")
Back to Journals » Drug Design, Development and Therapy » Volume 10
Nelfinavir is effective against human cervical cancer cells in vivo: a potential treatment modality in resource-limited settings
Authors Davis M, Delaney JR, Patel C, Storgard R, Stupack D
Received 10 December 2015
Accepted for publication 25 February 2016
Published 2 June 2016 Volume 2016:10 Pages 1837—1846
DOI https://doi.org/10.2147/DDDT.S102241
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Wei Duan
Mitzie-Ann Davis,* Joe R Delaney,* Chandni B Patel, Ryan Storgard, Dwayne G Stupack
Division of Gynecologic Oncology, Department of Reproductive Medicine, Rebecca and John UCSD Moores Cancer Center, La Jolla, CA, USA
*These authors contributed equally to this work
Objective: The standard treatment for cervical cancer in developed countries includes surgery and chemoradiation, with standard of care lagging in developing countries. Even in the former case, treatment frequently yields recalcitrant tumors and women succumb to disease. Here we examine the impact of nelfinavir, an off-patent viral protease inhibitor, which has shown promise as an antineoplastic agent.
Methods: We evaluated the morphological and proliferative effects of the autophagy-stressing drug nelfinavir in normal and cisplatin-resistant cervical cancer cells. Immunofluorescent validation of autophagy markers was performed and the impact of nelfinavir in an in vivo model of tumor growth was determined.
Results: Nelfinavir exhibits cytotoxicity against both cisplatin-sensitive and -resistant ME-180 human cervical cancer cells in vitro and in vivo. Immunoblotting and immunofluorescence showed an expression of the autophagy marker LC3-II in response to nelfinavir treatment.
Conclusion: Nelfinavir, now available as an inexpensive generic orally dosed agent (Nelvir), is cytotoxic against cervical cancer cells. It acts by burdening the autophagy pathway to impair tumor cell survival and a modest induction of apoptosis. While further studies are needed to elucidate the optimal method of application of nelfinavir, it may represent an appealing global option for the treatment of cervical cancer.
Keywords: cervical cancer, nelfinavir, cisplatin resistance, autophagy, responsible medicine
Introduction
In the US, it is estimated that in 2015, there were 12,900 new cervical cancer cases and 4,100 women died from this disease.1 Worldwide these numbers increased by a factor of 40, with 500,000 new cervical cancer cases and over 250,000 deaths, placing cervical cancer as the fourth most common malignancy in women following breast, colorectal, and lung cancers.2 Human papilloma virus (HPV) has been implicated as the leading causative factor for the development of cervical malignancy.3 Understanding the clinical sequelae of HPV integration in cervical epithelial cells has given rise to newer treatment options for cervical dysplasia and cancer. This includes tools targeting prevention through vaccination, as well as screening for early detection of HPV DNA. The slow-growing nature of cervical cancer renders feasible treatment options if detected early.4 Once it has been diagnosed, cervical cancer is treated with surgery and/or chemoradiation protocols.5 Despite undergoing treatment, 30%–40% of women die from locally advanced cervical cancer.
As a largely preventable disease, cervical carcinoma disproportionately affects women in resource-limited areas. This problem is compounded by poverty and limited access to screening and treatment, as well as a high prevalence of human immunodeficiency virus (HIV) in these regions.6 In 2011, 69% of the global HIV burden was in sub-Saharan Africa.2 The link between HIV and cervical dysplasia/invasive disease is well established,7 with HIV-positive women demonstrating significantly higher rates of cervical intraepithelial neoplasia than HIV-negative women.8 The introduction of highly active antiretroviral therapy has been associated with a marked decrease in the incidence of some HIV-associated cancers such as Kaposi sarcoma and some lymphomas.9 Yet, the results in cervical cancer have been somewhat variable. Some studies show efficacy while others do not.9,10 This is possibly due to differences in the specific highly active antiretroviral therapies, and even differences within the subclasses of drugs used.
Among the viral protease inhibitors, nelfinavir has demonstrated both anti-HIV and antineoplastic activity.11 The reasons for the latter are unclear, but may be from direct tumor cytotoxicity and not secondary to attenuation of immunosuppression. For example, nelfinavir is a pleiotropic, although not potent, kinase inhibitor.12 It is also an inducer of endoplasmic reticulum (ER) stress13 and autophagy.14,15 It is possible that autophagy is induced secondary to, and remedial for, ER stress. Nelfinavir is unique among antiretroviral therapies for cancer treatment,16 but has not been further developed for treatment of neoplasia at the pharmaceutical level. Given the oral availability of the drug, the recent expiration of its patent, and its newly depressed cost (<$1/pill in the US at the time of writing, for the generic, Nelvir), it is potentially promising from a global health perspective.
Autophagy, a process of recycling of cytoplasm and cellular organelles in lysosomes, has emerged as a potential biological target for treating cervical cancer.17 In response to stress, nutrient depletion, hypoxia, or cytotoxic agents, autophagy generally supports cell survival by alleviating cellular stress, but it can be associated with cell death.18 Autophagy may also govern tumor carcinogenesis and subsequent metastasis.19 The core autophagy-related proteins LC3 and Beclin-1 may confer prognostic significance in early-stage cervical squamous cell carcinoma, as the protein levels of Beclin-1 and LC3 were significantly lower in squamous cervical cancer cells than normal squamous epithelial cells.20 Sun et al21 showed that autophagy may enhance paclitaxel-induced CaSki and SiHa cell death through regulation of Beclin-1 expression. Therefore, impact on the complex autophagic signaling pathway may provide new therapeutic options in the treatment of cervical cancer. In the present study, we demonstrate the cytotoxicity of nelfinavir in ME-180 cervical tumor models in vitro and in vivo, illustrating a potential role of autophagy-modulating drugs in the affordable treatment of cervical cancer.
Materials and methods
Cell lines and culture, cell lines and reagents
ME-180 cells (cervical squamous carcinoma) were originally obtained from the American Type Culture Collection (Manassas, VA, USA). ME-180 cisplatin-resistant (CPR) cells were generated by continuous passaging in increasing doses of cisplatin, as described previously by Donato et al,22 and maintaining in 1 μM cisplatin media. All the cells were cultured in Roswell Park Memorial Institute (RPMI) medium (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 2% glucose, nonessential amino acids, sodium pyruvate, and 10% fetal bovine serum (Omega Scientific Inc., Tarzana, CA, USA). All the cells were incubated at 37°C in a humidified atmosphere containing 5% CO2. The cells were maintained at log phase. Nelfinavir mesylate (Viracept) tablets for in vivo experiments were purchased from the Moores Cancer Center pharmacy (Agouron Pharmaceuticals, Durham, NC, USA), and for in vitro experiments as a special synthesis order from Creative Dynamics (Shirley, NY, USA). Cisplatin was purchased from Sigma-Aldrich Co., St Louis, MO, USA. For immunofluorescence studies, LC3 (#NB100-2220, Novus Biologicals, Littleton, CO, USA) and p62 (BD Biosciences #610382; San Diego, CA, USA) were used. AlexaFluor secondary antibodies were used: 488 nm goat anti-mouse (Invitrogen #A11001) and 568 nm goat anti-rabbit (Invitrogen #A11036; Carlsbad, CA, USA). For Western blots, actin (Sigma #A5441-.2ML), LC3B (Novus Biologicals #NB100-2220), p62 (BD Biosciences #610832), caspase-7 (Cell Signaling #9492; Beverly, MA, USA), Grp78 (Abcam #ab21685; Cambridge, MA, USA), Ripk1 (Cell Signaling #3493), Ripk3 (Cell Signaling #12107), and DyLight secondary antibodies were used: 800 nm for anti-rabbit (VWR #PI35571) and 680 nm for anti-mouse (VWR # PI35518; Radnor, PA, USA).
In vitro cell viability
Approximately 10,000 cells were seeded on to 96-well tissue culture-treated plates, allowed to adhere for 30 minutes, and then treated with drugs or control vehicle for a total volume of 100 μL. Plates were placed at 37°C for 48 hours. Media was removed and the cells were washed once with 125 μL phosphate-buffered saline (PBS). PBS was then removed and 50 μL crystal violet stain (0.11% crystal violet, 0.17 M NaCl, 22% MeOH, in water) was added. Stain was removed and 125 μL PBS was added as a wash. Supernatant was removed carefully to minimize cell disturbance, while permitting maximal removal of unbound crystal violet. Plates were then dried. Dye was solubilized with 85 μL methanol and absorbance was read at 600 nm to determine cell density. The percent cell loss was calculated using the formula: 100 - (100*DrugA600/ControlA600); where A600 denotes absorbance at a wavelength of 600 nm. All human cell line and mouse experiments were approved by the University of California Institutional Review Board ethics committee.
Immunofluorescence
Coverslips coated with 2 μg/mL fibronectin overnight in PBS were placed into non-tissue culture-treated six-well plates. Excess fibronectin was removed with PBS washes, 10k cells were seeded directly on to the coverslip and allowed to adhere for 30 minutes at 37°C. Media with drugs or vehicle was then added to a total volume of 3 mL. The cells were incubated at 37°C for 24 hours. Media was removed and 1 mL 4% paraformaldehyde in PBS was added (15 minutes) at ambient temperature. Liquid was aspirated and the cells were washed once with PBS, then 2 mL 0.1% Triton in PBS was added for intracellular permeabilization. After 2 minutes, the supernatant was aspirated and coverslips washed in PBS. Coverslips were blocked in 2% bovine serum albumin (BSA) for 30 minutes at ambient temperature, washed, and primary antibodies were then added in 2% BSA (1:1,000 dilution for LC3; 1:500 dilution for p62) and allowed to incubate for 90 minutes at room temperature. Three PBS washes were performed. Secondary antibodies were added in 2% BSA with DAPI (1.5 μg/mL) at a 1:1,000 dilution for 90 minutes, three quick gentle PBS washes were performed, followed by a 10-minute wash. Liquid was aspirated, coverslips were mounted on glass slides with 20 μL Vectashield (#H-1400, Vector Laboratories, Burlingame, CA, USA), then imaged on a Nikon confocal microscope.
Western blotting
Cells at 50%–80% confluency on 10 cm plates were treated with drugs or control for 24 hours at 37°C. After treatment, the cells were quickly washed with PBS and incubated with radioimmunoprecipitation assay buffer (Sigma-Aldrich) on ice for 5 minutes. Samples were centrifuged at 12,000× g for 15 minutes at 4°C. The supernatants were collected to perform Western blots. The protein concentration was measured using micro BCA (bicinchoninic acid) protein assay. Thirty micrograms of protein was loaded per well of a 15% SDS-PAGE (sodium dodecyl sulfate polyacrylamide gel electrophoresis) gel and transferred onto a PVDF (polyvinylidene difluoride) membrane. The membrane was blocked in PBS and 0.1% casein. The membranes were incubated with primary antibody for 1 hour at room temperature. The blots were washed with tris-buffered saline and Tween-20 and incubated with secondary antibody for 1 hour at room temperature. Primary antibodies were used at 1:1,000 dilution and secondary antibodies at 1:10,000 dilution. Secondary antibodies were visualized using a LI-COR Odyssey scanner (LI-COR Biosciences, Lincoln, NE, USA).
Mouse models
All animal protocols were approved by the Institutional Animal Care and Use Committee of the University of California at San Diego, and the rules and regulations of the committee were followed during experimentation on animals. Tumors were generated by implanting cells dissolved in 100 μL sterile saline subcutaneously into the abdominal flanks of 8-week-old female athymic nude mice. To account for differential growth rates of the ME-180 and ME-180 CPR cell lines, 4×106 ME-180 cells were inoculated into the left flank and 2.5×106 ME-180 CPR cells into the right flank. Tumor volume was monitored in two dimensions using a manual caliper and calculated as [(length) × (width)2×0.5]. Once ME-180 CPR tumor reached a volume of at least 100 mm3, the mice were started on daily treatment for 21 days with nelfinavir or control (50% polyethylene glycol-400 in water)gastric gavages. It should be noted that the quantitative focus in this experiment was therefore on ME-180 CPR, since the ME-180 tumors reached a stochastic range of sizes at the time of initial treatment; only ME-180 CPR was well controlled at 100 mm3. The volume of the drug administered depended on daily weight and ranged between 125–200 μL (25 g mouse set at 200 μL dose to attain 250 mg/kg/day). During the 21-day treatment period, we noted that one mouse died of unknown etiology in the control group, while another did not attain the minimum tumor volume to begin treatment and thus was not included in the analysis. However, mice treated with nelfinavir tolerated the daily treatment well. The mice were sacrificed according to the Guidelines for Euthanasia of Rodents using carbon dioxide and cervical dislocation. Six mice were randomized to each group. Mice weight was monitored daily.
Statistical analysis
A two-tailed Student’s t-test was used for statistical analysis, unless otherwise indicated as a Wilcoxon rank-sum test. P-values of ≤0.05 were considered statistically significant. Asterisks indicated a significant difference between experimental groups (*, indicates P≤0.05; **, indicates P≤0.01; ***, indicates P≤0.001, and nonsignificant (ns) indicates P>0.05).
Results
Nelfinavir suppresses growth of cervical cancer cell lines
We examined the morphology of ME-180 cervical cancer cells and platinum resistant ME-180 CPR cells 20 hours after treatment with cisplatin or nelfinavir. The overall morphology of cells treated with nelfinavir did not initially exhibit any significant differences from untreated cells, but within 20 hours, the ME-180 cells treated with nelfinavir exhibited a failure to thrive, with regions of clearing/cell death appearing (Figure 1). A closer examination of the cells revealed the appearance of prominent, clear vacuoles present in the cell, just prior to rounding and release from the substratum. The results were similar in the CPR daughter line. By contrast, only the sensitive parental cells underwent cell death in the presence of cisplatin. The CPR cells changed morphology somewhat, “flattening” and growing more slowly in the presence of cisplatin, and exhibited less cell death.
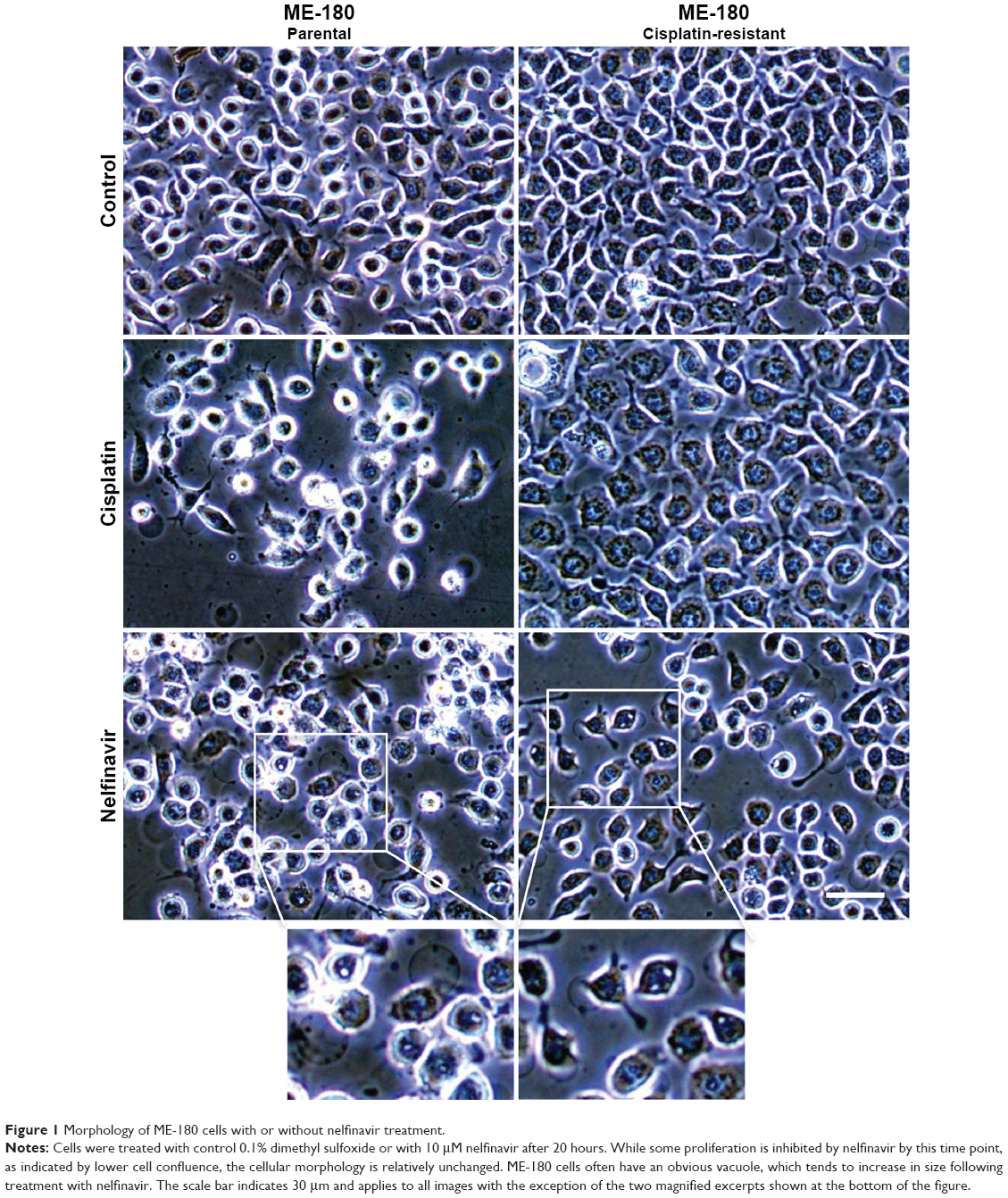 | Figure 1 Morphology of ME-180 cells with or without nelfinavir treatment. |
To evaluate the degree of growth inhibition in response to nelfinavir, the treatment was further evaluated in ME-180 and ME-180 CPR cervical cancer lines. We chose a time point of 48 hours to capture types of death, which may be independent of apoptosis,23 which can occur within 24 hours of treatment. A clear impact on tumor cell growth was observed in nelfinavir-treated cells compared to controls (Figure 2A and B). Interestingly, we noted a significant increase in sensitivity to nelfinavir when comparing the CPR form of ME-180 to the parental cells (70.5%±4.2% ME-180 CPR cells, 45.3%±3.2% ME-180 cells at 10 μM nelfinavir, P<0.001, Figure 2A). By comparison, peak levels of nelfinavir23 and cisplatin24 in peripheral blood plasma of treated patients are 10 and 5 μM, respectively, and trough levels are 0.3 and 1.5 μM, respectively. At peak doses, nelfinavir eliminated 70.5% of ME-180 CPR cells: far more than cisplatin (P<0.001 by t-test).
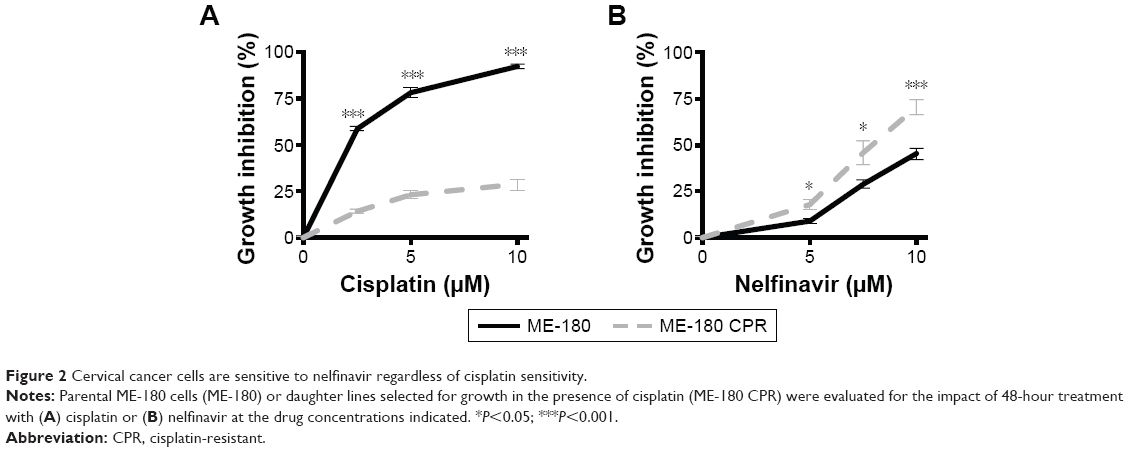 | Figure 2 Cervical cancer cells are sensitive to nelfinavir regardless of cisplatin sensitivity. |
Autophagy is altered by nelfinavir
Cytotoxic drugs induce stresses, which can frequently induce an autophagy response as a cell survival mechanism. Due to the observation of vacuoles during the death process, we wondered if the cells were inducing autophagy as a (failed) survival mechanism in response to nelfinavir. We blotted for autophagy markers prior to maximal cell death to better resolve key signaling events, choosing a time point of 24 hours following treatment. Autophagy was initially assayed by immunoblotting for LC3-II, the autophagosomal form of LC3. Nelfinavir was observed to upregulate LC3-II (Figure 3A). A trend was noted in which ME-180 CPR cells appeared to have more autophagosomes than ME-180 cells, but this failed to achieve statistical significance. As a general measure of stress, p62 was also evaluated by immunoblotting. The p62 protein is cleared by autophagy and thus can serve as a marker of decreased autophagic flux, but steady-state levels may be increased during stress.25,26 Increases in p62 were not consistently observed (ns, comparisons quantified in Figure 3A), in contrast to the reproducible increases in LC3-II upon nelfinavir treatment. Since localization of these autophagy markers into punctate patterns is indicative of their function, we followed the western study with immunofluorescence assays. Immunofluorescence demonstrated punctate patterns of LC3 and p62, indicative of autophagosome and sequestosome formation (Figure 3B), which increased with nelfinavir treatment. Interestingly, baseline p62 is significantly higher in ME-180 CPR cells compared to ME-180 cells, suggesting a higher baseline “stress” level or a preinduced capacity to deal with stress. An increase in p62 punctae in the presence of increased LC3-II is a general indication of an inability of the cell to appropriately clear proteotoxic stress.
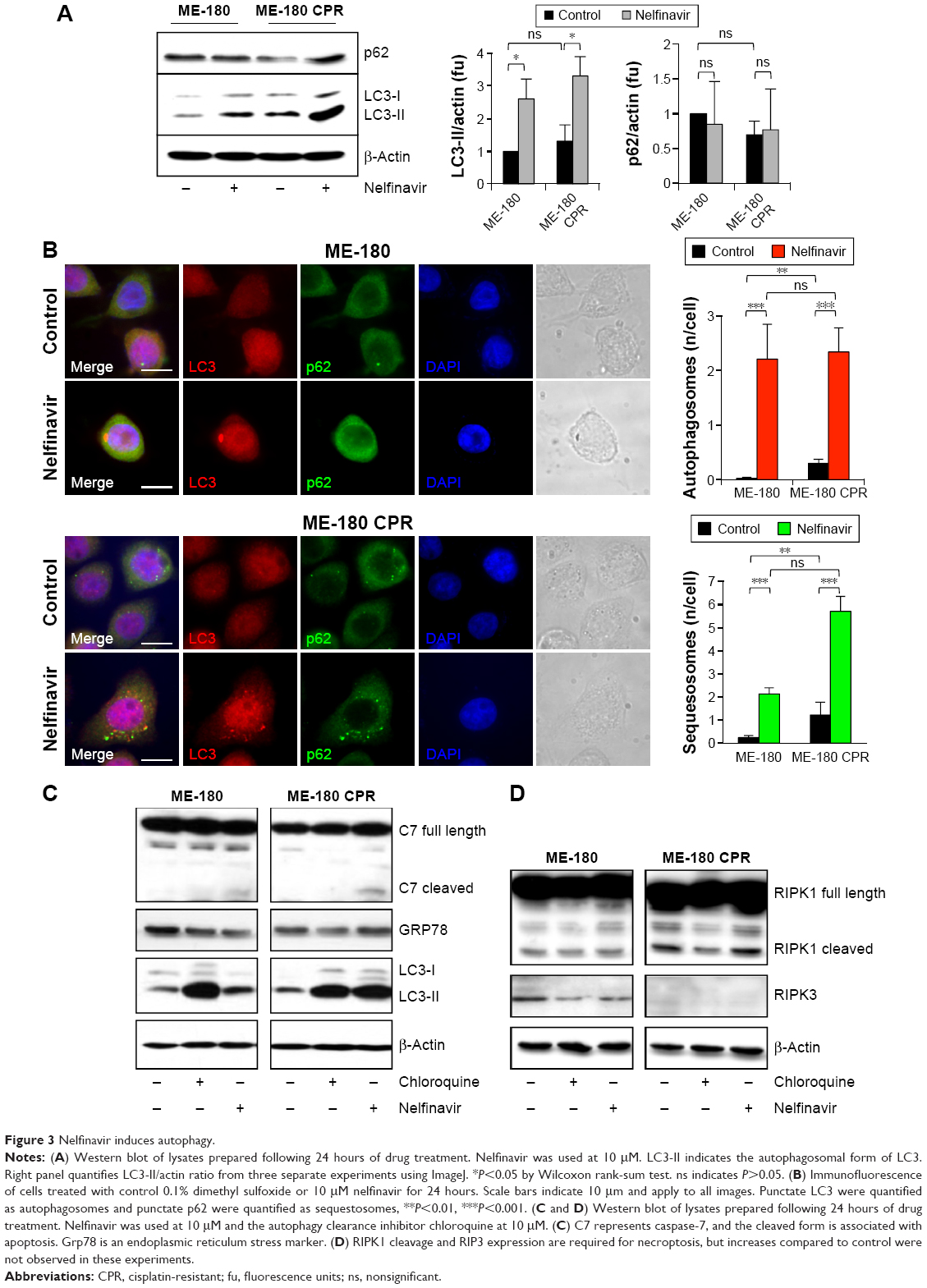 | Figure 3 Nelfinavir induces autophagy. |
An overwhelming of the autophagic recycling system would lead to an “unprogrammed cell death”. In order to assess programmed cell death, we evaluated apoptosis and necroptosis, which can co-occur with proteotoxic stress. We observed a slight cleavage of caspase-7, indicative of apoptosis, in nelfinavir-treated ME-180 and ME-180 CPR cells, with a much stronger induction of LC3-II (Figure 3C). Interestingly, treatment with the autophagy clearance inhibitor chloroquine permitted the accumulation of LC3-II as expected, but did not lead to any cleavage of caspase-7. This result would suggest nelfinavir is acting both as an agent that stresses the autophagy system in addition to activating apoptosis in some fraction of cells. However, we did not observe increases in markers of necroptosis, such as cleaved RIPK1 and RIPK3 following treatment with nelfinavir or chloroquine (Figure 3D), nor ER stress under treatment as assayed by Grp78 levels (Figure 3C).
In vivo efficacy and safety of nelfinavir
While nelfinavir has been implicated in cell death in vitro for cervical cancer,28,29 in vivo preclinical efficacy has not previously been demonstrated. To test for preclinical efficacy, we injected nude mice subcutaneously. ME-180 cells (4×106) were inoculated onto the left flank and ME-180 CPR (2.5×106) cells were inoculated into the right flank, based on normalization from in vitro growth rates. The mice were randomized into treatment groups of following tumor cell inoculation. As CPR forms of the disease are more critical and challenging to develop new therapies for, we prioritized the start of treatment based on the ME-180 CPR tumors; treatment was begun when ME-180 CPR tumors reached 100 mm3. None of the mice required euthanasia: the average mouse weight between the two groups was not significantly different (Figure 4A). Nelfinavir treatment significantly reduced both cisplatin-sensitive (P<0.023) and CPR (P<0.046) tumor volume by the end of the 21-day cycle of treatment (Figure 4B and C).
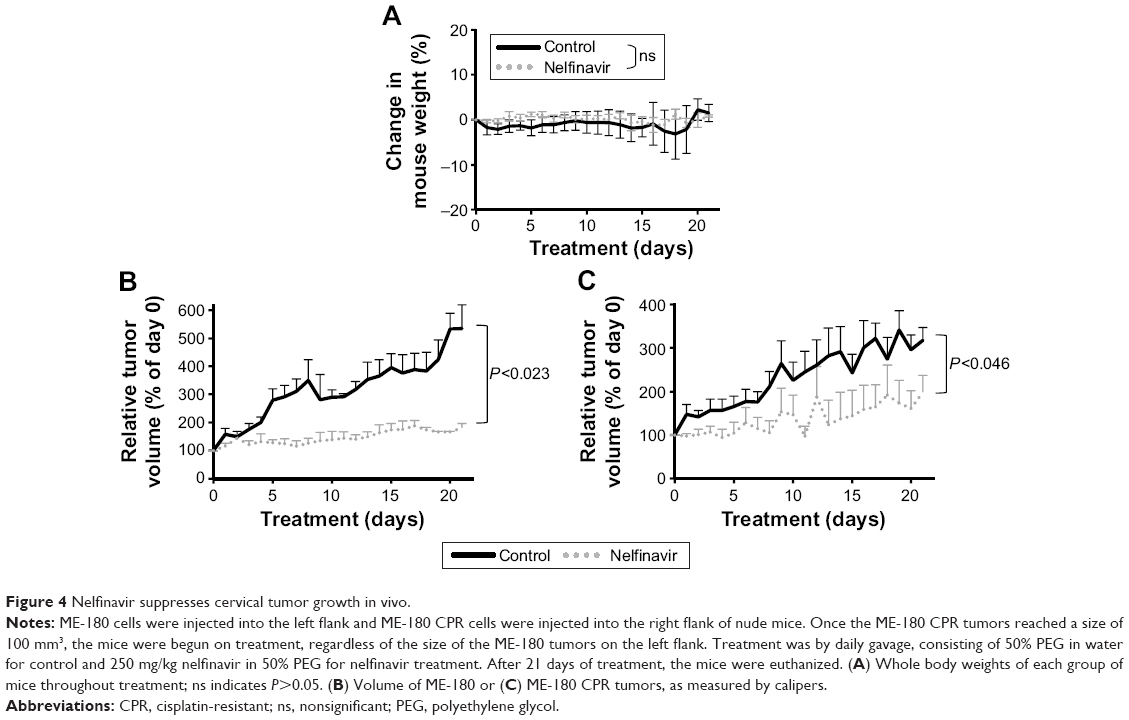 | Figure 4 Nelfinavir suppresses cervical tumor growth in vivo. |
Common deletions of core autophagy genes in patient biopsies
To gain insight as to why autophagy did not rescue the cervical cancer cells following the treatment with nelfinavir, we queried the The Cancer Genome Atlas dataset for possible cervical cancer genomic alterations,30,31 particularly those which further disrupt autophagy and would therefore allow for treatment with autophagy manipulating drugs. We noted that cervical cancers frequently exhibited deletions of core autophagy initiation genes ATG7 (36% of patients) and ATG10 (25% of patients) (Figure 5A), key enzymes regulating autophagosome maturation. These losses were concordant with a reduction in messenger RNA (Figure 5B) and in the case of ATG10, the loss of an allele may contribute to disease progression (Figure 5C). The observations here are in agreement with immunohistochemistry studies showing a reduction in LC3 staining of cervical cancer compared to normal squamous cervical epithelial cells.20 It is reasonable to predict that without a fully competent core autophagy system, cervical cancer cells may be impaired in their response to the autophagic stress induced by nelfinavir. Future studies will be required to test if these autophagy gene deletions directly influence nelfinavir sensitivity.
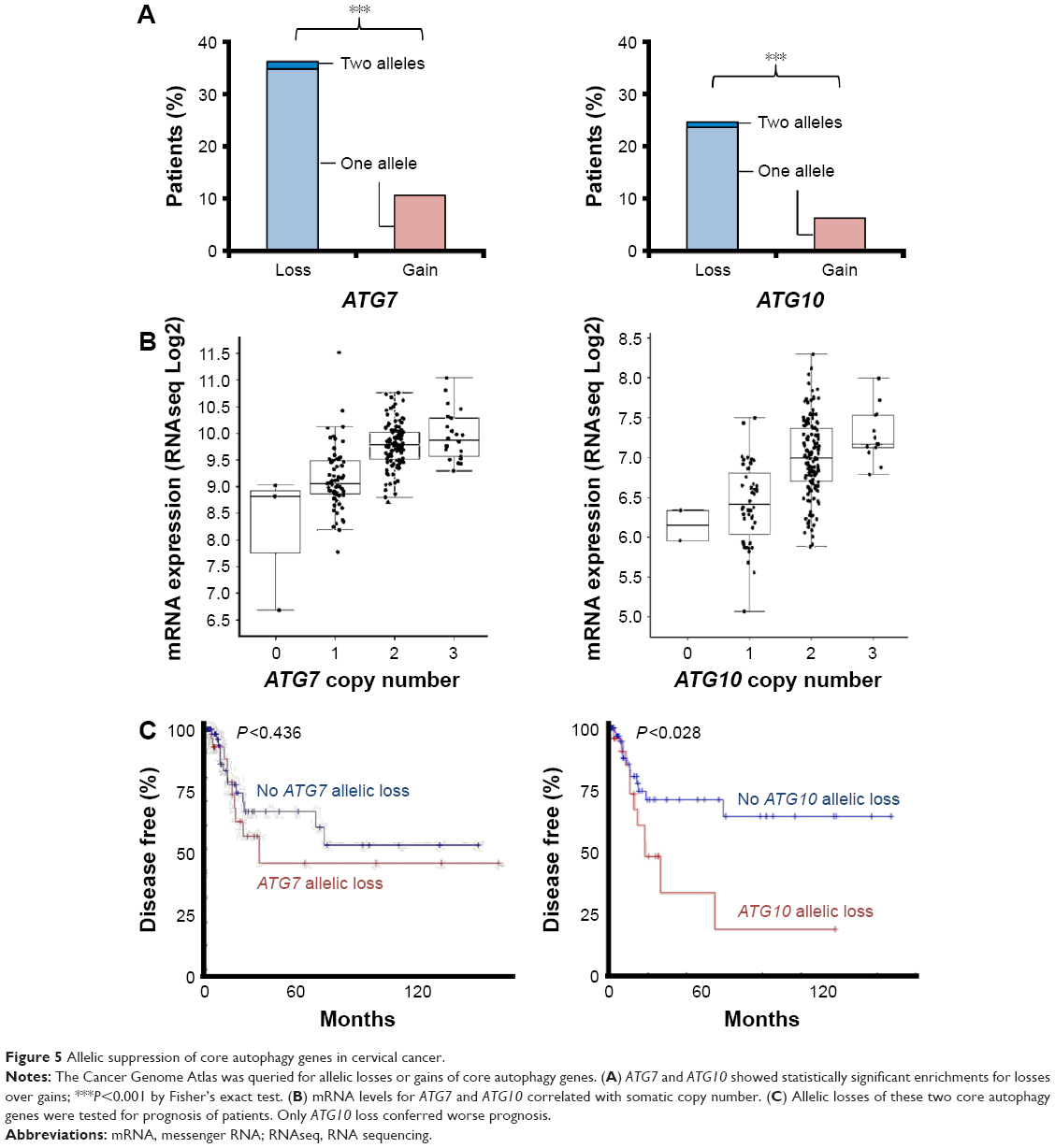 | Figure 5 Allelic suppression of core autophagy genes in cervical cancer. |
Discussion
Here we demonstrate for the first time that nelfinavir can effectively slow the growth of human cervical cancer cells in an in vivo mouse model. Importantly, this slowing of tumor growth occurs independent of cisplatin resistance status. We found that markers of autophagy and apoptosis increase upon treatment with nelfinavir treatment in vitro, which is consistent with a proteotoxic mode of action of nelfinavir.
In addition to demonstrating antiviral activity, HIV protease inhibitors such as nelfinavir have also shown antineoplastic activity against a wide range of cancers.32,33 Protease inhibitors are effective against HIV, as they inhibit the cleavage of polyproteins by viral aspartyl protease34 and thus block the maturation of HIV virus. Nelfinavir is no longer used in the cocktail to treat HIV due to high pill burden, which reduces adherence,35 and a 5% rate of controllable but uncomfortable adverse events when combined with other antiretroviral drugs.36 Nelfinavir antitumor activity is thought to occur via several mechanisms including autophagy disruption, apoptosis, induction of ER stress, and the inhibition of Akt signaling, angiogenesis, and cell invasion.12–16,37,38 We found evidence for a minority of cells to be sensitized to apoptosis and a majority to have an increased autophagosome accumulation. We did not find evidence for ER stress or necroptosis. Nelfinavir has been reported to exhibit antiviral activity against specific HPV-transformed cervical carcinoma cells, apparently via inhibition of E6-mediated proteasomal degradation of mutant p53.39 In our cell viability assays, we observed that nelfinavir was cytotoxic against cervical carcinoma cells by inducing autophagic stress, as evidenced by the expression of LC3-II protein and the cumulative p62 puncta observed via immunofluorescence.
Some caution in interpretation is warranted. Despite reaching statistical significance, the sample sizes in the in vivo models were relatively small and our mechanistic observations are based on the use of a single cisplatin-sensitive/CPR cell line pair. Thus, the results of our study are not necessarily generalizable to all cervical cancer histology types. Furthermore, additional studies are necessary to more precisely elucidate the trigger for the autophagy-relieved stress(es) induced by nelfinavir: damaged organelles,40 inhibition of clearance, ER stress,16 or other possible routes.
Currently, locally advanced cervical cancer is treated with cisplatin and chemoradiation. The published cure rates of 40%–100% are highly dependent on stage,41–43 with approximately 35% of women having recurrence of their cancer. This recurrence, the global nature of the disease, and the infrastructure required for primary care underscore the need for new modalities of treatment.44 The findings here support the use of nelfinavir against tumors independent of their response to platinum-based agents. As nelfinavir has both cytotoxic and radiation-sensitizing effects, it is an ideal agent to use in combination with cisplatin-based chemoradiation in locally advanced cervical cancers. Indeed, nelfinavir has been evaluated as a radiosensitizer in lung, head and neck, and pancreatic cancers in Phase I/II clinical trials.45–47 Those studies have supported the notion that nelfinavir is a viable option with a low toxicity profile.
From the global health perspective, nelfinavir is off patent and has the potential to become an affordable therapy in both wealthy nations and resource-limited areas where little or no treatment would otherwise be possible. A generic nelfinavir is now available under the brand name of Nelvir (Cipla, Mumbai, India). Further studies will be necessary to determine if nelfinavir can simply be combined with standard of care in cervical cancer, but such an approach appears to have very few issues related to toxicity23 or (currently) cost.
Acknowledgments
This work was supported by NCI grants CA107263 (DGS), T32CA121938 (JRD), and by funding from the Nine Girls Ask Foundation. We thank the members of the Stupack and Schlaepfer labs for technical help and expertise as well as for their scientific advice. The study sponsors played no role in any aspect of the study.
Disclosure
The authors report no conflicts of interest in this work.
References
Society AC. Cervical Cancer Key Statistics. Cancer Research UK; 2015. | ||
World Health Organization. World Cancer Report 2014. Geneva: World Health Organization; 2014. | ||
Walboomers JM, Jacobs MV, Manos MM, et al. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J Pathol. 1999;189(1):12–19. | ||
Holowaty P, Miller AB, Rohan T, To T. Natural history of dysplasia of the uterine cervix. J Natl Cancer Inst. 1999;91(3):252–258. | ||
Eifel PJ, Winter K, Morris M, et al. Pelvic irradiation with concurrent chemotherapy versus pelvic and para-aortic irradiation for high-risk cervical cancer: an update of radiation therapy oncology group trial (RTOG) 90-01. J Clin Oncol. 2004;22(5):872–880. | ||
Ahdieh L, Klein RS, Burk R, et al. Prevalence, incidence, and type-specific persistence of human papillomavirus in human immunodeficiency virus (HIV)-positive and HIV-negative women. J Infect Dis. 2001;184(6):682–690. | ||
Wright TC Jr, Ellerbrock TV, Chiasson MA, Van Devanter N, Sun XW. Cervical intraepithelial neoplasia in women infected with human immunodeficiency virus: prevalence, risk factors, and validity of Papanicolaou smears. New York Cervical Disease Study. Obstet Gynecol. 1994;84(4):591–597. | ||
Conti M, Agarossi A, Parazzini F, et al. HPV, HIV infection, and risk of cervical intraepithelial neoplasia in former intravenous drug abusers. Gynecol Oncol. 1993;49(3):344–348. | ||
Nasti G, Martellotta F, Berretta M, et al. Impact of highly active antiretroviral therapy on the presenting features and outcome of patients with acquired immunodeficiency syndrome-related Kaposi sarcoma. Cancer. 2003;98(11):2440–2446. | ||
Minkoff H, Ahdieh L, Massad LS, et al. The effect of highly active antiretroviral therapy on cervical cytologic changes associated with oncogenic HPV among HIV-infected women. AIDS. 2001;15(16):2157–2164. | ||
Bernstein WB, Dennis PA. Repositioning HIV protease inhibitors as cancer therapeutics. Curr Opin HIV AIDS. 2008;3(6):666–675. | ||
Xie L, Evangelidis T, Xie L, Bourne PE. Drug discovery using chemical systems biology: weak inhibition of multiple kinases may contribute to the anti-cancer effect of nelfinavir. PLoS Comput Biol. 2011;7(4):e1002037. | ||
Bruning A, Burger P, Vogel M, et al. Nelfinavir induces the unfolded protein response in ovarian cancer cells, resulting in ER vacuolization, cell cycle retardation and apoptosis. Cancer Biol Ther. 2009;8(3):226–232. | ||
Gills JJ, Lopiccolo J, Dennis PA. Nelfinavir, a new anti-cancer drug with pleiotropic effects and many paths to autophagy. Autophagy. 2008;4(1):107–109. | ||
Bruning A, Rahmeh M, Friese K. Nelfinavir and bortezomib inhibit mTOR activity via ATF4-mediated sestrin-2 regulation. Mol Oncol. 2013;7(6):1012–1018. | ||
Gills JJ, Lopiccolo J, Tsurutani J, et al. Nelfinavir, A lead HIV protease inhibitor, is a broad-spectrum, anticancer agent that induces endoplasmic reticulum stress, autophagy, and apoptosis in vitro and in vivo. Clin Cancer Res. 2007;13(17):5183–5194. | ||
Wang ZH, Li L, Peng ZL, Duan ZL. [Effect of autophagy gene Beclin 1 on the growth of cervical cancer HeLa cells in vitro and vivo]. Zhonghua Zhong Liu Za Zhi. 2011;33(11):804–809. | ||
Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140(3):313–326. | ||
Qu X, Yu J, Bhagat G, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest. 2003;112(12):1809–1820. | ||
Zhu W, Pan X, Li F, Zhang Y, Lu X. Expression of Beclin 1 and LC3 in FIGO stage I-II cervical squamous cell carcinoma and relationship to survival. Tumour Biol. 2012;33(5):1653–1659. | ||
Sun Y, Zhang J, Peng ZL. Beclin1 induces autophagy and its potential contributions to sensitizes SiHa cells to carboplatin therapy. Int J Gynecol Cancer. 2009;19(4):772–776. | ||
Donato NJ, Perez M, Kang H, Siddik ZH, Ling YH, Perez-Soler R. EGF receptor and p21WAF1 expression are reciprocally altered as ME-180 cervical carcinoma cells progress from high to low cisplatin sensitivity. Clin Cancer Res. 2000;6(1):193–202. | ||
Delaney JR, Patel C, McCabe KE, et al. A strategy to combine pathway-targeted low toxicity drugs in ovarian cancer. Oncotarget. 2015;6(31):31104–31118. | ||
Urien S, Lokiec F. Population pharmacokinetics of total and unbound plasma cisplatin in adult patients. Br J Clin Pharmacol. 2004;57(6):756–763. | ||
Su H, Wang X. p62 Stages an interplay between the ubiquitin-proteasome system and autophagy in the heart of defense against proteotoxic stress. Trends Cardiovasc Med. 2011;21(8):224–228. | ||
Yu H, Su J, Xu Y, et al. p62/SQSTM1 involved in cisplatin resistance in human ovarian cancer cells by clearing ubiquitinated proteins. Eur J Cancer. 2011;47(10):1585–1594. | ||
McCabe KE, Bacos K, Lu D, et al. Triggering necroptosis in cisplatin and IAP antagonist-resistant ovarian carcinoma. Cell Death Dis. 2014;5:e1496. | ||
Xiang T, Du L, Pham P, Zhu B, Jiang S. Nelfinavir, an HIV protease inhibitor, induces apoptosis and cell cycle arrest in human cervical cancer cells via the ROS-dependent mitochondrial pathway. Cancer Lett. 2015;364(1):79–88. | ||
Bruning A, Vogel M, Mylonas I, Friese K, Burges A. Bortezomib targets the caspase-like proteasome activity in cervical cancer cells, triggering apoptosis that can be enhanced by nelfinavir. Curr Cancer Drug Targets. 2011;11(7):799–809. | ||
Cancer Genome Atlas Research N, Weinstein JN, Collisson EA, Mills GB, et al. The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet. 2013;45(10):1113–1120. | ||
Gao J, Aksoy BA, Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269):1. | ||
Esposito V, Palescandolo E, Spugnini EP, et al. Evaluation of antitumoral properties of the protease inhibitor indinavir in a murine model of hepatocarcinoma. Clin Cancer Res. 2006;12(8):2634–2639. | ||
Lebbe C, Blum L, Pellet C, et al. Clinical and biological impact of antiretroviral therapy with protease inhibitors on HIV-related Kaposi’s sarcoma. AIDS. 1998;12(7):F45–F49. | ||
Blumenstein JJ, Copeland TD, Oroszlan S, Michejda CJ. Synthetic non-peptide inhibitors of HIV protease. Biochem Biophys Res Commun. 1989;163(2):980–987. | ||
Besch CL. Antiretroviral therapy in drug-naive patients infected with human immunodeficiency virus. Am J Med Sci. 2004;328(1):3–9. | ||
Jarvis B, Faulds D. Nelfinavir. A review of its therapeutic efficacy in HIV infection. Drugs. 1998;56(1):147–167. | ||
Johnson CE, Hunt DK, Wiltshire M, et al. Endoplasmic reticulum stress and cell death in mTORC1-overactive cells is induced by nelfinavir and enhanced by chloroquine. Mol Oncol. 2015;9(3):675–688. | ||
Mathur A, Abd Elmageed ZY, Liu X, et al. Subverting ER-stress towards apoptosis by nelfinavir and curcumin coexposure augments docetaxel efficacy in castration resistant prostate cancer cells. PLoS One. 2014;9(8):e103109. | ||
Hampson L, Kitchener HC, Hampson IN. Specific HIV protease inhibitors inhibit the ability of HPV16 E6 to degrade p53 and selectively kill E6-dependent cervical carcinoma cells in vitro. Antivir Ther. 2006;11(6):813–825. | ||
Utkina-Sosunova IV, Niatsetskaya ZV, Sosunov SA, Ratner VI, Matsiukevich D, Ten VS. Nelfinavir inhibits intra-mitochondrial calcium influx and protects brain against hypoxic-ischemic injury in neonatal mice. PLoS One. 2013;8(4):e62448. | ||
Peters WA 3rd, Liu PY, Barrett RJ 2nd, et al. Concurrent chemotherapy and pelvic radiation therapy compared with pelvic radiation therapy alone as adjuvant therapy after radical surgery in high-risk early-stage cancer of the cervix. J Clin Oncol. 2000;18(8):1606–1613. | ||
Duenas-Gonzalez A, Zarba JJ, Patel F, et al. Phase III, open-label, randomized study comparing concurrent gemcitabine plus cisplatin and radiation followed by adjuvant gemcitabine and cisplatin versus concurrent cisplatin and radiation in patients with stage IIB to IVA carcinoma of the cervix. J Clin Oncol. 2011;29(13):1678–1685. | ||
Rose PG, Degeest K, McMeekin S, Fusco N. A phase I study of gemcitabine followed by cisplatin concurrent with whole pelvic radiation therapy in locally advanced cervical cancer: a Gynecologic Oncology Group study. Gynecol Oncol. 2007;107(2):274–279. | ||
Disaia PJ, Creasman WT. Clinical Gynecologic Oncology. 8th ed. Philadelphia, PA: Elsevier; 2007. | ||
Rengan R, Mick R, Pryma D, et al. A phase I trial of the HIV protease inhibitor nelfinavir with concurrent chemoradiotherapy for unresectable stage IIIA/IIIB non-small cell lung cancer: a report of toxicities and clinical response. J Thorac Oncol. 2012;7(4):709–715. | ||
Brunner TB, Geiger M, Grabenbauer GG, et al. Phase I trial of the human immunodeficiency virus protease inhibitor nelfinavir and chemoradiation for locally advanced pancreatic cancer. J Clin Oncol. 2008;26(16):2699–2706. | ||
Blumenthal GM, Gills JJ, Ballas MS, et al. A phase I trial of the HIV protease inhibitor nelfinavir in adults with solid tumors. Oncotarget. 2014;5(18):8161–8172. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.