")
Back to Journals » Neuropsychiatric Disease and Treatment » Volume 12
Neurobiology of hedonic tone: the relationship between treatment-resistant depression, attention-deficit hyperactivity disorder, and substance abuse
Received 2 May 2016
Accepted for publication 7 June 2016
Published 25 August 2016 Volume 2016:12 Pages 2149—2164
DOI https://doi.org/10.2147/NDT.S111818
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Roger Pinder
Tia Sternat,1,2 Martin A Katzman1–4
1START Clinic for Mood and Anxiety Disorders, 2Department of Psychology, Adler Graduate Professional School, Toronto, 3Division of Clinical Sciences, The Northern Ontario School of Medicine, 4Department of Psychology, Lakehead University, Thunder Bay, ON, Canada
Abstract: Anhedonia, defined as the state of reduced ability to experience feelings of pleasure, is one of the hallmarks of depression. Hedonic tone is the trait underlying one’s characteristic ability to feel pleasure. Low hedonic tone represents a reduced capacity to experience pleasure, thus increasing the likelihood of experiencing anhedonia. Low hedonic tone has been associated with several psychopathologies, including major depressive disorder (MDD), substance use, and attention-deficit hyperactivity disorder (ADHD). The main neural pathway that modulates emotional affect comprises the limbic–cortical–striatal–pallidal–thalamic circuits. The activity of various components of the limbic–cortical–striatal–pallidal–thalamic pathway is correlated with hedonic tone in healthy individuals and is altered in MDD. Dysfunction of these circuits has also been implicated in the relative ineffectiveness of selective serotonin reuptake inhibitors used to treat anxiety and depression in patients with low hedonic tone. Mood disorders such as MDD, ADHD, and substance abuse share low hedonic tone as well as altered activation of brain regions involved in reward processing and monoamine signaling as their features. Given the common features of these disorders, it is not surprising that they have high levels of comorbidities. The purpose of this article is to review the neurobiology of hedonic tone as it pertains to depression, ADHD, and the potential for substance abuse. We propose that, since low hedonic tone is a shared feature of MDD, ADHD, and substance abuse, evaluation of hedonic tone may become a diagnostic feature used to predict subtypes of MDD, such as treatment-resistant depression, as well as comorbidities of these disorders.
Keywords: dopamine, catecholamines, noradrenaline, anhedonia, treatment-resistance, prefrontal cortex
Introduction
Emotions, mood, and affect are the underlying phenomena of psychological states and disorders. Emotions represent complex psychological states that are elicited as a response to one’s external environment and involve the interplay between behavioral and physiological responses. Unlike emotions, which are specific and usually occur in response to a stimulus, mood is a general feeling of one’s psychological state that is longer lasting than specific emotions.1 Core affect is defined as a neurophysiological state that underlies the general feelings of “good” or “bad”, “drowsy” or “energized”.2 It has been suggested that the core affect shapes hedonic valence of one’s experiences.
Anhedonia, defined as the state of reduced ability to experience feelings of pleasure, is one of the hallmarks of depression.3,4 However, the term anhedonia is insufficient to summarize the intricate and multidimensional reward-associated deficits displayed by patients with neuropsychiatric disorders.2,5 Deficits in reward-related processing may present as loss of interest or pleasure and may impede an individual’s ability to engage in goal-directed behavior.2,5 These behaviors may include the lack of anticipation or prediction of expected rewards, lack of ability to evaluate the perceived values and costs associated with anticipated rewards, inability to gauge the amount of effort required to attain rewards, inability to evaluate whether the effort is sufficiently rewarded, and lack of motivation to execute actions required to attain rewards.5
If anhedonia is the state of reduced ability to feel pleasure, then hedonic tone, also referred to as hedonic capacity or hedonic responsiveness, is the trait or genetic predisposition underlying one’s baseline range and lifelong characteristic ability to feel pleasure. Low hedonic tone represents a reduced capacity to experience pleasure at any given time, thus increasing the likelihood of experiencing anhedonia.6,7
Low hedonic tone has been associated with several psychopathologies, including major depressive disorder (MDD), substance use, and schizophrenia,8,9 although variations in hedonic tone can also be observed in healthy individuals.10
It can be hypothesized that individuals suffering with genetically lower set point, or lower hedonic tone, will need to do more to feel neutral or euthymic, resulting in an increased need for stimulation.11 In part, this may manifest as seeking external stimulation (eg, dangerous or risky behavior and substance abuse) or internal stimulation (eg, fantasy) that will raise their hedonic tone.12 Regardless of the choice to externally or internally raise their hedonic tone, patients with this trait would consistently attempt to cope by finding ways to maximize pleasure and raise mood from their low baseline tone. Furthermore, when stimulation is absent, individuals with low hedonic tone would be hypothesized to experience a shift toward that individual’s more usual lower hedonic tone and therefore suffer a drop in their mood to their more common baseline dysphoric state (Figure 1).12
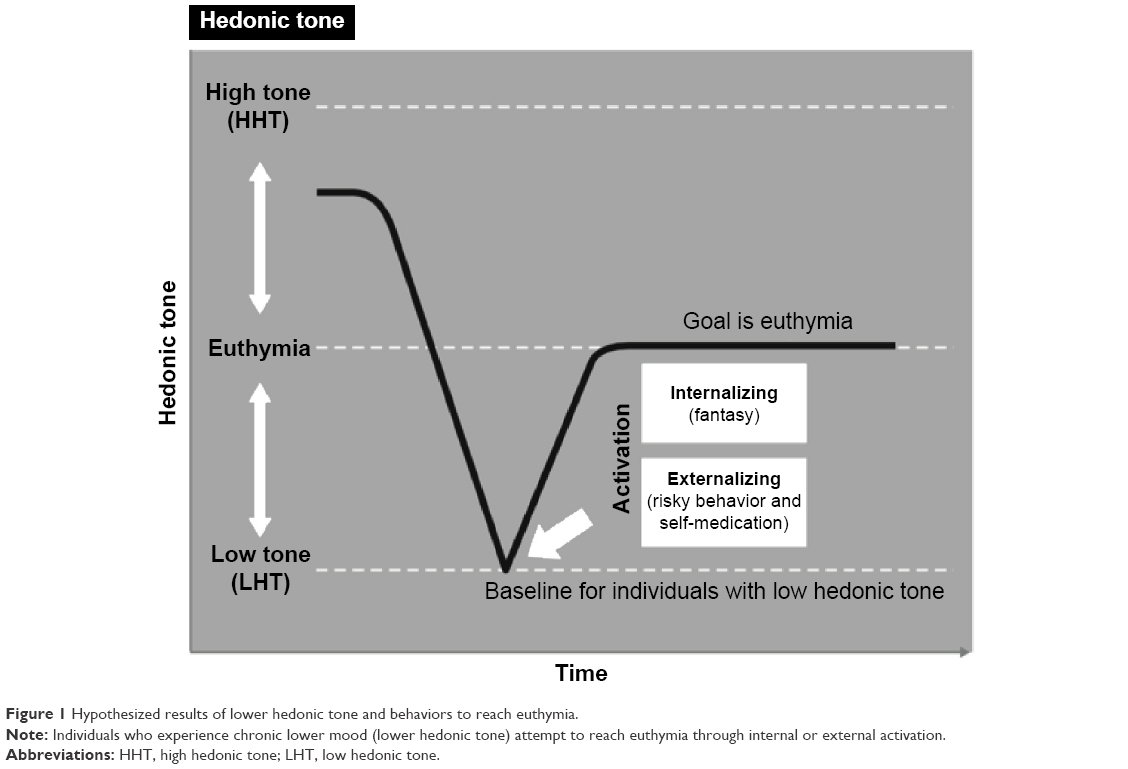 | Figure 1 Hypothesized results of lower hedonic tone and behaviors to reach euthymia. |
Thus, it is not surprising that a large body of evidence points to the involvement of the reward system, particularly the mesolimbic dopaminergic system, in mediating the degree of anhedonia at any given time6,7 as well as the risk of developing dysthymia and depression.13,14 Recent research into the neurobiology of anhedonia has focused on neurobiology of reward and motivation and has identified associated dopaminergic circuits as playing a key role in the maintenance of hedonic tone,2,4,15,16 specifically neural circuits that contain bottom-up and top-down projections into the prefrontal cortex (PFC), lateral habenula, and the ventral tegmental area (VTA) dopamine (DA) system.17 In association, specific deficits in a variety of PFC areas have been implicated in anhedonia (Figure 2).5
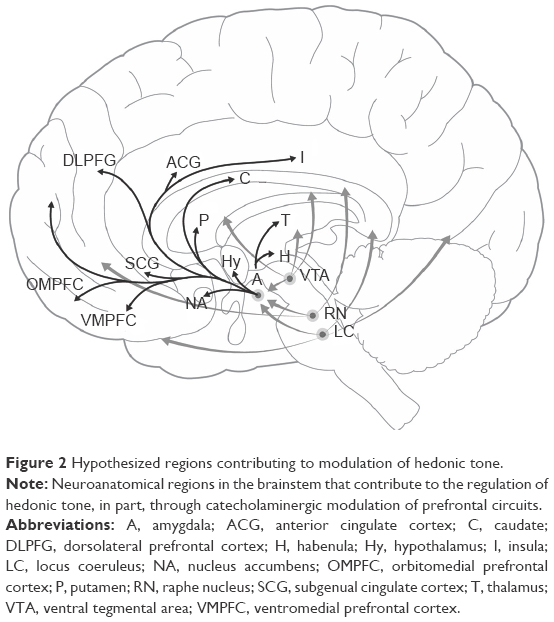 | Figure 2 Hypothesized regions contributing to modulation of hedonic tone. |
The purpose of this article is to review the neurobiology of hedonic tone as it pertains to depression, attention-deficit hyperactivity disorder (ADHD), and the potential for substance abuse. We propose the hypothesis that the experience of living for these individuals would best be understood through awareness of their low hedonic tone as they endeavor to attain a euthymic state by modulating the neurobiology of these areas. Furthermore, we propose that a dysfunction in the reward circuitry that comprises the ventromedial prefrontal cortex (VMPFC), subgenuate, and the nucleus accumbens (NA) is the underlying biological mechanism to this low hedonic tone and, therefore, the various mood disorders (Figure 2).
Methods
To identify studies relevant to our review, we performed a literature search on PubMed, PsychNET, and Medline databases until January 2016 using keywords “hedonic tone” AND “mood disorder” AND any of the following terms: “reward pathways”, “dopamine”, “neurobiology”, “depression”, “ADHD”, or “substance abuse”. Inclusion criteria for studies were 1) English language, 2) articles published in peer-reviewed journals, and 3) original research or review articles. Human and nonhuman primate studies were included in the neurobiology of mood and hedonic tone section. All other sections of this review included only human studies.
Neurobiology of mood disorders
Mood and emotional expression are regulated by a complex neural network, which involves the interplay between numerous regions of the central nervous system (CNS). Regions that modulate mood and emotions are closely intertwined with feelings of reward, pleasure, motivation, as well as our internal state and external environment. These regions and the associated neural pathways that regulate these functions span from the cerebral cortex to the brain stem and include the peripheral nervous system via the vagus nerve.
The main neural pathway that modulates emotional affect comprises the limbic–cortical–striatal–pallidal–thalamic (LCSPT) circuits, which consist of connections between the orbital and medial prefrontal cortex (OMPFC), ventromedial striatum, ventral pallidum, hippocampal subiculum, mediodorsal and midline thalamic nuclei, and amygdala.18 Through reciprocal connections with cortical regions that control higher cognitive functions as well as regions involved in the regulation of autonomic functions, including the periaqueductal gray and the hypothalamus, these circuits integrate higher cognitive functions with visceral information and external environmental conditions to affect mood and emotional states.19 Furthermore, the connections between the OMPFC and the dorsolateral prefrontal cortex (DLPFC) connect mood dysregulation with deficits in working memory and cognitive flexibility features that are dysfunctional in mood disorders (Figure 3).20–22
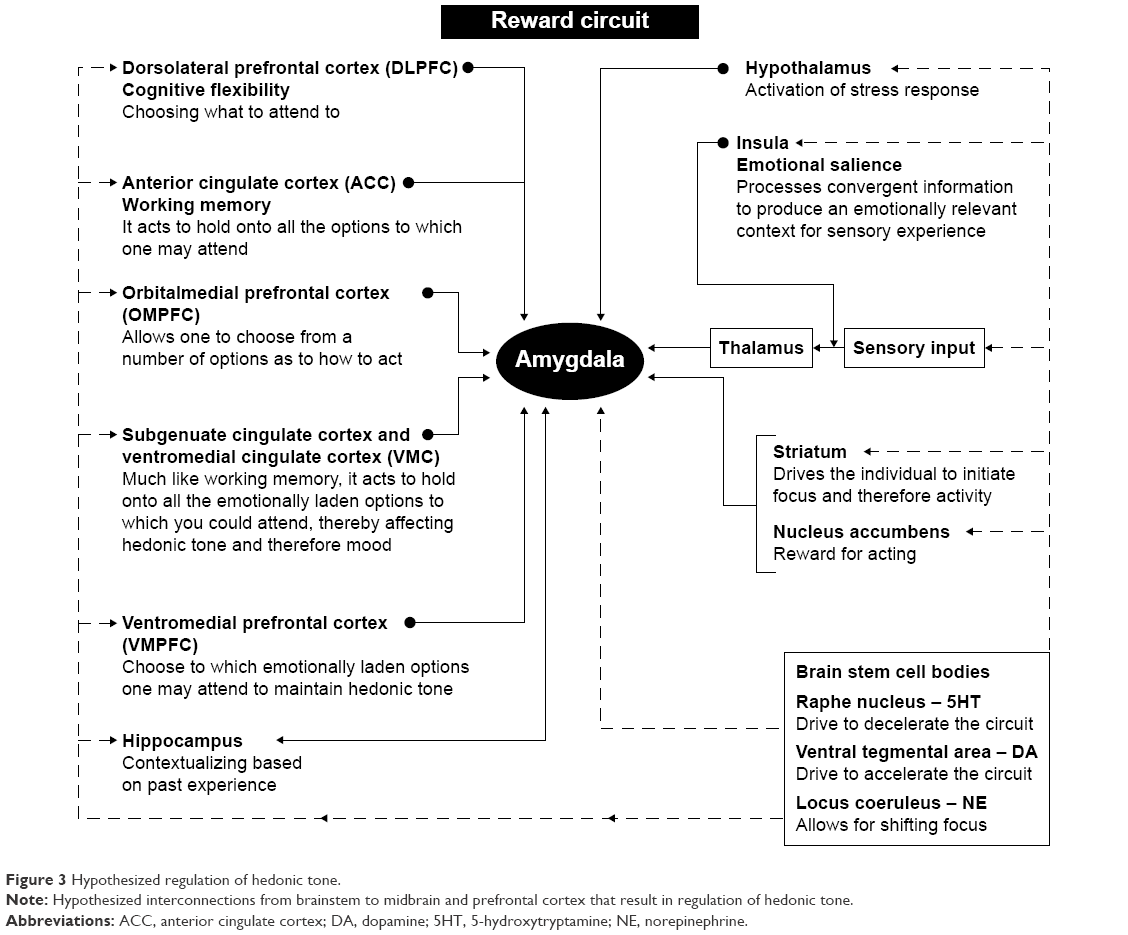 | Figure 3 Hypothesized regulation of hedonic tone. |
The OMPFC includes a significant portion of the cerebral cortex and is specifically involved in decision making and emotional- and reward-driven behaviors.18 Functional studies have identified two prefrontal networks within the OMPFC:23,24 the orbital network having been implicated in sensing food-related information and the anticipation of reward24 and the medial network projecting to visceral control centers in the hypothalamus and the periaqueductal gray25,26 and modulating visceral activity in response to affective stimuli.24 Thus, the orbital network is responsible for associating stimuli with a reward and specifically with the reward value of the stimulus.27 On the other hand, the medial network is involved with visceral responses to emotions, such as increased respiratory and heart rate, changes in blood pressure, and changes in the digestive activities (Figure 3).24
As well, the OMPFC has also been implicated in the inhibition of impulsive behavior,28 with reductions in dopaminergic input to the OMPFC having been shown to diminish the ability to inhibit impulsive behavior.29 Since impulsive behavior is one of the features of several mood disorders, dysfunction in dopaminergic inputs to OMPFC may represent a precursor to developing these conditions. Furthermore, in healthy individuals, activation of the medial network is associated with an increase in galvanic skin conductance, which represents an indirect measure of emotional arousal through increased activity in the sympathetic nervous system,30–32 further supporting the regions’ role in the regulation of emotional affect (Figure 3).
The amygdala as a part of the limbic system plays a crucial role in various forms of emotional learning, including fear conditioning,33 and mediates emotional responses to stress.34 Functional imaging studies have shown that the amygdala is activated by fear conditioning,35 and patients with lesions to the amygdala are unable to recognize fearful stimuli.36 The amygdala receives information about the external environment via the thalamus and the sensory cortex and is reciprocally connected with the OMPFC, the hippocampus, and sensory association areas. The amygdala also sends efferent projections to the striatum, both the dorsal (caudate nucleus and the putamen) and the ventral striatum, and the NA.36 Through projections to brainstem regions that control visceral functions, such as cardiovascular and respiratory functions, the amygdala plays a role in coordinating higher cognitive functions and emotions with the physiological state, including heart rate and respiratory rate observed during stress. Furthermore, increased reactivity of the amygdala to stressful stimuli has been implicated as a precursor to depression in adolescents.37
The amygdala, particularly the central nucleus, coordinates the fear response via connections to autonomic and cortical regions. Sensory stimuli, such as visual, auditory, and other stimuli, are perceived by the visual regions of the thalamus, namely, the lateral geniculate, which sends these messages to the central nucleus of the amygdala. Activation of this nucleus, in turn, coordinates the autonomic response to fear, which includes an increase in the respiratory rate via connections to the parabrachial nucleus, increased arousal via connections to the lateral hypothalamus, increased autonomic activation via projections to the paraventricular nucleus of the hypothalamus, increased catecholamine release via projections to the locus coeruleus, as well as activation of defensive behaviors via connections to the periaqueductal gray.38 In addition, the amygdala also has reciprocal connections with cortical regions involved in the processing and evaluation of sensory information. These regions integrate sensory information and decide whether fear is warranted.38 For example, the hippocampus is involved in contextualizing fear and can either increase or decrease fear reaction based on prior experiences and memories, whereas the medial PFC is involved in fear extinction.39,40 In individuals who have been conditioned to fear, or those with panic disorders, dysfunction in this circuitry leads to an inability to extinguish or contextualize fear.41 Furthermore, the central nucleus of the amygdala is connected to the insular cortex, which is involved in contextualizing sensory inputs of various modalities, including nociceptive, auditory, and visual.42–44 This notion is supported by the findings that lesions in the insular cortex are associated a reduced sensitivity to the pain of others (Figure 3).45
Emotional responses to stressful stimuli are partly mediated by the reciprocal connections of the amygdala, the thalamus, and the hippocampus.46 Whereas the dorsal part of the hippocampus is primarily involved in memory consolidation, the ventral hippocampus has been shown to modulate emotions and mood.46,47 Interestingly, neurons in the hippocampus of adults display a high degree of neuroplasticity and neurogenesis and are easily influenced by life experiences and medications.48
The basal ganglia, including the dorsal striatum (eg, caudate nucleus, putamen), the ventral striatum (eg, NA, olfactory tubercle, globus pallidus, ventral pallidum), the subthalamic nucleus, and the substantia nigra, are also closely interconnected with the PFC and the amygdala.49 The basal ganglia are involved in motor control, as well as motivation and reward processing.49 It has been demonstrated that in healthy individuals, volume of the pallidum50 and the caudate51 are correlated with anhedonia scores, suggesting that these structures may be involved in the regulation of hedonic tone. More specifically, the caudate nucleus is thought to be involved in evaluating the magnitude of a reward and in driving appropriate behaviors required to obtain that reward.52,53 Based on these functions, individuals with low activity in the caudate nucleus would have a reduced capacity to estimate the value of a reward or to initiate actions needed to obtain a reward. These are also the characteristics of low hedonic tone.12
The DLPFC circuit is involved in working memory and cognitive flexibility and has been linked to the pathogenesis of mood disorders (Figure 3).20,21 This circuit originates in the DLPFC and corresponds to Brodmann’s areas 9 and 10. The major efferent connections of DLPFC are composed of pyramidal neurons, which project to the dorsolateral caudate nucleus, which, in turn, projects to the globus pallidus and substantia nigra. These basal ganglia regions project to the ventral anterior and mediodorsal thalamic nuclei, which send afferent projections back to the DLPFC.21 Dysfunction of this circuit has been linked to impaired reasoning ability and cognitive inflexibility.21 Patients with DLPFC syndrome exhibit poor working memory and organizational abilities as well as inability to shift their attention between tasks.21 It has also been shown that depression is associated with hypoactivity in the DLPFC (Figure 3).22
Neural circuits regulating mood and emotional affect release a wide range of neurotransmitters and are also affected by circulating hormones, particularly those involved in the stress response.19 As is the case throughout the CNS, neuronal activity within LCSPT circuits is predominantly glutamatergic and is locally modulated through the gamma-aminobutyric acid (GABA) system.54 Glutamate is the main excitatory neurotransmitter in the CNS and activates all regions of the LCSPT circuits. Furthermore, disruptions in glutamate signaling have been implicated in disorders of reward processing, including substance abuse.55 On the other hand, GABA acts as the main inhibitory neurotransmitter/modulator of the glutaminergic synapses and is present in interneurons situated throughout and acts to specifically inhibit neuronal activity.56 In corticostriatal circuits, GABA neurons are responsible for the selection and inhibition of motivated behaviors.57 Although glutamate and GABA are the predominant neurotransmitters in the LCSPT circuits, their activity can be modulated by a variety of other neurotransmitters and neuromodulators. For example, endocannabinoids have been shown to inhibit the release of both glutamate and GABA in the VTA.58 Similarly, glutamate and GABA release can be inhibited by opioids in various regions of the CNS, including the regions involved in reward processing, such as the VTA, amygdala, and the hippocampus.58–60 Furthermore, the activity of glutamate and GABA-containing neurons can also be modulated by monoamines. In fact, serotonergic modulation of glutamate release has been implicated as one of the strategies for treating MDD.61
With respect to mood regulation, monoamine neurotransmitters (including serotonin, DA, norepinephrine (NE), and epinephrine) have been the main focus of research and have been the primary targets in pharmaceutical treatments of depression.19 The monoamine hypothesis of depression postulates that depression is caused by decreased modulating function of serotonin, NE, or DA in the brain.62 Nevertheless, this theory does not explain the apparent lack of efficacy in a relatively large subset of patients with treatment-resistant depression (TRD) who continue to suffer with residual symptoms, including anhedonia. Nonetheless, the extent to which monoamines influence neuronal activity is dependent on the expression level of the neurotransmitter itself as well as on the number of available receptors and transporters on the postsynaptic membrane. For example, serotonin can bind to 14 different receptor types,63–65 and the effect of serotonin on neurons can be region-specific.61 As such, one can imagine that this variability may explain the individual differential response to the various antidepressants. For example, it has been proposed that vortioxetine, which is described as a mixed multimodal antidepressant, likely acts as an atypical serotonin modulator, enhancing GABA activity in the striatum but reducing GABA activity in the PFC.61 Vortioxetine has also been shown to increase glutamate activity via antagonism of serotonin receptors 6 and 7 and agonism of serotonin receptor 1a.66 Given the complexity of neurotransmitter systems regulating mood and emotional states, it is not surprising that mood disorders resulting from a disrupted balance of these circuits can take on various forms and manifest as highly variable symptoms (Figure 3).67
While the role of monoamine antidepressants in modulation of reward circuits has been the most extensively investigated, recent studies have demonstrated that substances that modulate other neurotransmitter systems, particularly N-nitrosodimethylamine receptors (glutamate), endogenous opioids, and acetylcholine, also affect reward pathways and hedonic tone. It has been demonstrated that symptoms of depression can be relieved with ketamine, an N-nitrosodimethylamine receptor antagonist,68 scopolamine, a muscarinic cholinergic receptor antagonist,69 and buprenorphine, a partial mu opioid receptor agonist,70 and these pharmacological agents have also been shown to alter reward-processing circuits. A single infusion of ketamine was shown to reduce anhedonia levels in patients with TRD. The reduction in anhedonia was accompanied by an increase in glucose metabolism in the dorsal anterior cingulate cortex (ACC) and putamen,71 suggesting that antidepressant properties of ketamine are mediated by its actions on reward processing. Clinical response to scopolamine is correlated with a greater neural activity in response to happy versus sad faces in the subgenual anterior cingulate cortex (sgACC).72 Furthermore, opioid dependence is associated with a reduced hedonic response to positive stimuli and reduced neural activation in the rostral PFC and ventrolateral prefrontal cortex (VLPFC),73 and buprenorphine and samidorphan exert their antidepressant effects by affecting the levels of hedonic tone and altering the circuitry of the PFC. Taken together, these data indicate that nonmonoamine antidepressants, such as ketamine, scopolamine, and buprenorphine, induce antidepressant effects by restoring hedonic tone to control levels. A better understanding of the effects of these agents on neural circuits that regulate mood and hedonic tone is an important strategy for identifying novel effective treatments of depression.
Dysfunction in LCSPT circuits and the associated neurotransmitter systems have been implicated as playing a key role in MDD.19 Neuroimaging studies among depressed patients have demonstrated changes in the activity in LCSPT circuits in response to various treatments. For example, it has been shown that depression is associated with frontal hypometabolic activity accompanied by hypermetabolic activity in regions associated directly with the limbic system, including the VMPFC and sgACC.19,74–76 Furthermore, it has been shown that in depressed patients, the activity in the sgACC is positively correlated with the severity of depression in response to sad stimuli and negatively correlated in response to happy stimuli. At the end of the study, following various antidepressant treatments, patients who showed the greatest reduction in the activity in the sgACC in response to sad stimuli experienced the greatest reductions in Hamilton Rating Scale for Depression scores.77 For clarity in this review, it should be noted that the VMPFC overlaps with the region referred to as the ACC, particularly the pregenual and sgACC parts of the ACC. Therefore, the term “VMPFC” is used here for the region as a whole, and the term “sgACC” is used in describing studies that have focused on this particular area. The involvement of the PFC, the VMPFC, and the sgACC in depression is further highlighted by studies demonstrating normalization of functional activity to levels observed in nondepressed subjects following successful therapy, including treatment with antidepressants, deep brain stimulation (DBS), and vagus nerve stimulation (VNS) (Figure 2).
Selective serotonin reuptake inhibitors (SSRIs) fluoxetine and paroxetine have been shown to increase glucose metabolism in the PFC, particularly the dorsolateral, ventrolateral, and medial aspects, parietal cortex, and the sgACC.75,78 In addition, fluoxetine reduced glucose metabolism in the sgACC, hippocampus, insula, and pallidum.78 These findings suggest that dysfunction in serotonin reuptake in these regions accounts for symptoms of depression.
In patients with TRD, VNS has been shown to improve symptoms of depression.79,80 Imaging studies using either single photon emission computed tomography or blood oxygen level-dependent methods revealed that improvement in depression symptoms induced by VNS was associated with decreased activity in the sgACC, VMPFC, and ACC and increased activity of the superior temporal gyrus.81,82
DBS has also been shown to be useful in TRD with associated modulated activity of the sgACC and PFC in treatment-resistant patients. Mayberg et al31 investigated whether chronic DBS could modulate the activity of the sgACC over 6 months among patients with TRD, defined as failure to respond to a minimum of four different antidepressant treatments during the current major depressive episode lasting at least 1 year. Positron emission tomography measures of regional blood flow at baseline identified increased blood flow in the sgACC and decreased blood flow in the PFC in depressed patients compared to healthy controls. After 6 months of treatment with DBS, blood flow in the DLPFC and VLPFC among responders increased to levels observed in healthy controls, while blood flow in the sgACC decreased to levels lower than those measured in healthy controls.
Taken together, the results of these studies show that hypoactivity in the VLPFC and DLPFC and increased activity in the VMPFC and sgACC are common findings among patients with depression and that reversal of activity in these regions to levels observed among healthy individuals is associated with improvement of depressive symptoms. Furthermore, the activity of these regions can be modulated not only by antidepressants in the responsive population but also by VNS and DBS in those who do not respond to pharmaceutical treatment.
Neurobiology of hedonic tone
It can be hypothesized that hedonic tone is closely related to mood, reward, and motivation and is modulated by LCSPT. Imaging studies have identified components of the LCSPT pathway that are involved in the regulation of hedonic tone. These studies have shown that the activity of various components of the LCSPT pathway is correlated with hedonic tone in healthy individuals and is altered in MDD.
In healthy subjects, presentation of positive stimuli increases the activation of regions involved in reward processing, including the caudate, putamen, NA, basal forebrain, medial frontal region, ACC, inferior parietal area, right fusiform, and lingual gyrus.83–85 When asked to suppress a positive emotion, healthy subjects show activation of the right VLPFC.86 These studies show that these regions are involved in the perception of positive and rewarding stimuli.
Several studies have shown that anhedonia alters the activation of these regions, as it is negatively correlated with the activation of the NA, basal forebrain, and the hypothalamus51,84,87 and positively correlated with the activity in the VMPFC.51 It has also been shown that subjects with higher anhedonia scores have a decreased NA volume and a decreased resting activity in the rostral sgACC.87 These findings suggest that anhedonia may at least partly be due to the insufficient activation of neural circuits that regulate feelings of pleasure. We propose that continued low activity of these circuits, particularly in depressed individuals, would suggest chronic low hedonic tone, an increased likelihood of developing anhedonia, and an increased probability of TRD. Furthermore, when presented with positive stimuli, patients with MDD show attenuated activation of the ventral striatum, medial frontal cortex, and the NA83,85,88,89 and increased activation in the inferior frontal cortex, sgACC, thalamus, putamen, and the insula.85 As such, it has also been reported that depression is correlated with a difficulty in sustaining the connectivity between the NA and the DLPFC, as well as between the NA and the middle temporal gyrus,88,90 showing that depressive state is associated with an inability to activate regions responsible for feelings of reward.90,91
Further in support are data examining the CNS differences in activity of adult females with both ADHD and MDD in comparison with those with pure MDD.92 Gardner et al reported increased severity of depressive symptoms in patients with MDD + ADHD (vs MDD) in association with significantly enhanced activity in bilateral frontal regions (Brodmann areas 8, 9, 10, 32) in the “Depression + ADHD” subgroup compared to those in “the Depression group”. They also reported significantly decreased activity within the bilateral cerebellum in the “Depression + ADHD” subgroup, perhaps suggesting that chronic depression can be subtyped by the presence of ADHD and perhaps an associated low hedonic tone.92
Furthermore, studies have also shown that the dysfunction in the activity and connections of these circuits observed in depression can be regulated by antidepressant treatment. After treatment with fluoxetine or venlafaxine, depressed patients reported increases in positive affect; interestingly, those patients demonstrating the largest increases in positive affect also demonstrated the largest increases in frontal activity.88 Furthermore, depressed patients who did not exhibit positive emotions also showed decreased PFC activity, which changed with improvement in anhedonia in response to treatment with fluoxetine and venlafaxine.86 Thus, given the similarities between activation of these regions in response to positive experiences in healthy populations with anhedonia and in patients with MDD, it has been postulated that anhedonia, or hedonic tone, may be predictive of development of MDD. The results of these studies demonstrate that depressed patients show decreased activation in the VMPFC and NA and increased activation in the sgACC, compared to healthy controls. Normalization of activity in these regions to levels observed among healthy individuals is associated with improvement of anhedonia. Thus, it has been postulated that patients with low hedonic tone look for ways to raise their hedonic tone by manipulating the neurobiology of the mood system.12 Studies published in the literature showing changes in the activation of different regions are summarized in Table 1.
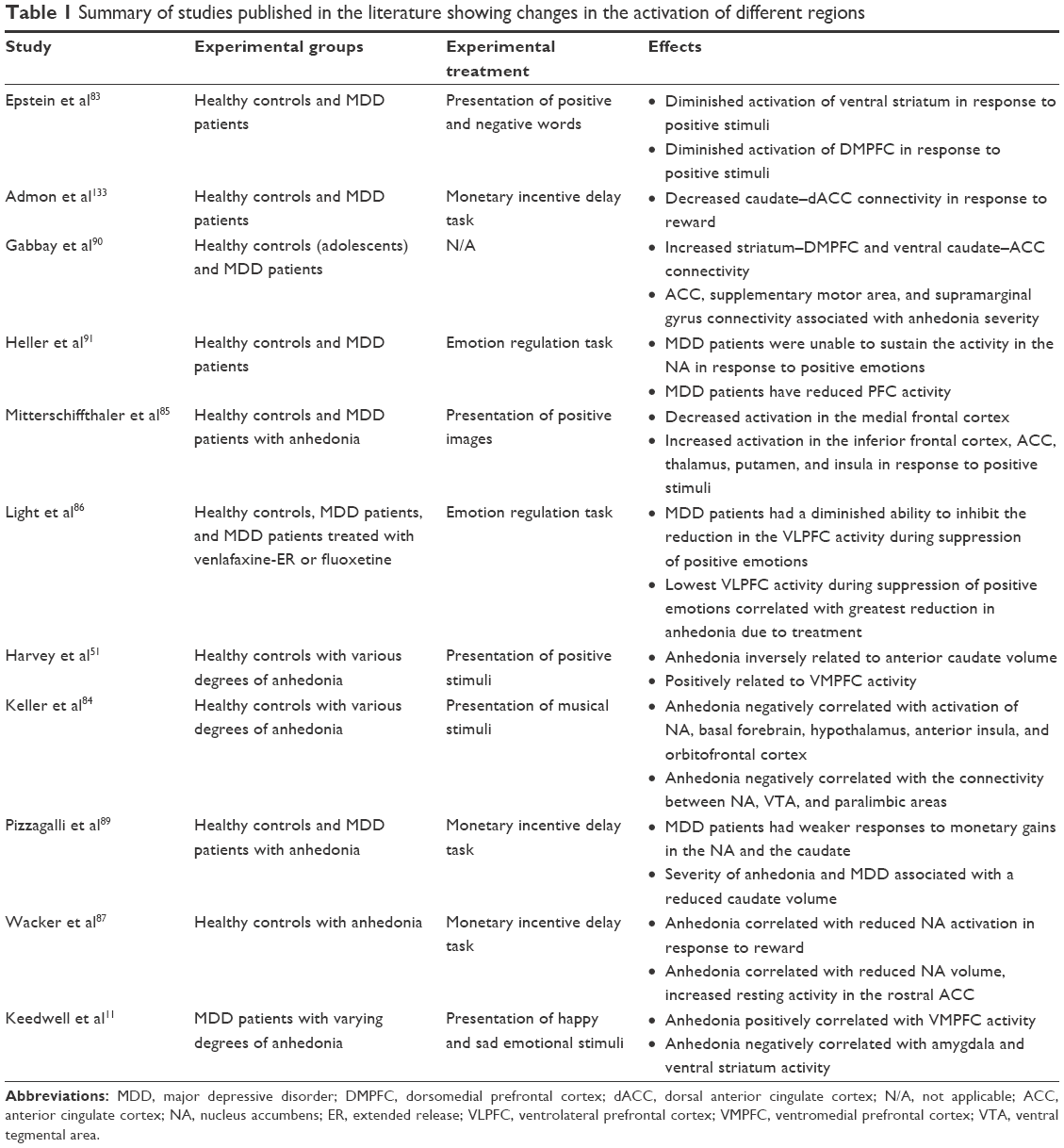 | Table 1 Summary of studies published in the literature showing changes in the activation of different regions |
Symptoms of low hedonic tone
Hedonic tone, also referred to as hedonic capacity or hedonic responsiveness, is the trait or genetic predisposition underlying one’s baseline range and lifelong ability to feel pleasure. High hedonic tone is associated with an increased likelihood of experiencing happiness, whereas low hedonic tone manifests as an increased risk and therefore a general feeling of sadness.12 Low hedonic tone is suggestive of a reduced lifetime capacity to experience euthymia and a lower likelihood of experiencing pleasure at any given time. Thus, this results in the increased likelihood of experiencing anhedonia, depression, and/or dysthymia.6,7 Individuals with low hedonic tone may be unable to anticipate expected reward, to ascertain the amount of effort required to attain the anticipated reward, and to engage in goal-directed behaviors required to attain a reward.5 In addition, it has been proposed that those with a low hedonic tone require higher volumes of positive experiences to raise their natural hedonic tone to a more euthymic tone, which is often accomplished through substance abuse or risky behaviors.12 This phenomenon is commonly seen in conditions of low hedonic tone and in particular in MDD and ADHD (Figure 1).9,12 Thus, this likely explains the findings reported by Gardner et al92 of differences in severity between those with MDD + ADHD versus pure MDD as well as differences in activity in the frontal regions and the cerebellum.
The assessment of hedonic tone is complicated by the lack of reliable methods for its evaluation. Currently used methods include the Snaith-Hamilton Pleasure Scale, the Dimensional Anhedonia Rating Scale, and the Scale for the Assessment of Negative Symptoms. Of these, the Dimensional Anhedonia Rating Scale has the best reliability and validity.93 However, currently available assessments evaluate the current state of hedonic tone and do not take into account its long-term aspects. Therefore, development of novel assessment methods is required in order to properly assess the life-long trait of hedonic tone.
Antidepressant side effects and low hedonic tone
Dysfunction of the neural circuits that regulate reward processing and motivation has also been implicated in the poor effectiveness of SSRIs used to treat anxiety and depression in patients with low hedonic tone.94 Patients undergoing SSRI treatment frequently report having low energy levels and emotional blunting,95 and symptoms of low hedonic tone often persist during SSRI treatment even if other clinical symptoms have been alleviated.96 For example, it has also been demonstrated that citalopram, a commonly used SSRI, diminishes the neural activation of the striatum to both aversive and rewarding stimuli, suggesting that efficacy of SSRIs may be blunted due to their effects on reward processing.94 This blunting may be related to the inhibitory effects of serotonin on dopamine and noradrenaline through the activity of serotonin on the serotonin 2C and 2A receptors, respectively.97 This may well explain the commonly understood side effect of SSRI-induced emotional numbing98 or indifference99 as well as what has been described as an amotivational frontal lobe syndrome, which is accompanied by apathy and lack of motivation, as well as in some cases behavioral disinhibition (all of which were symptoms that were dose related and reversible).100
Thus, this would suggest a specific differential beneficial role for norephinephrine reuptake inhibitors, serotonin–norephinephrine reuptake inhibitors, and stimulants in patients who suffer with SSRI-induced flat affect to directly elevate the extracellular DA and NA in areas such as the VMPFC and DLPFC.101–104 Furthermore, this would suggest a unique sensitivity to this side effect in those already having pretreatment lower catecholaminergic modulation of the PFC and potential higher risk of low hedonic tone.95
This relationship between lowered noradrenergic and dopaminergic activity and depression has been supported most recently by Harmer et al105 who reported that acute administration of a catecholaminergic acting antidepressant could increase positive affective processing. The authors reported that depressed patients receiving placebo showed reduced, 1) recognition of positive facial expressions, 2) decreased speed in responding to positive self-relevant personality adjectives, and 3) reduced memory for this positive information in comparison with healthy volunteers receiving placebo.105 They then reported that this effect was reversed in depressed patients who received a single dose of reboxetine, which as a noradrenergic reuptake inhibitor raises synaptic catecholaminergic levels.105 Interestingly, these effects were noted prior to changes in subjective ratings of mood or anxiety. Thus, the authors concluded that catecholaminergic enhancing antidepressant drug administration modulates emotional processing in depressed patients very early in treatment.105 In fact, this effect happens even before changes occur in mood symptoms, resulting in the amelioration of the negative biases in information processing that characterize the mood disorders.105
Hedonic tone and mood disorders
Low hedonic tone is a characteristic feature of several psychopathologies, including MDD, substance use, and schizophrenia,8,9 although variations in hedonic tone can also be observed in healthy individuals.10 All these disorders are characterized by changes in reward processing, and altered monoamine signaling has been implicated as their underlying mechanism. These observations raise the possibility that low hedonic tone can be used to predict subtypes of these disorders as well as their comorbidities. While the possibility that low hedonic tone may predict other subtypes of MDD cannot be eliminated, this review focuses on TRD, as well as depression comorbid with ADHD and substance abuse as representative MDD subtypes that share low hedonic tone as their feature.
Dysfunctional reward processing in MDD has been well characterized,106 with depression having been shown to affect various aspects of reward processing, such as evaluating the value of rewards and motivation to obtain a reward. Pizzagalli et al107 have shown that depressed patients have a reduced capacity to integrate reinforcement over time and to respond to more frequently rewarded cues. Similar observations were reported by studies in those with history of depression.108,109 Depressed patients are also less able to assess the value of the reward and exhibit a dissociation between perceived effort required to obtain a reward and reward value, suggesting that low hedonic tone in depression manifests as an impaired ability to modify behavior in response to a reward.106 Taken together, these findings suggest that in depressed patients, low hedonic tone and anhedonia are associated with an altered ability to feel pleasure and a reduced motivation for goal-directed behaviors (Figure 1).
Anhedonia has also been shown to be a predictor of TRD.110,111 In addition, DBS of the regions hypoactive in individuals with low hedonic tone, such as the NA,112 the PFC,113 and the sgACC,78 has been shown to improve symptoms in TRD. These observations raise the possibility that hedonic tone can be used as a tool to predict treatment outcomes and select best treatments for MDD.
In ADHD, low hedonic tone is manifested as an altered sensitivity to reinforcement.12 Children with ADHD have been reported to respond only to immediate rewards but not when the rewards are delayed and therefore only exhibit conditioning to immediate rewards.114 It has also been reported that children with ADHD have an increased sensitivity to rewards and therefore suffer from a heightened frustration when the reward is not presented.115 Yet another theory, proposed by Quay,116,117 is that children with ADHD are less responsive to punishment or nonreward. Although these studies report different manifestations of reduced hedonic tone in ADHD, the common finding is that ADHD is associated with altered feelings of pleasure. These behavioral features of ADHD are paralleled by changes in the neural pathways that regulate reward and motivation. A meta-analysis published by Dickstein et al118 found significant hypoactivity in several frontal regions in individuals with ADHD compared with control subjects, specifically the orbitofrontal cortices, inferior prefrontal, dorsolateral prefrontal, and the anterior cingulate. In addition, hypoactivity was detected in related regions, such as areas of the thalamus, basal ganglia, and parietal cortices.109 The meta-analysis also determined that some areas (eg, hypoactivated areas of the frontal and parietal regions) showed hyperactivation in individuals with ADHD. While this may suggest a compensatory mechanism, it has been postulated that this pattern may reflect “invasion” in areas where activation would be expected to diminish during a cognitive task.119 Furthermore, highly correlated brain activity can be seen during rest and a reduction in activation throughout tasks that require attention. Others have suggested that the performance deficit in individuals with ADHD may be related to the persistence of spontaneous and low-frequency activation of the default-mode network, which compete with task-specific processing.120
Alterations in the reward circuitry are also observed in substance abuse, and addictive substances are known to alter hedonic tone.121 As dependence on a given substance develops, an increasing dose of that substance is needed to elicit the same pleasurable effect. For example, heroin addicts often report that after being addicted for some time, they no longer administer the drug to get high, but simply to “get straight”, meaning that the goal of the drug is to return their baseline hedonic tone back toward normal levels.121 In addition, low hedonic tone is considered to be one of the risk factors for substance abuse, since many addictive substances, including prescription painkillers, work by raising the hedonic tone.121 In adolescents, increased attentional processing of pleasant stimuli has been shown to increase the likelihood of alcohol use later in life.122 Anhedonia is also positively correlated with drug cravings in detoxified alcohol- and opioid-dependent users123 and smokers.124,125 Positron emission tomography studies have shown that alcohol intake induces dopamine release in the ventral striatum of human subjects,126,127 suggesting that alcohol use alters the function on dopaminergic reward pathways. It has also been shown that reduced VLPFC activation, during shifting in the probabilistic reversal learning task, may be a marker of cocaine addiction and pathological gambling.128 This finding further supports the notion that drug abuse is associated with alterations in reward processing, which underlie low hedonic tone.
In addition to sharing low hedonic tone as a key feature, MDD, ADHD, and substance abuse also share a dysfunction in monoamine signaling, particularly in the ventral striatum. Neurotransmitter studies have revealed abnormalities in DA and NE signaling in both MDD and ADHD, suggesting a potentially shared underlying pathophysiology, at least in some individuals.129–132 Imaging studies have reported decreased striatal activation in depressed patients.106 More specifically, depressed patients display a stronger striatal–ACC connectivity in response to negative stimuli, but a weaker connectivity of the same pathway in response to positive stimuli.133 Support for the dopaminergic theory of ADHD, which states that ADHD results from DA deficits and therefore lowered activity in the frontal cortex and striatum,134 comes from a variety of pharmacological and imaging studies that have revealed hypoperfusion and lowered activations in those areas.135–138 Interestingly, with treatment with methylphenidate, normalization of the hypoperfusion of prefrontal areas is associated with the corresponding improvement in ADHD symptoms.135,139–142
Furthermore, the ability of methylphenidate to elevate DA levels in the ventral striatum is attenuated in alcoholics, suggesting that DA plays a role in the alterations in reward pathways in alcoholism.143 It has also been shown that the severity of anhedonia is negatively correlated with the ventral striatal activity and volume.11,51,95,144 Observations that dysfunction of this circuitry is also shared by MDD, ADHD, and substance abuse further support the idea that hedonic tone is the common link between MDD, ADHD, and depression.
Given that MDD, ADHD, and substance abuse share the characteristic of low hedonic tone and underlying neurobiology, it is not surprising that these disorders also have high levels of comorbidities. For example, most individuals with depression experience psychiatric comorbidity, according to the National Comorbidity Survey Replication.145,146 It has been reported that depression occurs in 9%–50% of patients with ADHD,147 and STAR*D study has reported that approximately a third of patients with MDD also have a substance use disorder. When MDD is comorbid with ADHD, it is associated with a much more severe course, an earlier age of illness onset, more complex psychiatric comorbidities, decreased quality of life, greater burden of illness, greater illness complexity, lower social functioning, lower work productivity, and lower employment rates than patients with MDD alone.148,149 As well, individuals with MDD and ADHD experience higher levels of substance use and psychosocial impairment than each disorder in isolation,150–153 suggesting a potential variant of MDD, and therefore explaining the treatment resistance in this population when treated with traditional SSRIs. Among respondents with ADHD, there is significant comorbidity with a wide range of Diagnostic and Statistical Manual of Mental Disorders, fourth edition disorders, with odds ratios of 2.7–7.5 for mood disorders, 1.5–5.5 for anxiety disorders, 1.5–7.9 for substance use disorders, and 3.7 for intermittent explosive disorder.145 Sternat et al reported that in patients referred to a tertiary care center, 32.5% of TRD met criteria for ADHD.154 Furthermore, the best predictors of comorbid ADHD with TRD were the number of SSRIs and less specifically the number of antidepressants that the patient had failed.
Thus, it has been proposed that low hedonic tone is a predictor of comorbidities of various mood disorders.12 Neuroimaging studies have identified morphological and functional features common to both MDD and ADHD, including decreased brain volumes and altered activity of frontal lobe structures (and specifically the DLPFC and ACC), areas that are associated with attention regulation, behavior selection, and emotion155,156 and significantly worsened outcome. In ADHD, other than hyperactivity and inattention, impulsivity is one of the most prevalent diagnostic symptoms. Impulse control has been associated with prefrontal functioning, specifically in the DLPFC, VMPFC, VLPFC, inferior frontal gyrus, the rostral and dorsal ACC, and the insula.157–159 Dysregulation in these regions is associated with the impulsivity observed in patients with ADHD and overlaps with the dysregulation observed among depressed individuals, as well as individuals with substance abuse disorders. Interestingly, altered activity of these regions is also observed in people with low hedonic tone, suggesting that low hedonic tone may be a behavioral manifestation of dysregulation of these circuits and consequent mood disorders. Thus, these patient populations share low hedonic tone as a common endophenotype, which may lead to dysthymia and double depression. Furthermore, studies investigating the role of monoamines in these disorders point to a key role for DA dysfunction in the pathophysiology of ADHD and its comorbidities, including MDD. With evidence pointing to dysfunctional neuromodulation of the DLPFC in relation to executive dysfunction in ADHD,160–164 it is not too much of a stretch to consider low hedonic tone as an affective correlate in ADHD explaining the lack of responsiveness to SSRIs in depression. Low dopaminergic activity has been reported in patients with MDD and ADHD, as well as those with low hedonic tone.134,165 Furthermore, SSRI treatment is commonly associated with symptoms of low hedonic tone, even in the absence of other MDD symptoms.67,96 Taken together, these observations suggest that low hedonic tone results from reduced baseline catecholaminergic activity in the regions involved in reward processing and executive functions, which may also be a predictive factor of resistance to SSRI treatment as well as a predictor of comorbidity of various disorders. In support of this suggestion, it has been reported in both the STAR*D and GENDEP studies that the percent reduction on the primary outcome scale over 12 weeks of treatment was best predicted by loss of interest and diminished activity (perhaps proxy measures for low hedonic tone) even after adjustment for overall depression severity and other clinical covariates such as age, sex, and center differences.166
Conclusion
Low hedonic tone is characterized by a dysfunction of neural circuits that regulate motivation and reward processing and is a common feature of MDD, ADHD, and substance abuse. Anhedonic, depressed patients consistently show decreased prefrontal activity and increased activity in the VMPFC and sgACC. These patients with low hedonic tone attempt to cope with their depression and enter into a euthymic state by raising their hedonic tone, thus achieving a shift toward a more neutral hedonic tone, and therefore a rise in their mood. In the absence of antidepressant therapy, or in TRD where altered activity in these regions cannot be normalized with pharmacologic therapy, patients will often choose ways to self-medicate, often through substance abuse, in order to modify the neurobiology of the mood system to correct their chronic low hedonic tone. Similarly, patients with ADHD share a similar functional dysregulation in the frontal cortex and similar abnormalities in DA and NE signaling. Perhaps, this explains the high comorbidity between MDD and ADHD. If one imagines then that the presence of anomalous activity of the PFC and the cingulate gyrus is a function of a chronic disturbance in catecholaminergic innervation (and specifically lower dopaminergic and noradrenergic tone),167,168 then activities that raise dopaminergic and noradrenergic tone, such as substance abuse, would be favored by patients suffering with this neurobiology.
Since low hedonic tone is a shared feature of these disorders, evaluation of hedonic tone may become a diagnostic feature used to predict subtypes of MDD, such as TRD, as well as comorbidities of MDD with ADHD and substance abuse.
Acknowledgment
The authors would like to thank Tanja Babic for her assistance in the preparation of this paper, specifically contributions to fact finding, writing, and editing in association with the development of this manuscript.
Disclosure
Tia Sternat reports no conflicts of interest in this work. Martin A Katzman has received grant support, participated in advisory boards, and/or received honorarium for giving lectures from the following: Canadian Foundation for Innovation, Lotte & John Hecht Memorial Foundation, Allergan, Astra Zeneca, Bedrocan, Biotics, Bristol-Myers Squibb, Eli Lilly, Genuine Health, Janssen, Lundbeck, Merck, Pfizer, Purdue, Shire, and Tweed.
References
Ekkekakis P. The Measurement of Affect, Mood, and Emotion: A Guide for Health-Behavioral Research. Cambridge, UK, New York: Cambridge University Press; 2013. | ||
Russel J. Emotion, core affect, and psychological construction. Cogn Emotion. 2009;23(7):1259–1283. | ||
Gorwood P. Neurobiological mechanisms of anhedonia. Dialogues Clin Neurosci. 2008;10(3):291–299. | ||
Stein DJ. Depression, anhedonia, and psychomotor symptoms: the role of dopaminergic neurocircuitry. CNS Spectr. 2008;13(7):561–565. | ||
Der-Avakian A, Markou A. The neurobiology of anhedonia and other reward-related deficits. Trends Neurosci. 2012;35(1):68–77. | ||
Di Nicola M, De Risio L, Battaglia C, et al. Reduced hedonic capacity in euthymic bipolar subjects: a trait-like feature? J Affect Disord. 2012;147(1–3):446–450. | ||
Loas G. Vulnerability to depression: a model centered on anhedonia. J Affect Disord. 1996;41(1):39–53. | ||
Snaith RP. Identifying depression: the significance of anhedonia. Hosp Pract (Off Ed). 1993;28(9A):55–60. | ||
Snaith RP, Hamilton M, Morley S, Humayan A, Hargreaves D, Trigwell P. A scale for the assessment of hedonic tone the Snaith-Hamilton Pleasure Scale. Br J Psychiatry. 1995;167(1):99–103. | ||
Jankowski KS, Ciarkowska W. Diurnal variation in energetic arousal, tense arousal, and hedonic tone in extreme morning and evening types. Chronobiol Int. 2008;25(4):577–595. | ||
Keedwell PA, Andrew C, Williams SC, Brammer MJ, Phillips ML. The neural correlates of anhedonia in major depressive disorder. Biol Psychiatry. 2005;58(11):843–853. | ||
Sternat T, Lodzinski A, Katzman MA. Hedonic tone: a bridge between the psychobiology of depression and its comorbidities. J Depress Anxiety. 2014;3:1. | ||
Berton O, Hahn CG, Thase ME. Are we getting closer to valid translational models for major depression? Science. 2012;338(6103):75–79. | ||
Nestler EJ, Carlezon WA Jr. The mesolimbic dopamine reward circuit in depression. Biol Psychiatry. 2006;59(12):1151–1159. | ||
Bevins RA, Besheer J. Novelty reward as a measure of anhedonia. Neurosci Biobehav Rev. 2005;29(4–5):707–714. | ||
Knutson B, Fong GW, Adams CM, Varner JL, Hommer D. Dissociation of reward anticipation and outcome with event-related fMRI. Neuroreport. 2001;12(17):3683–3687. | ||
Lammel S, Tye KM, Warden MR. Progress in understanding mood disorders: optogenetic dissection of neural circuits. Genes Brain Behav. 2013;13(1):38–51. | ||
Ongur D, Ferry AT, Price JL. Architectonic subdivision of the human orbital and medial prefrontal cortex. J Comp Neurol. 2003;460(3):425–449. | ||
Drevets WC, Price JL, Furey ML. Brain structural and functional abnormalities in mood disorders: implications for neurocircuitry models of depression. Brain Struct Funct. 2008;213(1–2):93–118. | ||
Grimm S, Beck J, Schuepbach D, et al. Imbalance between left and right dorsolateral prefrontal cortex in major depression is linked to negative emotional judgment: an fMRI study in severe major depressive disorder. Biol Psychiatry. 2008;63(4):369–376. | ||
Tekin S, Cummings JL. Frontal-subcortical neuronal circuits and clinical neuropsychiatry: an update. J Psychosom Res. 2002;53(2):647–654. | ||
Koenigs M, Grafman J. The functional neuroanatomy of depression: distinct roles for ventromedial and dorsolateral prefrontal cortex. Behav Brain Res. 2009;201(2):239–243. | ||
Carmichael ST, Price JL. Connectional networks within the orbital and medial prefrontal cortex of macaque monkeys. J Comp Neurol. 1996;371(2):179–207. | ||
Ongur D, Price JL. The organization of networks within the orbital and medial prefrontal cortex of rats, monkeys and humans. Cereb Cortex. 2000;10(3):206–219. | ||
An X, Bandler R, Ongur D, Price JL. Prefrontal cortical projections to longitudinal columns in the midbrain periaqueductal gray in macaque monkeys. J Comp Neurol. 1998;401(4):455–479. | ||
Ongur D, An X, Price JL. Prefrontal cortical projections to the hypothalamus in macaque monkeys. J Comp Neurol. 1998;401(4):480–505. | ||
Schultz W, Tremblay L, Hollerman JR. Reward prediction in primate basal ganglia and frontal cortex. Neuropharmacology. 1998;37(4–5):421–429. | ||
Sebastian A, Jung P, Krause-Utz A, Lieb K, Schmahl C, Tuscher O. Frontal dysfunctions of impulse control – a systematic review in borderline personality disorder and attention-deficit/hyperactivity disorder. Front Hum Neurosci. 2014;8:698. | ||
Kayser AS, Allen DC, Navarro-Cebrian A, Mitchell JM, Fields HL. Dopamine, corticostriatal connectivity, and intertemporal choice. J Neurosci. 2012;32(27):9402–9409. | ||
Patterson JC 2nd, Ungerleider LG, Bandettini PA. Task-independent functional brain activity correlation with skin conductance changes: an fMRI study. Neuroimage. 2002;17(4):1797–1806. | ||
Mayberg HS, Lozano AM, Voon V, et al. Deep brain stimulation for treatment-resistant depression. Neuron. 2005;45(5):651–660. | ||
Critchley HD, Elliott R, Mathias CJ, Dolan RJ. Neural activity relating to generation and representation of galvanic skin conductance responses: a functional magnetic resonance imaging study. J Neurosci. 2000;20(8):3033–3040. | ||
Kim JJ, Jung MW. Neural circuits and mechanisms involved in Pavlovian fear conditioning: a critical review. Neurosci Biobehav Rev. 2006;30(2):188–202. | ||
Phelps EA, LeDoux JE. Contributions of the amygdala to emotion processing: from animal models to human behavior. Neuron. 2005;48(2):175–187. | ||
Phelps EA, Delgado MR, Nearing KI, LeDoux JE. Extinction learning in humans: role of the amygdala and vmPFC. Neuron. 2004;43(6):897–905. | ||
Janak PH, Tye KM. From circuits to behaviour in the amygdala. Nature. 2015;517(7534):284–292. | ||
Swartz JR, Williamson DE, Hariri AR. Developmental change in amygdala reactivity during adolescence: effects of family history of depression and stressful life events. Am J Psychiatry. 2015;172(3):276–283. | ||
Davis M, Whalen PJ. The amygdala: vigilance and emotion. Mol Psychiatry. 2001;6(1):13–34. | ||
Maren S, Phan KL, Liberzon I. The contextual brain: implications for fear conditioning, extinction and psychopathology. Nat Rev Neurosci. 2013;14(6):417–428. | ||
Milad MR, Quirk GJ. Neurons in medial prefrontal cortex signal memory for fear extinction. Nature. 2002;420(6911):70–74. | ||
Gorman JM, Kent JM, Sullivan GM, Coplan JD. Neuroanatomical hypothesis of panic disorder, revised. Am J Psychiatry. 2000;157(4):493–505. | ||
Carlson JM, Greenberg T, Rubin D, Mujica-Parodi LR. Feeling anxious: anticipatory amygdalo-insular response predicts the feeling of anxious anticipation. Soc Cogn Affect Neurosci. 2011;6(1):74–81. | ||
Simmons A, Matthews SC, Stein MB, Paulus MP. Anticipation of emotionally aversive visual stimuli activates right insula. Neuroreport. 2004;15(14):2261–2265. | ||
Starr CJ, Sawaki L, Wittenberg GF, et al. Roles of the insular cortex in the modulation of pain: insights from brain lesions. J Neurosci. 2009;29(9):2684–2694. | ||
Gu X, Hof PR, Friston KJ, Fan J. Anterior insular cortex and emotional awareness. J Comp Neurol. 2013;521(15):3371–3388. | ||
Fanselow MS, Dong HW. Are the dorsal and ventral hippocampus functionally distinct structures? Neuron. 2010;65(1):7–19. | ||
Henke PG. Hippocampal pathway to the amygdala and stress ulcer development. Brain Res Bull. 1990;25(5):691–695. | ||
Kheirbek MA, Hen R. Dorsal vs ventral hippocampal neurogenesis: implications for cognition and mood. Neuropsychopharmacology. 2011;36(1):373–374. | ||
Ring HA, Serra-Mestres J. Neuropsychiatry of the basal ganglia. J Neurol Neurosurg Psychiatry. 2002;72(1):12–21. | ||
Wang Y, Deng Y, Fung G, et al. Distinct structural neural patterns of trait physical and social anhedonia: evidence from cortical thickness, subcortical volumes and inter-regional correlations. Psychiatry Res. 2014;224(3):184–191. | ||
Harvey PO, Pruessner J, Czechowska Y, Lepage M. Individual differences in trait anhedonia: a structural and functional magnetic resonance imaging study in non-clinical subjects. Mol Psychiatry. 2007;12(8):703, 767–775. | ||
Delgado MR, Locke HM, Stenger VA, Fiez JA. Dorsal striatum responses to reward and punishment: effects of valence and magnitude manipulations. Cogn Affect Behav Neurosci. 2003;3(1):27–38. | ||
Grahn JA, Parkinson JA, Owen AM. The cognitive functions of the caudate nucleus. Prog Neurobiol. 2008;86(3):141–155. | ||
Carlson PJ, Singh JB, Zarate CA Jr, Drevets WC, Manji HK. Neural circuitry and neuroplasticity in mood disorders: insights for novel therapeutic targets. NeuroRx. 2006;3(1):22–41. | ||
Roberts-Wolfe DJ, Kalivas PW. Glutamate transporter GLT-1 as a therapeutic target for substance use disorders. CNS Neurol Disord Drug Targets. 2015;14(6):745–756. | ||
Brambilla P, Perez J, Barale F, Schettini G, Soares JC. GABAergic dysfunction in mood disorders. Mol Psychiatry. 2003;8(8):721–737, 715. | ||
Hayes DJ, Jupp B, Sawiak SJ, Merlo E, Caprioli D, Dalley JW. Brain gamma-aminobutyric acid: a neglected role in impulsivity. Eur J Neurosci. 2014;39(11):1921–1932. | ||
Lupica CR, Riegel AC, Hoffman AF. Marijuana and cannabinoid regulation of brain reward circuits. Br J Pharmacol. 2004;143(2):227–234. | ||
Manzoni OJ, Williams JT. Presynaptic regulation of glutamate release in the ventral tegmental area during morphine withdrawal. J Neurosci. 1999;19(15):6629–6636. | ||
Terman GW, Drake CT, Simmons ML, Milner TA, Chavkin C. Opioid modulation of recurrent excitation in the hippocampal dentate gyrus. J Neurosci. 2000;20(12):4379–4388. | ||
Pehrson AL, Sanchez C. Serotonergic modulation of glutamate neurotransmission as a strategy for treating depression and cognitive dysfunction. CNS Spectr. 2014;19(2):121–133. | ||
Krishnan V, Nestler EJ. The molecular neurobiology of depression. Nature. 2008;455(7215):894–902. | ||
Glennon RA, Dukat M, Westkaemper RB. Serotonin Receptor Subtypes and Ligands. American College of Neuropsychopharmacology; 2000. Available from: http://www.acnp.org/g4/GN401000039/Ch039.html. Accessed August 2, 2016. | ||
Lucki I. Serotonin receptor specificity in anxiety disorders. J Clin Psychiatry. 1996;57(suppl 6):5–10. | ||
Nestler EJ, Hyman SE, Malenka RC. Molecular Neuropharmacology: A Foundation for Clinical Neuroscience. 2nd ed. New York: McGraw-Hill Medical; 2009. | ||
Stahl SM. Modes and nodes explain the mechanism of action of vortioxetine, a multimodal agent (MMA): modifying serotonin’s downstream effects on glutamate and GABA (gamma amino butyric acid) release. CNS Spectr. 2015;20(4):331–336. | ||
Price JL, Drevets WC. Neural circuits underlying the pathophysiology of mood disorders. Trends Cogn Sci. 2012;16(1):61–71. | ||
Ionescu DF, Rosenbaum JF, Alpert JE. Pharmacological approaches to the challenge of treatment-resistant depression. Dialogues Clin Neurosci. 2015;17(2):111–126. | ||
Drevets WC, Furey ML. Replication of scopolamine’s antidepressant efficacy in major depressive disorder: a randomized, placebo-controlled clinical trial. Biol Psychiatry. 2010;67(5):432–438. | ||
Ehrich E, Turncliff R, Du Y, et al. Evaluation of opioid modulation in major depressive disorder. Neuropsychopharmacology. 2015;40(6):1448–1455. | ||
Lally N, Nugent AC, Luckenbaugh DA, Ameli R, Roiser JP, Zarate CA. Anti-anhedonic effect of ketamine and its neural correlates in treatment-resistant bipolar depression. Transl Psychiatry. 2014;4:e469. | ||
Furey ML, Drevets WC, Szczepanik J, Khanna A, Nugent A, Zarate CA Jr. Pretreatment differences in BOLD response to emotional faces correlate with antidepressant response to scopolamine. Int J Neuropsychopharmacol. 2015;18(8):1–10. | ||
Huhn AS, Meyer RE, Harris JD, et al. Evidence of anhedonia and differential reward processing in prefrontal cortex among post-withdrawal patients with prescription opiate dependence. Brain Res Bull. 2016;123:102–109. | ||
Greicius MD, Flores BH, Menon V, et al. Resting-state functional connectivity in major depression: abnormally increased contributions from subgenual cingulate cortex and thalamus. Biol Psychiatry. 2007;62(5):429–437. | ||
Kennedy SH, Evans KR, Krüger S, et al. Changes in regional brain glucose metabolism measured with positron emission tomography after paroxetine treatment of major depression. Am J Psychiatry. 2001;158(6):899–905. | ||
Liotti M, Mayberg HS, Brannan SK, McGinnis S, Jerabek P, Fox PT. Differential limbic – cortical correlates of sadness and anxiety in healthy subjects: implications for affective disorders. Biol Psychiatry. 2000;48(1):30–42. | ||
Keedwell PA, Drapier D, Surguladze S, Giampietro V, Brammer M, Phillips M. Subgenual cingulate and visual cortex responses to sad faces predict clinical outcome during antidepressant treatment for depression. J Affect Disord. 2010;120(1–3):120–125. | ||
Mayberg HS, Liotti M, Brannan SK, et al. Reciprocal limbic-cortical function and negative mood: converging PET findings in depression and normal sadness. Am J Psychiatry. 1999;156(5):675–682. | ||
Oldani L, Dell’Osso B, Altamura AC. Long-term effects of vagus nerve stimulation in treatment-resistant depression: a 5-year follow up case series. Brain Stimulat. 2015;8(6):1229–1230. | ||
Yuan TF, Li A, Sun X, Arias-Carrión O, Machado S. Vagus nerve stimulation in treating depression: a tale of two stories. Curr Mol Med. 2016;16(1):33–39. | ||
Nahas Z, Teneback C, Chae JH, et al. Serial vagus nerve stimulation functional MRI in treatment-resistant depression. Neuropsychopharmacology. 2007;32(8):1649–1660. | ||
Zobel A, Joe A, Freymann N, et al. Changes in regional cerebral blood flow by therapeutic vagus nerve stimulation in depression: an exploratory approach. Psychiatry Res. 2005;139(3):165–179. | ||
Epstein J, Pan H, Kocsis JH, et al. Lack of ventral striatal response to positive stimuli in depressed versus normal subjects. Am J Psychiatry. 2006;163(10):1784–1790. | ||
Keller J, Young CB, Kelley E, Prater K, Levitin DJ, Menon V. Trait anhedonia is associated with reduced reactivity and connectivity of mesolimbic and paralimbic reward pathways. J Psychiatr Res. 2013;47(10):1319–1328. | ||
Mitterschiffthaler MT, Kumari V, Malhi GS, et al. Neural response to pleasant stimuli in anhedonia: an fMRI study. Neuroreport. 2003;14(2):177–182. | ||
Light SN, Heller AS, Johnstone T, et al. Reduced right ventrolateral prefrontal cortex activity while inhibiting positive affect is associated with improvement in hedonic capacity after 8 weeks of antidepressant treatment in major depressive disorder. Biol Psychiatry. 2011;70(10):962–968. | ||
Wacker J, Dillon DG, Pizzagalli DA. The role of the nucleus accumbens and rostral anterior cingulate cortex in anhedonia: integration of resting EEG, fMRI, and volumetric techniques. Neuroimage. 2009;46(1):327–337. | ||
Heller AS, Johnstone T, Light SN, et al. Relationships between changes in sustained fronto-striatal connectivity and positive affect in major depression resulting from antidepressant treatment. Am J Psychiatry. 2013;170(2):197–206. | ||
Pizzagalli DA, Holmes AJ, Dillon DG, et al. Reduced caudate and nucleus accumbens response to rewards in unmedicated individuals with major depressive disorder. Am J Psychiatry. 2009;166(6):702–710. | ||
Gabbay V, Ely BA, Li Q, et al. Striatum-based circuitry of adolescent depression and anhedonia. J Am Acad Child Adolesc Psychiatry. 2013;52(6):628–641.e13. | ||
Heller AS, Johnstone T, Shackman AJ, et al. Reduced capacity to sustain positive emotion in major depression reflects diminished maintenance of fronto-striatal brain activation. Proc Natl Acad Sci U S A. 2009;106(52):22445–22450. | ||
Gardner A, Salmaso D, Varrone A, et al. Differences at brain SPECT between depressed females with and without adult ADHD and healthy controls: etiological considerations. Behav Brain Funct. 2009;5:37. | ||
Rizvi SJ, Quilty LC, Sproule BA, Cyriac A, Michael Bagby R, Kennedy SH. Development and validation of the Dimensional Anhedonia Rating Scale (DARS) in a community sample and individuals with major depression. Psychiatry Res. 2015;229(1–2):109–119. | ||
McCabe C, Mishor Z, Cowen PJ, Harmer CJ. Diminished neural processing of aversive and rewarding stimuli during selective serotonin reuptake inhibitor treatment. Biol Psychiatry. 2010;67(5):439–445. | ||
Price J, Cole V, Goodwin GM. Emotional side-effects of selective serotonin reuptake inhibitors: qualitative study. Br J Psychiatry. 2009;195(3):211–217. | ||
Hasler G, Drevets WC, Manji HK, Charney DS. Discovering endophenotypes for major depression. Neuropsychopharmacology. 2004;29(10):1765–1781. | ||
Trivedi MH, Hollander E, Nutt D, Blier P. Clinical evidence and potential neurobiological underpinnings of unresolved symptoms of depression. J Clin Psychiatry. 2008;69(2):246–258. | ||
Arnsten AF, Rubia K. Neurobiological circuits regulating attention, cognitive control, motivation, and emotion: disruptions in neurodevelopmental psychiatric disorders. J Am Acad Child Adolesc Psychiatry. 2012;51(4):356–367. | ||
Sansone RA, Sansone LA. SSRI-induced Indifference. Psychiatry (Edgmont). 2010;7(10):14–18. | ||
Garland EJ, Baerg EA. Amotivational syndrome associated with selective serotonin reuptake inhibitors in children and adolescents. J Child Adolesc Psychopharmacol. 2001;11(2):181–186. | ||
Carboni E, Tanda GL, Frau R, Di Chiara G. Blockade of the noradrenaline carrier increases extracellular dopamine concentrations in the prefrontal cortex: evidence that dopamine is taken up in vivo by noradrenergic terminals. J Neurochem. 1990;55(3):1067–1070. | ||
Tanda G, Carboni E, Frau R, Di Chiara G. Increase of extracellular dopamine in the prefrontal cortex: a trait of drugs with antidepressant potential? Psychopharmacology (Berl). 1994;115(1–2):285–288. | ||
Millan MJ, Newman-Tancredi A, Audinot V, et al. Agonist and antagonist actions of yohimbine as compared to fluparoxan at alpha(2)-adrenergic receptors (AR)s, serotonin (5-HT)(1A), 5-HT(1B), 5-HT(1D) and dopamine D(2) and D(3) receptors. Significance for the modulation of frontocortical monoaminergic transmission and depressive states. Synapse. 2000;35(2):79–95. | ||
Linner L, Endersz H, Ohman D, Bengtsson F, Schalling M, Svensson TH. Reboxetine modulates the firing pattern of dopamine cells in the ventral tegmental area and selectively increases dopamine availability in the prefrontal cortex. J Pharmacol Exp Ther. 2001;297(2):540–546. | ||
Harmer CJ, O’Sullivan U, Favaron E, et al. Effect of acute antidepressant administration on negative affective bias in depressed patients. Am J Psychiatry. 2009;166(10):1178–1184. | ||
Admon R, Pizzagalli DA. Dysfunctional reward processing in depression. Curr Opin Psychol. 2015;4:114–118. | ||
Pizzagalli DA, Iosifescu D, Hallett LA, Ratner KG, Fava M. Reduced hedonic capacity in major depressive disorder: evidence from a probabilistic reward task. J Psychiatr Res. 2008;43(1):76–87. | ||
Pechtel P, Dutra SJ, Goetz EL, Pizzagalli DA. Blunted reward responsiveness in remitted depression. J Psychiatr Res. 2013;47(12):1864–1869. | ||
Pizzagalli DA, Jahn AL, O’Shea JP. Toward an objective characterization of an anhedonic phenotype: a signal-detection approach. Biol Psychiatry. 2005;57(4):319–327. | ||
McMakin DL, Olino TM, Porta G, et al. Anhedonia predicts poorer recovery among youth with selective serotonin reuptake inhibitor treatment-resistant depression. J Am Acad Child Adolesc Psychiatry. 2012;51(4):404–411. | ||
Rubin DH. Joy returns last: anhedonia and treatment resistance in depressed adolescents. J Am Acad Child Adolesc Psychiatry. 2012;51(4):353–355. | ||
Eggers AE. Treatment of depression with deep brain stimulation works by altering in specific ways the conscious perception of the core symptoms of sadness or anhedonia, not by modulating network circuitry. Med Hypotheses. 2014;83(1):62–64. | ||
Rea E, Rummel J, Schmidt TT, et al. Anti-anhedonic effect of deep brain stimulation of the prefrontal cortex and the dopaminergic reward system in a genetic rat model of depression: an intracranial self-stimulation paradigm study. Brain Stimulat. 2014;7(1):21–28. | ||
Sagvolden T, Aase H, Zeiner P, Berger D. Altered reinforcement mechanisms in attention-deficit/hyperactivity disorder. Behav Brain Res. 1998;94(1):61–71. | ||
Luman M, Oosterlaan J, Sergeant JA. The impact of reinforcement contingencies on AD/HD: a review and theoretical appraisal. Clin Psychol Rev. 2005;25(2):183–213. | ||
Quay HC. Theories of ADDH. J Am Acad Child Adolesc Psychiatry. 1988;27(2):262–263. | ||
Quay HC. Inhibition and attention deficit hyperactivity disorder. J Abnorm Child Psychol. 1997;25(1):7–13. | ||
Dickstein SG, Bannon K, Castellanos FX, Milham MP. The neural correlates of attention deficit hyperactivity disorder: an ALE meta-analysis. J Child Psychol Psychiatry. 2006;47(10):1051–1062. | ||
Raichle ME, MacLeod AM, Snyder AZ, Powers WJ, Gusnard DA, Shulman GL. A default mode of brain function. Proc Natl Acad Sci U S A. 2001;98(2):676–682. | ||
Sonuga-Barke EJ, Castellanos FX. Spontaneous attentional fluctuations in impaired states and pathological conditions: a neurobiological hypothesis. Neurosci Biobehav Rev. 2007;31(7):977–986. | ||
Gardner EL. Addiction and brain reward and antireward pathways. Adv Psychosom Med. 2011;30:22–60. | ||
Garfield JB, Allen NB, Cheetham A, Simmons JG, Lubman DI. Attention to pleasant stimuli in early adolescence predicts alcohol-related problems in mid-adolescence. Biol Psychol. 2015;108:43–50. | ||
Martinotti G, Cloninger CR, Janiri L. Temperament and character inventory dimensions and anhedonia in detoxified substance-dependent subjects. Am J Drug Alcohol Abuse. 2008;34(2):177–183. | ||
Cook JW, Spring B, McChargue D, Hedeker D. Hedonic capacity, cigarette craving, and diminished positive mood. Nicotine Tob Res. 2004;6(1):39–47. | ||
Leventhal AM, Waters AJ, Kahler CW, Ray LA, Sussman S. Relations between anhedonia and smoking motivation. Nicotine Tob Res. 2009;11(9):1047–1054. | ||
Aalto S, Ingman K, Alakurtti K, et al. Intravenous ethanol increases dopamine release in the ventral striatum in humans: PET study using bolus-plus-infusion administration of [(11)C]raclopride. J Cereb Blood Flow Metab. 2015;35(3):424–431. | ||
Boileau I, Assaad JM, Pihl RO, et al. Alcohol promotes dopamine release in the human nucleus accumbens. Synapse. 2003;49(4):226–231. | ||
Verdejo-Garcia A, Clark L, Verdejo-Roman J, et al. Neural substrates of cognitive flexibility in cocaine and gambling addictions. Br J Psychiatry. 2015;207(2):158–164. | ||
Dunlop BW, Nemeroff CB. The role of dopamine in the pathophysiology of depression. Arch Gen Psychiatry. 2007;64(3):327–337. | ||
Garnock-Jones KP, Keating GM. Spotlight on atomoxetine in attention-deficit hyperactivity disorder in children and adolescents. CNS Drugs. 2010;24(1):85–88. | ||
Nigg JT, Casey BJ. An integrative theory of attention-deficit/hyperactivity disorder based on the cognitive and affective neurosciences. Dev Psychopathol. 2005;17(3):785–806. | ||
Sergeant JA, Geurts H, Huijbregts S, Scheres A, Oosterlaan J. The top and the bottom of ADHD: a neuropsychological perspective. Neurosci Biobehav Rev. 2003;27(7):583–592. | ||
Admon R, Nickerson LD, Dillon DG, et al. Dissociable cortico-striatal connectivity abnormalities in major depression in response to monetary gains and penalties. Psychol Med. 2015;45(1):121–131. | ||
Levy F. The dopamine theory of attention deficit hyperactivity disorder (ADHD). Aust N Z J Psychiatry. 1991;25(2):277–283. | ||
Kim BN, Lee JS, Cho SC, Lee DS. Methylphenidate increased regional cerebral blood flow in subjects with attention deficit/hyperactivity disorder. Yonsei Med J. 2001;42(1):19–29. | ||
Kim BN, Lee JS, Shin MS, Cho SC, Lee DS. Regional cerebral perfusion abnormalities in attention deficit/hyperactivity disorder. Statistical parametric mapping analysis. Eur Arch Psychiatry Clin Neurosci. 2002;252(5):219–225. | ||
Lou HC, Henriksen L, Bruhn P. Focal cerebral hypoperfusion in children with dysphasia and/or attention deficit disorder. Arch Neurol. 1984;41(8):825–829. | ||
Lou HC, Henriksen L, Bruhn P, Børner H, Nielsen JB. Striatal dysfunction in attention deficit and hyperkinetic disorder. Arch Neurol. 1989;46(1):48–52. | ||
Lee JS, Kim BN, Kang E, et al. Regional cerebral blood flow in children with attention deficit hyperactivity disorder: comparison before and after methylphenidate treatment. Hum Brain Mapp. 2005;24(3):157–164. | ||
Matochik JA, Liebenauer LL, King AC, Szymanski HV, Cohen RM, Zametkin AJ. Cerebral glucose metabolism in adults with attention deficit hyperactivity disorder after chronic stimulant treatment. Am J Psychiatry. 1994;151(5):658–664. | ||
Matochik JA, Nordahl TE, Gross M, et al. Effects of acute stimulant medication on cerebral metabolism in adults with hyperactivity. Neuropsychopharmacology. 1993;8(4):377–386. | ||
Vaidya CJ, Austin G, Kirkorian G, et al. Selective effects of methylphenidate in attention deficit hyperactivity disorder: a functional magnetic resonance study. Proc Natl Acad Sci U S A. 1998;95(24):14494–14499. | ||
Volkow ND, Wang GJ, Telang F, et al. Profound decreases in dopamine release in striatum in detoxified alcoholics: possible orbitofrontal involvement. J Neurosci. 2007;27(46):12700–12706. | ||
Dowd EC, Barch DM. Anhedonia and emotional experience in schizophrenia: neural and behavioral indicators. Biol Psychiatry. 2010;67(10):902–911. | ||
Kessler RC, Adler L, Barkley R, et al. The prevalence and correlates of adult ADHD in the United States: results from the National Comorbidity Survey Replication. Am J Psychiatry. 2006;163(4):716–723. | ||
Kessler RC, Berglund P, Demler O, et al. The epidemiology of major depressive disorder: results from the National Comorbidity Survey Replication (NCS-R). JAMA. 2003;289(23):3095–3105. | ||
McIntosh D, Kutcher S, Binder C, Levitt A, Fallu A, Rosenbluth M. Adult ADHD and comorbid depression: a consensus-derived diagnostic algorithm for ADHD. Neuropsychiatr Dis Treat. 2009;5:137–150. | ||
Bond DJ, Hadjipavlou G, Lam RW, et al. The Canadian Network for Mood and Anxiety Treatments (CANMAT) task force recommendations for the management of patients with mood disorders and comorbid attention-deficit/hyperactivity disorder. Ann Clin Psychiatry. 2012;24(1):23–37. | ||
McIntyre RS, Kennedy SH, Soczynska JK, et al. Attention-deficit/hyperactivity disorder in adults with bipolar disorder or major depressive disorder: results from the international mood disorders collaborative project. Prim Care Companion J Clin Psychiatry. 2010;12(3):PCC.09m00861. | ||
Biederman J, Ball SW, Monuteaux MC, et al. New insights into the comorbidity between ADHD and major depression in adolescent and young adult females. J Am Acad Child Adolesc Psychiatry. 2008;47(4):426–434. | ||
Biederman J, Mick E, Faraone SV. Depression in attention deficit hyperactivity disorder (ADHD) children: “true” depression or demoralization? J Affect Disord. 1998;47(1–3):113–122. | ||
Chronis-Tuscano A, Molina BS, Pelham WE, et al. Very early predictors of adolescent depression and suicide attempts in children with attention-deficit/hyperactivity disorder. Arch Gen Psychiatry. 2010;67(10):1044–1051. | ||
Warden D, Riggs PD, Min SJ, et al. Major depression and treatment response in adolescents with ADHD and substance use disorder. Drug Alcohol Depend. 2012;120(1–3):214–219. | ||
Sternat T, Mohamed M, Furtado M et al. Attention deficit hyperactivity disorder and depression: Sequential and concurrent disorders. Poster presented at: Annual Meeting of the American Society of Clinical Psychopharmacology; June 1; 2016; Phoenix, AZ. | ||
Konarski JZ, McIntyre RS, Kennedy SH, Rafi-Tari S, Soczynska JK, Ketter TA. Volumetric neuroimaging investigations in mood disorders: bipolar disorder versus major depressive disorder. Bipolar Disord. 2008;10(1):1–37. | ||
Seidman LJ, Valera EM, Makris N, et al. Dorsolateral prefrontal and anterior cingulate cortex volumetric abnormalities in adults with attention-deficit/hyperactivity disorder identified by magnetic resonance imaging. Biol Psychiatry. 2006;60(10):1071–1080. | ||
Alvarez JA, Emory E. Executive function and the frontal lobes: a meta-analytic review. Neuropsychol Rev. 2006;16(1):17–42. | ||
Aron AR, Robbins TW, Poldrack RA. Inhibition and the right inferior frontal cortex: one decade on. Trends Cogn Sci. 2014;18(4):177–185. | ||
Laird AR, McMillan KM, Lancaster JL, et al. A comparison of label-based review and ALE meta-analysis in the Stroop task. Hum Brain Mapp. 2005;25(1):6–21. | ||
Cunha PJ, Gonçalves PD, Ometto M, et al. Executive cognitive dysfunction and ADHD in cocaine dependence: searching for a common cognitive endophenotype for addictive disorders. Front Psychiatry. 2013;4:126. | ||
Fuermaier AB, Tucha L, Koerts J, et al. Complex prospective memory in adults with attention deficit hyperactivity disorder. PLoS One. 2013;8(3):e58338. | ||
Madhoo M, Keefe RS, Roth RM, et al. Lisdexamfetamine dimesylate augmentation in adults with persistent executive dysfunction after partial or full remission of major depressive disorder. Neuropsychopharmacology. 2013;39(6):1388–1398. | ||
Missonnier P, Hasler R, Perroud N, et al. EEG anomalies in adult ADHD subjects performing a working memory task. Neuroscience. 2013;241:135–146. | ||
Qian Y, Shuai L, Chan RC, Qian QJ, Wang Y. The developmental trajectories of executive function of children and adolescents with attention deficit hyperactivity disorder. Res Dev Disabil. 2013;34(5):1434–1445. | ||
Booij L, Van der Does AJ, Haffmans PM, Riedel WJ, Fekkes D, Blom MJ. The effects of high-dose and low-dose tryptophan depletion on mood and cognitive functions of remitted depressed patients. J Psychopharmacol. 2005;19(3):267–275. | ||
Uher R, Perlis RH, Henigsberg N, et al. Depression symptom dimensions as predictors of antidepressant treatment outcome: replicable evidence for interest-activity symptoms. Psychol Med. 2012;42(5):967–980. | ||
Rajkowska G. Histopathology of the prefrontal cortex in major depression: what does it tell us about dysfunctional monoaminergic circuits? Prog Brain Res. 2000;126:397–412. | ||
Harrison PJ. The neuropathology of primary mood disorder. Brain. 2002;125(pt 7):1428–1449. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.