")
Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 11 » Issue 1
Neutral sphingomyelinase-2, acid sphingomyelinase, and ceramide levels in COPD patients compared to controls
Authors Lea S, Metcalfe H, Plumb J, Beerli C, Poll C, Singh D, Abbott-Banner K
Received 2 September 2015
Accepted for publication 11 April 2016
Published 6 September 2016 Volume 2016:11(1) Pages 2139—2147
DOI https://doi.org/10.2147/COPD.S95578
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Richard Russell
Simon R Lea,1,* Hannah J Metcalfe,1,* Jonathan Plumb,1 Christian Beerli,2 Chris Poll,3 Dave Singh,1 Katharine H Abbott-Banner3
1Centre for Respiratory Medicine and Allergy, Institute of Inflammation and Repair, Manchester Academic Health Science Centre, The University of Manchester and University Hospital of South Manchester, NHS Foundation Trust, Manchester, UK; 2Novartis Pharma AG, Postfach, Basel, Switzerland; 3Respiratory Diseases, Novartis Institute for Biomedical Research, Horsham, West Sussex, UK
*These authors contributed equally to this work
Background: Increased pulmonary ceramide levels are suggested to play a causative role in lung diseases including COPD. Neutral sphingomyelinase-2 (nSMase-2) and acid SMase (aSMase), which hydrolyze sphingomyelin to produce ceramide, are activated by a range of cellular stresses, including inflammatory cytokines and pathogens, but notably cigarette smoke appears to only activate nSMase-2. Our primary objective was to investigate nSMase-2 and aSMase protein localization and quantification in lung tissue from nonsmokers (NS), smokers (S), and COPD patients. In addition, various ceramide species (C16, C18, and C20) were measured in alveolar macrophages from COPD patients versus controls.
Materials and methods: Patients undergoing surgical resection for suspected or confirmed lung cancer were recruited, and nSMase-2 and aSMase protein was investigated in different areas of lung tissue (small airways, alveolar walls, subepithelium, and alveolar macrophages) by immunohistochemistry. Ceramide species were measured in alveolar macrophages from COPD patients and controls by mass spectrometry.
Results: nSMase-2 and aSMase were detected in the majority of small airways. There was a significant increase in nSMase-2 immunoreactivity in alveolar macrophages from COPD patients (54%) compared with NS (31.7%) (P<0.05), and in aSMase immunoreactivity in COPD (68.2%) and S (69.5%) alveolar macrophages compared with NS (52.4%) (P<0.05). aSMase labeling was also increased in the subepithelium and alveolar walls of S compared with NS. Ceramide (C20) was significantly increased in alveolar macrophages from COPD patients compared with controls.
Conclusion: nSMase-2 and aSMase are both increased in COPD alveolar macrophages at the protein level; this may contribute toward the elevated ceramide (C20) detected in alveolar macrophages from COPD patients.
Keywords: sphingomyelinase, COPD, ceramide, cigarette smoke
Background
COPD is characterized by airflow limitation that is in most cases progressive and associated with an abnormal inflammatory response of the lung to the inhalation of noxious particles.1 Oxidative stress generated as a consequence of cigarette smoke exposure is thought to contribute toward the inflammatory processes and tissue destruction observed in COPD patients.2 Current anti-inflammatory therapies for COPD have limited effects, and there is a pressing need for new and more effective therapies.3
Ceramide is an important bioactive molecule, as well as a precursor of other bioactive sphingolipids; these molecules play essential roles in a broad range of physiological processes including cell growth, survival, and death.4 There are different species of intracellular cell ceramides with long N-acyl chains typically ranging from 16 to 26 carbons in length. There is growing evidence to suggest that ceramide levels are elevated in the lungs of patients with a variety of respiratory diseases including COPD,5–7 cystic fibrosis,8 and idiopathic pulmonary fibrosis.5 Furthermore, ceramide levels are associated with the severity of COPD.6,7
Ceramide may contribute to tissue destruction and remodeling that occurs in COPD9 as excessive ceramide accumulation causes apoptosis of structural cells in the airways, such as epithelial cells.4,6 Furthermore, some COPD patients suffer from bacterial colonization and infectious exacerbations, which are the causes of high morbidity and mortality,10 and ceramide may also play a role here as it causes increased infection susceptibility.11
The sphingomyelinase (SMase) family of enzymes regulates the formation of ceramide. Diverse stimuli including inflammatory cytokines, pathogens, and oxidative stress activate neutral sphingomyelinase-2 (nSMase-2) and acid SMase (aSMase).12,13 nSMase activity, and consequently ceramide generation, is upregulated in bronchial epithelial cells exposed to oxidative stress.14 Furthermore, oxidative stress-induced human bronchial epithelial cell apoptosis is attenuated by siRNA silencing of nSMase-2.14
Alveolar macrophage numbers are increased in COPD. Cigarette smoking appears to cause alveolar macrophage dysfunction, as COPD macrophages display reduced phagocytosis and efferocytosis.15–17 Ceramide can modulate macrophage function. The expression of nSMase-2 and aSMase in COPD macrophages has not been properly explored. Furthermore, it is not known whether overproduction of ceramide species by these cells occurs.
It has been reported that nSMase-2 protein is upregulated in lung tissue of smokers with emphysema and is co-localized with ceramide and apoptotic cells.18 However, this study had a limited sample size of six emphysema patients and six controls. This is a potentially important finding, but these protein studies require confirmation using a larger sample size. We therefore sought to enhance these findings by; 1) comparing nSMase-2 and aSMase protein levels in lung tissue from COPD patients of both smoking and non-smoking controls using a larger sample size than previously reported; 2) identifying specific cell types that express these proteins; and 3) measuring various ceramide species (C16, C18, and C20) in alveolar macrophages from patients with COPD compared to controls.
Materials and methods
Subjects
In total, 89 patients undergoing surgical resection for suspected or confirmed lung cancer were recruited at the University Hospital of South Manchester; the demography of patients in each experiment is shown in Table 1. COPD was diagnosed based on a history of smoking for >10 pack-years (1 pack-year equals 20 cigarettes smoked per day for 1 year), typical symptoms (productive cough, breathlessness, or wheeze) and airflow obstruction, defined as forced expiratory volume in 1 second <80% predicted, and forced expiratory volume in 1 second/forced vital capacity ratio <70%. Controls with normal lung function were categorized as either smokers (S) (≥1 pack-year history) or nonsmokers (NS) (<1 pack-year history). All subjects gave written informed consent. The study was approved by the local research ethics committee (South Manchester Research Ethics Committee).
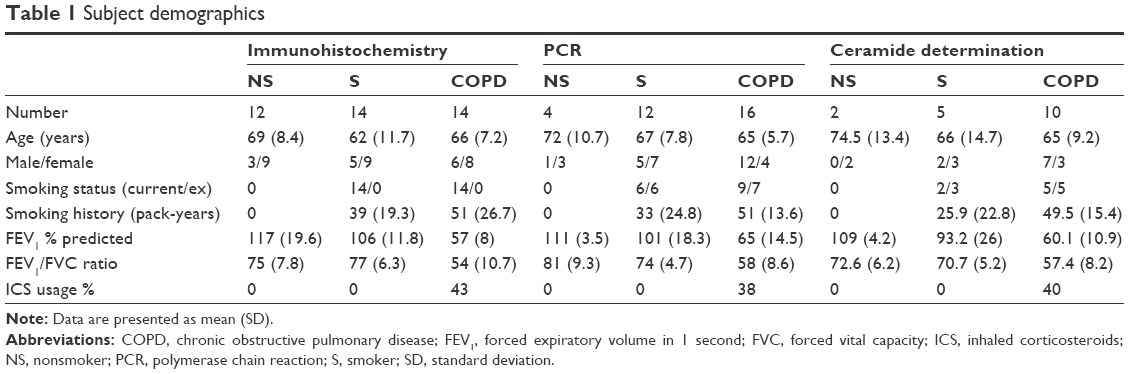 | Table 1 Subject demographics |
Immunohistochemistry
Tissue blocks were obtained from an area of the lung as far distal to the tumor as possible and were processed as described previously.19 Histological samples from a subset of patients were analyzed by pathologists at the University Hospital of South Manchester, and an emphysema score had been generated. Blocks were labeled using anti-nSMase-2 and anti-aSMase primary antibodies. Further details of methods and antibodies are described in the Supplementary materials. Labeling in each patient was quantified and reported for small airways, subepithelium, alveolar walls, and alveolar macrophages. Alveolar macrophages were identified using morphology (mononuclear cells with a clearly defined cytoplasm) and location (situated within the alveolar space) as the distinctive morphology of these cells easily allows identification without the need for counterstaining.20–23 Staining of nSMase-2 and aSMase in alveolar macrophages was also confirmed by the presence of cluster of differentiation 68 (CD68) on these morphologically distinct cells in serial sections (Figure S1). Antibody specificity was confirmed by analyzing alveolar macrophage cell lysates by Western blot, for the presence of a band of the appropriate size. Predicted molecular weights for aSMase and nSMase-2 were 65 and 71 kDa, respectively (Figure S2).
Alveolar macrophage culture
Table 1 summarizes the details of the patients from which macrophages were obtained for the nSMase-2/aSMase gene expression study. Alveolar macrophages were isolated from patient lung samples as described previously24 and were seeded at 4×105 per well of 24-well plate for gene expression analysis or 1×106 per well of six-well plate for ceramide determination. Further details are in the Supplementary materials.
nSMase-2 and aSMase gene expression
nSMase-2 and aSMase gene expression levels were compared between macrophages from COPD patients and controls using real-time polymerase chain reaction (PCR) as described in the Supplementary materials; the endogenous housekeeping control glyceraldehyde-3-phosphate dehydrogenase.
Ceramide determination
Alveolar macrophages were prepared for ceramide determination as described in the Supplementary materials. For quantitative determination of C16-, C18-, and C20-ceramides in cell samples, the methods25,26 were modified and adapted accordingly. The analysis was performed on an Agilent 1290 Infinity UHPLC-System (Agilent Technologies, Santa Clara, CA, USA), directly coupled to the AP_ESI source of an Agilent 6490 triple quadrupole mass spectrometer (Agilent Technologies) run in MRM. For detailed methods, see the Supplementary materials.
Statistical analysis
All data were analyzed using GraphPad PRISM version 5 (GraphPad Software, Inc., La Jolla, CA, USA) and InStat Version 3.06 (GraphPad Software). Immunohistochemistry and ceramide data were analyzed using the one-way analysis of variance and Dunnett’s multiple comparison test. Quantitative PCR data were analyzed using the Kruskal–Wallis test. Differences were considered significant when P<0.05.
Results
nSMase-2 immunohistochemistry
Distinct brown labeling of nSMase-2 expression in NS (n=12), S (n=14), and COPD patients (n=14) was visualized as shown in Figure 1, with quantitative analysis shown in Figure 2. nSMase-2 immunoreactivity was observed within the small airway epithelium in all the samples. nSMase-2 was observed within the cytoplasm of the epithelial cells with a more pronounced expression toward the apical surface (Figure 1A–F). There was no difference in epithelial nSMase-2 immunoreactivity between groups.
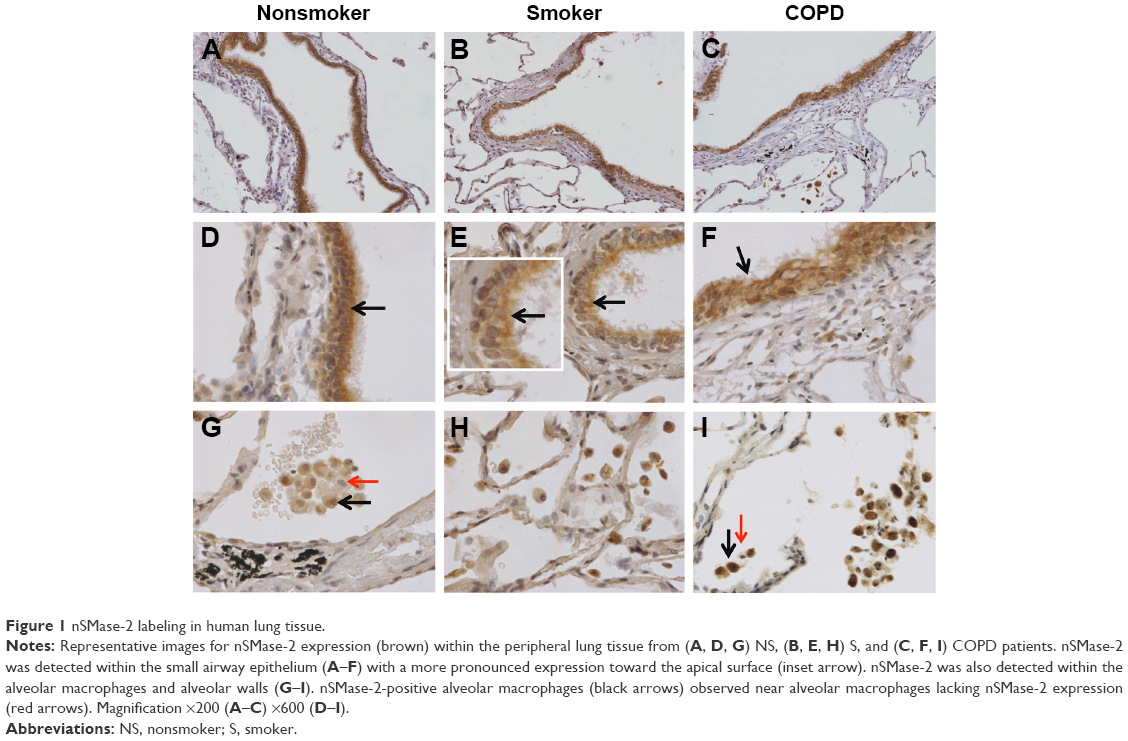 | Figure 1 nSMase-2 labeling in human lung tissue. |
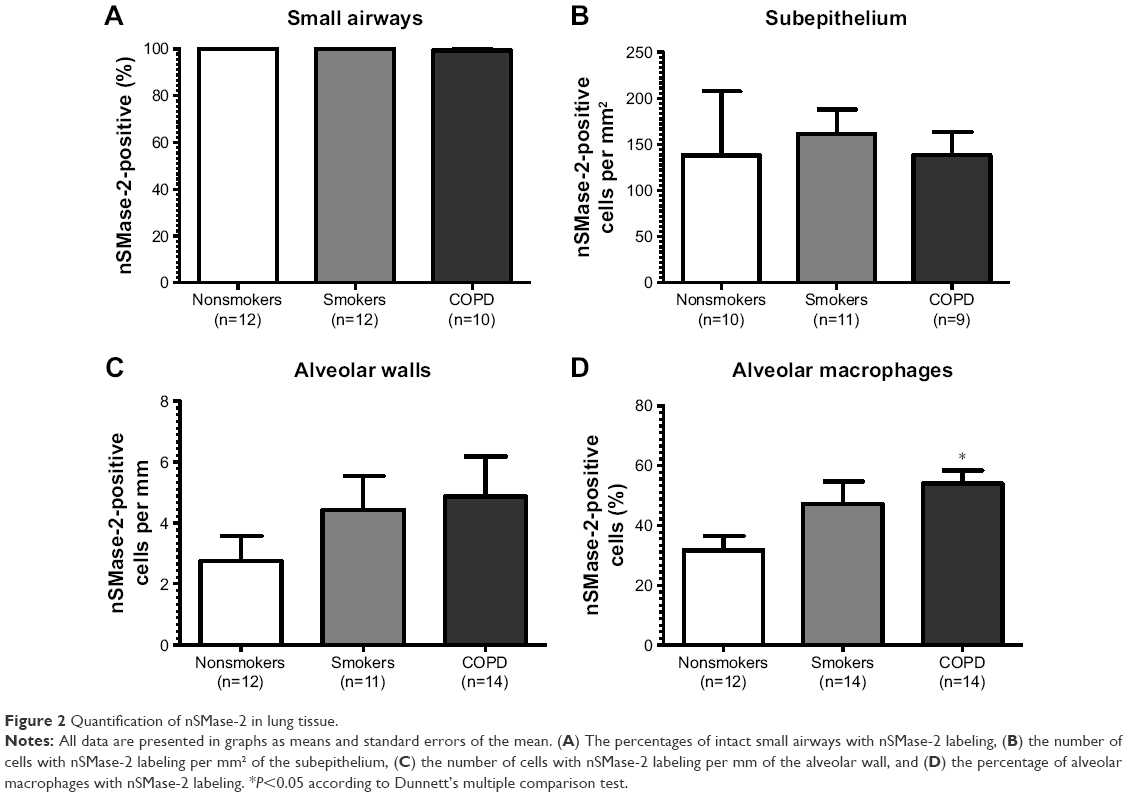 | Figure 2 Quantification of nSMase-2 in lung tissue. |
nSMase-2 immunoreactivity was observed within the alveolar macrophages and alveolar walls in all the samples (Figure 1G–I). There was a significant increase in the percentage of alveolar macrophages expressing nSMase-2 in COPD (54%) compared with NS (31.7%); P<0.05 (Figure 2D). No difference was detected between NS and S (47%) or S and COPD (P>0.05 for both comparisons), and there was no difference between groups in nSMase-2 labeling in the alveolar walls. There was a trend toward an association between nSMase-2 expression levels within the alveolar walls and the emphysema score (P=0.09, r=0.4087; Figure S3).
There was little nSMase-2 expression within the subepithelium, with no difference between groups. nSMase-2 immunoreactivity was observed in larger blood vessels with staining observed within occasional endothelial cells and the subjacent vascular smooth muscle (Figure S4). nSMase-2 immunoreactivity was not observed within the airway smooth muscle or within lymphocytic follicles (Figure S4).
aSMase immunohistochemistry
Representative pictures illustrating distinct brown labeling of aSMase in NS, S, and COPD patients are shown in Figure 3, with quantification shown in Figure 4. aSMase immunoreactivity was observed within the small airway epithelium in all the samples. Unlike nSMase-2, aSMase was observed within what morphologically appears as a nuclear location in epithelial cells (Figure 3A–F). There was no difference in epithelial aSMase immunoreactivity between the groups.
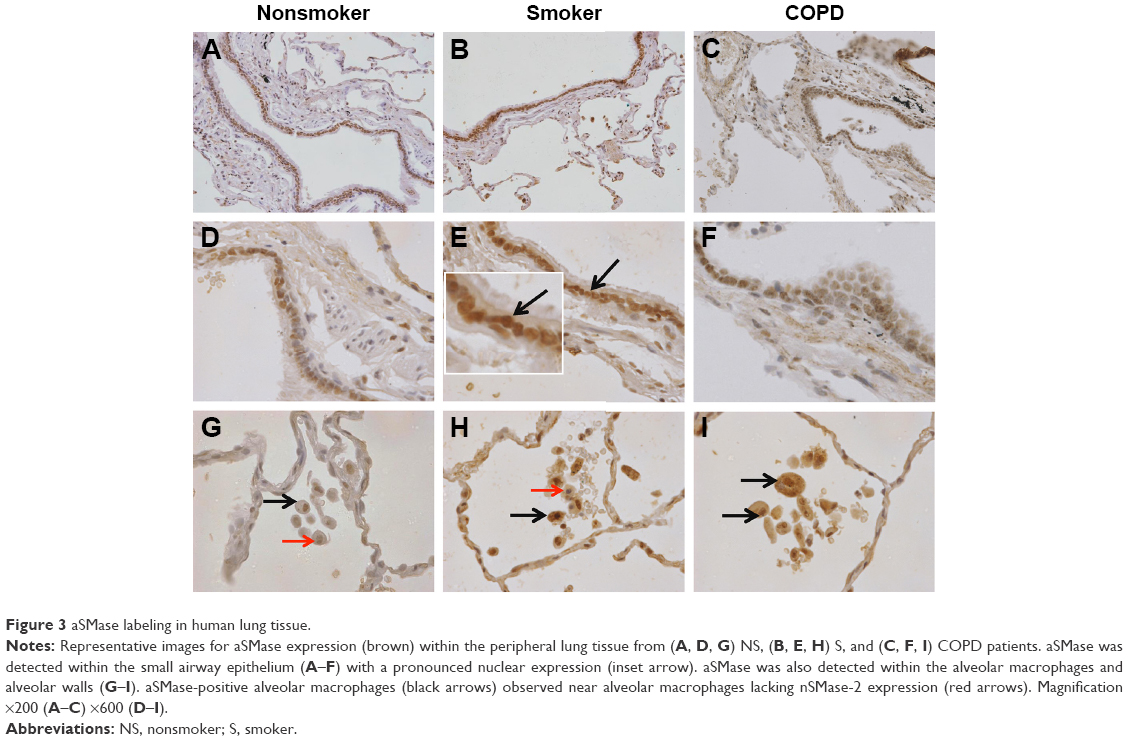 | Figure 3 aSMase labeling in human lung tissue. |
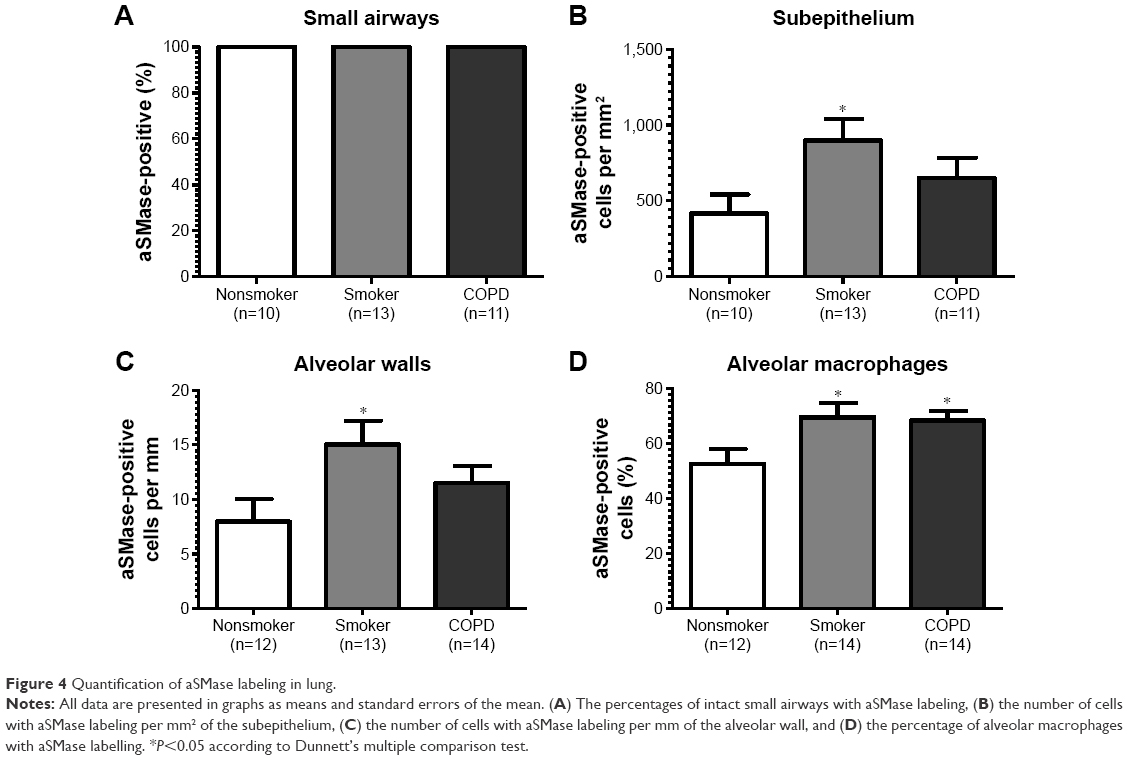 | Figure 4 Quantification of aSMase labeling in lung. |
aSMase immunoreactivity was observed within the alveolar macrophages and alveolar walls in all the samples (Figure 3G–I). There was a significant increase in the percentage of alveolar macrophages expressing aSMase in both S (69.5%) and COPD patients (68.2%) compared with NS (52.4%); P<0.05 for both comparisons (Figure 4D). aSMase was significantly increased within the alveolar walls of S compared with NS (P<0.05; Figure 4C) but no difference between COPD patients compared to the other groups. There was a trend toward an association between aSMase expression levels within the alveolar walls and the emphysema score (P=0.117, r=0.3148; Figure S3).
There was significantly increased aSMase labeling in the subepithelium of S compared with NS P<0.05 (Figure 4B). aSMase immunoreactivity was observed in larger blood vessels with staining observed within the endothelial cells and occasionally within the subjacent vascular smooth muscle (Figure S4). aSMase immunoreactivity was observed within the nuclei of lymphocytic follicles but not in the airway smooth muscle (Figure S4).
nSMase-2/aSMase gene expression in alveolar macrophages
Having observed increased nSMase-2 and aSMase protein expression in COPD alveolar macrophages, a gene expression analysis was performed for both of these enzymes using alveolar macrophages from NS (n=4), S (n=13), and COPD patients (n=15) (Figure 5). No significant difference could be detected in nSMase-2 or aSMase gene expression between the groups, and no difference could be detected in nSMase-2 or aSMase gene expression in macrophages isolated following 1 or 16 hours adherence (P>0.05, Figure S5).
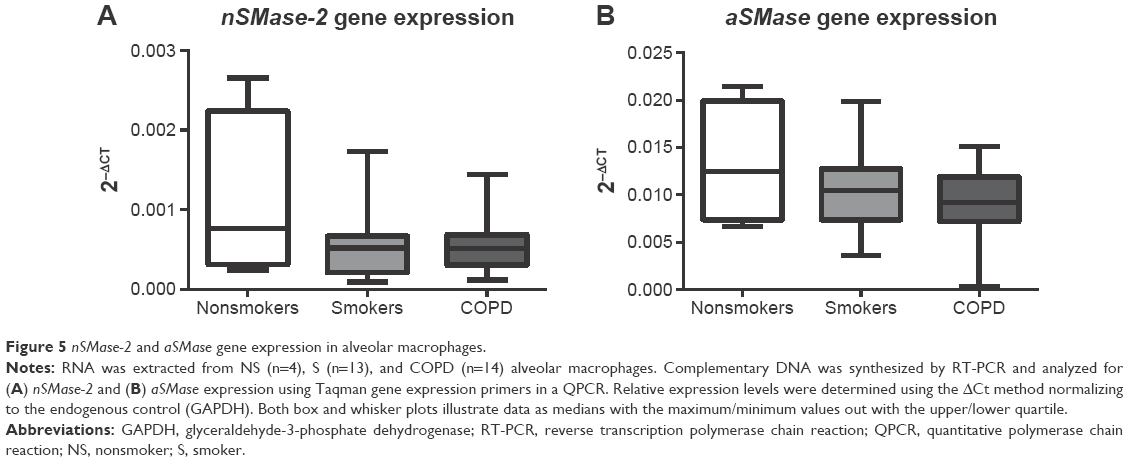 | Figure 5 nSMase-2 and aSMase gene expression in alveolar macrophages. |
Ceramide species in alveolar macrophages isolated
Alveolar macrophages from controls (n=2 NS and n=5 S) and COPD patients (n=10) were analyzed for C16, C18, and C20. There were numerical increases in the levels of all three species in COPD patients, with C20 ceramide levels significantly increased in COPD patients compared with controls (P=0.024) (Figure 6). The increase in C16 just failed to reach statistical significance (P=0.054). There were no differences in ceramide levels based on emphysema score (Figure S6).
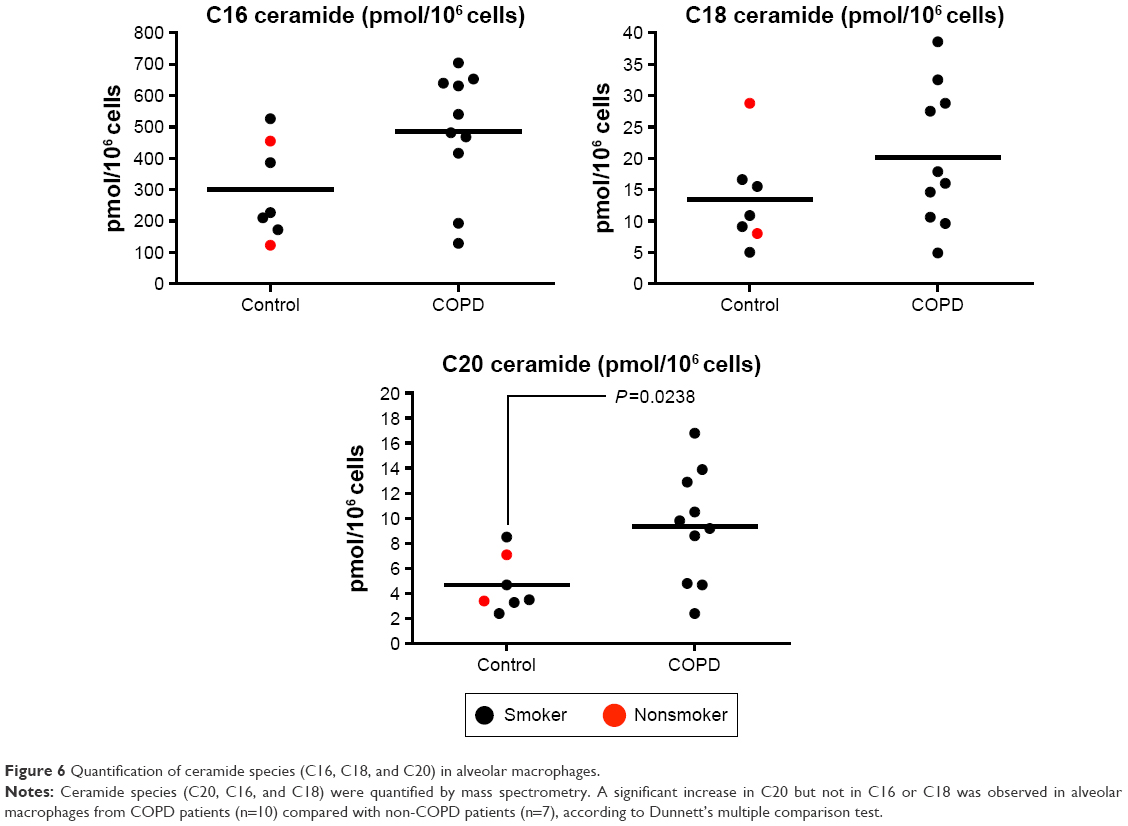 | Figure 6 Quantification of ceramide species (C16, C18, and C20) in alveolar macrophages. |
Discussion
It has been shown that nSMase-2 and aSMase protein levels are increased in alveolar macrophages from patients with COPD. A small study conducted by Filosto et al18 also demonstrated that nSMase-2 was increased in lung tissue examined from emphysema patients. Protein expression findings using a larger sample size have been reproduced, and the increase of aSMase expression in the lungs of COPD patients has been shown. A more detailed analysis of the localization and quantification of these enzymes in lung tissue were performed, and it is reported that COPD alveolar macrophages have increased nSMase-2 and aSMase protein expression. This may be a contributor to the elevated ceramide (C20) levels detected in alveolar macrophages.
Modulation of SMase activity can affect murine macrophage function. For example, a nonselective aSMase/nSMase inhibitor prevented lipopolysaccharide-induced macrophage activation27 and also inhibited oxidative stress-induced cell injury;28 in the latter study, a reduction in C16 and C18 ceramide levels was observed. Our findings in human alveolar macrophages suggest that abnormal SMase expression in COPD macrophages results in elevated ceramide C20 levels. Further investigations would be needed to prove this link, although this is hard to prove in humans in vivo and more easily explored in in vitro experiments. C16 also appeared to be elevated in COPD macrophages, but this just failed to reach statistical significance (P=0.054) possibly due to the relatively small sample size. C16, C18, and C20 but not C22 are all elevated in cystic fibrosis lung tissue;8 however, people are not aware of studies that have investigated the levels of these specific ceramide species in COPD tissue or indeed their relative potential importance in COPD. However, other ceramide species not investigated in this study should be considered in the context of COPD.
A potential role for ceramide in emphysema has been proposed, but recent work has shown only a weak correlation between ceramide levels and caspase-3 expressing cells, suggesting that ceramide does not induce apoptosis in all cells.5 In vitro activation or de novo synthesis of SMase enzymes leads to increased ceramide-induced apoptosis in murine pulmonary endothelial cells,29 a potential key mechanism in emphysema development.6 Understanding the roles of individual ceramide species could therefore be fundamental in unravelling the cellular mechanisms driving emphysema. Ceramide levels are elevated in other respiratory diseases such as lung fibrosis and cystic fibrosis.5,8 It is not clear whether the observed increase of a specific ceramide species is specific to COPD or also occurs in other lung diseases.
The cellular location of nSMase-2 in epithelial cells observed here suggests an active role in ceramide production. Under basal conditions, nSMase-2 resides in lysosomes within the cytoplasm. It has been shown that oxidative stress causes nSMase-2 accumulation at the plasma membrane where it regulates ceramide production and promotes apoptosis;14,30 a similar cellular location is observed in the present study. Conversely, under antioxidative conditions, nSMase-2 translocates to a perinuclear location preventing ceramide production and subsequent apoptosis.14,30
During normal membrane recycling, aSMase resides within the lysosomes converting sphingomyelin into phosphocholine and ceramide. Upon membrane damage or microbial insult, lysosomal aSMase translocates to the plasma membrane, where it cleaves surface-exposed sphingomyelin.31 Predominant nuclear aSMase-2 protein expression in epithelial cells was observed; the functional implication of this finding is unclear and warrants further investigation to determine its relevance.
Altered macrophage efferocytosis has been observed in macrophages from S and patients with COPD,15,17 and excessive lung ceramide levels have been implicated.9 As observed, increased nSMase-2 and aSMase in COPD macrophages is associated with elevated macrophage ceramide levels; then one could hypothesize that an inhibitor of aSMase and/or nSMase-2 could reduce the elevated ceramide levels and thus have the potential to modulate and potentially restore normal macrophage function.
Cigarette smoke-induced ceramide generation has been connected to nSMase-2 and not aSMase activity.14,18 Results clearly show that the expression of both the proteins is increased in macrophages from COPD patients compared with NS. Interestingly, there was also evidence that smoking alone increases aSMase expression, as S had higher levels of this protein compared with NS in the alveolar walls, alveolar macrophages, and subepithelium. Studies in mice have shown that nSMase-2 protein expression is elevated in lung tissue by acute cigarette smoke exposure.14 The reason for this difference may be species differences or a difference in the chronicity of the exposure to cigarette smoke.
There is also evidence to suggest that nSMase-2 but not aSMase is specifically activated by cigarette smoke in isolated airway epithelial cells in culture.14,18 It was observed that aSMase, rather than nSMase-2, levels were increased in S epithelium compared with NS. The differences in results between the acute epithelial cell culture model and our lung immunohistochemistry may again be due to chronicity of smoke exposure. Increased aSMase expression was observed in S epithelium and alveolar walls, but not COPD patients; this may be due to a feedback mechanism associated with the pathological development of COPD. Increased aSMase expression in smokers may signify an involvement of this enzyme in the early stages of COPD, but this activity then becomes blunted with disease progression and a change in macrophage phenotype. It is recognized that macrophages change their phenotype toward an alternative activation (M2) state with progression from being a smoker with normal lung function to developing established COPD.32 The gene expression analysis for nSMase and aSMase did not reproduce the differences between groups observed for protein expression. This suggests that the immunohistochemistry results are due to post-transcriptional mechanisms. The sample size of NS in this experiment was small, as the majority of patients undergoing lung surgery have a history of smoking, making it practically difficult to recruit NS. The negative result may be related to the small sample size for NS. While the effect of medicines used to lower lipid levels (eg, statins) on lipid composition within the lung is unknown, ceramide monohexoside and sphingomyelin are elevated within the brains of Wistar rats following statin treatment.33 The possibility that some of our subjects were on statins, due to cardiovascular comorbidities, may influence the aSMase, nSMase-2, and ceramide levels observed.
A limitation of the studies reported here is that the gene expression and ceramide species identification studies (shown in Figures 5 and 6, respectively) had limited numbers of samples from NS. The majority of patients undergoing surgery for lung cancer have a significant smoking history, so within the timeframe of this study there were few new samples from NS samples. Fresh (not stored) samples were needed for macrophage gene expression and ceramide species identification. In contrast, stored tissue could be used for immunohistochemistry, so a greater number of NS can be obtained for these experiments. For ceramide identification, the NS samples with S were pooled to avoid statistical comparisons between groups with low sample numbers.
Conclusion
In conclusion, both nSMase-2 and aSMase protein levels are increased in COPD lung tissue; this increase was observed in alveolar macrophages and was associated with elevated ceramide (C20) levels in COPD alveolar macrophages. Inhibition of aSMase and/or nSMase-2 may have therapeutic potential in COPD.
Acknowledgments
The authors would like to acknowledge Beatrice Urban for performing the bioanalytical work. This study is an independent research supported by National Institute for Health Research Respiratory and Allergy Clinical Research Facility at University Hospital of South Manchester NHS Foundation Trust. The views expressed in this publication are those of the author(s) and not necessarily those of the NHS, the National Institute for Health Research or the Department of Health. This work was partially funded by Novartis, and the article-processing charge will be paid by Novartis.
Disclosure
SRL, HJM, and JP have no competing interests. CB, CP, and KHAB all worked for Novartis at the time of this research study. KHAB now works for Verona Pharma plc. DS has received sponsorship to attend international meetings, honoraria for lecturing or attending advisory boards, and research grants from various pharmaceutical companies including Almirall, AstraZeneca, Boehringer Ingelheim, Chiesi, CIPLA, Forest, Genetech, GlaxoSmithKline, Merck, Novartis, Pfizer, and Takeda.
References
Decramer M, Janssens W, Miravitlles M. Chronic obstructive pulmonary disease. Lancet. 2012;379(9823):1341–1351. | ||
Yoshida T, Tuder RM. Pathobiology of cigarette smoke-induced chronic obstructive pulmonary disease. Physiol Rev. 2007;87(3):1047–1082. | ||
Soriano JB, Sin DD, Zhang X, et al. A pooled analysis of FEV1 decline in COPD patients randomized to inhaled corticosteroids or placebo. Chest. 2007;131(3):682–689. | ||
Arana L, Gangoiti P, Ouro A, Trueba M, Gómez-Muñoz A. Ceramide and ceramide 1-phosphate in health and disease. Lipids Health Dis. 2010;9:15. | ||
Scarpa MC, Baraldo S, Marian E, et al. Ceramide expression and cell homeostasis in chronic obstructive pulmonary disease. Respiration. 2013;85(4):342–349. | ||
Petrache I, Natarajan V, Zhen L, et al. Ceramide upregulation causes pulmonary cell apoptosis and emphysema-like disease in mice. Nat Med. 2005;11(5):491–498. | ||
Bodas M, Min T, Mazur S, Vij N. Critical modifier role of membrane-cystic fibrosis transmembrane conductance regulator-dependent ceramide signaling in lung injury and emphysema. J Immunol. 2011;186(1):602–613. | ||
Brodlie M, McKean MC, Johnson GE, et al. Ceramide is increased in the lower airway epithelium of people with advanced cystic fibrosis lung disease. Am J Respir Crit Care Med. 2010;182(3):369–375. | ||
Demedts IK, Demoor T, Bracke KR, Joos GF, Brusselle GG. Role of apoptosis in the pathogenesis of COPD and pulmonary emphysema. Respir Res. 2006;7(1):53. | ||
Sethi S, Murphy TF. Infection in the pathogenesis and course of chronic obstructive pulmonary disease. N Engl J Med. 2008;359(22):2355–2365. | ||
Teichgräber V, Ulrich M, Endlich N, et al. Ceramide accumulation mediates inflammation, cell death and infection susceptibility in cystic fibrosis. Nat Med. 2008;14(4):382–391. | ||
Clarke CJ, Hannun YA. Neutral sphingomyelinases and nSMase-2: bridging the gaps. Biochim Biophys Acta. 2006;1758(12):1893–1901. | ||
Uhlig S, Gulbins E. Sphingolipids in the lungs. Am J Respir Crit Care Med. 2008;178(11):1100–1114. | ||
Levy M, Khan E, Careaga M, Goldkorn T. Neutral sphingomyelinase 2 is activated by cigarette smoke to augment ceramide-induced apoptosis in lung cell death. Am J Physiol Lung Cell Mol Physiol. 2009;297(1):L125–L133. | ||
Donnelly LE, Barnes PJ. Defective phagocytosis in airways disease. Chest. 2012;141(4):1055–1062. | ||
Berenson CS, Kruzel RL, Eberhardt E, Sethi S. Phagocytic dysfunction of human alveolar macrophages and severity chronic obstructive pulmonary disease. J Infect Dis. 2013;208(12):2036–2045. | ||
Hodge S, Matthews G, Mukaro V, et al. Cigarette smoke-induced changes to alveolar macrophage phenotype and function are improved by treatment with procysteine. Am J Respir Cell Mol Biol. 2011;44(5):673–681. | ||
Filosto S, Castillo S, Danielson A, et al. Neutral sphingomyelinase 2: a novel target in cigarette smoke-induced apoptosis and lung injury. Am J Respir Cell Mol Biol. 2011;44(3):350–360. | ||
Plumb J, Smyth LJ, Adams HR, Vestbo J, Bentley A, Singh SD. Increased T-regulatory cells within lymphocyte follicles in moderate COPD. Eur Respir J. 2009;34(1):89–94. | ||
Renda T, Baraldo S, Pelaia G, et al. Increased activation of p38 MAPK in COPD. Eur Respir J. 2008;31(1):62–69. | ||
Gaffey K, Reynolds S, Plumb J, Kaur M, Singh D. Increased phosphorylated p38 mitogen-activated protein kinase in COPD lungs. Eur Respir J. 2013;42(1):28–41. | ||
Bazzan E, Saetta M, Turato G, et al. Expression of the atypical chemokine receptor D6 in human alveolar macrophages in COPD. Chest. 2013;143(1):98–106. | ||
Wallace AM, Loy LB, Abbound RT, et al. Expression of matrix metalloproteinase-1 in alveolar macrophages, type II pneumocytes, and airways in smokers: relationship to lung function and emphysema. Lung. 2014;192(4):467–472. | ||
Plumb J, Robinson L, Lea S, et al. Evaluation of glucocorticoid receptor function in COPD lung macrophages using beclomethasone-17-monopropionate. PLoS One. 2013;8(5):e64257. | ||
Merrill AH Jr, Sullards MC, Allegood JC, Kelly S, Wang E. Sphingolipidomics: high-throughput, structure-specific, and quantitative analysis of sphingolipids by liquid chromatography tandem mass spectrometry. Methods. 2005;36(2):207–224. | ||
Ejsing S, Moehring T, Bahr U, et al. Collision-induced dissociation pathways of yeast sphingolipids and their molecular profiling in total lipid extracts: a study by quadrupole TOF and linear ion trap-orbitrap mass spectrometry. J Mass Spectrom. 2006;41:372–389. | ||
Sakata A, Ochiai T, Shimeno H, et al. Acid sphingomyelinase inhibition suppresses lipopolysaccharide-mediated release of inflammatory cytokines from macrophages and protects against disease pathology in dextran sulphate sodium-induced colitis in mice. Immunology. 2007;122(1):54–64. | ||
Wang SH, Yang WB, Liu YC, et al. A potent sphingomyelinase inhibitor from Cordyceps mycelia contributes its cytoprotective effect against oxidative stress in macrophages. J Lipid Res. 2011;52(3):471–479. | ||
Medler TR, Petrusca DN, Lee PJ, et al. Apoptotic sphingolipid signaling by ceramides in lung endothelial cells. Am J Respir Cell Mol Biol. 2008;38(6):639–646. | ||
Levy M, Castillo SS, Goldkorn T. nSMase2 activation and trafficking are modulated by oxidative stress to induce apoptosis. Biochem Biophys Res Commun. 2006;344(3):900–905. | ||
Miller ME, Adhikary S, Kolokoltsov AA, Davey RA. Ebolavirus requires acid sphingomyelinase activity and plasma membrane sphingomyelin for infection. J Virol. 2012;86(14):7473–7483. | ||
Shaykhiev R, Krause A, Salit J, et al. Smoking-dependent reprogramming of alveolar macrophage polarization: implication for pathogenesis of chronic obstructive pulmonary disease. J Immunol. 2009;183(4):2867–2883. | ||
Vecka M, Tvrzická E, Stanková B, Novák F, Nováková O, Zák A. Hypolipidemic drugs can change the composition of rat brain lipids. Tohoku J Exp Med. 2004;204(4):299–308. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.