into baboons resulted in a proportional increase in circulating levels of baboon FIX analyzed using a species-specific enzyme-linked immunosorbent assay, suggesting displacement of the host protein from an accessible reservoir.18 Reanalysis of these data suggests that the extravascular component contains at least 3-fold more FIX than that present in the circulation.19 FIX (and FIXa) bind rapidly and reversibly to vascular endothelium and subendothelial extracellular matrix, largely mediated by the interaction of specific residues in the FIX GLA domain with collagen IV, located predominantly in the basement membrane.20,21 FIXa also demonstrates increased affinity for heparin/heparan sulfate relative to other coagulation factors.22 Immunohistochemical staining of both murine and human arteries demonstrate FIX (but not FX) binding on the endothelial surface. Remarkably, ~80% of injected FIX is sequestered in the liver within 2 minutes postinjection in hemophilia B mice, likely accounting for much of the rapid initial clearance.21 The extravascular pool contributed significantly to FIX hemostatic function in the mouse in two experiments. A knock-in mouse model expressing a FIX variant with reduced collagen IV affinity demonstrated delayed hemostasis, despite higher circulating FIX levels relative to wild-type protein.23 Similarly, bolus infusion of a FIX variant with enhanced collagen IV affinity demonstrated a prolonged hemostatic effect in the hemophilia B mouse, persisting days after circulating plasma levels reached <1%.19
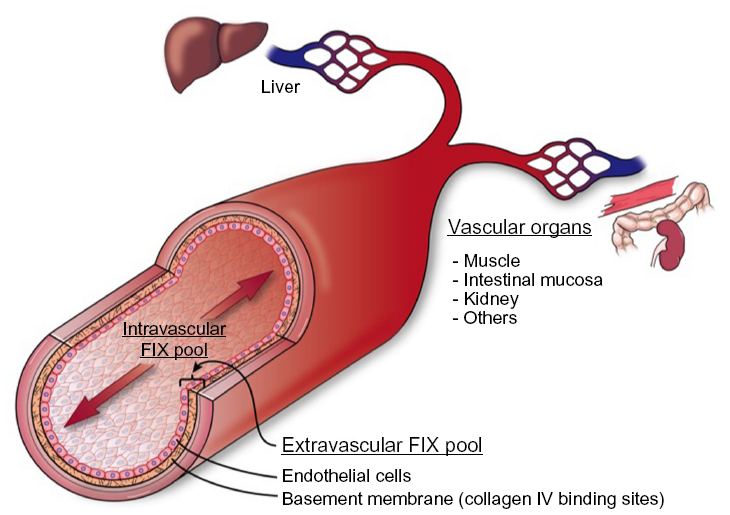 | Figure 2 Cross section of blood vessel indicating location of extravascular FIX pool. |
Recombinant FIX
The cellular origin of rFIX impacts posttranslational modifications that may contribute to differences in pharmacokinetic behavior. Recovery of FIX after bolus injection is significantly reduced for rFIX versus pdFIX.24–27 Currently approved formulations of rFIX expressed in Chinese hamster ovary (CHO) cells or HEK 293 cells are fully γ-carboxylated with variable Asp64 β-hydroxylation but demonstrate markedly lower levels of Tyr 155 sulfation and Ser 158 phosphorylation in the activation peptide.28 Similar differences in posttranslational modifications were found for myotubule-synthesized FIX during gene therapy.29 The rFIX from CHO cells and transgenic porcine milk have reduced sialic acid content in the N-glycans of the activation peptide, likely relevant given the demonstrated role of sialylation in glycoprotein clearance.30 Thus, differences in posttranslational modifications are likely responsible for the reduced recovery of rFIX relative to pdFIX, but the specific contribution of individual modifications remain incompletely defined.31
Evolution of hemophilia B therapy
Treatment of hemophilia B became available in the 1960s and 1970s with the development of fresh frozen plasma, followed by intermediate purity preparations such as prothrombin complex concentrates (PCC) or “FIX complex concentrates”.32 However, use of these intermediate purity preparations was complicated by significant infectious and thromboembolic risks. In the early 1980s, it was recognized that blood products for hemophilia A and B, pooled from multiple donors, were contaminated with blood-borne pathogens (particularly human immunodeficiency virus [HIV] and hepatitis B and C). Tragically, these pooled blood products resulted in HIV transmission to ~50% of all hemophilia patients and up to 90% of severe hemophilia patients.33 Methods for viral screening and inactivation were subsequently developed, which dramatically improved the safety of these products.34 Secondly, intermediate purity PCCs such as Bebulin (Baxter, Deerfield, IL, USA) and Profilnine (Grifols Biologicals Inc., Los Angeles, CA, USA) contain significant amounts of other vitamin K-dependent clotting factors (factors II, VII, and X) and low levels of activated coagulation factors. These products were associated with increased risk of thromboembolic complications, including venous thromboembolism, disseminated intravascular coagulation, and myocardial infarction.35 Highly purified pdFIX preparations were later developed using monoclonal-antibody-based purification that reduced the thromboembolic risks associated with earlier therapies.36 Finally, bioengineering of human FIX in CHO cells was undertaken, with the first purified rFIX product (Benefix, Pfizer, New York, NY, USA) receiving US Food and Drug Administration (FDA) approval in 1997.37
Factor replacement therapy for moderate or severe hemophilia B is based on two major approaches: “on-demand” versus prophylaxis. In the “on-demand” approach, FIX is infused in response to bleeding symptoms to target plasma FIX levels of 60%–80% for major bleeds and 20%–40% for minor bleeds. In the prophylaxis approach, plasma-derived or rFIX is infused 2–3 times per week with the goal of maintaining plasma FIX levels >1%, thereby changing the expected phenotype from severe to moderate hemophilia.38 Prophylactic infusion of replacement factor according to this strategy is superior to on-demand therapy for preventing clinical complications in severe hemophilia patients, particularly disabling hemophilic arthropathy.7,8 In patients with severe hemophilia who are negative for HIV and inhibitors, prophylaxis also reduces the risk of central nervous system hemorrhage by about half.39 Multiple studies suggest that the impact of prophylaxis on quality of life for hemophilia patients is substantial.40 Reduction in hemarthroses leads to less hemophilic arthropathy, fewer orthopedic interventions, and less disability. Despite the clear advantages of prophylaxis, barriers to broader use persist including prominent cost, the need for IV devices with their attending complications, and compliance with frequent factor infusions.41,42 Cost is particularly limiting for the use of prophylaxis in developing countries.43 Practically speaking, prolongation of the dosing interval would enhance compliance and reduce the need for central venous access devices.
Highly purified pdFIX
Highly purified pdFIX products are isolated from plasma or PCC by monoclonal-antibody-based affinity chromatography, which dramatically reduces contaminating vitamin K-dependent clotting factors (II, VII, and X).44 Thus, infusion of highly purified pdFIX in patients with hemophilia B is not associated with elevated in vivo markers of coagulation activation that are commonly seen with PCC products.45 Rigorous purification and viral inactivation processes, including immunoaffinity chromatography, dry heating, solvent/detergent treatment, sodium thiocyanate incubation, and membrane ultrafiltration have markedly reduced infectious risks. Indeed, there have been no documented transmissions of hepatitis B virus, hepatitis C virus, or HIV since the introduction of effective virus inactivation procedures.46 Rare reports describe contamination with infectious prions and parvovirus transmission, but the clinical implications of these findings remain unclear.47,48 In general, plasma-derived factor remains a very safe option for replacement therapy for most patients worldwide.49 Available high-purity pdFIX products in the USA include AlphaNine (Grifols) and MonoNine (CSL Behring, King of Prussia, PA, USA) (Table 1). In clinical studies, these products have demonstrated excellent hemostatic efficacy (~90%) and safety profiles in the prophylaxis, on-demand, and surgical settings.44,50–53 The estimated plasma half-life for highly purified pdFIX preparations is 16–19 hours,26,54,55 although other estimates are closer to 30 hours,56 likely due to methodological differences. Dosing 2–3 times weekly is appropriate for prophylaxis, although significant variation between individuals is observed.56,57
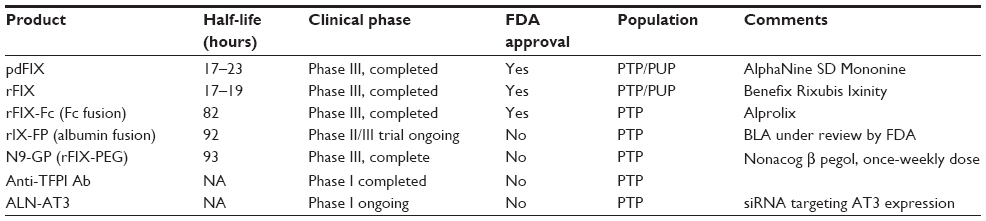 | Table 1 Therapeutic approaches to hemophilia B |
Recombinant FIX
The human FIX gene was identified and cloned in early 1980s,58 followed by insertion of the human FIX cDNA sequence into the CHO cell line and expression of rFIX in 1982.59 rFIX is purified by a four-step chromatographic process, including nanofiltration for viral reduction. Comparison of pharmacokinetics in patients with hemophilia B demonstrates that the recombinant protein has consistently lower (~30%–50%) in vivo recovery (peak levels) with a similar terminal half-life (17–19 hours) to pdFIX following bolus injection.26,54 Treatment of 56 previously treated patients (PTP) with rFIX demonstrated excellent hemostatic efficacy, with 80.9% of hemorrhages resolving with one dose, the majority of hemorrhages (61%) on prophylaxis occurring >72 hours after the last dose, and 27 surgical procedures in 20 patients demonstrating good or excellent hemostasis. Toxicity was limited to the development of a transient inhibitor in one patient, but no evidence of thromboembolism or viral transmission was observed.60 Similarly, treatment of 63 previously untreated patients (PUP) with rFIX also demonstrated excellent hemostatic efficacy, with 75% of hemorrhages resolving with one dose, 91% of prophylaxis responses in 32 patients rated as “excellent”, and 30 procedures on 23 patients with good or excellent hemostasis. Five patients demonstrated allergic reactions, and two of them demonstrated inhibitors (3%). No thrombotic or viral transmission events were observed.61 A French study on patients with moderate-to-severe hemophilia did not show any difference in quality of life between rFIX and pdFIX, although rFIX consumption was higher by a factor of 1.40. As expected, increased cost was associated with prophylaxis compared to on-demand therapy.62
Modified rFIX products with prolonged terminal half-lives
Clinical practice guidelines recommend that the goal of prophylaxis in hemophilia B is to maintain plasma FIX levels >1%,5 which has led to strategies designed to prolong the terminal plasma half-life of FIX. These products, which increase the feasibility of once-weekly FIX administration, have been recently approved or are in late clinical development stages (Table 1).
rFIXFc fusion protein
The Fc domain of the IgG molecule binds to the neonatal Fc receptor (FcRn), which is present on many adult cells, including endothelial cells, monocytes/macrophages, and epithelial cells.63 Following internalization by the endothelial cell, proteins that bind to the FcRn receptor in the acidified endosome are protected from lysosomal sorting and degradation, allowing recycling back to the cell surface with pH-dependent release into the circulation. Recycling via the FcRn is the mechanism responsible for the extended in vivo half-life of IgG, albumin, and therapeutic IgG1-Fc fusion proteins.63 Similarly, direct fusion (no linker region) of a monomeric Fc domain from human IgG1 to the carboxyl terminus of human FIX (rFIXFc) results in a long-acting rFIX (Figure 1A). rFIXFc demonstrates a 3- to 4-fold prolongation of terminal half-life relative to rFIX in multiple species (mouse, rat, dog, and monkey). Posttranslational modifications of rFIXFc expressed in HEK293 cells appear similar to rFIX expressed in CHO cells (Benefix, Pfizer). On a molar basis, the specific activity of rFIXFc is ~50% that of conventional rFIX, and the precise impact of the Fc domain on factor X activation kinetics was not reported.28
A Phase I–II clinical study of rFIXFc in 14 patients with hemophilia B demonstrated in vivo recovery ~20% higher than rFIX and confirmed an ~3-fold prolongation of terminal half-life.64 rFIXFc was subsequently examined in a Phase III study of 123 PTPs (median age: 30 years) divided into four treatment groups: weekly dose-adjusted prophylaxis, interval-adjusted prophylaxis, on-demand therapy, and perioperative treatment. As expected, prophylaxis groups demonstrated significantly lower median annualized bleeding rates of 3.1% and 2.4%, respectively, compared to 18.6% for on-demand therapy. The vast majority (90.4%) of bleeding episodes resolved with one injection. Hemostasis was rated as good or excellent in 14 major surgeries performed in 12 subjects. No inhibitors or anaphylactic reactions were observed, and the only thrombotic episode was an obstructive clot in the urinary collection system of a patient with previous hematuria. The terminal half-life for rFIXFc was 82 hours, compared to 33 hours for rFIX (Benefix).65 A post hoc analysis comparing patients who received prophylaxis for >6 months on this study with their prestudy prophylaxis suggested that rFIXFc markedly reduced infusion frequency and FIX consumption with fewer bleeding episodes.66 In 2014, rFIXFc (Alprolix, Biogen, Cambridge, MA, USA) was approved by the FDA for prophylaxis and treatment of bleeding episodes in individuals with hemophilia B, in both routine and perioperative settings. An ongoing clinical study (NCT02234310) will evaluate the safety and efficacy of rFIXFc in the PUP population with severe hemophilia B.
FIX-albumin fusion protein
The FIX-albumin (rIX-FP) fusion protein represents an alternative approach to prolongation of rFIX half-life that similarly takes advantage of recycling via the FcRn. Albumin interacts with the FcRn at a site independent of the IgG Fc domain but has similar pH-dependent binding affinity that allows recycling back to the circulation, resulting in a serum half-life of ~20 days.67 In rIX-FP, the albumin moiety has been fused to the C-terminus of FIX via a cleavable linker containing a FIX activation site. The design of this fusion protein allows proteolytic release of the FIXa molecule from albumin upon activation by TF–FVIIa or factor XIa (Figure 1B). The molar-specific activity of this fusion protein expressed in CHO cells is 20- to 30-fold higher than the fusion protein expressed with noncleavable linkers.68 In rats and rabbits, rIX-FP demonstrated a 1.6- to 1.7-fold increased recovery and ~4-fold increase in terminal half-life relative to rFIX, along with demonstrated efficacy in a tail-tip bleeding model in hemophilia B mice.68 Similar prolongation of therapeutic levels was observed in dogs with hemophilia B and nonhemophilic cynomolgus monkeys.69
In a multicenter, dose-escalation Phase I trial of 25 PTPs (mean age: 35 years), rIX-FP demonstrated a 44% increase in recovery and ~5-fold increase in half-life (mean: 92 hours) relative to rFIX, with no inhibitors or hypersensitivity events reported.70 A second Phase I–II study of 17 PTPs (mean age: 26 years) demonstrated similar pharmacokinetic results and included an 11-month safety and efficacy evaluation. During the extension study, 13 patients received weekly prophylaxis and four patients were treated on demand. Seven patients on prophylaxis were treated for 14 spontaneous bleeds during the prophylaxis period, while six had no bleeds, with a median annualized spontaneous bleeding rate (AsBR) of 1.13. For both on-demand and prophylaxis patients, 95% bleeding episodes were treated with single infusion and no inhibitors, antibodies to rIX-FP, or allergic reactions were detected.71 Preliminary results of multinational, Phase II–III studies evaluating the safety and efficacy of rIX-FP in PTP (adults and children) were reported at the 2015 International Society of Thrombosis and Haemostasis biannual meeting. These studies included 27 PTPs <12 years of age treated for 12 months with weekly prophylaxis or on demand. No inhibitors or antibodies to rIX-FP were reported, and 97% bleeding episodes were treated with one or two infusions.72 The adult study (age 12–61 years) enrolled 63 patients treated on demand (N=23) or with a 7-day prophylaxis interval (N=40) for 6 months, after which on-demand patients were switched to 7-day prophylaxis, and the therapy of selected prophylaxis patients was extended to 10- or 14-day intervals. As expected, a marked reduction in bleeding episodes was observed for prophylaxis (median AsBR: 0.00) versus on-demand. Additionally, 21 prophylaxis patients extended their treatment interval to 14 days, with no significant increase in bleeding and 50% reduction in factor consumption relative to historical use. Finally, ~94% of bleeding episodes resolved with one factor injection, and no serious adverse events were observed, including development of inhibitors or antibodies to rIX-FP.73 A surgical substudy reported 12 major operations in ten patients treated with preoperative rIX-FP, and the hemostatic response was characterized as good or excellent in all cases with 2–7 infusions during the 14-day postoperative period.74 The Biologics License Application for rIX-FP was accepted for review by the FDA in February 2015. An ongoing clinical study (NCT02053792) will address the safety and efficacy of rIX-FP in the PUP population.
GlycoPEGylated rFIX
Covalent modification of therapeutic proteins with polyethyelene glycol (PEG) chains is an established approach to prolonging protein half-life and in vivo activity.75 N9-GP (nonacog β-pegol) is a rFIX expressed in CHO cells and has been modified by site-directed glycoPEGylation with a 40-kDa PEG molecule on one (95%) or both (5%) N-glycans within the activation peptide (Figure 1C). Upon proteolytic the activation of FIX, the activation peptide containing the PEG moiety is removed, leaving native rFIXa. The kinetics of FIX activation by FXIa is identical to unmodified protein, while the activation by TF–FVIIa demonstrates a modest increase in Km for FIX. The specific activity of N9-GP is equivalent to plasma-derived or native rFIX. Preclinical studies of N9-GP in pigs and dogs with hemophilia B demonstrated a 2-fold increase in recovery and an ~5- to 7-fold increase in half-life relative to unmodified rFIX, along with demonstrated efficacy in a tail-tip bleeding model in hemophilia B mice.76
A Phase I trial of N9-GP in 16 PTPs with hemophilia B demonstrated markedly enhanced recovery and a half-life (93 hours) that was ~5-times longer than nonpegylated rFIX. No inhibitors were detected, but one patient experienced a hypersensitivity reaction.77 A multinational, randomized, single-blind Phase III trial of the safety and efficacy of N9-GP was conducted in 74 PTPs (mean age: 31 years) with hemophilia B. Patients chose between on-demand (N=15) and prophylaxis (N=59) therapy, with the latter group randomized between two different weekly doses. Pharmacokinetic analysis confirmed previous observations in the Phase I trial. Overall estimated success rate of achieving hemostasis was 92%, with best results in prophylaxis group. Median AsBR was 0.97 and <0.01 for 10 or 40 U/kg weekly prophylaxis, respectively, and 11.1 for on-demand therapy. No inhibitors, hypersensitivity reactions, or thromboembolic complications were reported.78 An extension of this trial (NCT01395810) and a study in patients undergoing major surgery (NCT01386528) have been completed but not reported. Additional trials are ongoing to evaluate N9-GP in both pediatric PTP (NCT01467427) and PUP (NCT02141074) populations.
Alteration in rFIX function by mutagenesis
Preclinical data exist for targeted mutagenesis of human FIX designed to enhance coagulant activity or in vivo therapeutic properties. These approaches include incorporation of the FIX Padua mutation or combined mutations to enhance coagulant activity,79 introduction of additional N-glycosylation sites to enhance pharmacokinetics and subcutaneous (SC) absorption,80 and disruption of antithrombin- and heparin-mediated regulation to enhance in vivo protease activity.81 Although the overall risk of inhibitors is lower in hemophilia B (see “Inhibitor formation and anaphylaxis” section), variant FIX sequences will require careful analysis for potential inhibitor formation.82
Gaps in knowledge
Although the potential advantages of rFIX proteins with extended half-lives for prophylaxis in hemophilia B are clear, there remain significant gaps in our knowledge with regard to the risk of inhibitor formation, safety in older hemophilic populations, and impact on the in vivo distribution of FIX.
Inhibitor formation and anaphylaxis
The incidence of inhibitors in hemophilia B (1%–5%) is significantly less than that in hemophilia A (25%–35%).4,83,84 This difference in inhibitor prevalence likely reflects the predominance of missense mutations in severe hemophilia B, as opposed to the large gene inversions associated with severe hemophilia A.85 The former mutations are much more likely to be associated with sufficient protein expression to confer immune tolerance, while the latter completely lacks protein expression, resulting in a substantially higher inhibitor risk. Consistent with this notion, the risk of inhibitor formation in hemophilia B is greatest in those patients with large gene deletions.86 This risk is highest in the PUP population during the first 5–6 years of life, occurring at a median of 11 exposure days to FIX product.87 Long-term safety and efficacy studies of rFIX demonstrate ~3% rate of inhibitor formation, consistent with rates seen with plasma-derived products.61 The Phase II–III studies for all three extended half-life products were performed in PTP populations with substantial previous FIX exposure and exclusion of patients with evidence of inhibitors.65,73,78 Clearly, the true incidence of inhibitor formation with these products will not be known until studies in PUP populations are completed. Patients with hemophilia B are uniquely at risk (compared to hemophilia A) for allergic reactions to factor infusion. These reactions occur in 1%–3% cases, with comparable incidence for both pdFIX and rFIX products.88 Allergic reactions are commonly associated with inhibitors and may actually precede inhibitor development.85 The exact mechanism for the allergic reactions remains unknown, but they are postulated to involve extravascular IgE-mediated responses or codeletion of adjacent immune-response loci with large FIX gene deletions.86 The true incidence of allergic reactions with the extended half-life agents will likewise not be known until clinical trials are completed in PUP populations.
Safety
As the dramatic impact of HIV infection on the hemophilia population fades, increasing life expectancy means that the thromboembolic risk associated with an aging hemophilic population may yield additional safety concerns. On the basis of the data reported in an Italian registry in 2009, it is estimated that approximately 15% of hemophiliacs are aged 45 years or above, and approximately 2% are older than 65 years.89 Mean subject age in recent clinical trials for the extended half-life agents range from 26 to 35 years of age, with additional exclusions for comorbid conditions.65,73,78 As additional risks may need to be considered in the older population, it will be important to include elderly patients with hemophilia B in future trials to the extent possible. For example, activation of rFIXFc (or rIX-FP with incomplete linker cleavage) may generate long-acting rFIXa forms that could potentially elevate thromboembolic risk in an older population. Likewise, although PEGylated protein therapies have a wide therapeutic index and substantial track record in the clinic, the life-long nature of hemophilia treatment raises concern about potential toxicity due to chronic PEG exposure.90 Conversely, PEGylated proteins tend to be less immunogenic than the unmodified protein, which could lead to a reduced rate of inhibitor formation.75
The role of extravascular FIX
The contribution of the extravascular pool to the hemostatic function of FIX in mouse models suggests that the access of modified rFIX forms to collagen IV binding sites in the subendothelium may be therapeutically relevant (Figure 2).19,23 Tritium-labeled rIX-FP and rFIX demonstrate similar overall tissue distribution, with early enhancement in liver levels and the expected prolonged half-life for the fusion protein in nonhemophilic rats.91 However, no direct analysis of binding to endothelium or tissue distribution in hemophilia B animals is available. N9-GP demonstrates 20-fold reduced affinity for binding to an endothelial monolayer relative to unPEGylated rFIX, suggesting that the PEG moiety may reduce access to, or affinity for, collagen IV binding sites.76 We are not aware of any published analysis of endothelial binding or in vivo tissue distribution for rFIXFc. It is not known whether reduced access or binding to extravascular sites by extended half-life forms impacts duration of the hemostatic effect or relative risk of bleeding at low trough levels compared to conventional FIX. However, the hemostatic contribution of extravascular FIX suggests that plasma FIX activity levels may not represent an ideal surrogate for therapeutic efficacy.
Cost-effectiveness of factor replacement
A major impetus behind the development of extended half-life FIX products was to enhance the feasibility of prophylaxis in hemophilia B. Thus, the contribution of these therapies will be judged, in part, on their ability to impact the cost-effectiveness of prophylaxis. Prophylaxis is clearly more expensive up-front than on-demand replacement therapy, although savings due to reduced joint bleeds and related complications potentially makes prophylaxis ultimately more cost-effective.92 Determination of indirect cost savings (hospitalizations, disability, and productivity) related to prophylaxis is both complex and critical to this cost-effectiveness analysis. Limitations in resources remain a major obstacle to prophylaxis in hemophilia, particularly in developing countries. At least, the costs associated with the extended half-life products will need to be justified by proportional reduction in factor use due to less frequent dosing for prophylaxis and surgical procedures.
Novel approaches
Although prophylactic factor replacement results in significant improvements in hemophilia outcomes, the need for vascular access and compliance with frequent infusions remain major challenges. Several long-acting strategies attempt to rebalance hemostasis in hemophilia by “inhibiting the inhibitor”. These strategies have the potential advantages of being effective in the presence of anti-FIX inhibitors and not requiring IV access.
Anti-tissue factor pathway inhibitor therapy
Inhibition of the TF–FVIIa complex by tissue factor pathway inhibitor (TFPI) shuts down the extrinsic pathway following initiation of coagulation, making thrombin generation in the propagation phase dependent on the FIXa–FVIIIa complex (defective in hemophilia). Antagonizing TFPI-mediated inhibition via inhibitory antibodies allows TF–FVIIa activity to persist, resulting in continued thrombin generation via the extrinsic pathway that may, in part, compensate for defective factor X activation by the FIXa–FVIIIa complex in hemophilia. Concizumab, a monoclonal antibody against the kunitz-2 inhibitor domain of TFPI, improves clot formation in hemophilic blood and plasma and substantially reduces cuticle bleeding in a rabbit hemophilia model.93 In a Phase I trial involving 28 healthy volunteers and 24 hemophilia patients (21 hemophilia A and 3 hemophilia B) without inhibitors, both IV and SC concizumab was well tolerated, demonstrating nonlinear pharmacokinetics with dose-dependent, target-mediated clearance. Concizumab demonstrated dose-dependent increases in d-dimer and prothrombin fragment 1+2 in both healthy volunteers and hemophilia patients, and antibody was detectable for up to 42 days at the highest doses tested.94 Additional humanized antibodies against TFPI are currently under development.
siRNA-mediated antithrombin gene knockdown
Reduction in antithrombin activity can restore normal levels of thrombin generation in hemophilic (A or B) plasma or whole blood. ALN-AT3 is an siRNA targeting a conserved region of the antithrombin (SERPINC1) transcript that has been conjugated with Gal-Nac to facilitate uptake by the asialoglyoprotein receptor in the liver. ALN-AT3 demonstrated dose-dependent reduction in antithrombin protein levels (up to >90%) in both mice and nonhuman primates, correcting clotting and bleeding phenotypes in the hemophilia A mouse. Similarly, ALN-AT3 rectified thrombin generation in nonhuman primates with antibody-induced hemophilia A.95 An ongoing Phase I trial has reported preliminary results in four healthy volunteers and 12 hemophiliacs (A and B) on ascending weekly dose levels, indicating that ALN-AT3 was generally well tolerated with no significant thromboembolic events. A maximal 86% reduction in plasma antithrombin was achieved with normalization of patient thrombin generation. A third arm will investigate the effects and tolerability of monthly dosing.96
Conclusion
Progress in the management of moderate-to-severe hemophilia B has been made possible by the availability of purified plasma-derived and rFIX with substantially reduced risks of infectious and thromboembolic complications. Further, the superior clinical outcomes associated with prophylaxis versus on-demand approaches to factor replacement have been broadly recognized. Current development efforts in hemophilia B therapy are focused on overcoming the barriers to broader use of prophylaxis, including requirement for IV access, compliance with frequent infusions, poor recovery of rFIX, and cost. Modified rFIX proteins with extended half-lives (FIXFc, rIX-FP, and N9-GP) may have a substantial impact on the feasibility of prophylaxis via significant prolongation of dosing intervals and potential for higher troughs that may have lifestyle implications for the active patient with hemophilia B. Unanswered questions regarding the extended half-life forms include the risk of inhibitor formation and allergic reactions in PUP populations, safety in elderly populations, and the potential therapeutic implications of altered in vivo distribution. Alternative approaches attempt to rebalance coagulation in hemophilia by inhibiting TFPI activity or antithrombin expression to normalize thrombin generation. These latter approaches represent long acting and potentially SC approaches to hemophilia therapy that address many of the barriers to prophylactic therapy. These novel, long-acting approaches to hemophilia B therapy have the potential to markedly reduce common complications such as hemophilic arthropathy, enhance the ability to lead a physically active lifestyle, and improve the quality of life for patients with moderate-to-severe hemophilia B.
Acknowledgments
This work was supported, in part, by funding from the University of Wisconsin Institute for Clinical and Translational Research (JPS) and the Pfizer US Hemophilia ASPIRE Program (JPS).
Disclosure
JPS has received previous research funding from the Bayer Hemophilia Awards Program and the Novo Nordisk Access to Insight Program. He has current research funding from a Pfizer US Hemophilia ASPIRE Hemophilia Grant. The authors report no other conflicts of interest in this work.
References
Srivastava A, Brewer AK, Mauser-Bunschoten EP, et al. Guidelines for the management of hemophilia. Haemophilia. 2013;19(1):e1–e47. | |
Bowen DJ. Haemophilia A and haemophilia B: molecular insights. Mol Pathol. 2002;55(2):127–144. | |
Gomez K, Klamroth R, Mahlangu J, Mancuso ME, Mingot ME, Ozelo MC. Key issues in inhibitor management in patients with haemophilia. Blood Transfus. 2014;12(Suppl 1):s319–s329. | |
Collins PW, Palmer BP, Chalmers EA, et al. Factor VIII brand and the incidence of factor VIII inhibitors in previously untreated UK children with severe hemophilia A, 2000–2011. Blood. 2014;124(23):3389–3397. | |
White GC 2nd, Rosendaal F, Aledort LM, Lusher JM, Rothschild C, Ingerslev J. Definitions in hemophilia. Recommendation of the scientific subcommittee on factor VIII and factor IX of the scientific and standardization committee of the International Society on Thrombosis and Haemostasis. Thromb Haemost. 2001;85(3):560. | |
Acharya SS. Exploration of the pathogenesis of haemophilic joint arthropathy: understanding implications for optimal clinical management. Br J Haematol. 2012;156(1):13–23. | |
Manco-Johnson MJ. Update on treatment regimens: prophylaxis versus on-demand therapy. Semin Hematol. 2003;40(3 Suppl 3):3–9. | |
Manco-Johnson MJ, Abshire TC, Shapiro AD, et al. Prophylaxis versus episodic treatment to prevent joint disease in boys with severe hemophilia. N Engl J Med. 2007;357(6):535–544. | |
Furie B, Furie BC. Molecular and cellular biology of blood coagulation. N Engl J Med. 1992;326(12):800–806. | |
Kaufman RJ. Post-translational modifications required for coagulation factor secretion and function. Thromb Haemost. 1998;79(6):1068–1079. | |
Mann KG. Biochemistry and physiology of blood coagulation. Thromb Haemost. 1999;82(2):165–174. | |
Hoffman M, Monroe DM, Oliver JA, Roberts HR. Factors IXa and Xa play distinct roles in tissue factor-dependent initiation of coagulation. Blood. 1995;86(5):1794–1801. | |
Rand MD, Lock JB, van’t Veer C, Gaffney DP, Mann KG. Blood clotting in minimally altered whole blood. Blood. 1996;88(9):3432–3445. | |
Björkman S, Berntorp E. Pharmacokinetics of coagulation factors: clinical relevance for patients with haemophilia. Clin Pharmacokinet. 2001;40(11):815–832. | |
Björkman S, Carlsson M, Berntorp E. Pharmacokinetics of factor IX in patients with haemophilia B. Methodological aspects and physiological interpretation. Eur J Clin Pharmacol. 1994;46(4):325–332. | |
Briet E. Three Problems of Hemophilia B. Leiden, The Netherlands: University of Leiden; 1977. | |
Uprichard J, Adamidou D, Goddard NJ, Mann HA, Yee TT. Factor IX replacement to cover total knee replacement surgery in haemophilia B: a single-centre experience, 2000–2010. Haemophilia. 2012;18(1):46–49. | |
Stern DM, Knitter G, Kisiel W, Nawroth PP. In vivo evidence of intravascular binding sites for coagulation factor IX. Br J Haematol. 1987;66(2):227–232. | |
Feng D, Stafford KA, Broze GJ, Stafford DW. Evidence of clinically significant extravascular stores of factor IX. J Thromb Haemost. 2013;11(12):2176–2178. | |
Cheung WF, van den Born J, Kühn K, Kjellén L, Hudson BG, Stafford DW. Identification of the endothelial cell binding site for factor IX. Proc Natl Acad Sci U S A. 1996;93(20):11068–11073. | |
Gui T, Lin HF, Jin DY, et al. Circulating and binding characteristics of wild-type factor IX and certain Gla domain mutants in vivo. Blood. 2002;100(1):153–158. | |
Bajaj SP, Rapaport SI, Prodanos C. A simplified procedure for purification of human prothrombin, factor IX and factor X. Prep Biochem. 1981;11(4):397–412. | |
Gui T, Reheman A, Ni H, et al. Abnormal hemostasis in a knock-in mouse carrying a variant of factor IX with impaired binding to collagen type IV. J Thromb Haemost. 2009;7(11):1843–1851. | |
White G, Shapiro A, Ragni M, et al. Clinical evaluation of recombinant factor IX. Semin Hematol. 1998;35(2 Suppl 2):33–38. | |
White GC 2nd, Shapiro AD, Kurczynski EM, Kim HC, Bergman GE. Variability of in vivo recovery of factor IX after infusion of monoclonal antibody purified factor IX concentrates in patients with hemophilia B. The Mononine Study Group. Thromb Haemost. 1995;73(5):779–784. | |
Ewenstein BM, Joist JH, Shapiro AD, et al. Pharmacokinetic analysis of plasma-derived and recombinant F IX concentrates in previously treated patients with moderate or severe hemophilia B. Transfusion. 2002;42(2):190–197. | |
Poon MC, Lillicrap D, Hensman C, Card R, Scully MF. Recombinant factor IX recovery and inhibitor safety: a Canadian post-licensure surveillance study. Thromb Haemost. 2002;87(3):431–435. | |
Peters RT, Low SC, Kamphaus GD, et al. Prolonged activity of factor IX as a monomeric Fc fusion protein. Blood. 2010;115(10):2057–2064. | |
Arruda VR, Hagstrom JN, Deitch J, et al. Posttranslational modifications of recombinant myotube-synthesized human factor IX. Blood. 2001;97(1):130–138. | |
Gil GC, Velander WH, Van Cott KE. Analysis of the N-glycans of recombinant human factor IX purified from transgenic pig milk. Glycobiology. 2008;18(7):526–539. | |
White GC 2nd, Beebe A, Nielsen B. Recombinant factor IX. Thromb Haemost. 1997;78(1):261–265. | |
Menache D. Prothrombin complex concentrates: clinical use. Ann N Y Acad Sci. 1981;370:747–756. | |
Evatt BL. The tragic history of AIDS in the hemophilia population, 1982–1984. J Thromb Haemost. 2006;4(11):2295–2301. | |
Mannucci PM. Clinical evaluation of viral safety of coagulation factor VIII and IX concentrates. Vox Sang. 1993;64(4):197–203. | |
Key NS, Negrier C. Coagulation factor concentrates: past, present, and future. Lancet. 2007;370(9585):439–448. | |
Limentani SA, Gowell KP, Deitcher SR. High-purity factor IX concentrates for treatment of hemophilia B: relative purity and thrombogenic potential. Acta Haematol. 1995;94(Suppl 1):12–17. | |
Pipe SW. Recombinant clotting factors. Thromb Haemost. 2008; 99(5):840–850. | |
Nilsson IM, Berntorp E, Löfqvist T, Pettersson H. Twenty-five years’ experience of prophylactic treatment in severe haemophilia A and B. J Intern Med. 1992;232(1):25–32. | |
Witmer C, Presley R, Kulkarni R, Soucie JM, Manno CS, Raffini L. Associations between intracranial haemorrhage and prescribed prophylaxis in a large cohort of haemophilia patients in the United States. Br J Haematol. 2011;152(2):211–216. | |
Oladapo AO, Epstein JD, Williams E, Ito D, Gringeri A, Valentino LA. Health-related quality of life assessment in haemophilia patients on prophylaxis therapy: a systematic review of results from prospective clinical trials. Haemophilia. 2015;21:e344–e358. | |
Coppola A, Tagliaferri A, Di Capua M, Franchini M. Prophylaxis in children with hemophilia: evidence-based achievements, old and new challenges. Semin Thromb Hemost. 2012;38(1):79–94. | |
Berntorp E. Joint outcomes in patients with haemophilia: the importance of adherence to preventive regimens. Haemophilia. 2009;15(6):1219–1227. | |
Chandy M. Management of hemophilia with minimal factor replacement in developing countries: role of ancillary therapy. Semin Thromb Hemost. 2005;31(5):501–506. | |
Kim HC, McMillan CW, White GC, Bergman GE, Horton MW, Saidi P. Purified factor IX using monoclonal immunoaffinity technique: clinical trials in hemophilia B and comparison to prothrombin complex concentrates. Blood. 1992;79(3):568–575. | |
Hampton KK, Preston FE, Lowe GD, Walker ID, Sampson B. Reduced coagulation activation following infusion of a highly purified factor IX concentrate compared to a prothrombin complex concentrate. Br J Haematol. 1993;84(2):279–284. | |
Tabor E. The epidemiology of virus transmission by plasma derivatives: clinical studies verifying the lack of transmission of hepatitis B and C viruses and HIV type 1. Transfusion. 1999;39(11–12):1160–1168. | |
Brown P, Will RG, Bradley R, Asher DM, Detwiler L. Bovine spongiform encephalopathy and variant Creutzfeldt-Jakob disease: background, evolution, and current concerns. Emerg Infect Dis. 2001;7(1):6–16. | |
Bartolomei Corsi O, Azzi A, Morfini M, Fanci R, Rossi Ferrini P. Human parvovirus infection in haemophiliacs first infused with treated clotting factor concentrates. J Med Virol. 1988;25(2):165–170. | |
Mannucci PM. The safety of plasma-derived versus recombinant concentrates. Occasional Papers. 2004;(5):1–4. | |
Lissitchkov T, Matysiak M, Zavilska K, et al. A clinical study assessing the pharmacokinetics, efficacy and safety of AlphaNine®, a high-purity factor IX concentrate, in patients with severe haemophilia B. Haemophilia. 2011;17(4):590–596. | |
White GC 2nd. Safety and recovery of Mononine in multiple-dose, high-dose regimens. Acta Haematol. 1995;94(Suppl 1):53–57; discussion 57–58. | |
Ménart C, Petit PY, Attali O, Massignon D, Dechavanne M, Négrier C. Efficacy and safety of continuous infusion of Mononine during five surgical procedures in three hemophilic patients. Am J Hematol. 1998;58(2):110–116. | |
Hoots WK, Leissinger C, Stabler S, et al. Continuous intravenous infusion of a plasma-derived factor IX concentrate (Mononine) in haemophilia B. Haemophilia. 2003;9(2):164–172. | |
Alamelu J, Bevan D, Sorensen B, Rangarajan S. Pharmacokinetic and pharmacodynamic properties of plasma-derived vs recombinant factor IX in patients with hemophilia B: a prospective crossover study. J Thromb Haemost. 2014;12(12):2044–2048. | |
Mauser-Bunschoten EP, Kleine Budde I, Lopaciuk S, et al. An ultrapure plasma-derived monoclonal antibody-purified factor IX concentrate (Nonafact(R)), results of phase III and IV clinical studies. Haemophilia. 2011;17(3):439–445. | |
Björkman S, Ahlén V. Population pharmacokinetics of plasma-derived factor IX in adult patients with haemophilia B: implications for dosing in prophylaxis. Eur J Clin Pharmacol. 2012;68(6):969–977. | |
National Hemophilia Foundation no 179. MASAC recommendation concerning prophylaxis (regular administration of clotting factor concentrate to prevent bleeding); 2007. | |
Choo KH, Gould KG, Rees DJ, Brownlee GG. Molecular cloning of the gene for human anti-haemophilic factor IX. Nature. 1982; 299(5879):178–180. | |
Kaufman RJ, Wasley LC, Furie BC, Furie B, Shoemaker CB. Expression, purification, and characterization of recombinant gamma-carboxylated factor IX synthesized in Chinese hamster ovary cells. J Biol Chem. 1986;261(21):9622–9628. | |
Roth DA, Kessler CM, Pasi KJ, et al. Human recombinant factor IX: safety and efficacy studies in hemophilia B patients previously treated with plasma-derived factor IX concentrates. Blood. 2001; 98(13):3600–3606. | |
Shapiro AD, Di Paola J, Cohen A, et al. The safety and efficacy of recombinant human blood coagulation factor IX in previously untreated patients with severe or moderately severe hemophilia B. Blood. 2005;105(2):518–525. | |
Polack B, Calvez T, Chambost H, et al. EQOFIX: a combined economic and quality-of-life study of hemophilia B treatments in France. Transfusion. 2015;55(7):1787–1797. | |
Kontermann RE. Strategies for extended serum half-life of protein therapeutics. Curr Opin Biotechnol. 2011;22(6):868–876. | |
Shapiro AD, Ragni MV, Valentino LA, et al. Recombinant factor IX-Fc fusion protein (rFIXFc) demonstrates safety and prolonged activity in a phase 1/2a study in hemophilia B patients. Blood. 2012; 119(3):666–672. | |
Powell JS, Pasi KJ, Ragni MV, et al. Phase 3 study of recombinant factor IX Fc fusion protein in hemophilia B. N Engl J Med. 2013; 369(24):2313–2323. | |
Powell J, Shapiro A, Ragni M, et al. Switching to recombinant factor IX Fc fusion protein prophylaxis results in fewer infusions, decreased factor IX consumption and lower bleeding rates. Br J Haematol. 2015;168(1):113–123. | |
Chaudhury C, Brooks CL, Carter DC, Robinson JM, Anderson CL. Albumin binding to FcRn: distinct from the FcRn-IgG interaction. Biochemistry. 2006;45(15):4983–4990. | |
Metzner HJ, Weimer T, Kronthaler U, Lang W, Schulte S. Genetic fusion to albumin improves the pharmacokinetic properties of factor IX. Thromb Haemost. 2009;102(4):634–644. | |
Nolte MW, Nichols TC, Mueller-Cohrs J, et al. Improved kinetics of rIX-FP, a recombinant fusion protein linking factor IX with albumin, in cynomolgus monkeys and hemophilia B dogs. J Thromb Haemost. 2012;10(8):1591–1599. | |
Santagostino E. PROLONG-9FP clinical development program – phase I results of recombinant fusion protein linking coagulation factor IX with recombinant albumin (rIX-FP). Thromb Res. 2013;131(Suppl 2):S7–S10. | |
Martinowitz U, Lissitchkov T, Lubetsky A, et al. Results of a phase I/II open-label, safety and efficacy trial of coagulation factor IX (recombinant), albumin fusion protein in haemophilia B patients. Haemophilia. 2015;21(6):784–790. | |
Kenet G, Chambost H, Male C, et al. Efficacy, pharmacokinetics (PK) and safety results of a phase 3 clinical study of recombinant fusion protein linking coagulation factor IX with albumin (RIX-FP) in previously treated children with hemophilia B. Paper presented at: International Society of Thrombosis and Haemostasis Meeting; 2015, Toronto, Canada. | |
Santagostino E, Martinowitz U, Lissitchkov T, et al. Efficacy and safety results of a phase 3 pivotal clinical study of recombinant fusion protein linking coagulation factor IX with albumin (RIX-FP) in previously treated patients with hemophilia B. Paper presented at: International Society of Thrombosis and Haemostasis Meeting, 2015, Toronto, Canada. | |
Negrier C, Lepatan LM, Lubetsky A, et al. Efficacy and safety of recombinant fusion protein linking coagulation factor IX with albumin (RIX-FP) in previously treated patients with hemophilia B undergoing a surgical procedure. Paper presented at: International Society of Thrombosis and Haemostasis Meeting, June 20–25, 2015, Toronto, Canada. | |
Caliceti P, Veronese FM. Pharmacokinetic and biodistribution properties of poly(ethylene glycol)-protein conjugates. Adv Drug Deliv Rev. 2003;55(10):1261–1277. | |
Østergaard H, Bjelke JR, Hansen L, et al. Prolonged half-life and preserved enzymatic properties of factor IX selectively PEGylated on native N-glycans in the activation peptide. Blood. 2011;118(8):2333–2341. | |
Negrier C, Knobe K, Tiede A, Giangrande P, Møss J. Enhanced pharmacokinetic properties of a glycoPEGylated recombinant factor IX: a first human dose trial in patients with hemophilia B. Blood. 2011;118(10):2695–2701. | |
Collins PW, Young G, Knobe K, et al. Recombinant long-acting glycoPEGylated factor IX in hemophilia B: a multinational randomized phase 3 trial. Blood. 2014;124(26):3880–3886. | |
Kao CY, Yang SJ, Tao MH, Jeng YM, Yu IS, Lin SW. Incorporation of the factor IX Padua mutation into FIX-Triple improves clotting activity in vitro and in vivo. Thromb Haemost. 2013;110(2):244–256. | |
Brooks AR, Sim D, Gritzan U, et al. Glycoengineered factor IX variants with improved pharmacokinetics and subcutaneous efficacy. J Thromb Haemost. 2013;11(9):1699–1706. | |
Westmark PR, Tanratana P, Sheehan JP. Selective disruption of heparin and antithrombin-mediated regulation of human factor IX. J Thromb Haemost. 2015;13(6):1053–1063. | |
Saini S, Hamasaki-Katagiri N, Pandey GS, et al. Genetic determinants of immunogenicity to factor IX during the treatment of haemophilia B. Haemophilia. 2015;21(2):210–218. | |
Puetz J, Soucie JM, Kempton CL, Monahan PE, Hemophilia Treatment Center Network I. Prevalent inhibitors in haemophilia B subjects enrolled in the Universal Data Collection database. Haemophilia. 2014;20(1):25–31. | |
Katz J. Prevalence of FIX Inhibitors among patients with hemophilia B: results of large scale North America survey. Haemophilia. 1996;2:28–31. | |
DiMichele D. Inhibitor development in haemophilia B: an orphan disease in need of attention. Br J Haematol. 2007;138(3):305–315. | |
Warrier I, Ewenstein BM, Koerper MA, et al. Factor IX inhibitors and anaphylaxis in hemophilia B. J Pediatr Hematol Oncol. 1997;19(1):23–27. | |
Ehrenforth S, Kreuz W, Scharrer I, et al. Incidence of development of factor VIII and factor IX inhibitors in haemophiliacs. Lancet. 1992;339(8793):594–598. | |
Recht M, Pollmann H, Tagliaferri A, Musso R, Janco R, Neuman WR. A retrospective study to describe the incidence of moderate to severe allergic reactions to factor IX in subjects with haemophilia B. Haemophilia. 2011;17(3):494–499. | |
Siboni SM, Mannucci PM, Gringeri A, et al. Health status and quality of life of elderly persons with severe hemophilia born before the advent of modern replacement therapy. J Thromb Haemost. 2009;7(5):780–786. | |
Webster R, Didier E, Harris P, et al. PEGylated proteins: evaluation of their safety in the absence of definitive metabolism studies. Drug Metab Dispos. 2007;35(1):9–16. | |
Herzog E, Harris S, Henson C, et al. Biodistribution of the recombinant fusion protein linking coagulation factor IX with albumin (rIX-FP) in rats. Thromb Res. 2014;133(5):900–907. | |
Gater A, Thomson TA, Strandberg-Larsen M. Haemophilia B: impact on patients and economic burden of disease. Thromb Haemost. 2011;106(3):398–404. | |
Hilden I, Lauritzen B, Sørensen BB, et al. Hemostatic effect of a monoclonal antibody mAb 2021 blocking the interaction between FXa and TFPI in a rabbit hemophilia model. Blood. 2012;119(24):5871–5878. | |
Chowdary P, Lethagen S, Friedrich U, et al. Safety and pharmacokinetics of anti-TFPI antibody (concizumab) in healthy volunteers and patients with hemophilia: a randomized first human dose trial. J Thromb Haemost. 2015;13(5):743–754. | |
Sehgal A, Barros S, Ivanciu L, et al. An RNAi therapeutic targeting antithrombin to rebalance the coagulation system and promote hemostasis in hemophilia. Nat Med. 2015;21(5):492–497. | |
Pasi KJ, Georgiev P, Mant T, et al. A subcutaneously administered investigational RNAi therapeutic (ALN-AT3) targeting antithrombin for treatment of hemophilia: interim weekly and monthly dosing results in patients with hemophilia A or B. Blood. 2015;126:551. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.