")
Back to Journals » Research and Reports in Neonatology » Volume 7
Neonatal erythroderma – clinical perspectives
Received 13 September 2016
Accepted for publication 19 January 2017
Published 22 June 2017 Volume 2017:7 Pages 1—9
DOI https://doi.org/10.2147/RRN.S104667
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Robert Schelonka
Christina L Boull, Kristen P Hook
Department of Dermatology, Division of Pediatric Dermatology, University of Minnesota, Minneapolis, MN, USA
Abstract: Neonatal erythroderma is rare, but significant as it may be the initial manifestation of an array of infectious, metabolic, and genetic conditions, some of which are life-threatening. Initial management should focus on identifying and treating life threatening etiololgies and complications, including infection, and fluid, electrolyte, and temperature disturbances. Often, the etiology of erythroderma is difficult to quickly identify in the neonate, as there is significant clinical overlap between causative entities. Furthermore, rapid definitive diagnostic tests are lacking. Herein we provide a review of the specific clinical features and diagnostic tests, which can aid in making a correct diagnosis. Skin care for the erythrodermic infant is also discussed. We encourage subspecialist consultation when appropriate to aid in the evaluation, especially when initial testing is nondiagnostic.
Keywords: psoriasis, atopic dermatitis, cutaneous candidiasis
Introduction
Erythroderma, defined as skin redness involving >90% of the body surface area, is a rare phenotype in neonates. When present, it requires an immediate evaluation for the underlying pathology. Neonatal erythroderma may be a feature of a wide range of conditions (Table 1). Inflammatory disorders and disorders of cornification are the most common etiologies based on data from the two largest case reviews.1,2 A smaller series, however, reports infection as the leading cause.3 Inherited disorders of metabolism and immunodeficiency are rarer etiologies, but important to recognize early to prevent harmful sequelae.
 | Table 1 Causes of infantile erythroderma |
The mortality rate of the erythrodermic infant is significant, estimated at 16%–26%, but varies widely based on the underlying diagnosis.1,2 It is therefore essential to diagnose the cause rapidly, ensuring optimal treatment. Diagnosis, however, is challenging. Clinical features often overlap, and definitive, expedient laboratory testing is lacking. The average time to diagnosis is reported to be 3–11 months with up to 10% of patients without a confirmed diagnosis at 3–5 years.1,2
Often, the sentinel features of erythrodermic disorders do not manifest until later in childhood, and there may be considerable overlap of clinical features in the neonate. Pruritus, a hallmark of atopic dermatitis, may be absent in affected infants.4 Alopecia which is suggestive of ectodermal dysplasias and syndromic ichthyoses, is also present in up to half of erythrodermic infants with atopic dermatitis.1 Nail plate changes are seen in erythrodermic infants with both ichthyosiform and inflammatory skin conditions.2 Poor weight gain or failure to thrive, classically associated with immunodeficiency syndromes, has also been described in infants with psoriasis and ichthyoses.1 Diarrhea, which is a symptom of primary immunodeficiency disorders, may also be difficult to assess in a newborn.
The value of laboratory testing in the erythrodermic infant is variable. Skin biopsy, while generally recommended in the initial evaluation, was diagnostic in only 41%–45% of cases in two case series.1,2 In a retrospective series, three dermatopathologists were able to correctly identify the diagnosis in 69% of the 72 cases of infantile erythroderma.5 The utility of skin biopsy likely varies based on the underlying diagnosis and the skill of the pathologist.
Immunologic studies may also be difficult to interpret in infants. Immunoglobulin G levels are not reliable until at least 6 months of age, prior to which time they reflect maternal values.6 Peripheral eosinophilia (>0.5×10/L) is noted in infants with inflammatory and ichthyosiform conditions, as well as those with severe combined immunodeficiency syndrome (SCID), and staphylococcal scalded skin syndrome (SSSS). Similarly, total Immunoglobulin E (IgE) level and eosinophil count, considered markers of atopic disease, are not specific for this condition, and levels do not correlate with the degree of erythroderma.1 Genetic testing is very sensitive and specific and is recommended to confirm the diagnosis of ichthyoses or immunodeficiency disorders. Results, however, may take weeks to months to return, and are therefore unlikely to aid in immediate management.
Despite the many challenges to diagnosis, several clinical factors are valuable in establishing the etiology of infantile erythroderma. The timing of onset is perhaps the most important feature, as erythroderma present at birth has fewer potential causes than later presentation. Likewise, the presence of a collodion membrane focuses the diagnostic investigation toward disorders of cornification. The finding of severe dehydration, while less specific, also limits the differential diagnosis. Blistering may be seen in a variety of rarer entities, but is usually caused by SSSS. Family history should be obtained in all cases, and while nondiagnostic (not specific), may indicate underlying risk factors for genetic diseases, atopy, or psoriasis.
The initial focus of the primary pediatrician or neonatologist should include evaluation for infectious triggers and management of other life-threatening complications including dehydration, electrolyte disturbances, and temperature dysregulation. Tertiary care center referral is usually indicated. Herein we discuss the most common causes of infantile erythroderma and their associated clinical features. We caution that rarer etiologies, especially the disorders of metabolism, are not within the scope of this review, but should be considered when the initial workup is unrevealing. As the full complement of potential laboratory testing is extensive, erythrodermic neonates are at risk of iatrogenic anemia. Subspecialty consultation is, therefore, suggested not only to assist in diagnosis but also to aid in careful selection of laboratory testing.
Erythroderma with congenital onset
Most erythrodermic infants present prior to 4 months of age, at an average of 7–9 weeks of life.1 Erythroderma at birth, known as congenital erythroderma, is less common, but is strongly suggestive of ichthyosis, Netherton syndrome, or immunodeficiency syndromes. In the two largest case series of erythrodermic infants, a combined total of 93 patients were described. Twenty-nine (31%) infants were erythrodermic at birth.1,2 Twenty-seven (93%) of these were later diagnosed with ichthyosis or Netherton syndrome, and two with primary immunodeficiency syndromes.
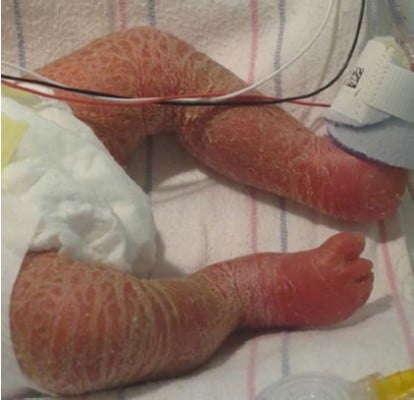
Both the nonsyndromic and syndromic ichthyoses can manifest with neonatal erythroderma. Nonsyndromic ichthyoses are isolated to the skin, while syndromic forms have multi-organ involvement.7 Congenital ichthyosiform erythroderma (CIE) is an autosomal recessive nonsyndromic ichthyosis that is most often associated with congenital erythroderma. Affected infants are often born with a collodion membrane, which is shed to reveal diffuse fine scale in a background of erythema. Skin may be thickened over the flexural surfaces.8 The CIE phenotype may also be seen in X-linked ichthyosis and in rare syndromic ichthyoses including Sjogren–Larsson syndrome, trichothiodystrophy, neutral lipid storage disease, and rarely metabolic syndromes (Figure 1).6,9,10
 | Figure 1 Infant with a metabolic disorder, widespread hyperkeratotic scale, but no collodion is present. |
More severe ichthyosis variants including harlequin ichthyosis and lamellar ichthyosis initially present with collodion membrane and diffuse erythema, but additional features of large plate-like scale and eversion of the eyelids and lips, referred to as ectropion and eclabion, respectively, are suggestive of the underlying diagnosis.
Genetic testing is available for syndromic and nonsyndromic ichthyoses. Multiple genetic mutations, including TGM-1, ALOX12B, ALOXE3, and ABCA12, have been implicated in the nonsyndromic ichthyoses.10 Gene panels are available commercially for testing, and many institutions perform their own genetic testing.
Congenital onset erythroderma with severe dehydration
Hypovolemic hypernatremia is the most common electrolyte disturbance seen in the erythrodermic neonate, but is usually mild and tends not to recur after initial treatment.
In the setting of congenital erythroderma with severe dehydration Netherton syndrome or immunodeficiency disorders should be suspected. Infectious triggers, such as SSSS, must also be considered, but congenital onset is rare.
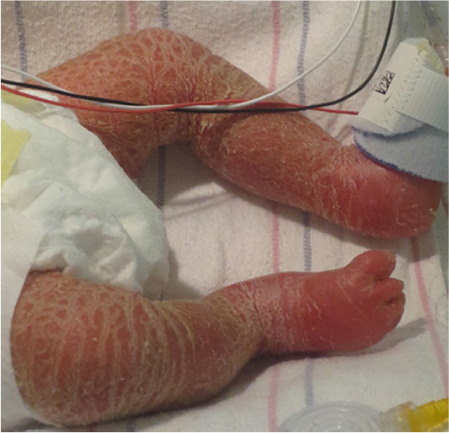
Netherton syndrome results from a mutation in the SPINK5 (serine protease inhibitor Kazal type 5) gene encoding the serine protease inhibitor lympho-epithelial Kazal-type-related inhibitor, which is expressed in epithelial tissue.11 Netherton patients often have failure to thrive with hypovolemic hypernatremia during the newborn period.1,2 Initially, the skin may be diffusely red and scaly (Figure 2). Hair is sparse, and examination of the hair shaft, especially the eyebrows, may reveal trichorrhexis invaginata or a ball-in-socket morphology.12 This characteristic hair change, however, may not be present until after 10 months of age.1 Skin may later develop a unique double-edged scale with a serpiginous border, referred to as ichthyosis linearis circumflexa. Growth restriction and frequent skin infections are common.13 Case series report increased atopic tendencies including increased IgE levels, environmental and food allergies, and itch.13,14 Immune deficiencies with abnormal immunoglobulins, increased IgE levels, or abnormal lymphocyte parameters have been reported, but are not consistent in all patients.10,15 Skin biopsy with immunohistochemical staining for lympho-epithelial Kazal-type-related inhibitor is helpful in making the diagnosis.5,16
 | Figure 2 Neonate with Netherton syndrome. Note: Skin is diffusely erythematous with fine scaling. |
Immunodeficiency disorders are also associated with congenital erythroderma and dehydration, and therefore carry a high risk of mortality.1,17 Affected infants develop diarrhea with failure to thrive and recurrent opportunistic viral, bacterial, or fungal infections. Pneumonia is especially common. SCID is a group of disorders that may be inherited in an autosomal recessive or X-linked recessive pattern. Multiple causative mutations have been identified and vary based on inheritance patterns. Patients have a deficiency in T lymphocytes with variable involvement of B lymphocytes and natural killer cells. Most affected infants do not have symptoms immediately at birth, but develop typical findings by 3–6 months of age.18 Infants with Omenn syndrome, a severe variant of SCID, will additionally show hepatosplenomegaly, lymphadenopathy, leukocytosis, and eosinophilia.19 Immunologic studies demonstrate increased numbers of oligoclonal T cells, decreased B cells and immunoglobulins, and elevated IgE level.18 Mutations in the recombination activating genes 1 and 2 (RAG1 and 2) are the most common cause.19,20
T-cell immunodeficiency syndromes like SCID and Omenn can be further complicated by neonatal graft-versus-host disease (GvHD) from maternal engraftment.21,22 A morbilliform eruption, which progresses to erythroderma, is characteristic, and skin biopsy shows features of GvHD. While maternal–fetal GvHD has been reported in immunocompetent infants due to intrauterine or postnatal exchange transfusion, this medical history should be apparent. In an infant without history of transfusion, the presence of GvHD should prompt investigation for T-cell immunodeficiencies.23
Infectious etiologies are rarer causes of congenital erythroderma. The history may be more likely to reflect erythroderma that developed within the first hours/days of life, but was not truly present at birth. Congenital cutaneous candidiasis arises from intrauterine exposure, often candidal chorioamnionitis. The rash is maculopapular with scattered pustules, and rarely vesicles. Lesions develop within the first 6 days of life, spreading from the trunk to acral sites, including the palms and soles.24 Mucous membrane involvement or thrush is uncommon. Premature infants are at increased risk for systemic infections with candida in the blood, urine, or cerebrospinal fluid, often triggering leukocytosis and respiratory distress. A severe burn-like erythema has been described in this group and is associated with a worse prognosis. In one series, the mortality rate for cutaneous candidiasis was 40% in infants <1000 g, which improved to 8% in infants >1000 g.25 A KOH scraping or yeast culture from the skin is helpful in establishing the diagnosis.
Herpes simplex virus infection is a rare cause of congenital erythroderma. In most cases, vesicles or erosions are localized, but in the intrauterine variant, infants may present with erythroderma and crusted erosions, sometimes without vesicles.6,26,27 Viral cultures, polymerase chain reaction, direct fluorescent antigen testing, or Tzanck smear will confirm the diagnosis. Central nervous system involvement is often present in widespread disease.6
While the differential diagnosis for congenital onset erythroderma, includes SSSS, erythrodermic congenital psoriasis, and diffuse cutaneous mastocytosis, all are exceptionally rare with only case reports described in the literature.12,15,28–30 Further discussion is provided below.
Noncongenital onset erythroderma
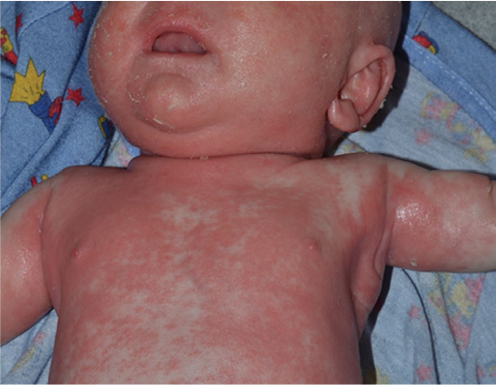
Atopic dermatitis and seborrheic dermatitis are the most common causes of noncongenital erythroderma.1,2,5 Onset is typically at 6–8 weeks of life.1,10 When widespread, the two conditions may be difficult to distinguish. The predilection for scalp and skin fold areas favors a diagnosis of seborrheic dermatitis, while sparing of the diaper area is more consistent with atopic dermatitis.5 The presence of a family history of atopy increases the likelihood of atopic dermatitis.2
Psoriasis may also first present in early infancy. The scalp and diaper area may be favored (Figure 3) as in seborrheic dermatitis, but periumbilical involvement is specific for psoriasis.5 Congenital psoriasis is uncommon with only nine case reports in the literature.31 In a series of patients with infantile psoriasis, 7/9 developed lesions at age 2–9 months with the other 2 infants developing lesions in the first week of life.32 Plaque psoriasis is the most typical infantile variant, with rare cases of erythroderma or pustular lesions.28,30–32 A positive family history of psoriasis was noted in 89% of patients with infantile psoriasis.32 The clinical appearance of erythrodermic psoriasis mimics CIE, and a biopsy may help to distinguish these entities. Skin biopsy may not be useful initially in differentiating erythrodermic psoriasis from dermatitis, but should eventually reveal the characteristic histologic findings.5
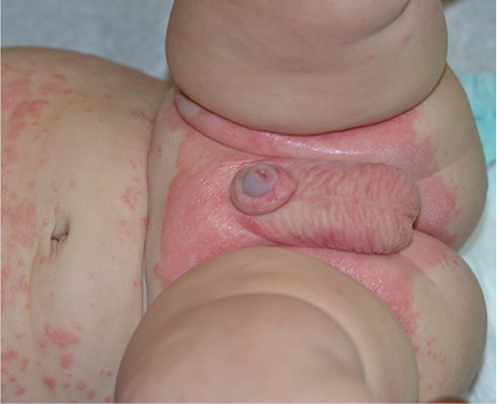 | Figure 3 Psoriasis in infants often localizes to the diaper area. Note: Periumbilical involvement is also common. Plaques are well demarcated. |
Collodion membrane
A collodion membrane is a tight, glossy encasement produced in utero by abnormal epidermal desquamation (Figure 4). It must be distinguished from a thick hyperkeratotic vernix, which may sometimes be noted in erythrodermic infants. The collodion is shed within the first 3 weeks of life to reveal the underlying skin phenotype. Associated diagnoses include autosomal recessive forms of ichthyosis, and rarely Netherton syndrome, trichothiodystrophy, Conradi–Hunermann, Gaucher disease type II, Sjogren–Larsson, and neutral lipid storage disease.9,33 When congenital erythroderma is seen in conjunction with a collodion, it is a very helpful clue to an ichthyosis diagnosis. Skin biopsy is usually not diagnostic in ichthyosiform conditions unless Netherton syndrome is suspected. A hearing screen and eye examination are recommended to evaluate for signs of keratitis–ichthyosis–deafness syndrome, although patients with ichthyoses often have abnormal hearing screens due to keratotic debris in the auditory canals. Fatty alcohol dehydrogenase activity level is used to screen for Sjogren–Larsson disease, which is associated with mental retardation and spasticity.34 A microscopic hair shaft examination under polarized light reveals characteristic light and dark banding in trichothiodystrophy. In neutral lipid storage disease, a peripheral blood smear detects lipids in granulocytes and monocytes.10
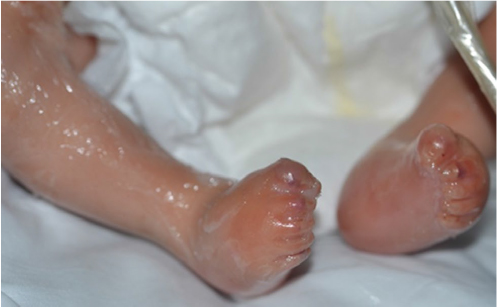 | Figure 4 A collodion membrane encases this infant’s body, including his feet and toes. |
Blistering
Neonatal erythroderma with blistering represents SSSS until proven otherwise. Rare cases of in utero SSSS have been described, producing congenital erythroderma and erosions.35 More commonly, onset is later, after maternal antibodies diminish, and is associated with widespread erythema and fever. The skin desquamates superficially, with most prominent involvement in skin folds. Nikolsky sign is positive. Skin detachment is caused by hematogenous spread of exfoliative toxins A or B, proteases that cleave the epidermis at the level of the stratum corneum. Blister fluid is sterile, but cultures of the nares, conjunctivae, umbilicus, or perirectal area often grow Staphylococcus aureus. Infants have a more severe course compared to toddlers due to high rates of sepsis, dehydration, and associated electrolyte abnormalities.35 It is uncertain as to why neonates are more susceptible to SSSS, but differences in barrier function between adult and infant skin, in addition to lower renal clearance of exfoliative toxins in infants have been suggested factors.36 Early hospital admission for antibiotics, fluid management, and skin cares is critical to decrease mortality.
Rarely, congenital syphilis produces diffuse erythema and bullae. The infection, contracted late in pregnancy or during delivery, can produce erosions, acral bullae, and dermatitis beginning at 6–8 weeks of age. Condyloma lata may be noted on mucous membranes. Low birth weight, hepatosplenomegaly, and anemia are other associated features.5,37 Serologies in mother and infant confirm the diagnosis.
Epidermolytic ichthyosis, previously referred to as bullous congenital ichthyosis or epidermolytic hyperkeratosis, may cause widespread blistering and erythema, which evolves to reveal scaling and hyperkeratotic plaques.10 Skin biopsy demonstrates the characteristic epidermolysis. Epidermolytic ichthyosis is usually inherited in an autosomal-dominant pattern as the result of a keratin 1 or 10 mutation.10
Diffuse cutaneous mastocytosis is a rare cause of neonatal erythroderma. Mast cell infiltrate the epidermis, giving it a “doughy” appearance. Unlike the other causes for neonatal erythroderma, scaling is not present. Infants are prone to urticaria, flushing, and diarrhea, and may develop bullae at sites of skin friction. Rubbing of the skin also produces raised wheals, known as Darier’s sign. Skin biopsy is diagnostic, revealing many mast cells in the skin. Management is aimed at mast cell stabilization with antihistamines. Mast cell degranulators including culprit medications and foods should be avoided, and the family should be counseled on the signs of anaphylaxis. Evaluation for systemic involvement includes a blood count, liver function tests, and bone scan.15 Bone marrow biopsy is not recommended unless there is persistence or worsening disease.38 Even in severe cases, systemic involvement is unusual. Bullae rarely persist past 1–3 years of age with resolution of all skin changes by puberty in 90% of patients.39
Metabolic and nutritional causes
Acrodermatitis enteropathica is a cutaneous manifestation of zinc deficiency in the neonate. It can be inherited due to a deficient zinc transporter in the intestine, or can be acquired. Low concentrations of zinc in breast milk or increased zinc requirements and low zinc stores, mainly in premature infants, are all potential causes. The source of the deficiency dictates the timing of onset. A scaling dermatitic eruption begins in the perioral and genital areas then spreads to produce erythroderma and erosions. If untreated, alopecia and diarrhea follow. Serum zinc levels should be assessed, but may not reliably detect a deficiency. Skin biopsy will show nonspecific features common to disorders of nutritional deficiency. Zinc supplementation corrects the symptoms within days to weeks.40
Holocarboxylase synthetase deficiency (HCSD) and biotinidase deficiency are uncommon causes of neonatal erythroderma. HCSD presents within the first week of life with ketoacidosis and coma. Biotinidase deficiency does not present until an average of 3 months of age, and is associated with lethargy and peeling scale similar to that seen in acrodermatitis enteropathica.15 Testing for blood and urine amino and organic acids is needed to confirm the diagnosis.10
Essential fatty acid deficiency is a reported etiology for neonatal erythroderma. It is typically triggered by gastrointestinal malabsorption, sometimes the result of cystic fibrosis.15
Leiner’s disease
Leiner’s disease is an eponym used historically to describe infants with erythroderma, diarrhea, and failure to thrive.41 Authors speculate as to the condition which afflicted Leiner’s initial series of patients, but biotin deficiency, Netherton syndrome, and immunodeficiency syndromes have all been suggested.42 As the term is nonspecific, it is no longer considered an adequate diagnosis, but may still be encountered in the literature.15,41,42
Family history
When the cause of erythroderma is uncertain, family history and, occasionally, parental examination may point toward the diagnosis. Parental consanguinity raises concern for autosomal recessive forms of ichthyosis, disorders of metabolism, and immunodeficiency syndromes. A family history of atopic tendencies or psoriasis increases the likelihood of these conditions in the infant. In one case series, a family history of erythroderma was noted in more than half of the infant ultimately diagnosed with ichthyosis, but it is unclear if these family members were similarly affected.2
Initial evaluation and management
There are no consensus guidelines for the evaluation and diagnosis of the erythrodermic infant. Suggestions are based on the authors’ clinical experience and other published recommendations on the topic. In Figure 5, we provide an algorithm to aid in diagnosis based on clinical features. We caution that while the most common etiologies are highlighted, additional rare causes of neonatal erythroderma are not included. Initial diagnostic workup is included in Table 2.
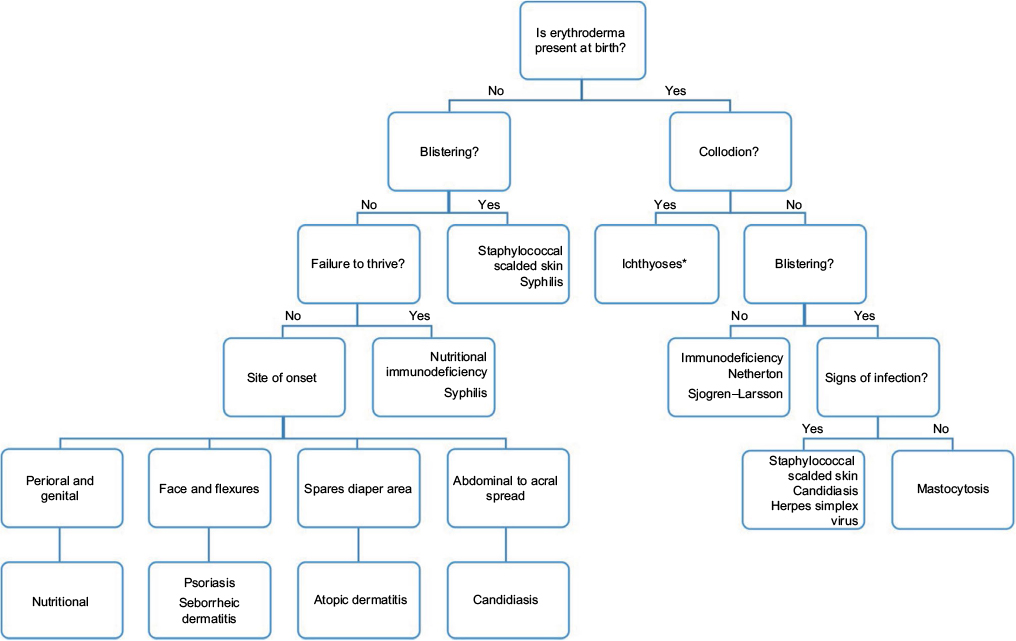 | Figure 5 Algorithm for diagnosis of the erythrodermic infant. Note: *Ichthyoses include congenital ichthyosiform erythroderma, lamellar ichthyosis, harlequin ichthyosis, and rarely Sjogren–Larsson, trichothiodystrophy, and neutral lipid storage disease. |
 | Table 2 Initial diagnostic testing for the erythrodermic infant Note: aSubspecialty consultation suggested. |
Baseline laboratory studies should be collected immediately upon presentation and include a complete blood count, comprehensive metabolic panel, and blood cultures. Bacterial and viral swabs collected from lesional skin and orifices help to identify infectious etiologies. Photographs obtained throughout the initial course can be very useful to consulting teams, especially if the infant is born in a community hospital and later transferred to a tertiary care center. Skin biopsies should be collected at the time of presentation, which are most informative when read by a dermatopathologist who is accustomed to diagnosing pediatric skin disease. If not available within the institution, specimens may be sent to outside laboratories for expert review. Additional tissue should be collected for bacterial and fungal culture. Imaging studies rarely aid in the diagnosis of the erythrodermic infant. Plain films of the chest, however, may detect a missing thymic shadow in the case of T-cell immunodeficiency syndromes. If an immunodeficiency or metabolic syndrome is suspected, the assistance of the appropriate subspecialist is of great value in both identifying the proper testing and interpreting the results. Testing for disorders of metabolism and zinc deficiency may be included as part of the initial laboratory series if neurological symptoms or appropriate lesion morphology is present. Management strategies for the erythrodermic infant are contingent on the primary etiology.
General measures
In all cases, life-threatening diagnoses and complications including dehydration, electrolyte imbalances, hyperthermia, and infection must be quickly identified and treated. Most erythrodermic infants receive an initial sepsis evaluation and empiric treatment with antimicrobial medications. Erythrodermic infants have high metabolic demands with failure to thrive noted in 69% of patients in one series.1 Increased transepidermal water is a source of energy expenditure, as it is accompanied by loss of heat.43,44 Proteins are also lost through the dysfunctional skin barrier. The early involvement of nutritional services is suggested to allow for adequate supplementation of protein and calories. Incubators decrease the loss of both heat and water from the skin, but the infant’s temperature must be monitored closely to prevent hyperthermia caused by hypohidrosis.43 Recommendations vary in regard to incubator humidification, but typically a humidity of at least 60% is suggested.43,45 When an inflammatory etiology is suspected, twice-daily application of topical low-to-mid potency corticosteroids in an ointment base is suggested, and may be both diagnostic and therapeutic.
Specific measures
Most infants with exfoliative erythroderma benefit from frequent bathing followed by application of thick, bland emollients, such as petrolatum, to prevent excessive transepidermal water loss. A Cochrane review found that the application of topical ointment in preterm infants increased the risk of coagulase-negative staphylococcal and nosocomial infections, but data and methodology from the compiled studies was variable.46 In collodion infants, xerosis increases skin fissuring, further worsening barrier function.
Precautions
The use of sterile petrolatum decreased the risk of infection in collodion infants in one study.43,45 Multi-use containers of topical agents may become contaminated with microorganisms leading to cutaneous or systemic infections, and should be avoided when possible.47 Nursing staff should not use wooden tongue depressors to apply topical agents, as these have been associated with invasive fungal infections in preterm infants.48
Contraindications
Topical calcineurin inhibitors and creams containing urea, silver sulfadiazine, and salicylic acid should not be applied to erythrodermic infants due to risk of systemic absorption and toxicity.49–52
Conclusion
Neonatal erythroderma, while rare, represents a heterogeneous collection of infectious, immunologic, inflammatory, ichthyosiform, and metabolic disorders. Sequelae may include neurologic compromise and death if the cause is not appropriately diagnosed and treated. As the underlying etiology is often difficult to determine promptly, immediate management should focus on identifying potential infections and treating life-threatening complications. Subspecialty consultation and tertiary care referral is indicated in most cases.
Disclosure
The authors report no conflicts of interest in this work.
References
Pruszkowski A, Bodemer C, Fraitag S, Teillac-Hamel D, Amoric JC, de Prost Y. Neonatal and infantile erythrodermas: a retrospective study of 51 patients. Arch Dermatol. 2000;136(7):875–880. | ||
Al-Dhalimi MA. Neonatal and infantile erythroderma: a clinical and follow-up study of 42 cases. J. Dermatol. 2007;34(5):302–307. | ||
Sarkar R. Neonatal and infantile erythroderma: ‘the red baby’. Indian J Dermatol. 2006;51(3):178. | ||
Yates VM, Kerr RE, MacKie RO. Early diagnosis of infantile seborrhoeic dermatitis and atopic dermatitis – clinical features. Br J Dermatol. 1983;108(6):633–638. | ||
Leclerc-Mercier S, Bodemer C, Bourdon-Lanoy E, et al. Early skin biopsy is helpful for the diagnosis and management of neonatal and infantile erythrodermas. J Cutan Pathol. 2010;37(2):249–255. | ||
Metz BJ, Levy ML. Erythrodermas, immunodeficiency, and metabolic disorders. In: Eichenfield LF, Frieden IJ, Zaenglein A, Mathes E, Esterly NB, editors. Neonatal Dermatology. Philadelphia, PA, USA: Elsevier Health Sciences; 2007:267–283. | ||
Oji V, Tadini G, Akiyama M, et al. Revised nomenclature and classification of inherited ichthyoses: results of the First Ichthyosis Consensus Conference in Soreze 2009. J Am Acad Dermatol. 2010;63(4):607–641. | ||
firstskinfoundation.org [homepage on the Internet]. First Foundation for ichythyosis and related skin types. Available from: http://www.firstskinfoundation.org. Accessed September 1, 2016. | ||
Craiglow BG. Ichthyosis in the newborn. Semin Perinatol. 2013;37(1):26–31. | ||
Fraitag S, Bodemer C. Neonatal erythroderma. Curr Opin Pediatr. 2010; 22(4):438–444. | ||
Chavanas S, Bodemer C, Rochat A, et al. Mutations in SPINK5, encoding a serine protease inhibitor, cause Netherton syndrome. Nat Genet. 2000;25(2):141–142. | ||
Griffiths WA. Leigh IM, Marks R. Disorders of Keratinization. London, Edinburgh, Boston, Melboume, Paris, Berlin,Vienna: Blackwell Scientific Publications; 1992;2:1362–1365. | ||
Komatsu N, Saijoh K, Jayakumar A, et al. Correlation between SPINK5 gene mutations and clinical manifestations in Netherton syndrome patients. J Invest Dermatol. 2008;128(5):1148–1159. | ||
Bitoun E, Chavanas S, Irvine AD, et al. Netherton syndrome: disease expression and spectrum of SPINK5 mutations in 21 families. J Invest Dermatol. 2002;118(2):352–361. | ||
Hoeger PH, Harper JI. Neonatal erythroderma: differential diagnosis and management of the “red baby”. Arch Dis Child. 1998;79(2):186–191. | ||
Ong C, O’Toole EA, Ghali L, et al. LEKTI demonstrable by immunohistochemistry of the skin: a potential diagnostic skin test for Netherton syndrome. Br J Dermatol. 2004;151(6):1253–1257. | ||
Smitt JH, Kuijpers TW. Cutaneous manifestations of primary immunodeficiency. Curr Opin Pediatr. 2013;25(4):492–497. | ||
Hague RA, Rassam S, Morgan G, Cant AJ. Early diagnosis of severe combined immunodeficiency syndrome. Arch Dis Child 1994;70(4):260–263. | ||
Pupo RA, Tyring SK, Raimer SS, Wirt DP, Brooks EG, Goldblum RM. Omenn’s syndrome and related combined immunodeficiency syndromes: diagnostic considerations in infants with persistent erythroderma and failure to thrive. J Am Acad Dermatol. 1991;25(2):442–446. | ||
Katugampola RP, Morgan G, Khetan R, Williams N, Blackford S. Omenn’s syndrome: lessons from a red baby. Clin Exp Dermatol. 2008; 33(4):425–428. | ||
Denianke KS, Frieden IJ, Cowan MJ, Williams ML, McCalmont TH. Cutaneous manifestations of maternal engraftment in patients with severe combined immunodeficiency: a clinicopathologic study. Bone Marrow Transplant. 2001;28(3):227–234. | ||
Alain G, Carrier C, Beaumier L, Bernard J, Lemay M, Lavoie A. In utero acute graft-versus-host disease in a neonate with severe combined immunodeficiency. J Am Acad Dermatol. 1993;29(5):862–865. | ||
Hentschel R, Broecker EB, Kolde G, et al. Intact survival with transfusion-associated graft-versus-host disease proved by human leukocyte antigen typing of lymphocytes in skin biopsy specimens. J Pediatr. 1995;126(1):61–64. | ||
Glassman BD, Muglia JJ. Widespread erythroderma and desquamation in a neonate. Congenital cutaneous candidiasis (CCC). Arch Dermatol. 1993;129(7):899–902. | ||
Darmstadt GL, Dinulos JG, Miller Z. Congenital cutaneous candidiasis: clinical presentation, pathogenesis, and management guidelines. Pediatrics. 2000;105(2):438–444. | ||
Hutto C, Arvin A, Jacobs R, et al. Intrauterine herpes simplex infections. J Pediatr. 1987;110(1):97–101. | ||
Honig PJ, Brown D. Congenital herpes simplex infection initially resembling epidermolysis bullossa. J Pediatr. 1982;101(6):958–960. | ||
Henriksen L, Zachariae H. Pustular psoriasis and arthritis in congenital psoriasiform erythroderma. Dermatologica. 1972;144(1):12–18. | ||
Oranje AP, Soekanto W, Sukardi A, Vuzevski VD, Willigen AV, Afiani HM. Diffuse cutaneous mastocytosis mimicking staphylococcal scalded-skin syndrome: report of three cases. Pediatric Dermatol. 1991;8(2):147–151. | ||
Salleras M, Sanchez-Regaña M, Umbert P. Congenital erythrodermic psoriasis: case report and literature review. Pediatric Dermatol. 1995; 12(3):231–234. | ||
Lehman JS, Rahil AK. Congenital psoriasis: case report and literature review. Pediatric Dermatol. 2008;25(3):332–338. | ||
Farber EM, Mullen RH, Jacobs AH, Nail L. Infantile psoriasis: a follow-up study. Pediatric Dermatol. 1986;3(3):237–243. | ||
Irvine AD, Paller AS. Disorders of cornification (ichthyosis). In: Eichenfield LF, Frieden IJ, Zaenglein A, Mathes E, Esterly NB, eds. Neonatal dermatology. Philadelphia, PA, USA: Elsevier Health Sciences; 2007:285–310. | ||
Alio AB, Bird LM, McClellan SD, Cunningham BB. Sjogren-Larsson syndrome: a case report and literature review. Cutis. 2006;78(1):61–65. | ||
Longhead JL. Congenital staphylococcal scalded skin syndrome. Pediatr Infect Dis J. 1992;11:413. | ||
Ladhani S, Joannou CL, Lochrie DP, Evans RW, Poston SM. Clinical, microbial, and biochemical aspects of the exfoliative toxins causing staphylococcal scalded-skin syndrome. Clin Microbiol Rev. 1999;12(2):224–242. | ||
Chawla V, Pandit PB, Nkrumah FK. Congenital syphilis in the newborn. Arch Dis Child. 1988;63(11):1393–1394. | ||
Heide R, Zuidema E, Beishuizen A, et al. Clinical aspects of diffuse cutaneous mastocytosis in children: two variants. Dermatology. 2009; 219(4):309–315. | ||
Willigen AH, Oranje AP. Diffuse cutaneous mastocytosis. Br J Dermatol. 1990;123(2):267–268. | ||
Maverakis E, Fung MA, Lynch PJ, et al. Acrodermatitis enteropathica and an overview of zinc metabolism. J Am Acad Dermatol. 2007;56(1):116–124. | ||
Leiner C. Erythroderma desquamativa (universal dermatitis of children at the breast). Br J Dis Child. 1908;5:244–251. | ||
Dhar S, Banerjee R, Malakar R. Neonatal erythroderma: diagnostic and therapeutic challenges. Indian J Dermatol. 2012;57(6):475–478. | ||
Prado R, Ellis LZ, Gamble R, Funk T, Arbuckle HA, Bruckner AL. Collodion baby: an update with a focus on practical management. J Am Acad Dermatol. 2012;67(6):1362–1374. | ||
Moskowitz DG, Fowler AJ, Heyman MB, et al. Pathophysiologic basis for growth failure in children with ichthyosis: an evaluation of cutaneous ultrastructure, epidermal permeability barrier function, and energy expenditure. J Pediatr. 2004;145(1):82–92. | ||
Taïeb A, Labrèze C. Collodion baby: what’s new. J Eur Acad Dermatol Venereol. 2002;16(5):436–437. | ||
Conner JM, Soll R, Edwards WH. Topical ointment for preventing infection in preterm infants. Cochrane Database Syst Rev. 2004;(1):CD001150. | ||
Gastmeier P, Groneberg K, Weist K, Rüden H. A cluster of nosocomial Klebsiella pneumoniae bloodstream infections in a neonatal intensive care department: identification of transmission and intervention. Am J Infect Control. 2003;31(7):424–430. | ||
Mitchell SJ, Gray J, Morgan ME, Hocking MD, Durbin GM. Nosocomial infection with Rhizopus microsporus in preterm infants: association with wooden tongue depressors. Lancet. 1996;348(9025):441–443. | ||
Allen A, Siegfried E, Silverman R, et al. Significant absorption of topical tacrolimus in 3 patients with Netherton syndrome. Arch Dermatol. 2001; 137(6):747–750. | ||
Beverley DW, Wheeler D. High plasma urea concentrations in collodion babies. Arch Dis Child. 1986;61(7):696–698. | ||
Yamamura S, Kinoshita Y, Kitamura N, Kawai S, Kobayashi Y. Neonatal salicylate poisoning during the treatment of a collodion baby. Clin Pediatr. 2002;41(6):451–452. | ||
Viala J, Simon L, Le Pommelet C, Philippon L, Devictor D, Huault G. Agranulocytosis after application of silver sulfadiazine in a 2-month old infant. Arch Pediatr. 1997;4(11):1103–1106. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.