")
Back to Journals » Neuropsychiatric Disease and Treatment » Volume 11
Neuroinflammation in Alzheimer’s disease
Received 8 October 2014
Accepted for publication 5 December 2014
Published 30 January 2015 Volume 2015:11 Pages 243—256
DOI https://doi.org/10.2147/NDT.S75546
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Wai Kwong Tang
Fengjin Zhang,1,2 Linlan Jiang1
1Department of Pharmacy, General Hospital of Guangzhou Military Command, Guangzhou City, People’s Republic of China; 2School of Bioscience and Bioengineering, South China University of Technology, Guangzhou City, People’s Republic of China
Abstract: Amyloid-β plaques and neurofibrillary tangles are the main neuropathological hallmarks in Alzheimer’s disease (AD), the most common cause of dementia in the elderly. However, it has become increasingly apparent that neuroinflammation plays a significant role in the pathophysiology of AD. This review summarizes the current status of neuroinflammation research related to AD, focusing on the connections between neuroinflammation and some inflammation factors in AD. Among these connections, we discuss the dysfunctional blood–brain barrier and alterations in the functional responses of microglia and astrocytes in this process. In addition, we summarize and discuss the role of intracellular signaling pathways involved in inflammatory responses in astrocytes and microglia, including the mitogen-activated protein kinase pathways, nuclear factor-kappa B cascade, and peroxisome proliferator–activated receptor-gamma transcription factors. Finally, the dysregulation of the control and release of pro- and anti-inflammatory cytokines and classic AD pathology (amyloid plaques and neurofibrillary tangles) in AD is also reviewed.
Keywords: inflammation, blood–brain barrier, glial cells, intracellular signaling pathways, inflammatory factors
Introduction
Neuroinflammation is defined as the brain’s activation of the innate immune system, and its main function is to protect the central nervous system (CNS) against infectious insults, injury, or disease.1 It has been well established that neuroinflammation is actively involved in neurological diseases and disorders such as Alzheimer’s disease (AD).2 It is a complex response involving a host of cellular and molecular changes, recruitment of peripheral immune cells, induction of some intracellular signaling pathways, and release of inflammatory mediators in the brain. All these factors can contribute to the occurrence of neuronal dysfunction and death in AD, either alone or in combination.3,4 These observations and other researches indicate that neuroinflammation is an early and continuous feature of AD. In the following contents, we will emphasize the role of the blood–brain barrier (BBB) and glial cells during neuroinflammation in the CNS and provide an outline of the involvement of intracellular signaling pathways and inflammatory mediators produced by glial cells and the main neuropathological hallmarks in the pathological cascade of events leading to AD.
BBB and neuroinflammation
BBB is a specialized barrier between blood and brain mainly consisting of specific endothelial cells, tight liner sheets formed by astrocytic end-feet and pericytes, and tight junctions (TJs) that are brought together.5 Because of the importance of the CNS, a physical and transport barrier for blood–borne cells and toxic solubles to invade the CNS parenchyma is indispensable.6 The BBB and blood–nerve barrier restrict exchanges of soluble factors and cells between the neural tissue and blood, thus playing a crucial role in the maintenance of the peripheral nervous system and CNS homeostasis.7
Increasing evidence indicates that amyloid-β (Aβ) and BBB disruption may mutually promote their effects on AD. P-glycoprotein, a transporter on the luminal surface of the BBB transports Aβ from the brain to the blood efficiently.8 The dysfunction of BBB can result in Aβ accumulation by inducing BBB leakage or insufficient clearance. Aβ42 is able to modify the expression of TJs and alter barrier properties functionally in vitro.9 Aβ accumulation can lead to BBB dysfunction by endothelial toxicity and increased monocyte adhesion.10 One cause of the BBB dysfunction is inflammatory cytokines. For example, it has been reported that tumor necrosis factor-α (TNF-α), interleukin-(IL)1β, and IL-17A could loosen the TJs and breach the BBB.11 Neuroinflammatory response is involved in the altered expression of TJs and loss of BBB integrity in capillary cerebral amyloid angiopathy,12 and BBB transcriptome changed in systemic inflammation.13 In an animal model of AD using an Alzheimer amyloid precursor protein (APP)-transgenic mouse (APP-Tg), the increase in brain inflammatory cytokine (IL-6) level is significantly higher in APP-Tg than in wild-type mice after peripheral lipopolysaccharide injection, and the BBB becomes more permeable during peripherally evoked inflammation. The results indicate the increased vulnerability of the BBB to inflammation in this animal model of AD.14 Research has usually focused on structural integrity rather than functionality, and BBB alteration has traditionally been explained as a consequence rather than a cause. But current knowledge suggests that BBB integrity plays a more important role than previously thought, and pharmacological modulation of the BBB may offer a new therapeutic target for AD treatment. For example, there is a patent showing that ultrasound neuromodulation can control the permeability of the BBB for the selective penetration of drugs or other substances into the target.15
Cell mediators of neuroinflammation
Astrocytes and microglia are the major types of glial cells in the CNS, and their activation involves various types of neurodegenerative processes. Reactive glial cells are closely associated with plaques and parallels tangles in AD.16 Once activated, their processes become hypertrophied; both astrocytes and microglia produce multiple inflammatory factors, including cytokines, prostanoids, chemokines, reactive oxygen species, and cyclooxygenase-(COX)2.17 Similarly, as indications of neuroinflammation, elevated levels of inflammatory cytokines are discovered in AD (see the section Cytokines and neuroinflammation).
Microglia
Due to the presence of the BBB, the lack of lymphatic drainage, and the reduced capability of leukocytes to invade the CNS, the need arises for a different approach to remove and eliminate injured tissue. Therefore, the CNS is equipped with high specificity of innate immune cells named microglia. They are the resident macrophages, accounting for approximately 5%–10% of the adult brain cell population in the CNS.18 These cells actually arise early from progenitors in the embryonic yolk sac and appear to persist into adulthood. Furthermore, microglia exist in all stages of brain development.19 Their main functions are to facilitate host defense by destroying invading pathogens, eliminating deleterious debris, and promoting tissue repair and homeostasis, partly by affecting the surrounding astrocytes and neurons.20 And inflammatory conditions have been shown to increase the supplement of circulating myeloid progenitors and differentiation into microglia.21 As the primary regulators of inflammation in the CNS, the activation of microglia can quickly lead to the release of inflammatory cytokines.22 In culture, it has been shown that microglia can release several potentially cytotoxic substances, such as cytokines, nitric oxide, arachidonic acid derivatives, proteases, reactive oxygen intermediates, excitatory amino acids, and various neurotrophic factors.23
Now, there is no doubt that activated microglia are involved in neuronal death and cognitive deficits in AD. Morales et al showed that Tau polymers induce activation of microglia and change their cellular morphology and the release of the inflammatory cytokine IL-6; furthermore, conditioned media of activated microglia induced neurofibrillary degeneration of hippocampal neurons in culture.24 In a triple-transgenic model of AD, activated microglia increased significantly compared with nontransgenic controls, and the activation of microglia was closely associated with Aβ plaque formation, smaller Aβ deposits, and Aβ plaques.25 Griciuc et al investigated the AD risk gene, CD33. AD brains have increased CD33 and CD33-positive microglia. Mice lacking the gene of CD33 have less AD pathology, revealing a role for microglia in Aβ clearance.26 Early in the emergence of senile plaques, microglia are strongly activated; they serve to limit the growth of senile plaques and then minimize local inflammatory damage to the brain components.27 Microglia express scavenger receptors of class A or B and receptors for advanced-glycosylation end products that promote Aβ phagocytosis.28,29 In addition, microglia also secrete proteolytic enzymes that degrade Aβ, such as insulin-degrading enzyme, matrix metalloproteinase, and plasminogen–plasmin complex.30,31 Moreover, microglia can secrete a number of soluble factors at the same time, such as the glia-derived neurotrophic factor, which plays a part in promoting the survival of neurons.32 It has been shown that bone marrow–derived microglia could delay or stop the progress of AD, and preventing their recruitment contributes to the pathology.33 Although there is a growing body of evidence to support a role for microglia in neuroprotection and Aβ clearance, why Aβ continues to accumulate and why AD pathology progresses despite continued microglia activation and recruitment remain to be answered. Possible explanations for the failure of microglia to slow AD pathology progresses would be that microglia become overloaded by the large amount of Aβ produced, cannot keep pace with the generation of Aβ, become more proinflammatory,34 and their activation is gradually diminished in the later stages of plaque formation (>150 days).27 The ability of the microglia to interact with Aβ fibrils undergoes an age-dependent decrease and their ability to phagocytize them is lost;35 thus, the uptake and degradation of Aβ are reduced. Major microglial functions progressively decline with the appearance of Aβ plaques in AD, and lowering Aβ burden could reverse this functional impairment.36 Microglia interact with Aβ, overproduction of inflammatory cytokines (IL-1β, TNF-α, IL-6) and substances released by dying neurons, activated β-secretase-1 (BACE1) and γ-secretases to reduce Aβ clearance, thereby promoting Aβ production,34 further proliferation and activation of microglia and neuronal damage, resulting in a vicious cycle in AD.37 Above all, the role of microglia in neuroinflammation of AD could be seen as a double-edged sword. Therefore, it is important to understand the activation of microglia in different stages of AD to be able to estimate the effect of potential therapies. A seminal study conducted by Heneka et al revealed that microglia-specific inflammasome is a promising type of cell-specific molecular target for therapeutic intervention of AD in the CNS.38 It could be proposed that pharmacological agents targeting microglia represent a novel angle for the therapeutic intervention of AD.
Astrocytes
Astrocytes are specialized glial cells accounting for approximately 35% of the total CNS cell population and are found in all the CNS regions.39 Like neurons and oligodendrocytes, they are derived from the neuroectoderm, which is in contrast to microglia.40 Astrocytes are morphologically heterogeneous. Fibrous astrocytes are shown in the white matter, and protoplasmic astrocytes are shown in the gray matter.41 The reaction of fibrous astrocytes and protoplasmic astrocytes is very different. Following mechanical injury, both of them undergo the process of hypertrophy of the cell bodies and processes. Fibrous astrocytes simplify and retract their branching structure, but not protoplasmic astrocytes.42 Astrocytes assume a variety of roles in maintaining an optimally suited milieu for neuronal function. Select astrocytic functions include the removal of debris and toxins from the cerebrospinal fluid, the regulation of neurotransmitter, the production of trophic factors and ion concentrations, and the maintenance of redox potential. These functional impairments associated with the injury of astrocytes during physiological reactions could trigger or exacerbate neuronal dysfunction.43
Glial cells are abundant in the CNS and play multiple roles in brain structure and function. In response to Aβ oligomers, reactive astrocytes are closely associated with the activation of the transcriptional factor nuclear factor-kappa B (NF-κB) and upregulation of TNF-α, IL-1β, and COX-2 in the rat cortex in vivo.44 Some scientists think that astrocytes are conductors of the AD neuroinflammatory symphony45 and astrocyte senescence as a component of AD.46 On the one hand, it has been demonstrated that primary human astrocytes or astrocytoma cells could be induced by interferon-γ in combination with IL-1β or TNF-α to produce Aβ.47 The astrocyte-secreted proinflammatory factors could contribute to the expression level of secretases, thereby enhancing the conversion of APP into neurotoxic, insoluble, fibrillary Aβ on the membrane of neurons.48,49 Reactive astrocytes express BACE, the enzyme responsible for the generation of Aβ, suggesting that they may promote Aβ accumulation in aged transgenic AD mice model.50,51 Reactive astrocytes from sporadic AD patients also have increased levels of presenilin-1, the catalytic component of the γ-secretase complex that is involved in the formation of Aβ.52 As the largest number of brain cells, activated astrocytes may represent a very important source of Aβ during neuroinflammation in AD.53 On the other hand, by providing trophic support to neurons and forming a protective barrier between neurons and Aβ deposits, astrocytes are known to promote the Aβ clearance and degradation.51 Reactive astrocytes could express Aβ-degrading enzymes, mainly matrix metalloproteinases-2, -3, and -9 and neprilysin.54,55 Aβ increases apoE protein levels, which is associated with neuroinflammatory responses and Aβ clearance in astrocytes. It may be a neuroprotective response to Aβ-induced cytotoxicity, in accordance with apoE’s role in cytoprotection.56 Thus, astrocytes undergoing modifications and chronic inflammation might suffer a deleterious transformation: acquire the capacity to generate Aβ and lose the ability to remove and degrade them.57
Astrocytes are closely involved in inflammatory and immunological events occurring in the CNS due to their ability to secrete and respond to a very large number of inflammatory cytokines, such as TNF-α, IL-1β, IL-6, IL-8, IL-10, transforming growth factor (TGF)-β, interferon-γ, and interferon-β.58 The mechanisms of astrocyte activation in response to Aβ may include NF-κB–mediated pro- and/or inflammatory gene expression.44 Aβ induces astrocytic glutamate release, synaptic loss, and extrasynaptic N-methyl-d-aspartic acid receptor activation.59 In vitro, Aβ42 triggered senescence, driving the expression of positive senescent astrocytes. Senescent astrocytes produce a number of inflammatory cytokines including IL-6, and an accumulation of senescent astrocytes may be associated with increased risk of sporadic AD with advancing age.60 Astrocytes exposure to increased amounts of Aβ for a long time, these cells must to clear large amounts of Aβ, hence, neglect their other functions, at last impairs the transmitter recycling and metabolic support of astrocytes in AD patients.61 Recently, the potential of astrocyte-targeted therapeutics in an intact animal model of AD was tested. When adeno-associated virus vectors containing the promoter of astrocyte-specific Gfa2 was used to target hippocampal astrocytes in APP/PS1 mice, APP/PS1 mice exhibited improved synaptic function, cognitive, lower amyloid levels, and reduced glial activation.62 The results lay the foundation to explore other novel astrocyte-based therapies.
Intracellular signaling pathway and neuroinflammation
The regulation of inflammatory cytokine production in activated microglia and astrocytes is under the control of intracellular signaling pathways, including the mitogen-activated protein kinase (MAPK) signaling pathway, the NF-κB signaling cascade, and peroxisome proliferator–activated receptor-γ (PPAR-γ).
Role of MAPK
MAPK signaling pathways are widespreading and highly evolutionarily conserved regulation mechanisms of the eukaryotic cell. MAPKs include extracellular signal–regulated kinase (ERK1/2), c-Jun N-terminal kinase (JNK1/2/3), and p38 kinase (p38αβγδ).63,64 They orchestrate the regulation of gene transcription, protein biosynthesis, cell cycle control, differentiation, and apoptosis.65 Loss of MAPK signaling regulation is implicated in diseases and cancer affecting the brain and the immune system.66,67
The ERKs were the first identified mammalian MAPKs. In many cases, the ERKs can be triggered by inflammatory cytokines including the TNF family. These mechanisms of ERK activation play important roles in inflammation and innate immunity.65
The JNKs are multifunctional signaling molecules that are activated in response to a wide range of inflammatory mediators and cellular stresses. JNKs regulate various processes, such as neuroinflammation, neuronal death, memory formation, brain development, and repair. These findings suggest that the JNKs play a role in Tau pathology and cognitive deficits68 and regulate soluble Aβ oligomers in AD mouse model.69 Activation of JNK in the brain triggered by intracellular Aβ accumulation leads to neuronal death in AD animal models and patients.70 The generation of proinflammatory cytokine induced by Aβ42 might be related to the toll-like receptor 2–dependent JNK/NF-κB signal pathway, at least in part. Blockade of this pathway could be beneficial in the AD pathogenesis.71
The first p38 MAPK isoform, α isoform, isolated from extracts of endotoxin-treated cells, could play multiple roles in AD pathophysiology. Microglial p38 MAPK leads to the inflammation of the AD brains, and astrocytes p38 MAPK modulates the excitotoxicity and expands the inflammatory response, while the neuronal p38 MAPK contributes to depression of synaptic plasticity, Tau phosphorylation, and neurofibrillary tangle (NFT) formation.72 Some effects on neuroinflammatory processes are summarized in Table 1.
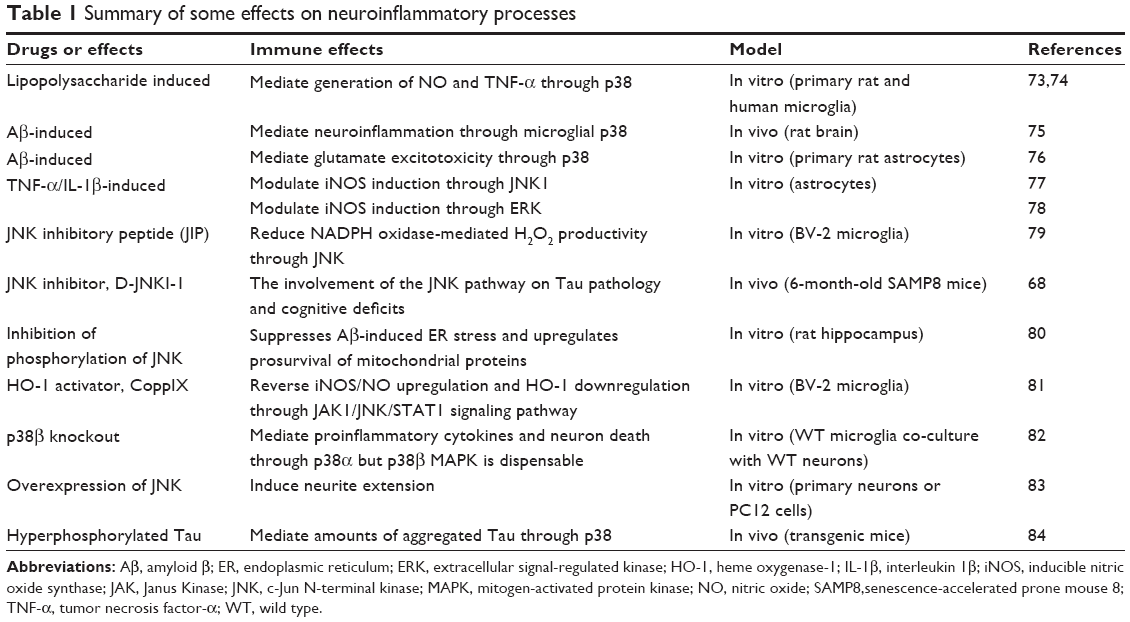 | Table 1 Summary of some effects on neuroinflammatory processes |
Among the effects, the MAPK signaling pathways play a pivotal role in lipopolysaccharide-, cytokine-, or Aβ-induced neuroinflammation and highlight the evidence of the potential inhibition of neuroinflammation through MAPK signaling pathways. Targeting this single kinase might have the potential to change the disease process. However, there are two unsolved problems: first, a single kinase inhibitor might not provide therapeutic efficacy, and whether three kinase common inhibitors possess the ability to have better effects is still unknown; second, the same potential drug targets the different MAPK signaling pathways in vivo and in vitro. For example, potential drug epigallocatechin gallate has different targets, mediated inflammation via ERK and NF-κB in vivo,1,85 but via NF-κB, p38, and JNK inhibition in vitro (U373MG).86 The same potential drug targets the different MAPK signaling pathways in vivo and in vitro. The pharmacodynamics of potential drugs is still missing, and clinical application of drugs targeting the MAPK in the brain still has a long way to go.
Besides the MAPK pathways, various transcription factors, including NF-κB87–90 and PPAR-γ91,92 have been shown to be involved in inflammatory responses in astrocytes and microglia.
Role of NF-κB
The NF-κB pathway was discovered in 1986 as a modulator of transcription in the light chain of B lymphocyte immunoglobulins.93 NF-κB is a redox transcription factor located within the cytoplasm and responsible for regulation in cytokine production. It is a dimer composed of members of the Rel family transcription factors: RelA (p65), RelB, c-Rel, p52, and p50. Normally, NF-κB is kept inactive by interaction with its inhibitors, which are family members of the inhibitor κB (IκB) protein (IκBα, IκBβ, IκBγ, IκBε, IκBδ, and Bcl-3) in the cytoplasm. IκB kinases (IKKs), components of a serine–threonine kinase complex, catalyze IκB phosphorylation. It is composed of three elements: a regulatory subunit, NEMO (also called IKK-γ), and two catalytic subunits, IKK-α and IKK-β.94,95 When the pathway is activated, phosphorylated NF-κB dimers are released and translocated to the nucleus, bind to the DNA sequences κB, and induce transcription of target genes. The effects caused by NF-κB activity are usually based on the control of neurite outgrowth, synaptic plasticity, and neuronal apoptosis in the CNS.95 In terms of neuroinflammation, NF-κB activation has been shown to mediate cytokine expression87,88 and inducible nitric oxide synthase (iNOS) induction and nitric oxide production89 in microglia. Furthermore, its activation is also involved in COX-2 expression in activated astrocytes,90 a molecule that mediates the formation of prostaglandin and seems to play a significant role in the processes of neuroinflammation. It has also been suggested that NF-κB plays an important role in sustaining the vicious cycle of inflammatory response and in neuroglial interactions in neurons and astrocytes.96
Several molecules have the ability to activate NF-κB, including TNF-α,97 IL-1β,98 Aβ, and secreted APP.99,100 In turn, the NF-κB is involved in Aβ42 oligomer production.101,102 NF-κB signaling upregulates BACE1 gene expression and facilitates APP processing, and the levels of NF-κB/p65 and BACE1 in the brains of AD patients were significantly increased.103 In mice models of AD (Tg2576), the increase of NF-κB activity has been associated with neuronal apoptosis and the process of the disease.104 Using different NF-κB kinase modulators in pharmacological approach, the results indicate that NF-κB pathways are involved in Aβ production.105 In the promoter region of human BACE1, two functional NF-κB–binding elements were identified, and disruption of NF-κB/p65 decreased the expression of BACE1 gene in RelA(p65) knockout cells and expression of NF-κB/p65 upregulated BACE cleavage and Aβ production.103 Paris et al have also shown that different kinds of NF-κB inhibitors reduce Aβ generation in 7W CHO cells overexpressing AβPP.106 Moreover, nonsteroidal anti-inflammatory drugs such as indomethacin effectively decreased Aβ production through the inactivation of NF-κB activity.107 Now a new study suggests that the expression level of the NF-κB1 (p105/50) gene in peripheral mononuclear cells of patients with AD was significantly higher than adult age-matched controls; furthermore, various NF-κB target genes expression and NF-κB/p65 and NF-κB/p50 DNA-binding activity were also increased.108 So it may be a promising pathogenic research tool for early diagnosis of AD at the peripheral levels. Taking all these reports into consideration, NF-κB can be viewed as a potential therapeutic target for AD treatment by directly inhibiting Aβ production.
Role of PPAR-γ
The PPARs belong to the superfamily of nuclear hormone receptors, a ligand-inducible transcription factor. Its activities are regulated by lipid and steroids metabolites.109 At present, three different PPAR subtypes (PPAR-α, PPAR-β [PPAR-δ], and PPAR-γ) have been identified.110 PPARs bind to PPAR response elements in target DNA sequences as heterodimers with the retinoid X receptor, leading to the transcription of target genes. It has been reported that they control the expression of genes related to inflammatory responses and glucose and lipid homeostasis.111
Under physiological conditions, PPAR-γ is expressed at low levels in the CNS.112 However, in some pathological conditions, PPAR-γ expression and mRNA levels, rather than other subtypes, were shown to be elevated in AD patients.113 These findings suggested that PPAR-γ may play a regulatory role in pathophysiological features of AD. In addition, PPAR-γ activation in microglia decreased the expression of inflammatory cytokines, iNOS, and nitric oxide (NO) production.91 These latter effects resulting from the PPAR-γ activation suppress the expression of proinflammatory genes by inhibiting NF-κB–mediated inflammatory signaling at multiple sites.114 Besides microglia, emerging data indicate neurons and astrocytes as basic cell type targets for the beneficial effects of PPAR-γ ligands.92 PPAR-γ activation has been reported to promote neurogenesis, and agonists regulate the proliferation and differentiation of neuronal stem cell.115
PPAR-γ target drugs have been largely described to have neuroprotective effects in neuropathological conditions, including Aβ-induced neurodegeneration and neuroinflammation.116 For example, cannabidiol reduces neuroinflammation induced by Aβ and promotes hippocampal neurogenesis through PPAR-γ.117 PPAR-γ agonists could suppress Aβ-mediated microglia activation, inhibit inflammatory gene expression, alter Aβ homeostasis, exhibit neuroprotective effects,91,118–120 and protect neurons through modulation of the mitochondrial fusion–fission events,121 thereby ameliorating the pathogenesis and progression of AD. Pioglitazone treatment decreased BACE1 and lowered the plaque burden in an animal model of AD.122 It protects cortical neurons against inflammatory mediators by improving peroxisomal function123 and reduces the levels of brain Aβ42 in Tg2576 mice.122 Furthermore, PPAR-γ agonists could reduce Tau protein phosphorylation, thereby inhibiting the accumulation of NFT in AD.124
At present, the main evidence suggests that PPAR-γ play a critical role in regulating pathophysiological features of AD. But there is evidence revealing that PPAR-α/δ have been involved in the pathogenesis of neuroinflammation.125,126 PPAR-δ deficiency leads to cognitive impairment associated with enhanced astrogliosis, inflammation, and Tau hyperphosphorylation in the cortex.127 These two isoforms are attracting more and more attention.
Cytokines and neuroinflammation
Cytokines are small, nonstructural proteins secreted from various cell types with molecular weights ranging from 8,000 Da to 40,000 Da that play varied biological activities. Chronic neuroinflammation induced by cytokines released from activated microglia and astrocytes has been recognized as one of the major mechanisms of AD neuropathology. Key to this review, we introduce both anti- and pro-inflammatory responses mediated by cytokines. Among these factors, we shall focus on cytokines and their interaction with Aβ and AD pathologies.
Tumor necrosis factor-α
TNF-α is a pleiotropic cytokine regulating numerous physiological and pathological processes, including differentiation, inflammation, and cell death. TNF-α expression levels in the healthy brain are low under physiological conditions. TNF is thought to play a central role in inflammatory or disease states. More and more studies present TNF as a neuromodulator in the process of AD pathologies.128 The dysregulation of TNF-α production has been linked to AD.129 Compared to the age-matched healthy controls, serum TNF-α levels significantly increased in the early-onset AD patients (age of onset <65) and late-onset AD patients (age of onset >65) groups.130 It has been shown that TNF-α regulate AβPP in mouse model of AD131 and induce Aβ production by increasing the expression of BACE1132 and the activity of γ-secretase.133 TNF-α synthesis inhibitor (3,6′-dithiothalidomide) suppresses Aβ-induced microglial activation, neuronal degeneration, and memory dysfunction and attenuates inflammation markers and AD pathology in the setting of neuroinflammation and AD.134 However, there are some opposing results. In their study, Uslu et al observed a correlation of Aβ42 levels with mini-mental state examination scores and body mass indexes in AD patients, but not with IL-6 and TNF-α levels in serum.135 The relationship between TNF-α and AD needs urgent solution and further research.
Interleukin-1
IL-1 is a kind of inflammatory cytokine that is usually produced by microglia in the brain and seems to play a significant role in the AD pathogenesis.136 A 10-year follow-up study of 133 institutionalized elderly patients in a nursing home indicates that systemic inflammatory IL-1 during agitation stage are risk factors in the development of AD.137 A recent study suggests that the polymorphism of IL-1 gene clusters may play a relevant role in AD pathogenesis in Brazilians.138 IL-1β can promote Aβ production by modulating γ-secretase activity in neurons.133 Enhanced message translation of IL-1β promotes the increase of AβPP synthesis in astrocytes.139 Likewise, IL-1β increased the levels of soluble amyloid precursor protein (sAPP) in a dose-dependent manner in the culture medium of primary neurons.140 Some studies have also demonstrated that IL-1β induces Tau protein phosphorylation and triggers the formation of paired-helical filaments, thus aggregating into NFTs.141,142 Sustained IL-1β overexpression increases in Tau phosphorylation and exacerbates Tau pathology in an IL-1β overexpression (IL-1βXAT) AD mouse model.143 In response to Aβ deposition, IL-1β released from astrocytes may be an important trigger of Tau pathology in the AD brain.45 Chronic hippocampal IL-1β overexpression and peripheral and intracerebral injection of IL-1β directly also impair long-term memory.144,145 However, one study indicates that IL-1β may play a beneficial role in limiting AD pathology. In the study by Matousek et al, regardless of the duration of cytokine expression or age, IL-1β overexpression leads to plaque clearance, and sustained IL-1β did not cause overt apoptosis in the hippocampus of an APPswe/PS1dE9 AD mouse model.146
Above all, there is no doubt that IL-1β causes AD pathologies in neuroinflammation, but its mechanism remains unclear and some key points have to be solved.
Interleukin-6
IL-6 is a major cytokine in the CNS. It plays an important role in the brain.147 IL-6 gene maps to chromosome 7p21 and has been assumed to be a good candidate for AD genetic risk factor.148 Recent studies showed that IL-6 polymorphisms are associated with risk of AD.149,150 Through a 10-year follow-up study on 133 elderly patients, they also found that systemic inflammatory IL-6 during the agitation stage are risk factors in AD development.137 In AD brain, IL-6 stimulates and promotes the recruitment of microglia and astrocytes to release proinflammatory cytokines,151 and it also promotes the Tau phosphorylation in neurons.152 But a meta-analysis suggested that IL-6-174G/C polymorphism would be a protective factor for AD in Asians but not in Caucasians. It may contribute to a decreased risk of AD.153 Taking into account the multifactorial etiology of AD, more complete case–control studies of stratified analysis of different environmental exposure, ethnic background, or other risk factors should be carried out to evaluate possible roles of IL-6 in the AD pathogenesis in the future.
Interleukin-10
IL-10 is one of the main anti-inflammatory cytokines and plays an important role in cell survival and neuronal homeostasis. Preexposure of glial cells to IL-10 inhibits the production of proinflammatory cytokines induced by Aβ or lipopolysaccharide.154 However, the stimulation of IL-10 did not suppress Aβ deposition in the mouse brain.155 It is inefficient in Aβ42 degradation and could potentially increase the ratio of Aβ42/Aβ40 (Aβ42 promotes amyloid deposition but Aβ40 inhibits it) in Tg2576 mice.156 A recent study shows that IL-10 in the periphery obviously associated with brain atrophy in AD.157 The relationship between IL-10 and AD needs more compelling evidence.
Transforming growth factor-β
TGF-βs are pleiotropic cytokines produced by various cell types throughout the body. They play an important role in some biological effects, such as brain development, neuroendocrine roles, synaptogenesis, synaptic functions, adult neurogenesis, and neuroprotection.158 In the brains of AD patients, increased levels of TGF-β1 in plasma and cerebrospinal fluid were demonstrated.159,160 Furthermore, increased levels of TGF-β1 mRNA expression was found before the manifestation of obvious dementia in AD patients, suggesting that it is mobilized in the process of AD pathology.161 The levels of TGF-β1 expression correlate with the degree of cerebrovascular amyloid deposition in AD.162 In contrast, some scientists found decreased levels of TGF-β1 in AD patients compared with the control group in serum163 and plasma.164
Peripheral mononuclear phagocyte TGF-β signaling in deficient AD mice increased the accumulation of microglia around Aβ plaques and reduced AD-like pathology.165 Reduced TGF-β1 signaling contributes to microglial activation and ectopic cell-cycle reactivation in neurons; both lead to neurodegeneration in the AD brain.166 TGF-β secreted by IL-34–treated microglia enhances the neuroprotective property of microglia and suppresses their proliferation.167 TGF-β1 modulates the activation of microglia partially through the Smad pathway, leading to reduced cytotoxicity and increased Aβ clearance.168 However in a transgenic AD model mice, it was observed that TGF-β1 promotes amyloidogenesis and Aβ deposition.169 Chronic overexpression of TGF-β1 negatively impacts hippocampal structure and impairs hippocampus-dependent learning.170 Due to methodological problems of individual studies and several underpowered studies, the effects of TGF-βs in AD pathology remain controversial.
Aβ plaques and NFTs as causes and consequences of neuroinflammation
It is critical to note in this review that both Aβ plaques and NFTs may represent causes and consequences of neuroinflammation in AD.
Aβ peptides, the main constituents of amyloid plaques, are derived from the proteolytic cleavage of APP.171,172 Diverse lines of evidence suggest that Aβ and APP contribute to the pathogenesis of AD causally. The conversion from preclinical AD to clinical AD is not only associated with widespread Aβ plaque deposition and NFT pathology but also with the appearance of various soluble/dispersible Aβ aggregates in the neuropil.173 Aβ regulates synaptic and neuronal activities, and Aβ accumulation in the brain leads to an interesting combination of synaptic depression and aberrant network activity.174 For the neuroinflammation, Aβ plaques are frequently associated with the activation of microglia and astrocytes (see the section “Cell mediators”). Aβ induces the expression of inflammatory cytokines and inflammatory enzymes such as COX-2 and iNOS. Inflammatory cytokines (such as TNF-α and IL-1β), in turn, enhance APP production and the process of APP proteolytic cleavage to increase the production of Aβ42 peptide. Aβ is able to stimulate NF-κB and MAPK signaling pathways. Both these signaling pathways are associated with the transcription of inflammatory mediators (see the section “Intracellular signaling pathway”). Also NF-κB participates in Aβ production through regulation of APP and BACE and γ-secretases; NF-κB/p65 is closely related to BACE1 expression, cleavage and Aβ production.103,105 Aβ can disrupt gliotransmitter release and astrocytic calcium signaling, and alter synaptic plasticity, which are vital processes for astrocyte–neuron communication.175 So, Aβ can be viewed both as a cause and consequence of neuroinflammation in AD. However, Aβ has also shown some antioxidant and neuroprotective effects under certain experimental conditions.176 A recent work suggests that Aβ immunotherapy leads to downregulation of microglial activation and reduction of Aβ and Tau pathology.177 Joseph-Mathurin et al used old lemurs to study Aβ immunotherapy. Worsening of iron accumulation is the side effect of Aβ immunization treatment at prodromal stages of AD.178 Now there are a growing number of studies on Aβ immunotherapy, such as the active anti-Aβ vaccine (AN1792), bapineuzumab and solanezumab, but the side effects were obvious and the clinical trials were disappointing. Second-generation anti-Aβ immunotherapies (crenezumab and gantenerumab) and Aβ vaccines (CAD106, ACC-001, and Affitope AD02) are being tested in AD patients.179
NFTs are the major neuropathological hallmarks in the brains of AD patients. They form intracellularly, aggregated and reorganized through hyperphosphorylation and acetylation of Tau proteins.180–182 The microtubule-associated protein, Tau, plays an important role in the stabilization and assembly of microtubules, which are crucial for normal cellular morphology and trafficking. Its aggregation in AD leads to deficits through a loss-of-function mechanism.183 Synapse degeneration closely correlates with cognitive decline in AD, while soluble, oligomeric Tau, as a synaptotoxic species, may contribute to synapse degeneration.184 During the process of the AD, the mechanisms and the triggers of Tau proteins aggregating into NFTs are unclear, but neuroinflammation could play a role.185 The number of NFTs and the progression of neurodegeneration as well as dementia showed a significant positive correlation in AD,186 leading to a common assumption that NFTs are neurotoxic. The repeated immunization with phosphorylated-Tau peptides causes neuroinflammation in NFT and wild-type mice.187 Similar to Aβ, NFTs can also be viewed both as causes and consequences in the neuroinflammation of AD.
Thus, in line with the study by Anand et al the amyloid- and Tau-based therapeutics may be the main direction of AD research and therapy in the near future.188
Conclusion
In this review, we described the potential role of inflammation factors in the process of neuroinflammation associated with AD. Neuroinflammation is currently recognized as an important pathophysiological feature of this neurodegenerative disorder. At the same time, it seems that every factor that associates with neuroinflammation could be a target for AD treatment and every single factor represents an attractive therapeutic target. But which one is the main cause of the process? There is usually no simple solution to complex problems and AD is sadly a complex neurodegenerative disorder. To date, preventative and therapeutic researches on AD focus on a single target, and few drugs are available to effectively treat this disease. Combination therapies with drugs targeting different causal or modifying factors may be the most successful treatment strategies, such as interruption of deleterious inflammatory pathways and improved plaque clearance by activated microglia.
Epidemiological evidences suggest that long-term use of non-steroidal anti-inflammatory drugs has a sparing effect on AD, but it failed to slow the progression of disease or improve the symptoms in patients with early or moderate AD in randomized clinical trials. Possible reasons for the failure of anti-inflammatory drugs in clinical trials may be associated with the advanced state of the disease in patients and the dosing regimens of the trials. Currently, most of the available anti-inflammatory drugs, including non-steroidal anti-inflammatory drugs, are not really “anti-inflammatory”. They prevent the pro-inflammatory response, but do not trigger the anti-inflammatory response. Given that neuroinflammation is now being recognized as a complex process that has both beneficial and detrimental aspects, rather than suppressing inflammation, orchestrating specific elements of the neuroinflammation may be a more appropriate therapeutic objective.
Finally, it is essential to define early biomarkers that correctly indicate inflammation of the CNS for potential AD subtypes. Disappointments of anti-inflammation drugs in AD suggest that attention should be taken in the intervention strategy, which is likely a better way to prevent the incidence of AD. It is crucial to develop better diagnostics relying upon peripheral inflammatory biomarkers. For example, serum or plasma inflammatory cytokines closely related to the AD pathological process can be used as indicators of AD. Efforts to discover more sensitive and selective biomarkers should be continued and integrated with more innovative approaches to clinical trial design. Certainly, neuroinflammation remains a young discipline, and our basic understanding of the neuroinflammatory network into therapeutic interventions still has a long way to go.
Acknowledgment
We gratefully acknowledge research funding from the grand science and technology special project of Guangdong, People’s Republic of China (2011A080300004).
Author contributions
All authors contributed toward data analysis, drafting and revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Spencer JPE, Vafeiadou K, Williams RJ, Vauzour D. Neuroinflammation: modulation by flavonoids and mechanisms of action. Mol Aspects Med. 2012;33(1):83–97. | ||
Hensley K. Neuroinflammation in Alzheimer’s disease: mechanisms, pathologic consequences, and potential for therapeutic manipulation. J Alzheimers Dis. 2010;21(1):1–14. | ||
McGeer EG, McGeer PL. Neuroinflammation in Alzheimer’s disease and mild cognitive impairment: a field in its infancy. J Alzheimer Dis. 2010;19(1):355–361. | ||
Brown GC, Bal-Price A. Inflammatory neurodegeneration mediated by nitric oxide, glutamate, and mitochondria. Mol Neurobiol. 2003;27(3):325–355. | ||
Arima Y, Kamimura D, Sabharwal L, et al. Regulation of immune cell infiltration into the CNS by regional neural Inputs explained by the gate theory. Mediators Inflamm. 2013;2013:898165. | ||
Abbott NJ, Rönnbäck L, Hansson E. Astrocyte – endothelial interactions at the blood–brain barrier. Nat Rev Neurosci. 2006;7(1):41–53. | ||
Maeda T, Kanda T. Blood–brain barrier and blood-nerve barrier in neuroinflammatory diseases. Nippon Rinsho. 2013;71(5):789–794. | ||
Syvanen S, Eriksson J. Advances in PET imaging of P-glycoprotein function at the blood–brain barrier. ACS Chem Neurosci. 2013;4(2):225–237. | ||
Gheorghiu M, Enciu A-M, Popescu BO, Gheorghiu E. Functional and molecular characterization of the effect of amyloid-beta(42) on an in vitro epithelial barrier model. J Alzheimers Dis. 2014;38(4):787–798. | ||
Burgmans S, van de Haar HJ, Verhey FRJ, Backes WH. Amyloid-beta interacts with blood–brain barrier function in dementia: a systematic review. J Alzheimers Dis. 2013;35(4):859–873. | ||
Steinman L. Inflammatory cytokines at the summits of pathological signal cascades in brain diseases. Sci Signal. 2013;6(258):e3. | ||
Carrano A, Hoozemans JJM, van der Vies SM, van Horssen J, de Vries HE, Rozemuller AJM. Neuroinflammation and blood–brain barrier changes in capillary amyloid angiopathy. Neurodegene Dis. 2012;10(1–4):329–331. | ||
Rivest S, Lacroix S, Vallières L, Nadeau S, Zhang J, Laflamme N. How the blood talks to the brain parenchyma and the paraventricular nucleus of the hypothalamus during systemic inflammatory and infectious stimuli. Proc Soc Exp Biol Med. 2000;223(1):22–38. | ||
Takeda S, Sato N, Ikimura K, Nishino H, Rakugi H, Morishita R. Increased blood–brain barrier vulnerability to systemic inflammation in an Alzheimer disease mouse model. Neurobiol Aging. 2013;34(8):2064–2070. | ||
Mischelevich DJ, Inventor; MISCHELEVICH DJ, (MISC-Individual), assignee. Method for altering permeability of blood–brain barrier in patient for use in treatment of eg., Alzheimer disease, involves energizing transducer to deliver pulsed ultrasound energy to target in brain or spinal cord. US patent US2013079682-A1. 2013 Mar 28. | ||
Serrano-Pozo A, Mielke ML, Gómez-Isla T, et al. Reactive Glia not only associates with plaques but also parallels tangles in Alzheimer’s disease. Am J Pathol. 2011;179(3):1373–1384. | ||
Heneka MT, O’Banion MK. Inflammatory processes in Alzheimer’s disease. J Neuroimmunol. 2007;184(1):69–91. | ||
Czeh M, Gressens P, Kaindl AM. The yin and yang of microglia. Dev Neurosci. 2011;33(3–4):199–209. | ||
Ginhoux F, Lim S, Hoeffel G, Low D, Huber T. Origin and differentiation of microglia. Front Cell Neurosci. 2013;17:7. | ||
Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Mechanisms underlying inflammation in neurodegeneration. Cell. 2010;140(6):918–934. | ||
Greter M, Merad M. Regulation of microglia development and homeostasis. Glia. 2013;61(1):121–127. | ||
Block ML, Zecca L, Hong J-S. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8(1):57–69. | ||
Harry GJ, Kraft AD. Microglia in the developing brain: a potential target with lifetime effects. Neurotoxicology. 2012;33(2):191–206. | ||
Morales I, Jimenez JM, Mancilla M, Maccioni RB. Tau oligomers and fibrils induce activation of microglial cells. J Alzheimers Dis. 2013;37(4):849–856. | ||
Rodriguez JJ, Witton J, Olabarria M, Noristani HN, Verkhratsky A. Increase in the density of resting microglia precedes neuritic plaque formation and microglial activation in a transgenic model of Alzheimer’s disease. Cell Death Dis. 2010;1:e1. | ||
Griciuc A, Serrano-Pozo A, Parrado AR, et al. Alzheimer’s disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron. 2013;78(4):631–643. | ||
Scheffler K, Stenzel J, Krohn M, et al. Determination of spatial and temporal distribution of microglia by 230 nm-high-resolution, high-throughput automated analysis reveals different amyloid plaque populations in an APP/PS1 mouse model of Alzheimer’s disease. Curr Alzheimer Res. 2011;8(7):781–788. | ||
Du Yan S, Chen X, Fu J, et al. RAGE and amyloid-β peptide neurotoxicity in Alzheimer’s disease. Nature. 1996;382(6593):685–691. | ||
El Khoury J, Hickman S, Thomas C, Loike J, Silverstein S. Microglia, scavenger receptors, and the pathogenesis of Alzheimer’s disease. Neurobiol Aging. 1998;19(1):S81–S84. | ||
Leissring MA, Farris W, Chang AY, et al. Enhanced proteolysis of β-amyloid in APP transgenic mice prevents plaque formation, secondary pathology, and premature death. Neuron. 2003;40(6):1087–1093. | ||
Li W, Poteet E, Xie L, Liu R, Wen Y, Yang S-H. Regulation of matrix metalloproteinase 2 by oligomeric amyloid β protein. Brain Res. 2011;1387:141–148. | ||
Liu B, Hong J-S. Role of microglia in inflammation-mediated neurodegenerative diseases: mechanisms and strategies for therapeutic intervention. J Pharmacol Exp Ther. 2003;304(1):1–7. | ||
Naert G, Rivest S. The role of microglial cell subsets in Alzheimer’s disease. Curr Alzheimer Res. 2011;8(2):151–155. | ||
Hickman SE, Allison EK, El Khoury J. Microglial dysfunction and defective β-amyloid clearance pathways in aging Alzheimer’s disease mice. J Neurosci. 2008;28(33):8354–8360. | ||
Floden AM, Combs CK. Microglia demonstrate age-dependent interaction with amyloid-beta fibrils. J Alzheimers Dis. 2011;25(2):279–293. | ||
Krabbe G, Halle A, Matyash V, et al. Functional impairment of microglia coincides with beta-amyloid deposition in mice with Alzheimer-like pathology. PLoS One. 2013;8(4):e60921. | ||
Hanisch UK, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci. 2007;10(11):1387–1394. | ||
Heneka MT, Kummer MP, Stutz A, et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature. 2012;493(7434):674–678. | ||
Sherwood CC, Stimpson CD, Raghanti MA, et al. Evolution of increased glia – neuron ratios in the human frontal cortex. Proc Natl Acad Sci U S A. 2006;103(37):13606–13611. | ||
Chan WY, Kohsaka S, Rezaie P. The origin and cell lineage of microglia – new concepts. Brain Res Rev. 2007;53(2):344–354. | ||
Molofsky AV, Krencik R, Ullian EM, et al. Astrocytes and disease: a neurodevelopmental perspective. Genes Dev. 2012;26(9):891–907. | ||
Sun D, Jakobs TC. Structural remodeling of astrocytes in the injured CNS. Neuroscientist. 2012;18(6):567–588. | ||
Sidoryk-Wegrzynowicz M, Wegrzynowicz M, Lee E, Bowman AB, Aschner M. Role of astrocytes in brain function and disease. Toxicol Pathol. 2011;39(1):115–123. | ||
Carrero I, Gonzalo MR, Martin B, Sanz-Anquela JM, Arevalo-Serrano J, Gonzalo-Ruiz A. Oligomers of beta-amyloid protein (A beta 1–42) induce the activation of cyclooxygenase-2 in astrocytes via an interaction with interleukin-1beta, tumour necrosis factor-alpha, and a nuclear factor kappa-B mechanism in the rat brain. Exp Neurol. 2012;236(2):215–227. | ||
Medeiros R, LaFerla FM. Astrocytes: conductors of the Alzheimer disease neuroinflammatory symphony. Exp Neurol. 2013;239:133–138. | ||
Crowe E, Bhat R, Bitto A, et al. Astrocyte senescence as a component of Alzheimer’s disease. Exp Gerontol. 2013;48(7):695–696. | ||
Blasko I, Veerhuis R, Stampfer-Kountchev M, Saurwein-Teissl M, Eikelenboom P, Grubeck-Loebenstein B. Costimulatory effects of interferon-gamma and interleukin-1beta or tumor necrosis factor alpha on the synthesis of Abeta1–40 and Abeta1–42 by human astrocytes. Neurobiol Dis. 2000;7(6 pt B):682–689. | ||
Yu Y, He J, Zhang Y, et al. Increased hippocampal neurogenesis in the progressive stage of Alzheimer’s disease phenotype in an APP/PS1 double transgenic mouse model. Hippocampus. 2009;19(12):1247–1253. | ||
Tang BL. Neuronal protein trafficking associated with Alzheimer disease: from APP and BACE1 to glutamate receptors. Cell Adh Migr. 2009;3(1):118–128. | ||
Hartlage-Rübsamen M, Zeitschel U, Apelt J, et al. Astrocytic expression of the Alzheimer’s disease β-secretase (BACE1) is stimulus-dependent. Glia. 2003;41(2):169–179. | ||
Roßner S, Lange-Dohna C, Zeitschel U, Perez-Polo JR. Alzheimer’s disease β-secretase BACE1 is not a neuron-specific enzyme. J Neurochem. 2005;92(2):226–234. | ||
Weggen S, Diehlmann A, Buslei R, Beyreuther K, Bayer T. Prominent expression of presenilin-1 in senile plaques and reactive astrocytes in Alzheimer’s disease brain. Neuroreport. 1998;9(14):3279–3283. | ||
Zhao J, O’Connor T, Vassar R. The contribution of activated astrocytes to A beta production: implications for Alzheimer’s disease pathogenesis. J Neuroinflammation. 2011;2:8. | ||
Miners JS, Van Helmond Z, Kehoe PG, Love S. Changes with age in the activities of β-secretase and the Aβ-degrading enzymes neprilysin, insulin-degrading enzyme and angiotensin-converting enzyme. Brain Pathol. 2010;20(4):794–802. | ||
Apelt J, Ach K, Schliebs R. Aging-related down-regulation of neprilysin, a putative β-amyloid-degrading enzyme, in transgenic Tg2576 Alzheimer-like mouse brain is accompanied by an astroglial upregulation in the vicinity of β-amyloid plaques. Neurosci Lett. 2003;339(3):183–186. | ||
Rossello XS, Igbavboa U, Weisman GA, Sun GY, Wood WG. AP-2 beta regulates amyloid beta-protein stimulation of apolipoprotein E transcription in astrocytes. Brain Res. 2012;1444:87–95. | ||
Rossi D, Volterra A. Astrocytic dysfunction: insights on the role in neurodegeneration. Brain Res Bull. 2009;80(4):224–232. | ||
Qin H, Benveniste EN. ELISA methodology to quantify astrocyte production of cytokines/chemokines in vitro. Methods Mol Biol. 2012;2012(814):235–249. | ||
Talantova M, Sanz-Blasco S, Zhang X, et al. A beta induces astrocytic glutamate release, extrasynaptic NMDA receptor activation, and synaptic loss. Proc Natl Acad Sci U S A. 2013;110(27):E2518–E2527. | ||
Bhat R, Crowe EP, Bitto A, et al. Astrocyte senescence as a component of Alzheimer’s disease. PLoS One. 2012;7(9):e45069. | ||
Thal DR. The role of astrocytes in amyloid beta-protein toxicity and clearance. Exp Neurol. 2012;236(1):1–5. | ||
Furman JL, Sama DM, Gant JC, et al. Targeting astrocytes ameliorates neurologic changes in a mouse model of Alzheimer’s disease. J Neurosci. 2012;32(46):16129–16140. | ||
Krishna M, Narang H. The complexity of mitogen-activated protein kinases (MAPKs) made simple. Cell Mol Life Sci. 2008;65(22):3525–3544. | ||
Wang X, Tournier C. Regulation of cellular functions by the ERK5 signalling pathway. Cell Signal. 2006;18(6):753–760. | ||
Kyriakis JM, Avruch J. Mammalian MAPK signal transduction pathways activated by stress and inflammation: a 10-year update. Physiol Rev. 2012;92(2):689–737. | ||
Gerits N, Kostenko S, Moens U. In vivo functions of mitogen-activated protein kinases: conclusions from knock-in and knock-out mice. Transgenic Res. 2007;16(3):281–314. | ||
Wagner EF, Nebreda ÁR. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat Rev Cancer. 2009;9(8):537–549. | ||
Orejana L, Barros-Minones L, Aguirre N, Puerta E. Implication of JNK pathway on tau pathology and cognitive decline in a senescence-accelerated mouse model. Exp Gerontol. 2013;48(6):565–571. | ||
Sclip A, Antoniou X, Colombo A, et al. c-Jun N-terminal kinase regulates soluble A beta oligomers and cognitive impairment in AD mouse model. J Biol Chem. 2011;286(51):43871–43880. | ||
Shoji M, Iwakami N, Takeuchi S, et al. JNK activation is associated with intracellular β-amyloid accumulation. Mol Brain Res. 2000;85(1):221–233. | ||
Lin W, Ding M, Xue J, Leng W. The role of TLR2/JNK/NF-kappa B pathway in amyloid beta peptide-induced inflammatory response in mouse NG108-15 neural cells. Int Immunopharmacol. 2013;17(3):880–884. | ||
Munoz L, Ammit AJ. Targeting p38 MAPK pathway for the treatment of Alzheimer’s disease. Neuropharmacology. 2010;58(3):561–568. | ||
Lee YB, Schrader JW, Kim SU. p38 MAP kinase regulates TNF-α production in human astrocytes and microglia by multiple mechanisms. Cytokine. 2000;12(7):874–880. | ||
Xing B, Xin T, Hunter RL, Bing G. Pioglitazone inhibition of lipopolysaccharide-induced nitric oxide synthase is associated with altered activity of p38 MAP kinase and PI3K/Akt. J Neuroinflammation. 2008;5(4):1–11. | ||
Giovannini MG, Scali C, Prosperi C, et al. β-amyloid-induced inflammation and cholinergic hypofunction in the rat brain in vivo: involvement of the p38MAPK pathway. Neurobiol Dis. 2002;11(2):257–274. | ||
Matos M, Augusto E, Oliveira C, Agostinho P. Amyloid-beta peptide decreases glutamate uptake in cultured astrocytes: involvement of oxidative stress and mitogen-activated protein kinase cascades. Neuroscience. 2008;156(4):898–910. | ||
Pawate S, Bhat NR. C-Jun N-terminal kinase (JNK) regulation of iNOS expression in glial cells: predominant role of JNK1 isoform. Antioxid Redox Signal. 2006;8(5–6):903–909. | ||
Marcus JS, Karackattu SL, Fleegal MA, Sumners C. Cytokine-stimulated inducible nitric oxide synthase expression in astroglia: role of Erk mitogen-activated protein kinase and NF-κB. Glia. 2003;41(2):152–160. | ||
Han JE, Choi JW. Control of JNK for an activation of NADPH oxidase in LPS-stimulated BV2 microglia. Arch Pharm Res. 2012;35(4):709–715. | ||
Yenki P, Khodagholi F, Shaerzadeh F. Inhibition of phosphorylation of JNK suppresses A beta-induced ER stress and upregulates prosurvival mitochondrial proteins in rat hippocampus. J Mol Neurosci. 2013;49(2):262–269. | ||
Lu D-Y, Leung Y-M, Su K-P. Interferon-alpha induces nitric oxide synthase expression and haem oxygenase-1 down-regulation in microglia: implications of cellular mechanism of IFN-alpha-induced depression. Int J Neuropsychopharmacol. 2013;16(2):433–444. | ||
Xing B, Bachstetter AD, Van Eldik LJ. Deficiency in p38beta MAPK fails to inhibit cytokine production or protect neurons against inflammatory insult in in vitro and in vivo mouse models. PloS One. 2013;8(2):e56852–e56852. | ||
Chang L, Jones Y, Ellisman MH, Goldstein LS, Karin M. JNK1 is required for maintenance of neuronal microtubules and controls phosphorylation of microtubule-associated proteins. Dev Cell. 2003;4(4):521–533. | ||
Kelleher I, Garwood C, Hanger DP, Anderton BH, Noble W. Kinase activities increase during the development of tauopathy in htau mice. J Neurochem. 2007;103(6):2256–2267. | ||
Lee JW, Lee YK, Ban JO, et al. Green tea (-)-epigallocatechin-3-gallate inhibits beta-amyloid-induced cognitive dysfunction through modification of secretase activity via inhibition of ERK and NF-kappa B pathways in mice. J Nutr. 2009;139(10):1987–1993. | ||
Kim SJ, Jeong HJ, Lee KM, et al. Epigallocatechin-3-gallate suppresses NF-kappa B activation and phosphorylation of p38 MAPK and JNK in human astrocytoma U373MG cells. J Nutr Biochem. 2007;18(9):587–596. | ||
Jana M, Dasgupta S, Liu X, Pahan K. Regulation of tumor necrosis factor-α expression by CD40 ligation in BV-2 microglial cells. J Neurochem. 2002;80(1):197–206. | ||
Nakajima K, Matsushita Y, Tohyama Y, Kohsaka S, Kurihara T. Differential suppression of endotoxin-inducible inflammatory cytokines by nuclear factor kappa B (NFκB) inhibitor in rat microglia. Neurosci Lett. 2006;401(3):199–202. | ||
Bhat NR, Feinstein DL, Shen Q, Bhat AN. p38 MAPK-mediated transcriptional activation of inducible nitric-oxide synthase in glial cells roles of nuclear factors, nuclear factor κB, cAMP response element-binding protein, CCAAT/enhance-binding protein-β, and activating transcription factor-2. J Biol Chem. 2002;277(33):29584–29592. | ||
Dai Y-Q, Jin D-Z, Zhu X-Z, Lei D-L. Triptolide inhibits COX-2 expression via NF-kappa B pathway in astrocytes. Neurosci Res. 2006;55(2):154–160. | ||
Combs CK, Johnson DE, Karlo JC, Cannady SB, Landreth GE. Inflammatory mechanisms in Alzheimer’s disease: inhibition of β-amyloid-stimulated proinflammatory responses and neurotoxicity by PPARγ agonists. J Neurosci. 2000;20(2):558–567. | ||
Landreth G, Jiang Q, Mandrekar S, Heneka M. PPARγ agonists as therapeutics for the treatment of Alzheimer’s disease. Neurotherapeutics. 2008;5(3):481–489. | ||
Sen R, Baltimore D. Multiple nuclear factors interact with the immunoglobulin enhancer sequences. Cell. 1986;46(5):705–716. | ||
Hayden MS, Ghosh S. Shared principles in NF-κB signaling. Cell. 2008;132(3):344–362. | ||
Mincheva-Tasheva S, Soler RM. NF-κB signaling pathways role in nervous system physiology and pathology. Neuroscientist. 2013;19(2):175–194. | ||
Pugazhenthi S, Zhang Y, Bouchard R, Mahaffey G. Induction of an inflammatory loop by interleukin-1 beta and tumor necrosis factor-alpha involves NF-kappa B and STAT-1 in differentiated human neuroprogenitor cells. PLoS One. 2013;8(7):e69585. | ||
Mir M, Tolosa L, Asensio VJ, Lladó J, Olmos G. Complementary roles of tumor necrosis factor alpha and interferon gamma in inducible microglial nitric oxide generation. J Neuroimmunol. 2008;204(1):101–109. | ||
Koo JW, Russo SJ, Ferguson D, Nestler EJ, Duman RS. Nuclear factor-κB is a critical mediator of stress-impaired neurogenesis and depressive behavior. Proc Natl Acad Sci U S A. 2010;107(6):2669–2674. | ||
Yamamoto Y, Gaynor RB. Therapeutic potential of inhibition of the NF-κB pathway in the treatment of inflammation and cancer. J Clin Invest. 2001;107(2):135–142. | ||
Barger SW, Mattson MP. Induction of neuroprotective κB-dependent transcription by secreted forms of the Alzheimer’s β-amyloid precursor. Mol Brain Res. 1996;40(1):116–126. | ||
Buggia-Prevot V, Sevalle J, Rossner S, Checler F. NFκB-dependent control of BACE1 promoter transactivation by Aβ42. J Biol Chem. 2008;283(15):10037–10047. | ||
Valerio A, Boroni F, Benarese M, et al. NF-κB pathway: a target for preventing β-amyloid (Aβ)-induced neuronal damage and Aβ42 production. Eur J Neurosci. 2006;23(7):1711–1720. | ||
Chen CH, Zhou W, Liu S, et al. Increased NF-kappa B signalling up-regulates BACE1 expression and its therapeutic potential in Alzheimer’s disease. Int J Neuropsychopharmacol. 2012;15(1):77–90. | ||
Niu YL, Zhang WJ, Wu P, et al. Expression of the apoptosis-related proteins caspase-3 and NF-κB in the hippocampus of Tg2576 mice. Neurosci Bull. 2010;26(1):37–46. | ||
Chami L, Buggia-Prévot V, Duplan E, et al. Nuclear factor-kappa B regulates beta APP and beta- and gamma-secretases differently at physiological and supraphysiological A beta concentrations. J Biol Chem. 2012;287(29):24573–24584. | ||
Paris D, Patel N, Quadros A, et al. Inhibition of Aβ production by NF-κB inhibitors. Neurosci Lett. 2007;415(1):11–16. | ||
Sung S, Yang H, Uryu K, et al. Modulation of nuclear factor-κB activity by indomethacin influences Aβ levels but not Aβ precursor protein metabolism in a model of Alzheimer’s disease. Am J Pathol. 2004;165(6):2197–2206. | ||
Ascolani A, Balestrieri E, Minutolo A, et al. Dysregulated NF-kappa B pathway in peripheral mononuclear cells of Alzheimer’s disease patients. Curr Alzheimer Res. 2012;9(1):128–137. | ||
Kummer MP, Heneka MT. PPARs in Alzheimer’s disease. PPAR Res. 2008;2008:403896. | ||
Kersten S. Peroxisome proliferator activated receptors and lipoprotein metabolism. PPAR Res. 2007;2008:132960. | ||
Bensinger SJ, Tontonoz P. Integration of metabolism and inflammation by lipid-activated nuclear receptors. Nature. 2008;454(7203):470–477. | ||
Bookout AL, Jeong Y, Downes M, Yu RT, Evans RM, Mangelsdorf DJ. Anatomical profiling of nuclear receptor expression reveals a hierarchical transcriptional network. Cell. 2006;126(4):789–799. | ||
de la Monte SM, Wands JR. Molecular indices of oxidative stress and mitochondrial dysfunction occur early and often progress with severity of Alzheimer’s disease. J Alzheimer Dis. 2006;9(2):167–181. | ||
Chen YC, Wu JS, Tsai HD, et al. Peroxisome proliferator-activated receptor gamma (PPAR-gamma) and neurodegenerative disorders. Mol Neurobiol. 2012;46(1):114–124. | ||
Wada K, Nakajima A, Katayama K, et al. Peroxisome proliferator-activated receptor γ-mediated regulation of neural stem cell proliferation and differentiation. J Biol Chem. 2006;281(18):12673–12681. | ||
Bright JJ, Kanakasabai S, Chearwae W, Chakraborty S. PPAR regulation of inflammatory signaling in CNS diseases. PPAR Res. 2008;2008:658520. | ||
Esposito G, Scuderi C, Valenza M, et al. Cannabidiol reduces Ab-induced neuroinflammation and promotes hippocampal neurogenesis through PPAR gamma involvement. PLoS One. 2011;6(12):e28668. | ||
Landreth G. Therapeutic use of agonists of the nuclear receptor PPAR in Alzheimers disease. Curr Alzheimer Res. 2007;4(2):159–164. | ||
Neumann KF, Rojo L, Navarrete LP, Farías G, Reyes P, Maccioni RB. Insulin resistance and Alzheimer’s disease: molecular links clinical implications. Curr Alzheimer Res. 2008;5(5):438–447. | ||
Jiang Q, Heneka M, Landreth GE. The role of peroxisome proliferator-activated receptor-γ PPARγ) in Alzheimer’s disease. CNS Drugs. 2008;22(1):1–14. | ||
Zolezzi JM, Silva-Alvarez C, Ordenes D, et al. Peroxisome proliferator-activated receptor (PPAR) gamma and PPAR alpha agonists modulate mitochondrial fusion-fission dynamics: relevance to reactive oxygen species (ROS)-related neurodegenerative disorders? PLoS One. 2013;8(5):e64019. | ||
Heneka MT, Sastre M, Dumitrescu-Ozimek L, et al. Acute treatment with the PPARγ agonist pioglitazone and ibuprofen reduces glial inflammation and Aβ1–42 levels in APPV717I transgenic mice. Brain. 2005;128(6):1442–1453. | ||
Gray E, Ginty M, Kemp K, Scolding N, Wilkins A. The PPAR-gamma agonist pioglitazone protects cortical neurons from inflammatory mediators via improvement in peroxisomal function. J Neuroinflammation. 2012;5:9. | ||
d’Abramo C, Ricciarelli R, Pronzato MA, Davies P. Troglitazone, a peroxisome proliferator-activated receptor-γ agonist, decreases tau phosphorylation in CHOtau4R cells. J Neurochem. 2006;98(4):1068–1077. | ||
Wang G, Namura S. Effects of chronic systemic treatment with peroxisome proliferator-activated receptor alpha activators on neuroinflammation induced by intracerebral injection of lipopolysaccharide in adult mice. Neurosci Res. 2011;70(2):230–237. | ||
Schnegg CI, Kooshki M, Hsu F-C, Sui G, Robbins ME. PPAR delta prevents radiation-induced proinflammatory responses in microglia via transrepression of NF-kappa B and inhibition of the PK alpha/MEK1/2/ERK1/2/AP-1 pathway. Free Radic Biol Med. 2012;52(9):1734–1743. | ||
Barroso E, del Valle J, Porquet D, et al. Tau hyperphosphorylation and increased BACE1 and RAGE levels in the cortex of PPAR beta/delta-null mice. Biochim Biophys Acta. 2013;1832(8):1241–1248. | ||
Sharma V, Thakur V, Singh SN, Guleria R. Tumor necrosis factor and Alzheimer’s disease: a cause and consequence relationship. Klinik Psikofarmakoloji B. 2012;22(1):86–97. | ||
Swardfager W, Lanctôt K, Rothenburg L, Wong A, Cappell J, Herrmann N. A meta-analysis of cytokines in Alzheimer’s disease. Biol Psychiatry. 2010;68(10):930–941. | ||
Gezen-Ak D, Dursun E, Hanağasi H, et al. BDNF, TNF alpha, HSP90, CFH, and IL-10 serum levels in patients with early or late onset Alzheimer’s disease or mild cognitive impairment. J Alzheimers Dis. 2013;37(1):185–195. | ||
McAlpine FE, Lee JK, Harms AS, et al. Inhibition of soluble TNF signaling in a mouse model of Alzheimer’s disease prevents pre-plaque amyloid-associated neuropathology. Neurobiol Dis. 2009;34(1):163–177. | ||
Yamamoto M, Kiyota T, Horiba M, et al. Interferon-γ and tumor necrosis factor-α regulate amyloid-β plaque deposition and β-secretase expression in Swedish mutant APP transgenic mice. Am J Pathol. 2007;170(2):680–692. | ||
Liao Y-F, Wang B-J, Cheng H-T, Kuo L-H, Wolfe MS. Tumor necrosis factor-α, interleukin-1β, and interferon-γ stimulate γ-secretase-mediated cleavage of amyloid precursor protein through a JNK-dependent MAPK pathway. J Biol Chem. 2004;279(47):49523–49532. | ||
Tweedie D, Ferguson RA, Fishman K, et al. Tumor necrosis factor-alpha synthesis inhibitor 3,6′-dithiothalidomide attenuates markers of inflammation, Alzheimer pathology and behavioral deficits in animal models of neuroinflammation and Alzheimer’s disease. J Neuroinflammation. 2012;29:9. | ||
Uslu S, Akarkarasu ZE, Ozbabalik D, et al. Levels of amyloid beta-42, interleukin-6 and tumor necrosis factor-alpha in Alzheimer’s disease and vascular dementia. Neurochem Res. 2012;37(7):1554–1559. | ||
Kornman KS. Interleukin 1 genetics, inflammatory mechanisms, and nutrigenetic opportunities to modulate diseases of aging. Am J Clin Nutr. 2006;83(2):475S–483S. | ||
Honma T, Hatta K, Hitomi Y, et al. Increased systemic inflammatory interleukin-1 beta and interleukin-6 during agitation as predictors of Alzheimer’s disease. Int J Geriatr Psychiatry. 2013;28(3):233–241. | ||
Payão SL, Gonçalves GM, de Labio RW, et al. Association of interleukin 1 beta polymorphisms and haplotypes with Alzheimer’s disease. J Neuroimmunol. 2012;247(1–2):59–62. | ||
Rogers JT, Leiter LM, McPhee J, et al. Translation of the Alzheimer amyloid precursor protein mRNA is up-regulated by interleukin-1 through 5′-untranslated region sequences. J Biol Chem. 1999;274(10):6421–6431. | ||
Liu L, Aboud O, Jones RA, Mrak RE, Griffin S, Barger SW. Apolipoprotein E expression is elevated by interleukin 1 and other interleukin 1-induced factors. J Neuroinflammation. 2011;8:175. | ||
Sheng JG, Jones RA, Zhou XQ, et al. Interleukin-1 promotion of MAPK-p38 overexpression in experimental animals and in Alzheimer’s disease: potential significance for tau protein phosphorylation. Neurochem Int. 2001;39(5):341–348. | ||
Griffin WST, Liu L, Li Y, Mrak RE, Barger SW. Interleukin-1 mediates Alzheimer and Lewy body pathologies. J Neuroinflammation. 2006;3(1):5. | ||
Ghosh S, Wu MD, Shaftel SS, et al. Sustained interleukin-1 beta overexpression exacerbates tau pathology despite reduced Amyloid burden in an Alzheimer’s mouse model. J Neurosci. 2013;33(11):5053–5064. | ||
Hein AM, Stutzman DL, Bland ST, et al. Prostaglandins are necessary and sufficient to induce contextual fear learning impairments after interleukin-1 beta injections into the dorsal hippocampus. Neuroscience. 2007;150(4):754–763. | ||
Hein AM, Stasko MR, Matousek SB, et al. Sustained hippocampal IL-1β overexpression impairs contextual and spatial memory in transgenic mice. Brain Behav Immun. 2010;24(2):243–253. | ||
Matousek SB, Ghosh S, Shaftel SS, Kyrkanides S, Olschowka JA, O’Banion MK. Chronic IL-1 beta-mediated neuroinflammation mitigates amyloid pathology in a mouse model of Alzheimer’s disease without inducing overt neurodegeneration. J Neuroimmune Pharmacol. 2012;7(1):156–164. | ||
Erta M, Quintana A, Hidalgo J. Interleukin-6, a major cytokine in the central nervous system. Int J Biol Sci. 2012;8(9):1254–1266. | ||
Serretti A, Olgiati P, De Ronchi D. Genetics of Alzheimer’s disease. A rapidly evolving field. J Alzheimer Dis. 2007;12(1):73–92. | ||
Chen SY, Chen TF, Lai LC, et al. Sequence variants of interleukin 6 (IL-6) are significantly associated with a decreased risk of late-onset Alzheimer’s disease. J Neuroinflammation. 2012;24:9. | ||
Qi H-P, Qu Z-Y, Duan S-R, Wei S-Q, Wen S-R, Bi S. IL-6-174 G/C and-572 C/G polymorphisms and risk of Alzheimer’s disease. PLoS One. 2012;7(6):e37858. | ||
Querfurth HW, LaFerla FM. Mechanisms of disease. N Engl J Med. 2010;362(4):329–344. | ||
Quintanilla RA, Orellana DI, González-Billault C, Maccioni RB. Interleukin-6 induces Alzheimer-type phosphorylation of tau protein by deregulating the cdk5/p35 pathway. Exp Cell Res. 2004;295(1):245–257. | ||
Hua Y, Guo X, Huang Q, Kong Y, Lu X. Association between interleukin-6-174G/C polymorphism and the risk of Alzheimer’s disease: a meta-analysis. Int J Neurosci. 2013;123(9):626–635. | ||
Szczepanik AM, Funes S, Petko W, Ringheim GE. IL-4, IL-10 and IL-13 modulate Aβ (1–42)-induced cytokine and chemokine production in primary murine microglia and a human monocyte cell line. J Neuroimmunol. 2001;113(1):49–62. | ||
Town T, Laouar Y, Pittenger C, et al. Blocking TGF-β–Smad2/3 innate immune signaling mitigates Alzheimer-like pathology. Nat Med. 2008;14(6):681–687. | ||
Kim J, Onstead L, Randle S, et al. Abeta40 inhibits amyloid deposition in vivo. J Neurosci. 2007;27(3):627–633. | ||
Leung R, Proitsi P, Simmons A, et al. Inflammatory proteins in plasma are associated with severity of Alzheimer’s disease. PLoS One. 2013;8(6):e64971. | ||
Dobolyi A, Vincze C, Pal G, Lovas G. The neuroprotective functions of transforming growth factor beta proteins. Int J Mol Sci. 2012;13(7):8219–8258. | ||
Rota E, Bellone G, Rocca P, Bergamasco B, Emanuelli G, Ferrero P. Increased intrathecal TGF-β1, but not IL-12, IFN-γ and IL-10 levels in Alzheimer’s disease patients. Neurol Sci. 2006;27(1):33–39. | ||
Motta M, Imbesi R, Di Rosa M, Stivala F, Malaguarnera L. Altered plasma cytokine levels in Alzheimer’s disease: correlation with the disease progression. Immunol Lett. 2007;114(1):46–51. | ||
Morimoto K, Horio J, Satoh H, et al. Expression profiles of cytokines in the brains of Alzheimer’s disease (AD) patients compared to the brains of non-demented patients with and without increasing AD pathology. J Alzheimer Dis. 2011;25(1):59–76. | ||
Lifshitz V, Weiss R, Benromano T, et al. Immunotherapy of cerebrovascular amyloidosis in a transgenic mouse model. Neurobiol Aging. 2012;33(2):e431–e432. | ||
Huang L, Jia J, Liu R. Decreased serum levels of the angiogenic factors VEGF and TGF-beta1 in Alzheimer’s disease and amnestic mild cognitive impairment. Neurosci Lett. 2013;550:60–63. | ||
Mocali A, Cedrola S, Della Malva N, et al. Increased plasma levels of soluble CD40, together with the decrease of TGFβ1, as possible differential markers of Alzheimer disease. Exp Gerontol. 2004;39(10):1555–1561. | ||
Khoury JE, Luster AD. Mechanisms of microglia accumulation in Alzheimer’s disease: therapeutic implications. Trends Pharmacol Sci. 2008;29(12):626–632. | ||
Caraci F, Spampinato S, Sortino MA, et al. Dysfunction of TGF-β1 signaling in Alzheimer’s disease: perspectives for neuroprotection. Cell Tissue Res. 2012;347(1):291–301. | ||
Ma D, Doi Y, Jin S, et al. TGF-beta induced by interleukin-34-stimulated microglia regulates microglial proliferation and attenuates oligomeric amyloid beta neurotoxicity. Neurosci Lett. 2012;529(1):86–91. | ||
Tichauer JE, von Bernhardi R. Transforming growth factor-beta stimulates beta amyloid uptake by microglia through Smad3-dependent mechanisms. J Neurosci Res. 2012;90(10):1970–1980. | ||
Weiss R, Lifshitz V, Frenkel D. TGF-β1 affects endothelial cell interaction with macrophages and T-cells leading to the development of cerebrovascular amyloidosis. Brain Behav Immun. 2011;25(5):1017–1024. | ||
Martinez-Canabal A, Wheeler AL, Sarkis D, et al. Chronic over-expression of TGF beta 1 alters hippocampal structure and causes learning deficits. Hippocampus. 2013;23(12):1198–1211. | ||
Citron M. Alzheimer’s disease: strategies for disease modification. Nat Rev Drug Discov. 2010;9(5):387–398. | ||
De Strooper B, Vassar R, Golde T. The secretases: enzymes with therapeutic potential in Alzheimer disease. Nat Rev Neurol. 2010;6(2):99–107. | ||
Thal DR, von Arnim C, Griffin WS, et al. Pathology of clinical and preclinical Alzheimer’s disease. Eur Arch Psychiatry Clin Neurosci. 2013;263:S137–S145. | ||
Palop JJ, Mucke L. Amyloid-[beta]-induced neuronal dysfunction in Alzheimer’s disease: from synapses toward neural networks. Nat Neurosci. 2010;13(7):812–818. | ||
Vincent AJ, Gasperini R, Foa L, Small DH. Astrocytes in Alzheimer’s disease: emerging roles in calcium dysregulation and synaptic plasticity. J Alzheimers Dis. 2010;22(3):699–714. | ||
Carrillo-Mora P, Luna R, Colin-Barenque L. Amyloid beta: multiple mechanisms of toxicity and only some protective effects? Oxid Med Cell Longev. 2014;2014:795375. | ||
Zotova E, Bharambe V, Cheaveau M, et al. Inflammatory components in human Alzheimer’s disease and after active amyloid-beta(42) immunization. Brain. 2013;136:2677–2696. | ||
Joseph-Mathurin N, Dorieux O, Trouche SG, et al. Amyloid beta immunization worsens iron deposits in the choroid plexus and cerebral microbleeds. Neurobiol Aging. 2013;34(11):2613–2622. | ||
Panza F, Logroscino G, Imbimbo BP, Solfrizzi V. Is there still any hope for amyloid-based immunotherapy for Alzheimer’s disease? Curr Opin Psychiatry. 2014;27(2):128–137. | ||
Cohen TJ, Guo JL, Hurtado DE, et al. The acetylation of tau inhibits its function and promotes pathological tau aggregation. Nat Commun. 2011;2:252. | ||
Iqbal K, Liu F, Gong C-X, Grundke-Iqbal I. Tau in Alzheimer disease and related tauopathies. Curr Alzheimer Res. 2010;7(8):656. | ||
Min SW, Cho SH, Zhou Y, et al. Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron. 2010;67(6):953–966. | ||
Morris M, Maeda S, Vossel K, Mucke L. The many faces of tau. Neuron. 2011;70(3):410–426. | ||
Pooler AM, Noble W, Hanger DP. A role for tau at the synapse in Alzheimer’s disease pathogenesis. Neuropharmacology. 2014;76:1–8. | ||
Arnaud L, Robakis NK, Figueiredo-Pereira ME. It may take inflammation, phosphorylation and ubiquitination to “tangle” in Alzheimer’s disease. Neurodegener Dis. 2006;3(6):313–319. | ||
Lee VM, Goedert M, Trojanowski JQ. Neurodegenerative tauopathies. Annu Rev Neurosci. 2001;24(1):1121–1159. | ||
Rozenstein-Tsalkovich L, Grigoriadis N, Lourbopoulos A, et al. Repeated immunization of mice with phosphorylated-tau peptides causes neuroinflammation. Exp Neurol. 2013;248:451–456. | ||
Anand R, Gill KD, Mahdi AA. Therapeutics of Alzheimer’s disease: past, present and future. Neuropharmacology. 2014;76:27–50. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.