")
Back to Journals » Clinical, Cosmetic and Investigational Dermatology » Volume 15
NEMO Gene Mutations in Two Chinese Females with Incontinentia Pigmenti
Authors Jiang J, Zeng J, He Q, Yang J, Wang S, Zhang Z
Received 1 March 2022
Accepted for publication 21 April 2022
Published 5 May 2022 Volume 2022:15 Pages 815—821
DOI https://doi.org/10.2147/CCID.S363683
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Jeffrey Weinberg
Jingjing Jiang,1 Junjie Zeng,2 Qi He,1 Jiao Yang,1 Shenglan Wang,1 Zhengzhong Zhang1
1Department of Dermatology, Affiliated Hospital of North Sichuan Medical College, Nanchong, Sichuan Province, People’s Republic of China; 2Department of Dermatology, Taikang Sichuan Southwest Hospital Company Limited, Chengdu, Sichuan Province, People’s Republic of China
Correspondence: Zhengzhong Zhang, Department of Dermatology, Affiliated Hospital of North Sichuan Medical College, 1 Maoyuan South Road, Shunqing District, Nanchong, Sichuan Province, 637000, People’s Republic of China, Tel +8618080339898, Email [email protected]
Purpose: To identify the mutations of the NEMO gene in two Chinese females with incontinentia pigmenti.
Patients and Methods: Patients were both from Nanchong, Sichuan Province. Genomic DNA was extracted from the peripheral blood of patients and patient 1ʹs father. The mutations of the NEMO gene in patient 1 by GAP polymerase chain reaction and Sanger sequencing and her father were detected. NEMO-specific polymerase chain reaction and Sanger sequencing were used to identify the NEMO gene mutation in patient 2.
Results: DNA analysis identified a rare frameshift mutation, c.723_c.724insCAGG(p.A242QfsX15) in exon 5 of the NEMO gene in patient 1 with a family history but not in her healthy father. The common deletion of exons 4– 10 of the NEMO gene was found in sporadic patient 2.
Conclusion: Our data revealed that the rare frameshift mutation, c.723_c.724insCAGG(p.A242QfsX15) in exon 5 of the NEMO gene in patient 1 and the deletion of exons 4– 10 of the NEMO gene in patient 2 could cause the occurrence of IP. Genetic testing is helpful for early diagnosis and genetic counseling for families.
Keywords: Chinese, incontinentia pigmenti, mutation, NEMO gene, NF-κB pathway
Introduction
Incontinentia pigmenti (IP, OMIM#308300) is a rare X-linked dominant genodermatosis resulting from the mutations of the NEMO gene.1 The NEMO gene is located on Xq28 structured in 10 exons and encodes a kappa light polypeptide gene enhancer in B cells, kinase gamma, which has a key role in the modulation of nuclear transcription factor –kappa B (NF-κB).2,3 There is a nonfunctional copy of the gene, NEMO pseudogene structured in exons 3–10, opposite in direction to the NEMO gene, existing downstream of the NEMO gene. Both of them have a common sequence NEMO 4–10. The recurrent genomic deletion of exons 4–10 within the NEMO gene accounted for approximately 90% of the mutations.2,4 Other mutations such as point mutations, insert mutations, and so on have also been reported but not many. Almost all types of mutations of the NEMO gene caused a frameshift and premature protein truncation, resulting in loss of NEMO function and creating a susceptibility to cell apoptosis in response to TNF-α.4,5
This disorder affects not only the skin but also other ectodermal tissues containing the eyes, teeth, hair, nail, and central nervous system (CNS).6,7 The prominent skin lesions begin in infancy and there are four progressive stages of the skin lesions but sometimes overlapping: stage I–vesicular, stage II–verrucous, stage III-hyperpigmented with linear hyperpigmentation following the Blaschko’s lines, and stage IV-hypopigmented with linear hypopigmentation and atrophy.1,6 The diagnosis of IP is based primarily on the characteristic skin lesions besides histology and genetic analysis could help confirm IP.8
In China, most reports of the NEMO gene mutations in IP were the NEMO gene rearrangement, the deletion of exons 4–10 of the NEMO gene. Minor mutations were rarely reported. In this study, we performed gene testing on two Chinese females with IP to expand our knowledge of this disease and explore more new mutations of the causative gene.
Materials and Methods
The two IP female patients were from Nanchong city, Sichuan province, and diagnosed by the Department of Dermatology, Affiliated Hospital of North Sichuan Medical College. The diagnoses of IP in the two patients were based mainly on the clinical manifestations (according to the clinical criterion of Landy and Donnai6) and one of them was confirmed by the histology of the typical skin lesions. Gene detection was used to find the mutations, also in some way to confirm the diagnosis. After we obtained written informed consent from their guardians, blood samples of patients and patient 1ʹs father were collected. Genomic DNA was extracted from peripheral blood leukocytes. The institutional approval was required to publish the case details and the study was approved by the Medical Ethics Committee of Affiliated Hospital of North Sichuan Medical College (2022ER133-1). Our study was carried out in complies with the declaration of Helsinki.
The blood samples of patient 1 and her father were sent to Chigene (Beijing) Translational Medical Research Center Co.Ltd. The rearrangement analysis in patient 1 was carried out by using GAP polymerase chain reaction (PCR). A NEMO-specific primer referred to as In2S was designed. JF3R and Rep3s as non-specific primers were completely complementary to the NEMO gene and the NEMO pseudogene. In2S and JF3R were used to amplify the deletion of exons 4–10 of the NEMO gene. The products after PCR amplification were identified by agarose gel electrophoresis. The product of Rep3s and JF3R was 733 bp as the internal reference band, which must be present in the tested sample, negative control, and positive control. The product of In2S and JF3R was 2243 bp, appearing in positive control. If 2243 bp showed in the tested sample, it suggested the deletion of exons 4–10 of the NEMO gene. For the tested sample without this deletion, specific primers were designed for all coding regions of the NEMO gene for PCR amplification to screen for other NEMO mutations. The PCR amplified products were identified by agarose gel electrophoresis. The product was consistent with the result of the targeted band and Sanger sequencing was performed in patient 1 and her father. The chromas 2.0 was used to compare with the coding region of the gene and analyze the sequence.
The blood sample of patient 2 was sent to Shanghai Gengsi Bio-technology co., LTD. NEMO-specific PCR was performed by using specific primers NEMO-E03-10del_F/NEMO-E03-10del_R to identify the exons 4–10 deletion of the NEMO gene. The NEMO-specific PCR product in patient 2 was identified by 1.5% agarose gel electrophoresis, purified, and isolated. Then Sanger sequencing was performed to verify as NEMO gene specific fragment. For the product indicating the common deletion, we used primers NEMO-E03-10del_F-1/NEMO-E03-10del_R-1 to verify the breaking points. The Applied Biosystems 3730xl DNA Analyzers was used to analyze the sequence.
Case Report
Patient 1 with a family history was a 15-year-old girl manifesting blisters around the trunk at birth. The skin lesions gradually developed typical liner whorled hyperpigmented and hypopigmented lesions following Blaschko’s lines on the trunk and extremities (Figure 1A and B). Besides she had sparse hair and many decayed teeth. Patient 2 age 2 months, a sporadic case, developed skin blisters and erythematous papules over the right side of the trunk and the extremities shortly after birth. Then the skin lesions developed verrucous and linear hyperpigmented lesions tracing Blaschko’s lines on the trunk and legs (Figure 1C and D). Histology of the lesion on the leg revealed the typical changes of IP in patient 1 (Figure 2).
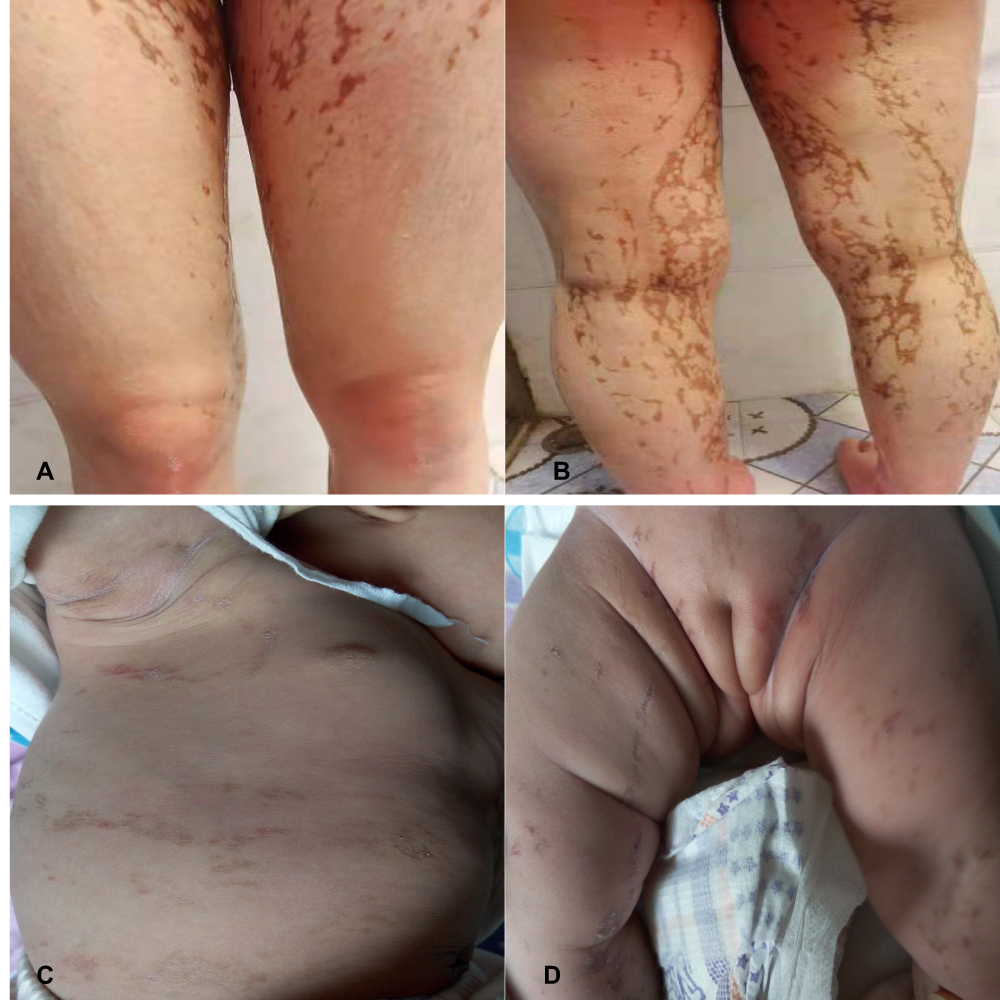 |
Figure 1 Clinical features. (A and B) Brownish linear and whorled hyperpigmentation along Blaschko’s lines on the legs in patient 1. (C and D) Verrucous and linear hyperpigmented lesions and tracing Blaschko’s lines on the trunk and legs in patient 2. |
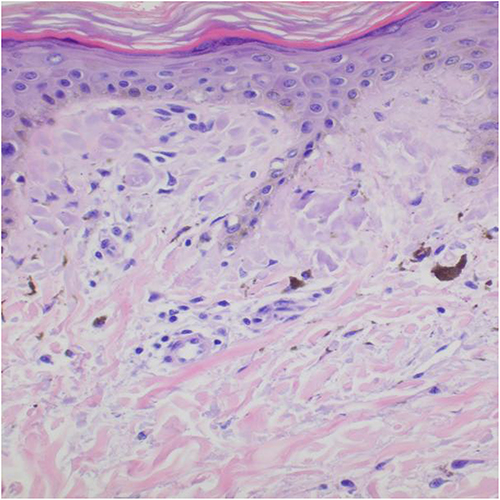 |
Figure 2 Histological image of patient 1. The net-like keratinized epidermis, epidermal hyperplasia, marked melanin incontinence with many melanophages in the dermis, and infiltration of a few lymphocytes around blood vessels (HE, x200). |
Results
GAP PCR demonstrated that there was no deletion of exons 4–10 of the NEMO gene in patient 1 (Figure 3A). While Sanger sequence analysis detected a rare heterozygous frameshift mutation, c.723_724insCAGG(p.A242QfsX15) in exon 5 of the NEMO gene (NM_001099856.2) in patient 1, which was not found in her healthy father (Figure 3B and C). The insertion of CAGG at the position between 723 and 724 caused the change of amino acid at position 242 from Ala (A) to Gln (Q) and caused the termination codon to appear earlier at position 256, resulting in NEMO truncated protein. It was a great pity that the mother of patient 1 who had the same skin lesions but milder did not participate in the study because of some contradictions in the family. NEMO-specific PCR showed a 2000 bp band in patient 2, indicating a common deletion of exons 4–10 of the NEMO gene (NM_001099856.3)(Figure 3D). Sequence analysis showed that the amplified fragment contained intron 2 of the NEMO gene, showing that the amplified product was NEMO gene specific fragments. The amplified sequence contained the partial sequence of exon 4 of the NEMO gene and the partial downstream sequence of exon 10 of the NEMO gene, confirming the deletion of exons 4–10 of the NEMO gene (Figure 3E). The large fragment deletion in patient 2 caused a NEMO protein truncation and loss of its function.
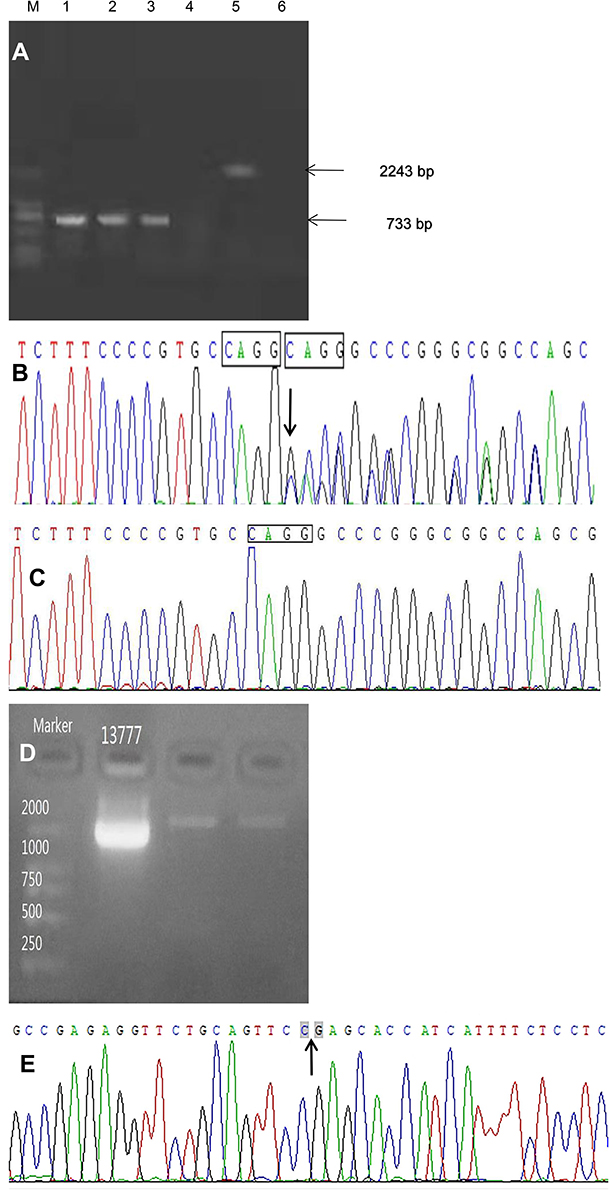 |
Figure 3 NEMO mutation analysis. (A) A normal copy number of the NEMO 4–10 exon in patient 1 according to GAP PCR: lane M: 2000 bp Marker; lane 1: 733 bp in patient 1; lane 2: 733 bp in the positive control; lane 3: 733 bp in the negative control; lane 4: no bands in patient 1; lane 5: 2243 bp, which was expected for exon 4–10 deletion, in the positive control, not in patient 1 (lane 4); lane 6: no bands in the negative control. (B) The sequencing result of patient 1: arrow indicates the position of mutation site, a frameshift mutation, c.723_724insCAGG(p.A242QfsX15) in exon 5 of the NEMO gene. (C) The wild type of patient 1ʹs father. (D) The deletion of exons 4–10 in patient 2 according to NEMO-specific PCR: Marker: DNA ladder 2000–1000-750-500-250 (top to bottom), lane 1 (13,777): 12377-NEMO-E03-10del product (2000 bp). (E) The sequencing result of patient 2: arrow indicates the gap on both sides, the partial sequence of exon 4 of the NEMO gene on the left of the arrow, and the partial downstream sequence of exon 10 of the NEMO gene, confirming the deletion of exons 4–10 of the NEMO gene. |
Discussion
To our knowledge, most IP occurrences result from a variety of mutations of the NEMO gene, leading to loss of its function. The most common mutation is the deletion of exons 4–10 of the NEMO gene.2 NEMO is an essential part of the IκB kinase (IKK) complex and can regulate the canonical NF-κB signaling pathway.9,10 The IKK complex consists of NEMO, which regulates activation of the kinase subunits, and two kinase subunits (IKKα and IKKβ).9 After being stimulated by stimuli like bacterial endotoxins and pro-inflammatory cytokines TNF-α and interleukin-1, the canonical NF-κB signaling pathway is activated.9 How it generally works is as follows. Once stimulated, signals are transmitted to the IKK complex. The IKK complex phosphorylates downstream effectors, finally resulting in translocation of NF-κB transcription factors into the nucleus, regulating gene expression during development, skin homeostasis, and immunity.9–12 In the whole process, the IKK complex could not work well without the regulation of the proper function of NEMO protein.
NEMO is composed of two HeLical domains, two Coiled-Coil domains, a NEMO Ubiquitin (Ub) Binding (NUB) domain, a Leucine Zipper domain, and a Zinc Finger domain.9,14 The activity of NEMO relies on its dimerization and on its ability to interact with linear or K63-linked polyubiquitin chains.10,13 To realize this function, two ubiquitin-binding sites, one in the NUB domain, participating in NEMO dimerization, and one in the ZF domain are required.10 All models of NEMO-regulated NF-κB activation rely on the ability of NEMO, through its ubiquitin-binding domains, to recognize proteins labeled with nondegradative ubiquitin chains.10 Hence, the CC2-ZF domains play a great regulating role.
There are two copies of the human NEMO sequence, the NEMO gene and a nonfunctional copy of the NEMO gene, NEMO pseudogene which is opposite in direction to the NEMO gene and exists downstream of the NEMO gene.4 In our study, we used NEMO-specific primers to distinguish the locations of the mutations. We identified this large fragments deletion of exons 4–10 of the NEMO gene in patient 2 by using NEMO-specific PCR and Sanger sequencing. This mutation resulted in a truncated structure of NEMO, which would only interact with IKK catalytic subunits if NEMO was stable Consequently, the activation of the canonical NF-κB signaling pathway was abolished and IP cells were more sensitive to TNF-induced apoptosis,14 causing the occurrence of IP in patient 2. In patient 1, we did rearrangement analysis by using GAP PCR. The GAP PCR and electrophoretogram showed a negative result. For patient 1 without this deletion, we used specific primers to screen for other NEMO mutations and performed Sanger sequencing in patient 1 and her father. A pathogenic point mutation, c.723_c.724insCAGG(p.A242QfsX15) was identified in patient 1. This point mutation was in exon 5 of the NEMO gene, causing the termination codon to appear earlier at position 256. Thus a NEMO truncated protein was produced. The truncated NEMO protein missed the CC2-ZF domains, which damaged the dimerization and its ability to interact with linear or K63-linked polyubiquitin chains. Moreover, the activation of NEMO and IKK catalytic subunits was influenced, affecting the NF-κB signaling pathway and influencing the related gene expressions. Meanwhile, the same frameshift mutation was only reported in a Chinese article.
The phenotypes of IP vary from different people even if the related persons with the same mutation may present different phenotypes. This phenomenon might be a result of X-chromosome inactivation.15 Although the classic phenotype is most common in females, sometimes 46, XY males present an IP phenotype. The reason why these rare cases show the typical skin lesions is that they are postzygotic genetic mosaics for the NEMO mutation.2 IP could also be found in males with a 47, XXY karyotype (Klinefelter syndrome). And IP males who have the combination of somatic and germ-line mosaicism for NEMO loss of function mutations also can result in the transmission of the disease to a female child, though it is rare.2,16,17 In our cases, the correlations of genotype-phenotype could not be made with NEMO mutations, because the functional effects of the mutations in the two patients were the same. Both the two mutations abolished NEMO activity and influenced the NF-κB activation.
In most cases, the diagnosis of IP is according to the clinical criteria6 and can be confirmed by molecular analyses. Skin lesions are major criteria.6,14 Dental, nail, hair, and retinal anomalies are IP minor criteria.6 Patient 1 had classical skin lesions, decayed teeth, and sparse hair, suggesting the disorder had already affected multiple neuroectodermal tissues. Patient 2 only presented the skin lesions at that time. They were diagnosed with IP based on clinical features and then confirmed by genetic testing. They both need further screening for other disorders associated with IP and should be closely followed up.
Conclusion
Our study presented a rare frameshift mutation, c.723_c.724insCAGG(p.A242QfsX15) in exon 5 of the NEMO gene in patient 1 with a family history and a common large fragments deletion exons 4–10 of the NEMO gene in sporadic patient 2. Genetic testing could not only help diagnose IP but also be helpful for early diagnosis and genetic counseling for families.
Consent for Publication
We have obtained written informed consent from the patients’ guardians for the publication of this case series and accompanying images. All authors have read and approved the final manuscript for submission.
Acknowledgments
We thank all the families and physicians for participating in our research. We also thank Affiliated Hospital of North Sichuan Medical College, Chigene (Beijing) Translational Medical Research Center Co.Ltd, and Shanghai Gengsi Bio-technology co., LTD.
Funding
There is no funding to report.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Fryssira H, Kakourou T, Valari M, Stefanaki K, Amenta S, Kanavakis E. Incontinentia pigmenti revisited. A novel nonsense mutation of the IKBKG gene. Acta Paediatr. 2011;100:128–133.
2. Fusco F, Conte MI, Diociauti A, et al. Unusual Father-to-Daughter Transmission of Incontinentia Pigmenti Due to Mosaicism in IP Males. Pediatrics. 2017;140:20162950.
3. Fusco F, Paciolla M, Conte MI, et al. Incontinentia pigmenti: report on data from 2000 to 2013. Orphanet J Rare Dis. 2014;9:93.
4. Aradhya S, Woffendin H, Jakins T, et al. A recurrent deletion in the ubiquitously expressed NEMO (IKK-gamma) gene accounts for the vast majority of incontinentia pigmenti mutations. Hum Mol Genet. 2001;10:2171–2179.
5. Has C, Danescu S, Volz A, Noh F, Technau K, Bruckner-Tuderman L. Incontinentia pigmenti in a newborn with a novel nonsense mutation in the NEMO gene. Br J Dermatol. 2007;156:378–410.
6. Landy SJ, Donnai D. Incontinentia pigmenti(Bloch-Sulzberger syndrome). J Med Genet. 1993;30:53–59.
7. Kawai M, Kato T, Tsutsumi M, Shinkai Y, Inagaki H, Kurahashi H. Molecular analysis of low‐level mosaicism of the IKBKG mutation using the X Chromosome Inactivation pattern in Incontinentia Pigmenti. Mol Genet Genomic Med. 2020;8:1531.
8. Okita M, Nakanishi G, Fujimoto N, et al. NEMO gene rearrangement (exon 4–10 deletion) and genotype–phenotype relationship in Japanese patients with incontinentia pigmenti and review of published work in Japanese patients. J Dermatol. 2013;40:272–276.
9. Olivera G, Mokina K, Stéphane D, et al. DARPin-Assisted Crystallography of the CC2-LZ Domain of NEMO Reveals a Coupling between Dimerization and Ubiquitin Binding. J Mol Biol. 2010;395:89–104.
10. Fusco F, Pescatore A, Conte MI, et al. EDA-ID and IP, Two Faces of the Same Coin: how the Same IKBKG/NEMO Mutation Affecting the NF-κB Pathway Can Cause Immunodeficiency and/ or Inflammation. Int Rev Immunol. 2015;34:445–459.
11. Maubach G, Naumann M. NEMO Links Nuclear Factor-κB to Human Diseases. Trends Mol Med. 2017;1285:1–18.
12. Rahighi S, Ikeda F, Kawasaki M, et al. Specific Recognition of Linear Ubiquitin Chains by NEMO Is Important for NF-κB Activation. Cell. 2009;136:1098–1109.
13. Courtois G, Gilmore TD. Mutations in the NF-κB signaling pathway: implications for human disease. Oncogene. 2006;25:6831–6843.
14. Courtois G, Israël A. IKK Regulation and Human Genetics. Berlin, Heidelberg: Springer Berlin Heidelberg; 2010:73–95.
15. Minić S, Trpinac D, Gabriel H, Gencik M, Obradović M. Dental and oral anomalies in incontinentia pigmenti: a systematic review. Clin Oral Investig. 2013;17:1–8.
16. Hull S, Arno G, Thomson P, et al. Somatic mosaicism of a novel IKBKG mutation in a male patient with incontinentia pigmenti. Am J Med Genet A. 2015;167:1601–1604.
17. Fusco F, Fimiani G, Tadini G, Michele D, Ursini MV. Clinical diagnosis of incontinentia pigmenti in a cohort of male patients. J Am Acad Dermatol. 2007;56:264–267.
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.