")
Back to Journals » Drug Design, Development and Therapy » Volume 13
Neuroprotective Effect of S-trans, Trans-farnesylthiosalicylic Acid via Inhibition of RAS/ERK Pathway for the Treatment of Alzheimer’s Disease
Authors Wang X, Wang Y , Zhu Y , Yan L , Zhao L
Received 3 October 2019
Accepted for publication 21 November 2019
Published 29 November 2019 Volume 2019:13 Pages 4053—4063
DOI https://doi.org/10.2147/DDDT.S233283
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Tin Wui Wong
Xiang Wang,1 Yu Wang,2 Yiyi Zhu,1 Luxia Yan,1 Liandong Zhao1
1Department of Neurology, Xuzhou Medical University Affiliated Hospital of Huaian, Huai’an, Jiangsu Province 223002, People’s Republic of China; 2Department of Neurology, The Fourth Affiliated Hospital of Nanjing Medical University, Nanjing Pukou Hospital, Nanjing, Jiangsu 210000, People’s Republic of China
Correspondence: Liandong Zhao
Department of Neurology, Xuzhou Medical University Affiliated Hospital of Huaian, No 62 Huaihainan Road, Huai’an, Jiangsu Province 223002, People’s Republic of China
Tel/Fax +86 517 80871724
Email [email protected]
Background: Alzheimer’s disease (AD), a leading cause of dementia, becomes a serious health issue for individuals and society around the world. AD is a neurodegenerative disease characterized by the deposition of amyloid-β (Aβ) peptides and neurofibrillary tangles (NFT) and the loss of large numbers of neurons. To date, there is no effective treatment for AD, and thus, to enhance neurogenesis in the AD brain may be a therapeutic strategy. RAS signaling pathway involves in synaptic plasticity and memory formation, which is overexpressed in brains with AD. This study used Aβ1-42-injected mice (Aβ1-42-mice) as the AD model to investigate the effects of S-trans, trans-farnesylthiosalicylic acid (FTS), a synthetic Ras inhibitor, on the impairment of neurogenesis and the spatial cognitive deficits.
Materials and methods: AD model mice were manufactured through intracerebroventricular injection of Aβ1-42. Morris water maze (MWM) was performed to evaluate the capacity of spatial memory, and Nissl staining was applied to assess neuronal damage in the hippocampus CA1. Immunohistochemistry of 5-bromo-2-deoxyuridine (BrdU), BrdU/neuronal nuclei (NeuN), and doublecortin (DCX) were used to detect progenitor cell proliferation, maturation, and neurite growth, respectively. And the expression levels of RAS, ERK/ERK phosphorylation (p-ERK) and CREB/CREB phosphorylation (p-CREB) were detected by Western blot.
Results: The results demonstrated that FTS could prevent Aβ1-42 to impair survival and neurite growth of newborn neurons in the hippocampal dentate gyrus (DG) in Aβ1-42-mice. Furthermore, behavioral indexes and morphological findings showed that FTS improved the learning and spatial memory abilities of Aβ1-42-mice. In addition, FTS could inhibit the levels of hippocampal p-ERK and p-CREB activated by Aβ, which is the underlying molecular mechanism.
Conclusion: In conclusion, these findings suggest that FTS as a RAS inhibitor could be a potential therapeutic agent for the treatment of AD.
Keywords: Alzheimer’s disease, S-trans, trans-farnesylthiosalicylic acid, spatial cognition, amyloid-β, neurogenesis, signaling pathway
Introduction
With the progressive aging of the population, Alzheimer’s disease (AD), a leading cause of dementia, shows the increasing prevalence and becomes a serious health issue for individuals and society around the world.1 Cognitive deficits of AD depend in part on adult neurogenic damage. As an important structure of the learning and memory system in the brain, dentate gyrus (DG) of the hippocampus can continuously produce new nerve cells.2 The newborn neurons could improve hippocampal-dependent learning and memory, while neurogenesis is blocked in AD patients and animal models that leads to memory loss.3–6
The deposition of amyloid-β (Aβ) is considered to be the central link in the pathogenesis of AD.7 Aβ1-42 could enhance the RAS/ERK signaling cascade, which implies a pathologic link between Aβ and altered RAS signaling.8 The RAS/RAF/MEK/ERK signaling pathway could transmit extracellular signals into the nucleus to affect cell fate, including cell proliferation, differentiation, survival, and transformation. Activation of this pathway under different conditions could trigger cell specificity and even the opposite response.9 Generally, sustained and well-intensity activation promotes cell proliferation by promoting protein synthesis, cyclin/cyclin-dependent kinase (CDK) complex formation.10 However, overactivation of the pathway blocks the cell cycle and accumulates intracellular cyclinD1, whereas overaccumulation of cyclinD1 binds to the cell cycle inhibitor p21cip1, preventing p21cip1 from degrading, causing cells to enter a resting state.11 Mitogenic overstimulation of neurons led to the dedifferentiation of cells, causing abnormal entry of the cell cycle, ultimately causing neurocyte death.12 In AD, Aβ oligomer-induced abnormal cell cycle activation and subsequent cell loss may be associated with RAS.13 RAS has to be anchored to the inner leaf of the cell membrane through farnesylation to receive and transmit signals.14 The level of RAS farnesylation in the brain of AD patients was significantly higher than that in the elderly with non-cognitive disorders.15 Numerous studies have shown that statins weaken RAS activity by reducing RAS farnesylation, thereby improving cognitive function in AD mice.11,14
S-trans, trans-farnesylthiosalicylic acid (FTS), a synthetic Ras inhibitor, acts directly on the saturated RAS anchor site in the cell membrane, preventing RAS from binding to these sites.16 The molecular formula of FTS is C22H30O2S 358.54 with molecular weight 493.58 Da, the chemical structure of which is shown in Figure 1. FTS has been shown to inhibit RAS-dependent cell growth.17 In vitro studies have shown that FTS can organize abnormal cell cycle re-entry induced by soluble Aβ oligomers.13 Recently, a study has shown that the inhibition of RAS by FTS could enhance NMDAr-dependent long-term potentiation by increasing Src activity, resulting in enhanced spatial memory.18 The study focused on the effects of FTS on the survival and neurite outgrowth of Aβ-impaired neonatal neurons and explored its mechanism against AD.
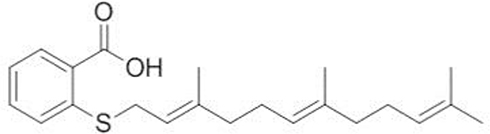 |
Figure 1 Chemical structure of FTS. Abbreviation: FTS, S-trans, trans-farnesylthiosalicylic. |
Materials and Methods
Aβ1-42-Mice as AD Model
Male mice (ICR, Xuzhou Medical University Animal Experiment Center), aged 3 months, were used in this study. The Aβ1-42-mice were obtained as previously described as AD model.19 The experimental programs were approved by the Ethics Committee of Animal Laboratory of Xuzhou Medical University and conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Drug Administration
FTS was obtained from Cayman chemical (USA). A daily dose of 5mg/kg of FTS was administered intraperitoneally 4 hrs after Aβ1-42 injection. This dose has proven to be both effective and safe for study.17,20
Morris Water Maze as Behavior Analysis
The test of Morris water maze was performed to assess the ability of spatial memory. In brief, with a pool made of black-colored plastic, the time mice taken to reach the hidden platform was recorded. In addition, the number of mice passing through the platform and the swimming time in the platform quadrant were also scored on all trials.
Immunohistochemistry of 5-Bromo-2-Deoxyuridine and Doublecortin
All mice received thrice intraperitoneal injection of 5-bromo-2-deoxyuridine (BrdU, Sigma) (50 mg/kg) with an interval of 6 hrs to label newborn cells. On day 1, 14, or 28 after BrdU injection, the sections of brains (40 mm) were obtained from the killed mice using a vibrating microtome. After treated with 0.3% Triton X-100 and 3% normal horse serum for 45 mins, the sections were probed with mouse monoclonal anti-BrdU antibody (1:1000, USA) as the primary antibody at 4 °C overnight. After washed thrice in phosphate buffer saline (PBS), the sections were incubated with biotin-labeled goat anti-mouse IgG antibody as the secondary antibody for 2 hrs. Finally, the sections were washed thrice in PBS and visualized by avidin-biotin horseradish peroxidase complex and 3,3ʹ-diaminobenzidine. Images were captured by an optical microscope (Olympus, Japan) with a 40× objective.
The above same procedure was used for the immunohistochemistry of doublecortin (DCX), in which the primary and secondary antibody was goat polyclonal anti-DCX antibody (USA) and rabbit anti-goat IgG antibody, respectively.
BrdU/Neuronal Nuclei Double Immunofluorescence
The sections were incubated with rat monoclonal anti-BrdU antibody (UK) and mouse monoclonal anti-neuronal nuclei (NeuN) antibody (USA) and then revealed by CY3-labeled anti-rat IgG antibody (USA) and fluorescein-labeled anti-mouse antibody (USA), respectively. Images were captured by a confocal laser-scanning microscope (Leica, Germany). Quantitative and image analyses refer to the previous description.17
Nissl Staining
Nissl staining is used to detect the nuclei of neurons in fixed, embedded, and frozen tissue. Preparation of brain slices and Nissl staining steps were obtained as previously described.21 Images were captured by an optical microscope (Olympus, Japan). Quantitative and image analyses refer to the previous description.22
Western Blot
After the different treatments, the expressions of RAS, ERK, and CREB were detected by Western blot. The entire hippocampus taken quickly from the killed mice was homogenized in a lysis buffer. After centrifuged, the supernatant was collected, and protein concentration was measured with BCA Protein Assay Kit (USA). Total proteins (20 mg) were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to a polyvinylidene fluoride membrane. The membrane incubated with RAS, ERK, CREB, or β-actin as primary monoclonal antibodies at 4°C overnight. Subsequently, the membranes were incubated with an HRP-labeled secondary antibody at room temperature for 1 hr. The protein bands were detected using an enhanced chemiluminescence detection system (Millipore).
Statistical Analysis
The data were expressed as the mean ± standard deviation. SPSS 22.0 (SPSS Inc., USA) was performed to analyze the data. Differences among treatments were analyzed by F-test. The difference was statistically significant with P < 0.05.
Results
Improvement of Spatial Cognitive Deficit in Aβ1-42- Mice by FTS
To assess whether FTS has an impact on the cognitive function, the Morris water maze (MWM) test was used, starting from 2 weeks of the FTS administration. In the visible platform tests used to examine search behavior, the escape latency of each group is similar (P > 0.05) (Figure 2A). In the hidden platform tests reflecting hippocampus-dependent spatial memory, compared to the control, Aβ1-42-mice took longer to reach the hidden platform on days 4–6 of training (P < 0.01). But, after the treatment with FTS, Aβ1-42-mice improved the escape latency (P < 0.05), while the control mice were not affected (P > 0.05) (Figure 2B). Although the number of Aβ1-42-mice passing through the platform and the swimming time in the platform quadrant were significantly less than that of the control mice (P < 0.01), FTS could correct the reduction in the number of crossings through the platform (Figure 2C), as well as swimming time in Aβ1-42-mice (Figure 2D). In the entire MWM test, each group of mice has no difference in swimming speed (P > 0.05) (Figure 2E).
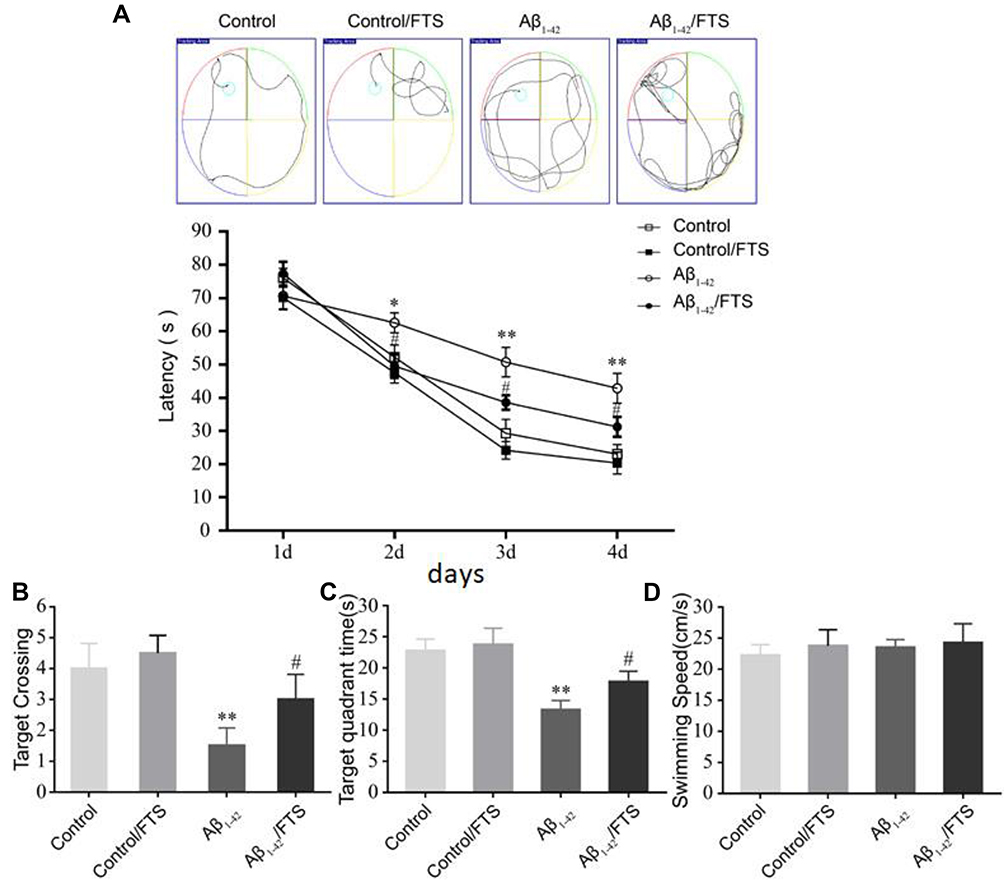 |
Figure 2 FTS improves cognitive deficits in Aβ1-42-mice. (A) The escape latency of the mice in the Morris water maze test. (B) Statistical analysis of the escape latency time. (C) The number of crossings through the platform. (D) The swimming time in the platform quadrant. (E) The swimming speed in the water maze. Data are expressed as mean ± standard deviation, *P < 0.05 and **P < 0.01 vs control mice; #P < 0.05 vs Aβ1-42-mice. Abbreviations: FTS, S-trans, trans-farnesylthiosalicylic; vs, versus. |
Neuronal Loss Blocked in Aβ1-42-Mice by FTS
Nissl staining was applied to assess the effects of FTS on Aβ-induced neuronal loss in the hippocampus. The morphological results demonstrated that, compared with control mice, the loss of Nissl bodies, neuron atrophy, and nucleus shrinkage happened in the hippocampal CA1 region of Aβ1-42-mice, and the neurons disappeared (Figure 3A). Further quantification indicated that neurons in the hippocampal CA1 area of the Aβ1-42-mice decreased on the first day (P < 0.05), and the difference was more obvious at 14 days (P < 0.01) and reached the maximum at 28 days (P < 0.01). But, Aβ-induced hippocampal neuron loss in Aβ1-42-mice could be blocked by FTS (P < 0.01), while control mice were not affected (P > 0.05) (Figure 3B)
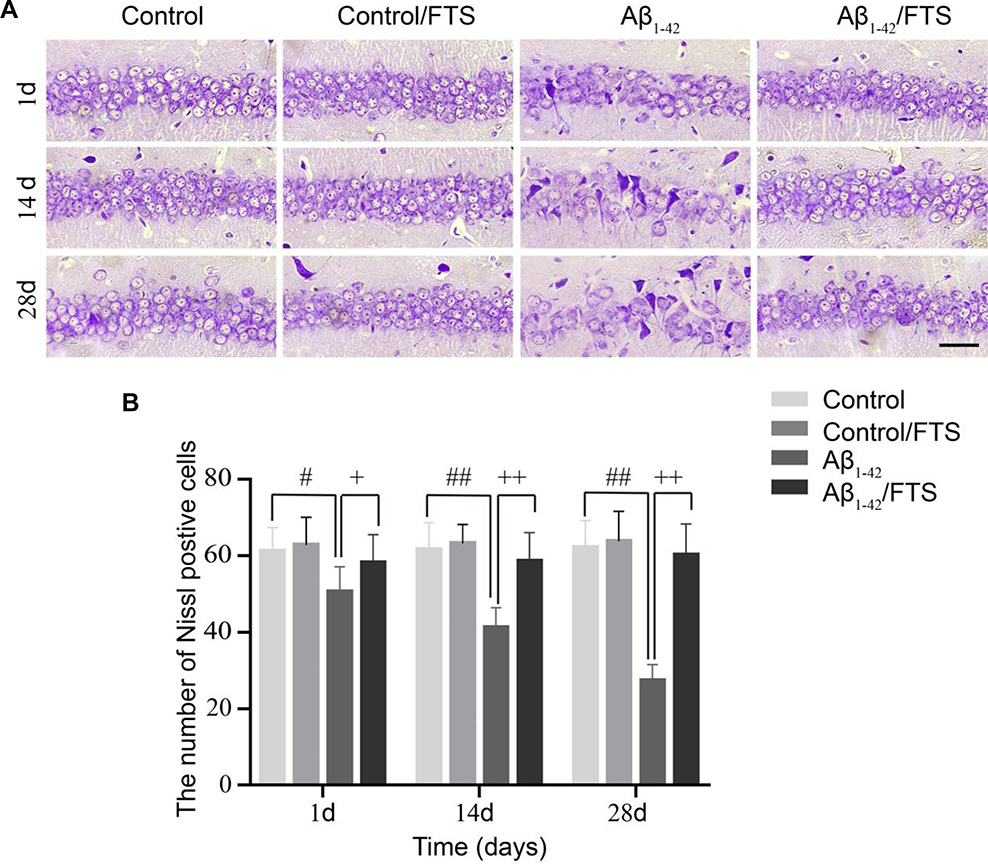 |
Figure 3 Effect of FTS on neuronal loss in hippocampal CA1 region of Aβ1-42-mice. (A) Typical morphology of Nissl staining across hippocampal CA1 region (magnification 400×, scale bar 50 µm). (B) Quantification of Nissl bodies (#p < 0.05, ##p < 0.01 vs control mice, +p < 0.05, ++p < 0.01 vs Aβ1-42-mice). Abbreviations: FTS, S-trans, trans-farnesylthiosalicylic; vs, versus. |
Protection Survival of Newborn Neurons in Aβ1-42-Mice by FTS
Although Aβ1-42 injection stimulates cell proliferation, regrettably, half of the newborn neurons die within 2 weeks.23 In this study, newborn cells were labeled by intraperitoneal injection of BrdU to examine the effect of FTS on their survival and maturation impaired from Aβ1-42, in which BrdU-positive (BrdU +) cells were counted on days 1, 14, and 28, respectively. Compared with the control mice, the number of 1-day-old BrdU+ cells in Aβ1-42-mice was increased (P < 0.05) (Figure 4A). However, compared with the control group, the number of BrdU+ cells was significantly reduced in Aβ1-42-mice on 14 (P < 0.01) and 28 days (P < 0.05), which could be rescued by FTS (P < 0.05) in a dose-dependent manner with EC50 of 3.68 mg/kg (P < 0.01) (Figure 4A–C). By contrast, FTS did not affect the number of 1-day-old, 14-day-old, and 28-day-old BrdU+ cells in control mice (P > 0.05). Similarly, FTS could prevent the loss of 28-day-old BrdU+/NeuN+ cells in Aβ1-42-mice (P < 0.01) (Figure 4D and E).
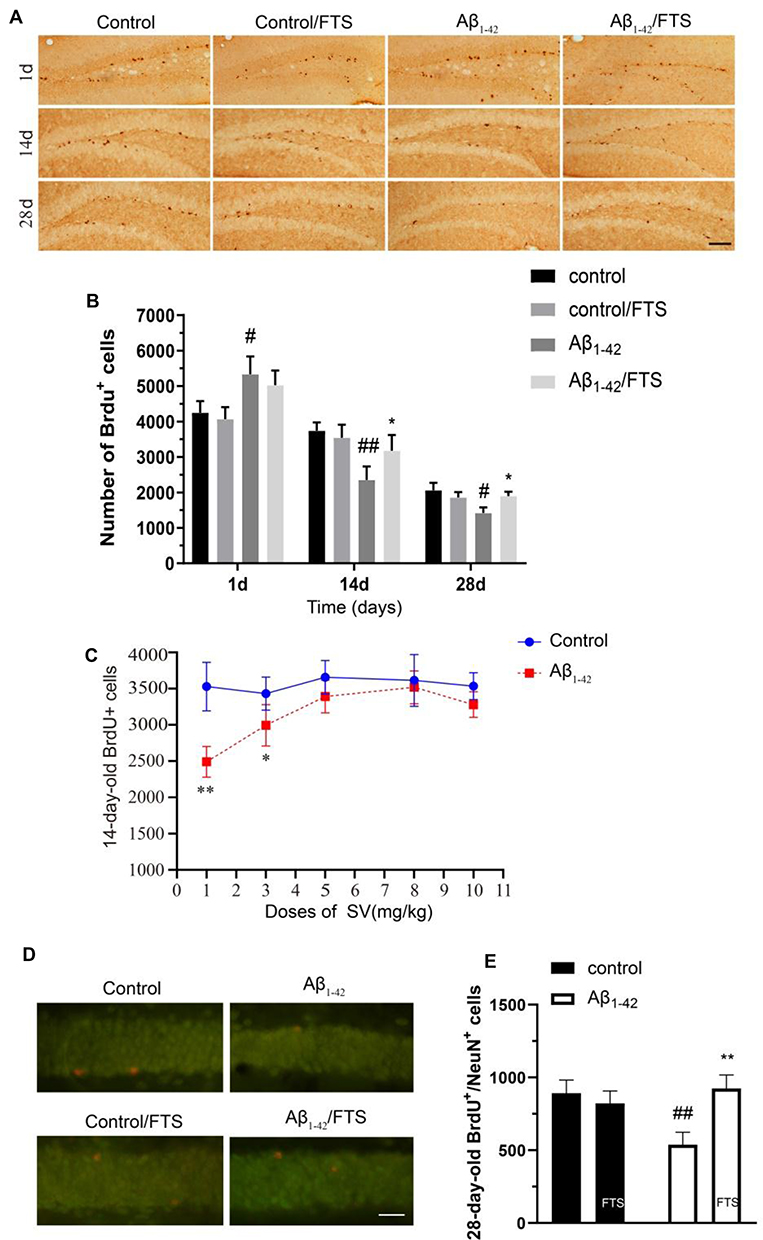 |
Figure 4 The effect of FTS on survival of newborn neurons in Aβ1-42-mice. (A) Immunohistochemistry of 1-, 14-, and 28-day-old BrdU+ cells in control mice (control), FTS-treated control mice (FTS), Aβ1-42-mice (Aβ1-42), and FTS-treated Aβ1-42-mice (Aβ1-42/FTS). Scale bar = 50 mm. (B) Comparison of the number of 1-day-old BrdU+ cells. #p < 0.05 and ##p < 0.01 vs control mice; *p < 0.05 vs Aβ1-42-mice. (C) Dose-dependency of FTS-protected 14-day-old BrdU+ cells in Aβ1-42-mice. *p < 0.05 and **P < 0.01 vs FTS-treated group. (D) Immunofluorescence of 28-day-old BrdU+/NeuN+ cells. Representative pictures of BrdU+/NeuN+ cells. Scale bar = 25 mm. (E) Comparison of the number of 28-day-old BrdU+/NeuN+ cells. ##p < 0.01 vs control mice; **p < 0.01 vs Aβ1-42-mice. Abbreviations: FTS, S-trans, trans-farnesylthiosalicylic; vs, versus; BrdU, 5-bromo-2-deoxyuridine. |
Protection of Neurite Growth of Newborn Neurons in Aβ1-42-Mice by FTS
DCX is a microtubule-associated protein that is specifically expressed in the soma and dendrites of immature neonatal neurons but not in glial cells.24 As shown in Figure 5, compared with control mice, not only the number of DCX-positive cells (DCX+) (P < 0.05) and the number of DCX+ fibers per DCX+ cell (P < 0.01) was reduced in Aβ1-42-mice due to the impairment of Aβ1-42. However, FTS could perfectly prevent the decrease in the number of DCX+ cells (P < 0.05) and DCX+ fibers (P<0.01) in Aβ1-42-mice.
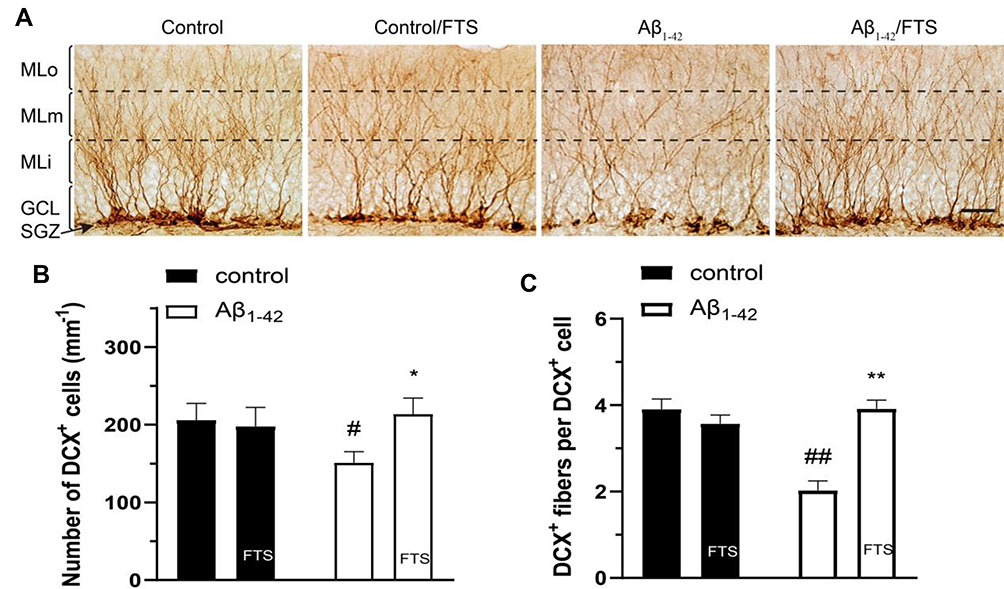 |
Figure 5 Effect of FTS on neurite growth of newborn neurons in Aβ1-42-mice. (A) Typical image of DCX+ cells and fibers in hippocampal DG. Molecular layer (ML) of DG was divided into 3 sub-regions of inner (MLi), middle (MLm), and outer (MLo). GCL indicates granule cell layer. Scale bar =25 mm. (B) Comparison of the number of DCX+ cells. (C) The number of DCX+ fibers per DCX+ cell. #P < 0.05 and ##P < 0.01 vs control mice; *P < 0.05 and **P < 0.01 vs Aβ1-42-mice. Abbreviations: FTS, S-trans, trans-farnesylthiosalicylic; vs, versus; DCX, doublecortin; DG, dentate gyrus. |
Inhibition of Aβ-Mediated Activation of RAS-ERK Signaling Pathway by FTS
Aβ exposure could increase the activation of RAS protein and RAS-ERK signaling cascade activation.8 In the study, compared with DMSO injection mice, the level of hippocampal RAS in Aβ1-42-mice was significantly increased on day 1 after Aβ1-42 injection (p < 0.01) (Figure 6A and B). The levels of hippocampal ERK phosphorylation (p-ERK) and CREB phosphorylation (p-CREB) were measured on day 13 after Aβ1-42 injection. In comparison with controls, the level of p-ERK in Aβ1-42-mice increased (P < 0.01) (Figure 6C and D), with a concomitant increase in the level of p-CREB (P < 0.01) (Figure 6E and F). Similar to the decrease in control mice by FTS as RAS inhibitor, the levels of p-ERK and p-CREB in Aβ1-42-mice were inhibited (P < 0.01) (Figure 6C and D). Altogether, these findings indicate that FTS could reverse Aβ-induced abnormal activation of ERK signaling pathway in Aβ1-42-mice.
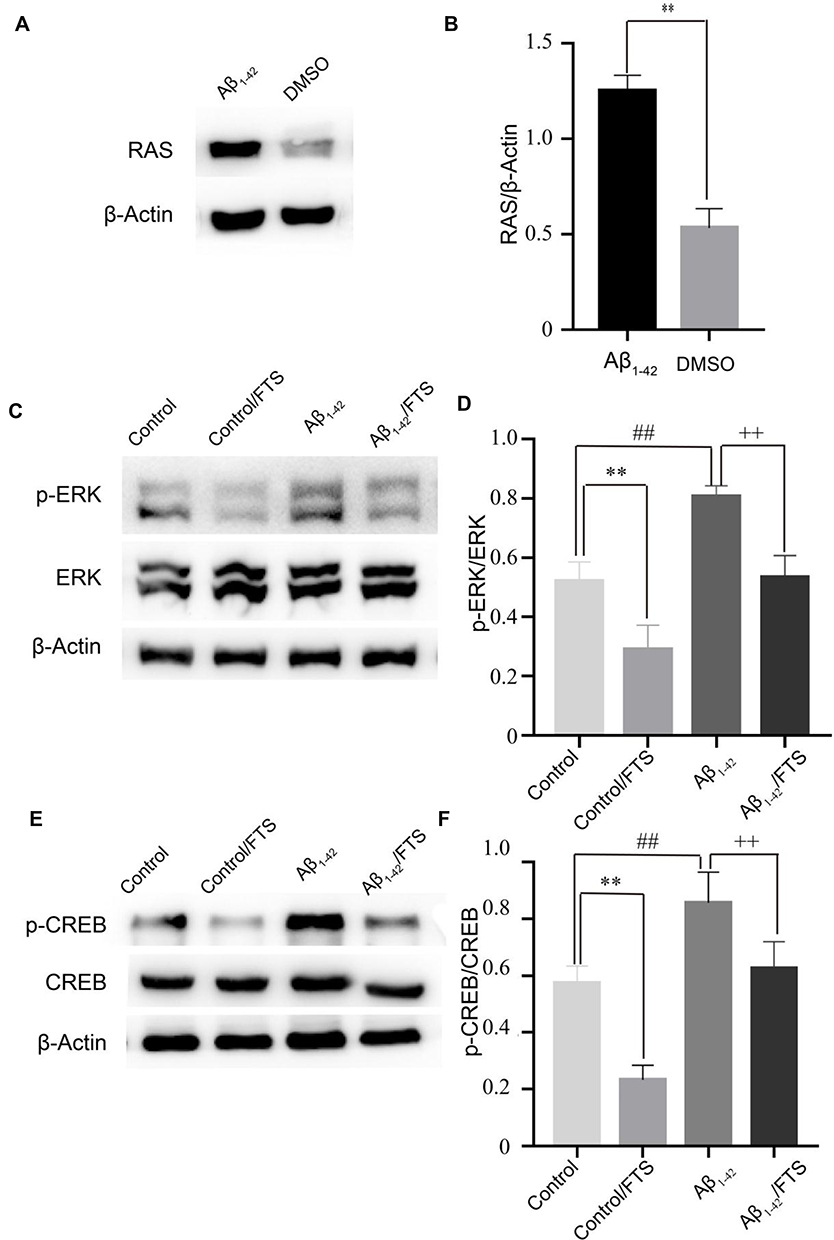 |
Figure 6 Inhibition of the activation of RAS-ERK signaling pathway in Aβ1-42-mice by FTS. (A) Western blot analysis of total RAS. β-Actin was used as an internal control. (B) Densitometric analysis of RAS. **p < 0.01 vs DMSO group. (C) Western blot analysis of total ERK and p-ERK. (D) Densitometric analysis of total ERK and p-ERK. **p < 0.01; ##p < 0.01 vs control group; ++p < 0.01 vs Aβ1-42. (E) Western blot analysis of total CREB and p-CREB. (F) Densitometric analysis of total CREB and p-CREB.**p < 0.01; ##p < 0.01 vs control; ++p < 0.01 vs Aβ1-42. Abbreviations: FTS, S-trans, trans-farnesylthiosalicylic; vs, versus; p-ERK, ERK phosphorylation; p-CREB, CREB phosphorylation. |
Discussion
In this study, we found that FTS, a RAS inhibitor, could prevent Aβ1-42 to impair survival and neurite growth of newborn neurons in hippocampal DG in Aβ1-42-mice as an AD model. Behavioral indexes and morphological findings also demonstrated that FTS could improve the learning and spatial memory abilities of Aβ1-42-mice. The underlying molecular mechanism should be that FTS inhibited the levels of hippocampal p-ERK and p-CREB activated by Aβ.
The lateral ventricle injection of Aβ1-42 oligomer is a mature AD model, which can well reflect the neurotoxicity of Aβ, and is often used in neurogenesis research.22,23,25 It is known that overproduction and subsequent Aβ aggregation limit the survival of new neurons in the DG of APP/PS1 mice.26 In our study, Aβ1-42 caused a significant decrease in the number of 14-day-old BrdU+ cells which represent newborn neurons27 and DCX+ cells which represent early immature neurons28 in hippocampal DG, indicating that many Aβ-induced neoplastic cells were apoptotic within 2 weeks, which is consistent with previous studies.23 However, the number of BrdU+ cells in Aβ1-42-mice of AD model was significantly increased after 14 days of continuous administration of FTS, compared to that of unadministered AD mice. Furthermore, in Aβ1-42-mice, FTS also increased the number of DCX+ cells. The above results indicate that FTS not only protects the survival of newborn neurons but also promotes the maturation of neurons. AD is the most common form of age-related dementia and still lacks effective treatments and preventive measures. Most of the therapeutic approaches aimed at reducing Aβ burden fail to improve the clinical symptoms of AD. More and more researchers focus on stimulating neurogenesis and vascularization.29,30 Here, we provided in vivo evidence that the FTS could not only protect the survival of newborn neurons but also promote the maturation in Aβ1-42-mice of the AD model. Neonatal neurons integrate into the hippocampal loop after 3–4 weeks of maturation, anatomically supporting the cause–result relationship between neurogenesis and cognitive performance.31 To some extent, neonatal neurons integrate into the hippocampal neural network, replacing the denatured and degenerate nerve cells in AD, thereby improving neuropsychiatric symptoms of learning and spatial memory.32
A recent study has reported for the first time that the RAS inhibitor, FTS, could improve the spatial memory of AD mice to some extent.18 Consistent with the previous study, our results show that FTS significantly improved spatial learning and memory detected by the MWM test in Aβ1-42 mice after 18 days of the treatment with FTS.18 Physical exercise can increase the tolerance of cells and tissues to oxidative stress, vascularization, energy metabolism, and neurotrophin synthesis, which are beneficial for neurogenesis, memory improvement, and brain plasticity.33 A multicenter, investigator-masked, random controlled trial showed that physical exercise improves the physical fitness of AD patients without slowing cognitive impairment in patients with mild-to-moderate dementia.34 A recent study indicated that promoting adult hippocampal neurogenesis could ameliorate AD pathology and cognitive deficits in the presence of a healthier brain environment stimulated by physical exercise.29 Further investigation of combination pharmacology of FTS and physical exercise based on our present study should be extended to seek a more effective treatment strategy for AD.
Ras is activated by a variety of extracellular stimuli and then activates the RAF–MEK–ERK cytokine kinase cascade and cAMP response element-binding protein (CREB) pathway.14 In APP-expressing B103 neuroblastoma cells, Aβ precursor protein (APP) and Aβ42 mediated RAS-MAPK signaling cascade and GSK-3 activation.8 In AD patients, RAS protein and major downstream elements of the MAPK-cascade reaction were elevated at an early stage of the disease.35 Interestingly, the RAS/ERK signaling cascade regulates so many different and even opposite cellular processes; it not only plays a regulatory role in a variety of cellular processes such as proliferation, differentiation, development, learning, survival, etc., but also can cause apoptosis if aberrantly activated.36 Mitogenic overstimulation of neurons is an important cause of abnormal activation of the cell cycle and subsequent cell death,12 which is an important component of the pathogenesis of AD and other degenerative diseases.37 Aβ42 oligomer preparation could also induce cell cycle activation and subsequent neuronal loss by activating RAS.13 RAS signaling pathway is involved in synaptic plasticity and memory formation.38 Long-term potentiation (LTP) impairment and learning and memory deficits were observed due to excessive RAS activity in the knock-out mice of Nf1.39 Neural plasticity is considered the neurophysiological correlate of learning and memory. Transcranial magnetic stimulation (TMS) studies have shown that increased cortical excitability and impaired brain plasticity are one of the pathophysiological mechanisms of AD and major depression.40,41 RAS as a small molecule guanosine triphosphate (GTPase) protein needs to be modified by farnesylation to localize on the cell membrane and bind to GTP under the action of guanylate exchange factor (GEF) to activate.42 Aβ could induce phosphorylation of the threonine (668 site) of the cytoplasmic domain of APP, which activates the adaptor protein growth factor receptor-binding protein 2 (Grb2) and then recruits the guanylate exchange factor SOS2, thereby promoting RAS farnesylation.8
Since RAS activity is associated with neurodegenerative pathways, FTS acts as a chemical RAS inhibitor, which was developed and tested as a cancer drug, and its role in degenerative diseases such as AD has been increasingly studied recently.13,17 Here, we determined that FTS attenuated Aβ-induced ERK, CREB hyperphosphorylation by inhibiting RAS. CREB is an important transcription factor of ERK, and its phosphorylation promotes the transcriptional expression of cyclin-dependent kinases and cyclin genes.43 Mitosis requires ERK activity,44 but if ERK activity in the G2/M phase is too high, it could block mitotic entry and subsequent cell death.45 In our study, RAS in the brain of Aβ1-42-mice was significantly higher than that of the control mice, and FTS could inhibit Aβ1-42-induced overactivation of the hippocampal ERK signal cascade and CREB. This may be the underlying molecular mechanism of which FTS could protect the survival and neurite growth of newborn neurons and improve spatial cognitive deficits for AD.
Admittedly, our study has some limitations. In the present study, we used mice lateral ventricle injection of Aβ as an AD model, which limits the neurotoxicity of Aβ but not the other pathological features of AD. The effects of adult hippocampal neurogenesis manipulation on tau phosphorylation levels should also be explored in a future study.
Conclusion
In summary, FTS, a synthetic Ras inhibitor, could prevent Aβ1-42 to impair survival and neurite growth of newborn neurons in hippocampal dentate gyrus in Aβ1-42-mice as the AD model and thus improve the learning and spatial memory via inhibition of RAS/ERK pathway. These findings suggest that FTS could be a potential therapeutic agent for the treatment of AD.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Lane CA, Hardy J, Schott JM. Alzheimer’s disease. Eur J Neurol. 2018;25(1):59–70. doi:10.1111/ene.2018.25.issue-1
2. Schmidt-Hieber C, Jonas P, Bischofberger J. Enhanced synaptic plasticity in newly generated granule cells of the adult hippocampus. Nature. 2004;429(6988):184–187. doi:10.1038/nature02553
3. Winocur G, Wojtowicz JM, Sekeres M, Snyder JS, Wang S. Inhibition of neurogenesis interferes with hippocampus-dependent memory function. Hippocampus. 2006;16(3):296–304. doi:10.1002/(ISSN)1098-1063
4. Anacker C, Hen R. Adult hippocampal neurogenesis and cognitive flexibility – linking memory and mood. Nat Rev Neurosci. 2017;18(6):335–346. doi:10.1038/nrn.2017.45
5. Hamilton LK, Aumont A, Julien C, Vadnais A, Calon F, Fernandes KJ. Widespread deficits in adult neurogenesis precede plaque and tangle formation in the 3xTg mouse model of Alzheimer’s disease. Eur J Neurosci. 2010;32(6):905–920. doi:10.1111/j.1460-9568.2010.07379.x
6. Lazarov O, Hollands C. Hippocampal neurogenesis: learning to remember. Prog Neurobiol. 2016;138–140:1–18. doi:10.1016/j.pneurobio.2015.12.006
7. Selkoe DJ. Deciphering the genesis and fate of amyloid beta-protein yields novel therapies for Alzheimer disease. J Clin Invest. 2002;110(10):1375–1381. doi:10.1172/JCI0216783
8. Kirouac L, Rajic AJ, Cribbs DH, Padmanabhan J. Activation of Ras-ERK signaling and GSK-3 by amyloid precursor protein and amyloid beta facilitates neurodegeneration in Alzheimer’s disease. eNeuro. 2017;4:2. doi:10.1523/ENEURO.0149-16.2017
9. Murphy LO, Blenis J. MAPK signal specificity: the right place at the right time. Trends Biochem Sci. 2006;31(5):268–275. doi:10.1016/j.tibs.2006.03.009
10. Lee MS, Kao SC, Lemere CA, et al. APP processing is regulated by cytoplasmic phosphorylation. J Cell Biol. 2003;163(1):83–95.
11. Coleman ML, Marshall CJ, Olson MF. Ras promotes p21(Waf1/Cip1) protein stability via a cyclin D1-imposed block in proteasome-mediated degradation. EMBO J. 2003;22(9):2036–2046. doi:10.1093/emboj/cdg189
12. Arendt T, Holzer M, Stöbe A, et al. Activated mitogenic signaling induces a process of dedifferentiation in Alzheimer’s disease that eventually results in cell death. Ann N Y Acad Sci. 2000;920:249–255. doi:10.1111/j.1749-6632.2000.tb06931.x
13. Koseoglu MM, Ozdilek BA, Djakbarova U, Gulusur A. Targeting Ras activity prevented amyloid beta-induced aberrant neuronal cell cycle re-entry and death. Curr Alzheimer Res. 2016;13(11):1267–1276. doi:10.2174/1567205013666160625074520
14. Shields JM, Pruitt K, McFall A, Shaub A, Der CJ. Understanding Ras: ‘it ain’t over ‘til it’s over’. Trends Cell Biol. 2000;10(4):147–154. doi:10.1016/S0962-8924(00)01740-2
15. Dineley KT, Westerman M, Bui D, Bell K, Ashe KH, Sweatt JD. Beta-amyloid activates the mitogen-activated protein kinase cascade via hippocampal alpha7 nicotinic acetylcholine receptors: in vitro and in vivo mechanisms related to Alzheimer’s disease. J Neurosci. 2001;21(12):4125–4133. doi:10.1523/JNEUROSCI.21-12-04125.2001
16. Niv H, Gutman O, Kloog Y, Henis YI. Activated K-Ras and H-Ras display different interactions with saturable nonraft sites at the surface of live cells. J Cell Biol. 2002;157(5):865–872. doi:10.1083/jcb.200202009
17. Kloog Y, Cox AD, Sinensky M. Concepts in Ras-directed therapy. Expert Opin Investig Drugs. 1999;8(12):2121–2140. doi:10.1517/13543784.8.12.2121
18. Wang Y, Chen T, Yuan Z, et al. Ras inhibitor S-trans, trans-farnesylthiosalicylic acid enhances spatial memory and hippocampal long-term potentiation via up-regulation of NMDA receptor. Neuropharmacology. 2018;139:257–267. doi:10.1016/j.neuropharm.2018.03.026
19. Jin H, Chen T, Li G, et al. Dose-dependent neuroprotection and neurotoxicity of simvastatin through reduction of Farnesyl Pyrophosphate in mice treated with intracerebroventricular injection of Aβ 1-42. J Alzheimers Dis. 2016;50(2):501–516. doi:10.3233/JAD-150782
20. Shohami E, Yatsiv I, Alexandrovich A, et al. The Ras inhibitor S-trans, trans-farnesylthiosalicylic acid exerts long-lasting neuroprotection in a mouse closed head injury model. J Cereb Blood Flow Metab. 2003;23(6):728–738. doi:10.1097/01.WCB.0000067704.86573.83
21. Robert D, Mays W, Mays RW, et al. Development of an allogeneic adherent stem cell therapy for treatment of ischemic stroke. J Exp Stroke Transl Med. 2010;3:34–46. doi:10.6030/1939-067X-3.1.34
22. Wang C, Chen T, Li G, Zhou L, Sha S, Chen L. Simvastatin prevents β-amyloid(25–35)-impaired neurogenesis in hippocampal dentate gyrus through α7nAChR-dependent cascading PI3K-Akt and increasing BDNF via reduction of farnesyl pyrophosphate. Neuropharmacology. 2015;97:122–132. doi:10.1016/j.neuropharm.2015.05.020
23. Li L, Xu B, Zhu Y, Chen L, Sokabe M, Chen L. DHEA prevents Aβ25-35-impaired survival of newborn neurons in the dentate gyrus through a modulation of PI3K-Akt-mTOR signaling. Neuropharmacology. 2010;59(4–5):323–333. doi:10.1016/j.neuropharm.2010.02.009
24. Rao MS, Shetty AK. Efficacy of doublecortin as a marker to analyse the absolute number and dendritic growth of newly generated neurons in the adult dentate gyrus. Eur J Neurosci. 2004;19(2):234–246. doi:10.1111/j.0953-816X.2003.03123.x
25. Yun HM, Kim HS, Park KR, et al. Placenta-derived mesenchymal stem cells improve memory dysfunction in an Aβ1-42-infused mouse model of Alzheimer’s disease. Cell Death Dis. 2013;4:e958. doi:10.1038/cddis.2013.490
26. Verret L, Jankowsky JL, Xu GM, Borchelt DR, Rampon C. Alzheimer’s-type amyloidosis in transgenic mice impairs survival of newborn neurons derived from adult hippocampal neurogenesis. J Neurosci. 2007;27(25):6771–6780. doi:10.1523/JNEUROSCI.5564-06.2007
27. Kempermann G, Gast D, Kronenberg G, Yamaguchi M, Gage FH. Early determination and long-term persistence of adult-generated new neurons in the hippocampus of mice. Development. 2003;130(2):391–399. doi:10.1242/dev.00203
28. Brown JP, Couillard-Després S, Cooper-Kuhn CM, Winkler J, Aigner L, Kuhn HG. Transient expression of doublecortin during adult neurogenesis. J Comp Neurol. 2003;467(1):1–10. doi:10.1002/cne.v467:1
29. Choi SH, Bylykbashi E, Chatila ZK, et al. Combined adult neurogenesis and BDNF mimic exercise effects on cognition in an Alzheimer’s mouse model. Science. 2018;361:6406. doi:10.1126/science.aan8821
30. Bordet R, Ihl R, Korczyn AD, et al. Towards the concept of disease-modifier in post-stroke or vascular cognitive impairment: a consensus report. BMC Med. 2017;15(1):107. doi:10.1186/s12916-017-0869-6
31. Zhao C, Teng EM, Summers RG
32. Haughey NJ, Liu D, Nath A, Borchard AC, Mattson MP. Disruption of neurogenesis in the subventricular zone of adult mice, and in human cortical neuronal precursor cells in culture, by amyloid beta-peptide: implications for the pathogenesis of Alzheimer’s disease. Neuromolecular Med. 2002;1(2):125–135. doi:10.1385/NMM:1:2:125
33. Radak Z, Hart N, Sarga L, et al. Exercise plays a preventive role against Alzheimer’s disease. J Alzheimers Dis. 2010;20(3):777–783. doi:10.3233/JAD-2010-091531
34. Lamb SE, Sheehan B, Atherton N; DAPA Trial Investigators, et al.. Dementia And Physical Activity (DAPA) trial of moderate to high intensity exercise training for people with dementia: randomised controlled trial. BMJ. 361:k1675.
35. Gärtner U, Holzer M, Arendt T. Elevated expression of p21ras is an early event in Alzheimer’s disease and precedes neurofibrillary degeneration. Neuroscience. 1999;91(1):1–5. doi:10.1016/S0306-4522(99)00059-7
36. Shaul YD, Seger R. The MEK/ERK cascade: from signaling specificity to diverse functions. Biochim Biophys Acta. 2007;1773(8):1213–1226. doi:10.1016/j.bbamcr.2006.10.005
37. Lee HG, Casadesus G, Zhu X, et al. Cell cycle re-entry mediated neurodegeneration and its treatment role in the pathogenesis of Alzheimer’s disease. Neurochem Int. 2009;54(2):84–88. doi:10.1016/j.neuint.2008.10.013
38. Ye X, Carew TJ. Small G protein signaling in neuronal plasticity and memory formation: the specific role of ras family proteins. Neuron. 2010;68(3):340–361. doi:10.1016/j.neuron.2010.09.013
39. Li W, Cui Y, Kushner SA, et al. The HMG-CoA reductase inhibitor lovastatin reverses the learning and attention deficits in a mouse model of neurofibromatosis type 1. Curr Biol. 2005;15(21):1961–1967. doi:10.1016/j.cub.2005.09.043
40. Pennisi G, Ferri R, Lanza G, et al. Transcranial magnetic stimulation in Alzheimer’s disease: a neurophysiological marker of cortical hyperexcitability. J Neural Transm (Vienna). 2011;118(4):587–598. doi:10.1007/s00702-010-0554-9
41. Cantone M, Bramanti A, Lanza G, et al. Cortical plasticity in depression. ASN Neuro. 2017;9(3):1759091417711512. doi:10.1177/1759091417711512
42. McTaggart SJ. Isoprenylated proteins. Cell Mol Life Sci. 2006;63(3):255–267. doi:10.1007/s00018-005-5298-6
43. Chambard JC, Lefloch R, Pouysségur J, Lenormand P. ERK implication in cell cycle regulation. Biochim Biophys Acta. 2007;1773(8):1299–1310. doi:10.1016/j.bbamcr.2006.11.010
44. Tamemoto H, Kadowaki T, Tobe K, et al. Biphasic activation of two mitogen-activated protein kinases during the cell cycle in mammalian cells. J Biol Chem. 1992;267(28):20293–20297.
45. Yan Y, Spieker RS, Kim M, Stoeger SM, Cowan KH. BRCA1-mediated G2/M cell cycle arrest requires ERK1/2 kinase activation. Oncogene. 2005;24(20):3285–3296. doi:10.1038/sj.onc.1208492
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.