")
Back to Journals » Neuropsychiatric Disease and Treatment » Volume 13
Necrostatin-1 attenuates early brain injury after subarachnoid hemorrhage in rats by inhibiting necroptosis
Authors Chen F, Su X, Lin Z, Lin Y, Yu L, Cai J, Kang D, Hu L
Received 1 May 2017
Accepted for publication 20 June 2017
Published 7 July 2017 Volume 2017:13 Pages 1771—1782
DOI https://doi.org/10.2147/NDT.S140801
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Wai Kwong Tang
Fuxiang Chen,1 Xingfen Su,1 Zhangya Lin,1 Yuanxiang Lin,1 Lianghong Yu,1 Jiawei Cai,1 Dezhi Kang,1 Liwen Hu2
1Department of Neurosurgery, The First Affiliated Hospital of Fujian Medical University, Fuzhou, Fujian, China; 2Department of Pathology, The First Affiliated Hospital of Fujian Medical University, Fuzhou, Fujian, China
Abstract: Necroptosis is programmed cell death that has been recently proposed and reported to be involved in several neurologic diseases. However, the role of necroptosis in early brain injury after subarachnoid hemorrhage (SAH) is still unknown. The purpose of this study was to investigate whether necroptosis was involved in SAH-induced early brain injury, and to assess the possible neuroprotective effect of necrostatin-1 using an endovascular perforation rat model of SAH. Our results showed that the expression levels of necroptosis-related proteins including RIP1, RIP3 and MLKL in the basal cortex all increased at 3 hours after SAH (P<0.05) and peaked at 48 hours after SAH (P<0.05). However, they were greatly reduced after treatment with necrostatin-1 (P<0.05). Concurrently, neurologic outcomes were significantly improved after necrostatin-1 treatment (P<0.05). Furthermore, brain edema, blood–brain barrier disruption, necrotic cell death and neuroinflammation were also greatly inhibited after necrostatin-1 treatment. These results indicate that necroptosis is an important mechanism of cell death involved in the early brain injury after experimental SAH. Necrostatin-1 perhaps can serve as a promising neuroprotective agent for SAH treatment.
Keywords: subarachnoid hemorrhage, necroptosis, receptor-interacting protein 1, cell death, neuroprotection
Introduction
Early brain injury (EBI) is the most important pathophysiologic event responsible for the unsatisfactory outcomes of subarachnoid hemorrhage (SAH) that occurs within the first 72 hours after SAH.1 Several mechanisms have been reported to participate in the occurrence of EBI, including blood–brain barrier (BBB) breakdown, brain edema, neuronal apoptosis, oxidative stress, inflammatory response, as well as elevation of intracranial pressure and reduction of cerebral blood flow.1 All of these pathologic processes are likely to cause a similar ending, cell death.2 Recent study efforts have primarily focused on SAH-induced apoptosis of neurons within the cerebral cortex or hippocampus, which together with neuroinflammation are considered the most important determinant of brain injury and prognosis after SAH.3 Moreover, antiapoptotic and anti-inflammatory treatments also succeed in improving the outcomes of experimental animals in several studies.4–6 However, these promising results have not brought benefits to clinical treatment. The disability and mortality in patients suffering from SAH still remain high, despite advances in monitoring methods and microsurgical techniques over recent decades.7 Therefore, it means that other mechanisms besides the aforementioned may also be involved in brain damage after SAH.
Programmed necrosis or necroptosis is a type of caspase-independent cell death that is activated when caspase signaling is inhibited or inactive.8,9 Caspase dysfunction leads to necroptosis initiation followed by necrotic death complex (necrosome) formation, which consists of a string of molecules. RIP1, RIP3 and MLKL are the three core components of the necrosome. RIP1/RIP3/MLKL signaling cascade has also been widely considered as a conventional process of necroptosis pathway, which is triggered by tumor necrosis factor-α (TNF-α) family of cytokines or other death signals.10 Similar to necrosis, typical features of necrosis, including disruption of cytosolic membrane integrity, swelling of organelles and dissolution of cellular structures, also appear in necroptosis. To date, necroptosis has been widely reported to be involved in a number of neurologic diseases, such as intracerebral hemorrhage,11 multiple sclerosis,12 ischemic brain injury8 and spinal cord injury.13,14 However, to the best of our knowledge, the contribution of necroptosis to EBI after SAH has not been elucidated.
The kinase activity of RIP1 is indispensable for the initiation of programmed necrosis and activation of downstream proinflammatory transcription nuclear factor kappa B (NF-κB).15 Genetic ablation of RIP1 displayed severe embryonic developmental defect and led to postnatal lethality.16 Necrostatin-1 (Nec-1), a specific inhibitor of RIP1, plays a necroptosis-inhibitor role by suppressing phosphorylation of RIP1 and interaction with RIP3. Accumulating evidence has demonstrated that Nec-1 can enter the brain by crossing the impaired BBB and shows protective effect against necroptosis both in vivo and in vitro when caspases are inhibited or are inactive.8,17,18
In this study, we first explored the role of necroptosis on EBI following experimental SAH and then evaluated the potential neuroprotective effect of Nec-1 and its possible mechanisms through an endovascular perforation model of SAH.
Materials and methods
Animals
The research protocols were approved by the Animal Care and Use Committee of Fujian Medical University and are in accordance with the guidelines of National Institutes of Health. Adult male Sprague Dawley rats (300–400 g) were purchased from the SLAC Laboratory Animal Co., Ltd. (Shanghai, China). The rats were kept in a 12-hour light/dark cycle and raised with free access to water and food under controlled temperature and humidity conditions.
SAH model
The procedure to establish a rat model of SAH has been described in detail in a previous report.19 Briefly, rats were anesthetized with an intraperitoneal injection of chloral hydrate (4 mL/kg) and kept in a supine position. The right common carotid artery and its bifurcations were separated. Then, a sharpened 4-0 monofilament nylon suture was inserted into the stump of external carotid artery and advanced through internal carotid artery until a resistance was felt (length about 1.8–2.2 mm). The suture was pulled back slightly and advanced 3 mm further to puncture the bifurcation of anterior and middle cerebral artery and then kept for 10–15 s. After that, the rats were returned to their cages to recover. Rectal temperature was maintained around 37°C with a heating pad during the surgery. The sham-operated group underwent the same procedure without endovascular perforation.
Drug administration
Nec-1 (25 mg, Sigma-Aldrich) was dissolved in dimethyl sulfoxide (DMSO, Sigma-Aldrich) to a final concentration of 40 μg/μL. Intracerebroventricular administration was performed as described previously.20 A single dose of Nec-1 (5 μL) was administered using a 10 μL syringe 30 min after the surgery. The detailed process was similar to that described by others (coordinates: 1.0 mm posterior, 1.8 mm lateral to bregma, 4 mm depth).21 The sham and vehicle group received an equal volume of DMSO in the same way.
Experimental design
In experiment 1, necroptosis-related proteins expressed in different time points after SAH were measured. Rats were divided into six groups: sham (n=9), SAH 3 hours (n=6), SAH 12 hours (n=6), SAH 24 hours (n=6), SAH 48 hours (n=9) and SAH 72 hours (n=6). The basal cortices of brain in each group (n=6) were obtained for Western blot (WB) analysis. Double immunofluorescence staining for necroptosis-related proteins (RIP1, RIP3 or MLKL) and neuronal nuclei (NeuN) were performed in the remaining three rats in the sham and SAH 48-hour groups. In experiment 2, the operated rats were randomly assigned to three groups: the sham-operated group (n=27), the SAH+DMSO group (n=27) and the SAH+Nec-1 group (n=27). All rats were sacrificed at 48 hours post-SAH according to the results of experiment 1. Neurologic functions, SAH grade, brain edema, BBB permeability, as well as transmission electron microscopy (TEM) and WB analysis were used for evaluating the protective effect of Nec-1 against EBI after SAH. In experiment 3, three groups of rats as mentioned in experiment 2 were used to determine the levels of NF-κB and its downstream inflammatory cytokines as well as the number of propidium iodide (PI)-positive cells. In this part, six rats in each group were required for each item, except for PI-positive cells (n=3).
Neurologic testing
At 48 hours post-SAH, rats in each group were assessed for neurologic performances with the modified Garcia scale and Rotarod test. Spontaneous activity, symmetry movements of limbs, forepaw outstretching, climbing, body proprioception and tentacle reaction were evaluated according to the scoring system of Garcia et al.22 For the Rotarod test, animals were tested 1 day before surgery and at 48 hours after SAH. Briefly, rats were placed on a cylinder and then the rotational speed was gradually accelerated from 4 to 40 rpm in 5 min. Latency to fall off in three repeated trials was recorded according to previous studies.23–25
SAH grade
The basal surface of brain was photographed immediately after removing the brain at 48 hours post-SAH. The picture was divided into six parts and further graded according to a grading system as previously reported.26 Briefly, each part was determined from grade 0 to 3 along with the increasing amount of blood clots: grade 0, no subarachnoid blood; grade 1, minimal subarachnoid blood; grade 2, moderate blood with recognizable arteries and grade 3, blood clot covering all arteries within the segment. A total score was obtained by adding the scores of the six segments. The severity of SAH was assessed by an investigator blinded to treatment allocation. A higher score represents a more severe SAH degree. Operated rats with an SAH score <8 were excluded from the study.
Brain edema analysis
Brain edema study was carried out through calculating the brain water content of rats in each group. Briefly, the brains were removed at 48 hours after surgery and rapidly divided into four parts including the left hemisphere, right hemisphere, cerebellum and brain stem. Each part was weighed immediately to obtain wet weight and then dried in a 100°C oven for 24 hours to get dry weight. The brain water content was determined as previously described: ([wet weight − dry weight]/wet weight) ×100%.27
BBB permeability
BBB permeability was measured by detecting the Evans blue (EB) content of extravasation into brain tissue at 48 hours after SAH, as described previously.4 Briefly, EB (2%, 5 mL/kg) was injected into the right femoral vein and circulated for at least 1 hour before the rats were decapitated. After intracardial perfusion with PBS, the brain samples were separated and weighed as mentioned above. Each sample was homogenized in 50% trichloroacetic acid and centrifuged at 15,000 rpm for 20 min. The supernatant was diluted with ethanol (1:3) and incubated overnight at 4°C. Next day, the samples were centrifuged again at 15,000 rpm for 20 min. The content of extravasated EB in the brain tissue was measured using a SpectraMax M5 microplate reader (Molecular Devices LLC, Sunnyvale, CA, USA) with the absorbance at 610 nm. EB content was expressed as μg/g brain tissue.
TEM analysis
Brain samples for TEM were prepared as described previously.28,29 After 48 hours of SAH, rats were transcardially perfused with PBS containing 1.5% paraformaldehyde and 3% glutaraldehyde. Basal cortical samples (1×1×1 mm3) were obtained and further fixed overnight at 4°C in the same buffer. For postfixation, tissue fragments were transferred to 1% osmium tetroxide and 1.5% potassium ferricyanide for 1.5 hours. Then, the samples were dehydrated with different concentrations of ethanol and acetone and further embedded in epoxy resin and sectioned (80 nm). After staining with uranyl acetate and lead citrate, ultrathin sections were observed and photographed under a TEM (EM 208; Philips, Amsterdam, Holland).
Protein expression analysis
WB assay was carried out as described previously.30 Tissue samples were collected at the indicated time in experiment 1 and at 48 hours post-SAH in experiments 2 and 3. For the whole cell protein extraction, brain tissues were homogenized in radioimmunoprecipitation assay lysis buffer containing the proteinase inhibitor phenylmethanesulfonyl fluoride and placed in ice for 20 min. Then, the supernatants were collected after centrifugation at 13,000 rpm for 15 min at 4°C. Additionally, the cytosolic and nuclear proteins were extracted using the specialized extraction reagent (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s protocol. Protein concentrations were determined using a bicinchoninic acid kit (Beyotime, Jiangsu, China). Equal amounts of protein were run on 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and then transferred onto nitrocellulose membranes. After blocking with 5% milk in PBS for 1 hour at room temperature, the membranes were incubated overnight at 4°C with the following primary antibodies: anti-RIP1 (1:1,000, Cell Signaling Technology), anti-RIP3 (1:500, Abcam), anti-MLKL (1:500, Abcam), anti-NF-κB (1:500, Bioworld), anti-GAPDH (1:2,500, Beyotime) and anti-Lamin B1 (1:1,000, Proteintech). Horseradish peroxidase–conjugated goat anti-rabbit (1:1,000, Beyotime) was used as the secondary antibody the next day. Immunoreactivity was detected by enhanced chemiluminescence (Beyotime). Band intensities were quantified by using ImageJ software (http://rsb.info.nih.gov/ij/). The expression levels of proteins were expressed as the relative densities to GAPDH or Lamin B1.
Double immunofluorescence staining
Anesthetized rats were perfused with precooled saline followed by 4% paraformaldehyde in PBS. Brains were removed and postfixed overnight and then dehydrated in 30% sucrose for 48 hours. Coronal brain sections (10 μm) were obtained in a cryostat (CryoStar NX70; Thermo Fisher Scientific) at 100–150 μm intervals and processed for double immunofluorescence staining based on the methods described previously.31 The sections were washed in PBS containing 0.1% Triton X-100 three times and blocked with 10% donkey serum for 90 min. The sections were incubated with anti-RIP1 (1:100), anti-RIP3 (1:200) or anti-MLKL (1:100) primary antibodies overnight separately. After rinsing with PBS containing 0.1% Triton X-100, the sections were further incubated for 1 hour at room temperature in the dark with Alexa 594-conjugated goat anti-rabbit IgG secondary antibody (1:400, Jackson ImmunoResearch). After this, the sections were incubated with anti-NeuN (1:50, Millipore) primary antibody and Alexa 488-conjugated donkey anti-mouse IgG secondary antibody. After double staining, sections were covered with 4′,6-diamidino-2-phenylindole and observed under a Nikon Eclipse Ti fluorescence microscope (Nikon Corporation, Tokyo, Japan). Images were further processed using Photoshop CS5 (Adobe Systems, San Jose, CA, USA).
PI staining
PI labeling was used to examine cytosolic membrane permeability according to previous studies with a slight modification.9,32 Briefly, PI (25 mg; Sigma-Aldrich) was diluted in normal saline and injected intraperitoneally (3 mg/kg) to rats 2 hours before death. Frozen brain sections were obtained as above and covered with 4′,6-diamidino-2-phenylindole, and then immediately photographed. For quantification of PI-positive cells, photographs were randomly taken from three nonoverlapping ×200 fields in each slice. Three sections of each brain were analyzed by a researcher who was blinded to the experimental design. PI-positive cells were expressed as PI+ cells/×200 field.
Enzyme-linked immunosorbent assay
At 48 hours post-SAH, animals were transcardially perfused with PBS and the basal surface of temporal lobe was dissected. Brain tissues were homogenized and the protein concentrations were determined as above. The levels of TNF-α and interleukin-1β (IL-1β) in the supernatant were quantified using the corresponding enzyme-linked immunosorbent assay kits (Westang, Shanghai, China) according to the manufacturer’s protocol. Values were expressed as ng/g protein.
Statistical analysis
Data were presented as mean ± standard error of mean. The statistical analyses were performed using GraphPad Prism software and SPSS 17.0. Normality of data was determined using the Shapiro–Wilk test. One-way analysis of variance followed by Tukey’s multiple comparisons test was used for comparison between groups. P<0.05 was considered statistically significant.
Results
General observation
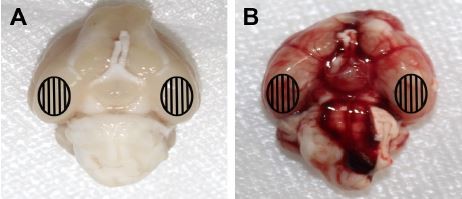
A total of 259 rats were subjected to surgery in the experimental processes in this study. Overall mortality rate was 0% (0/51) in the sham group, 20.4% (10/49) in the SAH group, 27.3% (21/77) in the SAH+DMSO group and 26.8% (22/82) in the SAH+Nec-1 group. Most of them died within 24 hours due to severe hemorrhage, neurogenic pulmonary edema and vitreous hemorrhage. Thirty-eight rats were excluded from the study as the SAH grade score was <8. The remaining 168 rats were included for the final analyses. No significant differences in body temperature, weight or heart rate were observed among the experimental groups (data not shown). When anesthetized rats were decapitated, blood clots were found mainly around the circle of Willis and the ventral surface of brain stem in rats subjected to SAH. As expected, there was no blood clot in the sham-operated rats (Figure 1).
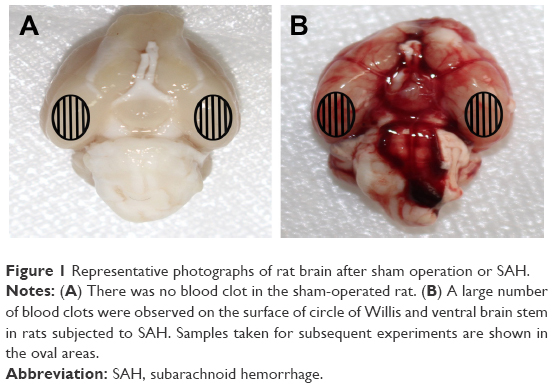 | Figure 1 Representative photographs of rat brain after sham operation or SAH. |
Increased expression of RIP1, RIP3 and MLKL within 72 hours after SAH
To explore induction of necroptosis at the early stage after SAH, we detected the expression levels of necroptosis-related proteins (RIP1, RIP3 and MLKL) in brains at different time points using WB analysis. As shown in Figure 2A–D, the expression of RIP1 was significantly enhanced beginning at 3 hours post-SAH as compared to the sham group (P<0.05) and was maintained at a high level for at least 72 hours. The level of RIP3 increased simultaneously with RIP1 and reached a peak at 48 hours (both P<0.05 versus SAH 24 hours and SAH 72 hours). In addition, a similar tendency was also observed in the expression of downstream kinase MLKL when compared with the sham group, although the differences at each time point were not always statistically significant. Consistent with the WB results, double immunofluorescence analyses further confirmed the increased expression of necroptosis-related proteins following SAH (Figure 3). Specifically, a small amount of RIP1 was expressed in the sham group, while almost no RIP3 and its substrate MLKL were found. On the contrary, these proteins were significantly enhanced at 48 hours after SAH, and most of them were colocalized with NeuN, a neuronal marker. These results indicated that RIP1/RIP3/MLKL-mediated necroptosis was activated in the early days after experimental SAH and appeared primarily in cortical neurons.
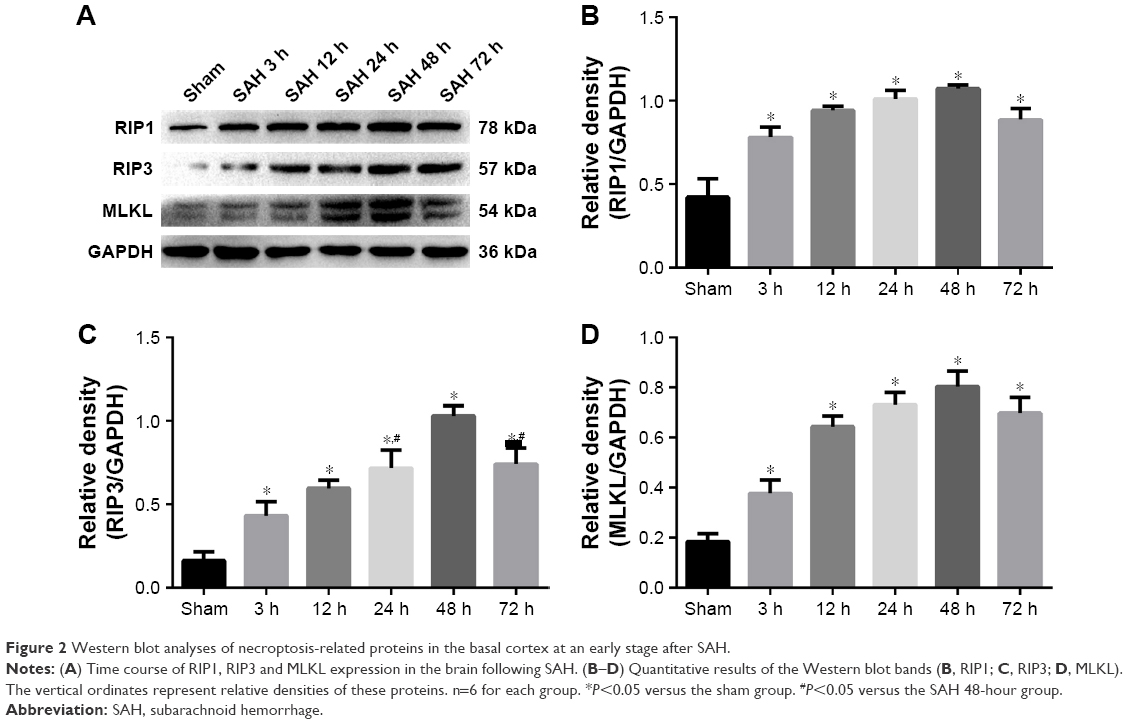 | Figure 2 Western blot analyses of necroptosis-related proteins in the basal cortex at an early stage after SAH. |
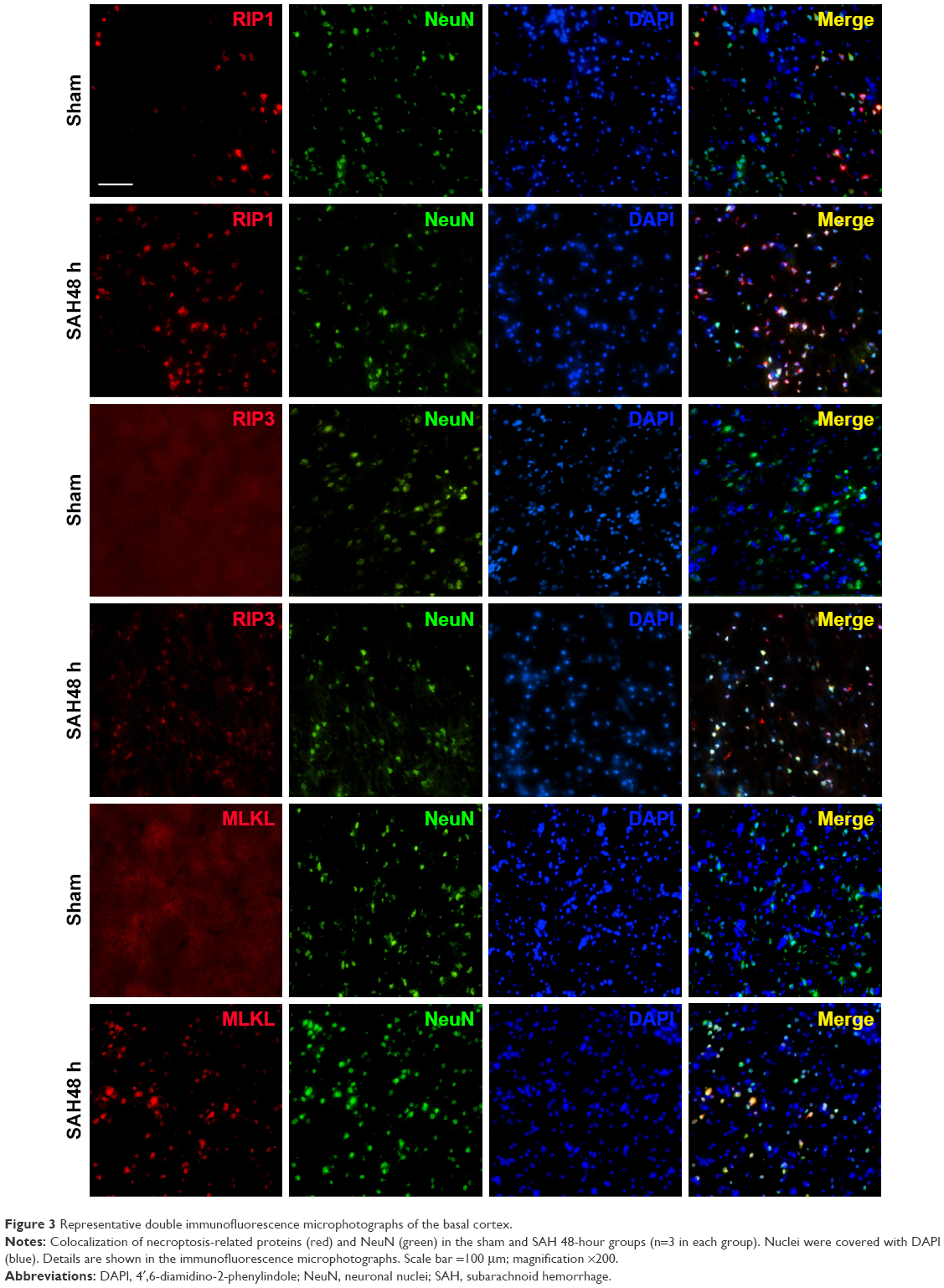 | Figure 3 Representative double immunofluorescence microphotographs of the basal cortex. |
Nec-1 alleviated neurologic deficits, BBB dysfunction and brain edema following SAH
To assess the neuroprotective effects of Nec-1 on SAH-induced EBI, we estimated the SAH grade, neurological outcomes, BBB permeability and brain edema at 48 hours after SAH based on the results of experiment 1. Our results indicated that rats in the SAH+DMSO group showed worse neurologic score and rotarod performance as compared to the rats in the sham group (both P<0.05). However, they were dramatically improved in the SAH+Nec-1 group (both P<0.05; Figure 4B and C). EB extravasation, a hallmark of BBB disruption, was heavily elevated in the left and right hemispheres and the brain stem after SAH induction (P<0.05; Figure 4D). However, the conditions were reversed by Nec-1 intracerebroventricular administration (P<0.05). Brain water content was often used to represent the severity of brain edema, which has been accepted as a crucial pathologic event contributing to SAH-induced brain injury. In this study, the brain water content was also markedly increased in both hemispheres (79.85% versus 78.54% in the left and 79.90% versus 78.39% in the right, both P<0.05), with no significant differences in cerebellum and brain stem (both P>0.05). Similarly, Nec-1 treatment reduced brain edema obviously (78.21% versus 79.85% in the left and 78.51% versus 79.90% in the right, both P<0.05; Figure 4E). However, there was no significant difference in SAH grade between the SAH groups (P>0.05; Figure 4A). These results suggested that Nec-1 could reduce brain injuries at an early stage after experimental SAH.
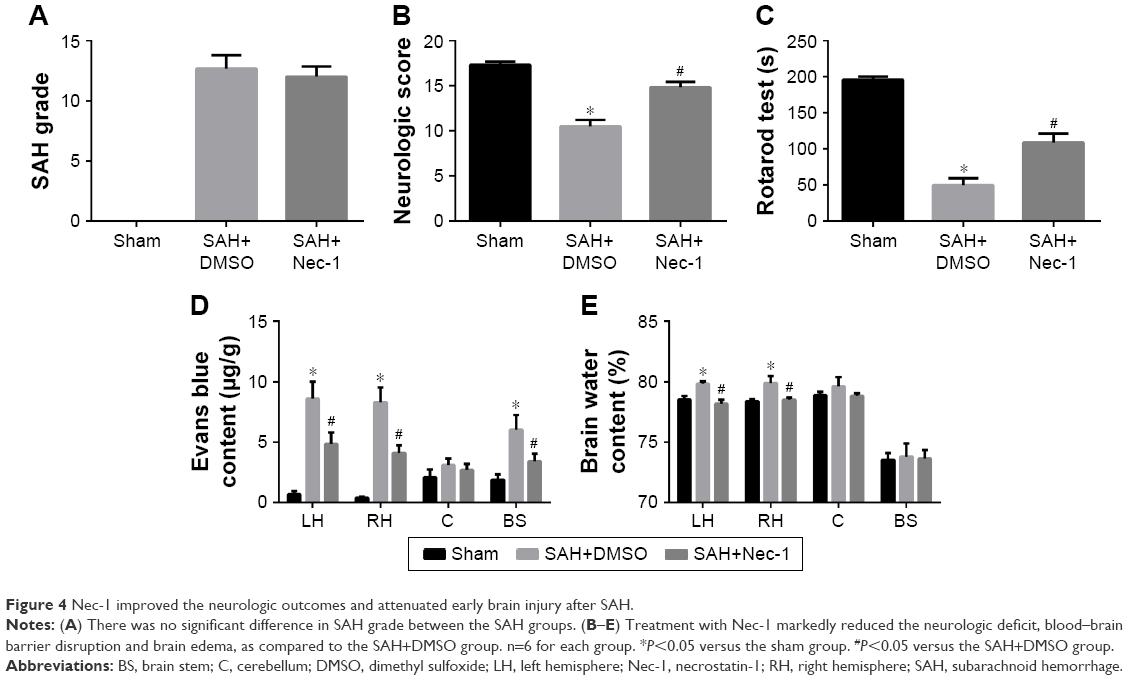 | Figure 4 Nec-1 improved the neurologic outcomes and attenuated early brain injury after SAH. |
Nec-1 inhibited RIP1/RIP3/MLKL-mediated necroptosis in rats after SAH
To demonstrate the inhibitory effect of Nec-1 on necroptosis, protein levels of RIP1, RIP3 and MLKL were examined after Nec-1 treatment in each group. Consistent with our assumptions, these proteins were all upregulated in the SAH+DMSO group, while they decreased significantly after Nec-1 treatment as compared to the SAH+DMSO group (P<0.05; Figure 5A–D). These results suggested that RIP1/RIP3/MLKL-mediated necroptosis could be significantly inhibited by Nec-1 treatment.
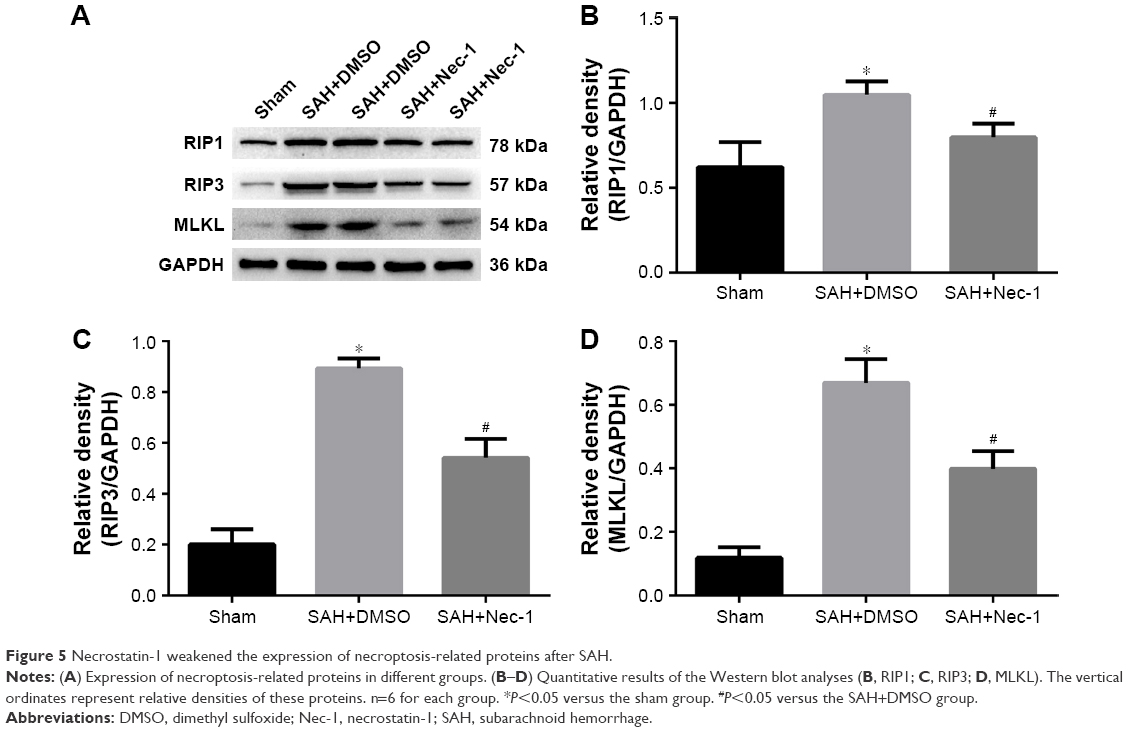 | Figure 5 Necrostatin-1 weakened the expression of necroptosis-related proteins after SAH. |
Nec-1 declined the number of PI-positive cells after SAH
PI was impermeable to the intact cell membrane and was used to label necrotic cell death. As expected, very few PI-positive cells were detected in the sham-operated group. Conversely, significantly increased PI-positive cells were observed in the brain following SAH, while they reduced remarkably after treatment with a single dose of Nec-1 (P<0.05; Figure 6A and B).
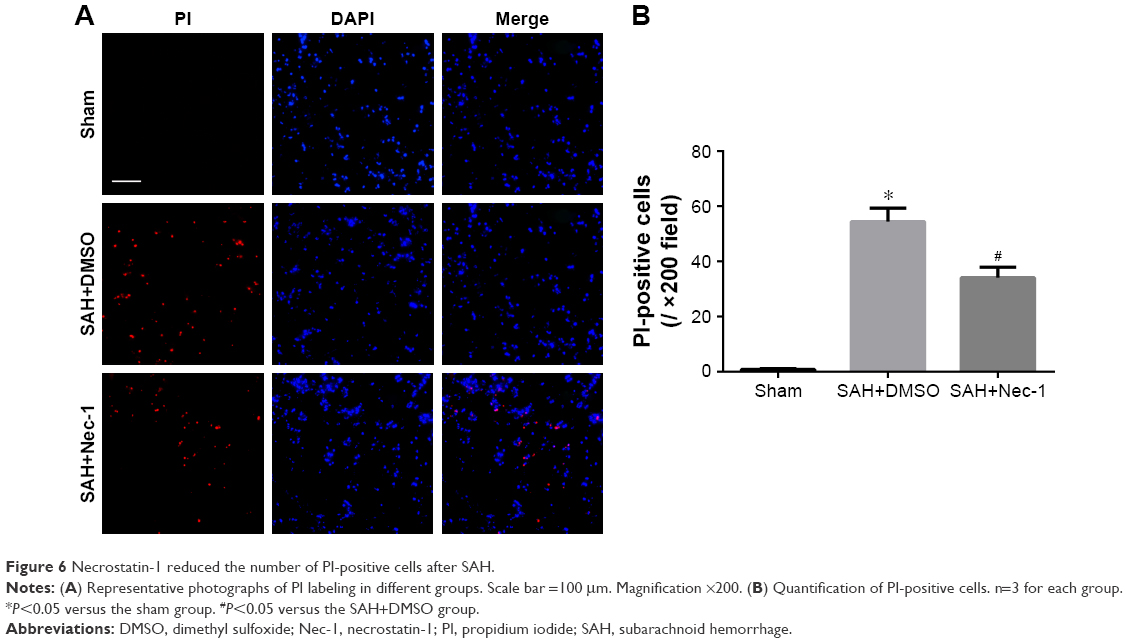 | Figure 6 Necrostatin-1 reduced the number of PI-positive cells after SAH. |
Nec-1 ameliorated necrotic cell death after SAH
Necroptosis was characterized by typical features of necrotic cell under TEM examination. As shown in Figure 7A, cortical neurons presented with normal nucleus, nuclear membrane and organelles in the sham group. In contrast, obvious necrotic changes, including breakdown of membrane integrity, swelling of organelles and dissolution of nuclei, as well as macrophage infiltration and astrocytes proliferation, were found in rats subjected to SAH (Figure 7B–E). However, these conditions were weakened in the SAH+Nec-1 group (Figure 7F). These together with PI results both supported that Nec-1 could ameliorate necrotic cell death in the basal cortex after SAH.
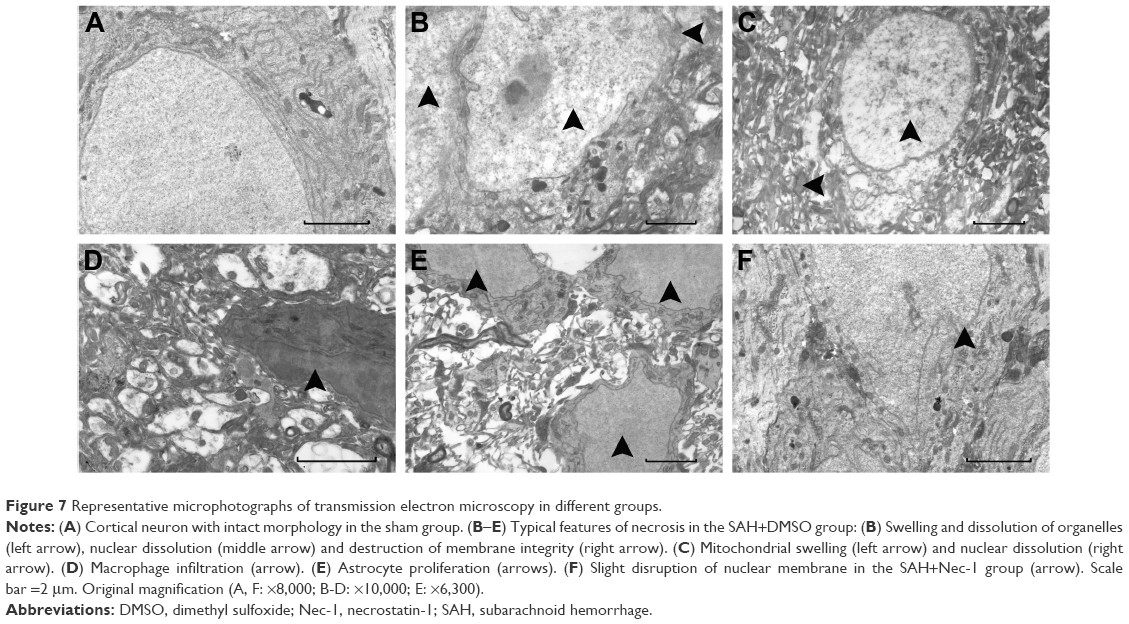 | Figure 7 Representative microphotographs of transmission electron microscopy in different groups. |
Nec-1 reduced the nuclear translocation of NF-κB and release of inflammatory cytokines after SAH
To further explore the effect of Nec-1 against neuroinflammation after SAH induction, in this study, we focused on its role on proinflammatory NF-κB signaling pathway. Physiologically, NF-κB is located mainly in the cytoplasm in a nonactivated status. Upon stimulation by SAH, NF-κB is activated and then subunit p65 is translocated to the nucleus, subsequently promoting the release of inflammatory mediators. As predicted, our results showed an increasing translocation of NF-κB p65 subunit to the nucleus and upregulation of inflammatory cytokines (TNF-α and IL-1β) after SAH. However, these inflammatory factors were reduced after Nec-1 treatment (P<0.05; Figure 8A–D), suggesting a significant decline of neuroinflammation post-SAH.
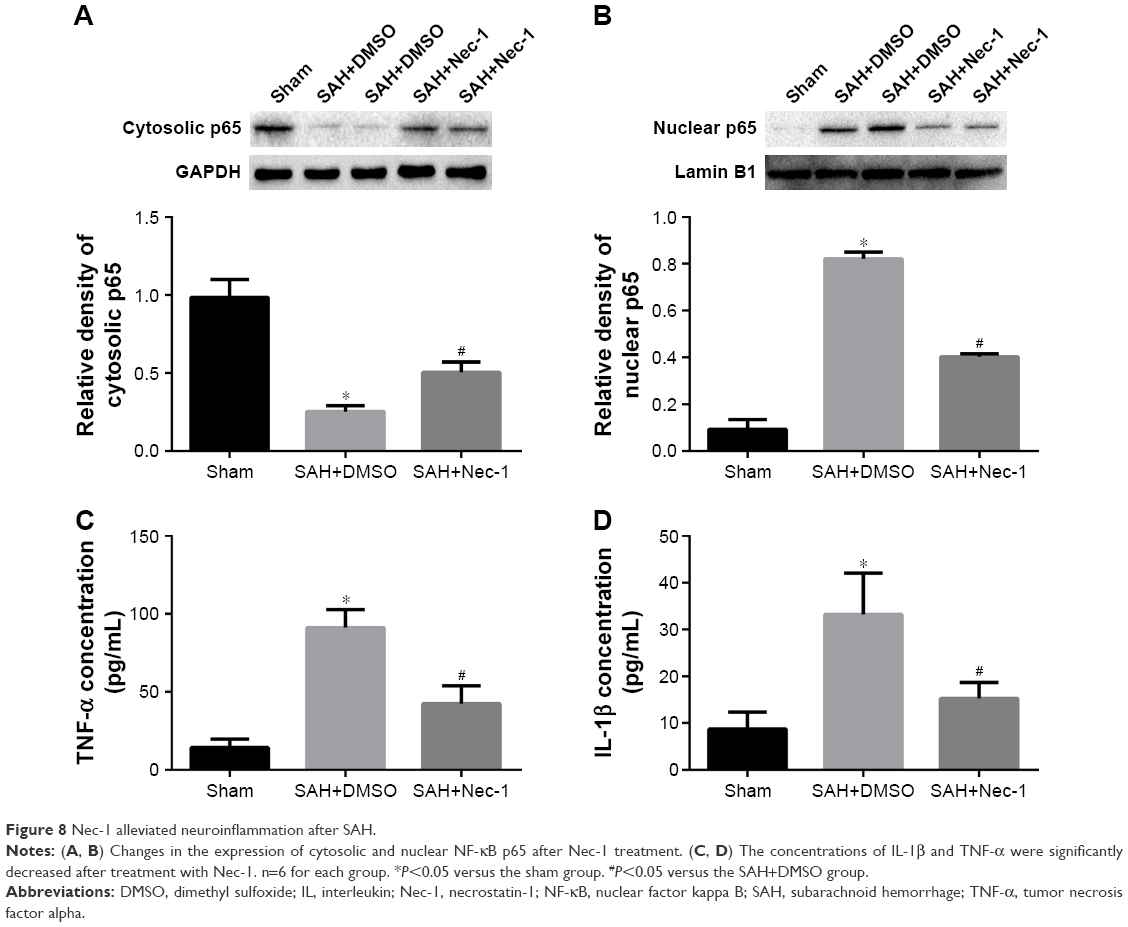 | Figure 8 Nec-1 alleviated neuroinflammation after SAH. |
Discussion
In this study, we first found that there was an activation of RIP1/RIP3/MLKL-mediated necroptosis at an early stage after SAH, which was accompanied by neurologic deficits, BBB disruption, brain edema, as well as inflammatory response and necrotic cell death. Moreover, the neural damages induced by SAH could be attenuated by treating with a single dose of Nec-1. These results suggest that necroptosis might be an important pathologic mechanism responsible for brain injuries in early days after experimental SAH, which could be effectively reduced by administration of Nec-1 through the inhibition of necroptosis and possible inflammation.
Cascade events occurred immediately after SAH, such as elevation of intracranial pressure, reduction of cerebral blood flow, suppression of cerebral perfusion pressure and fall in brain oxygenation.2 All of these pathogenic events can lead to cerebral ischemia and hypoxia, which have been reported to participate in the brain injury by triggering brain neuronal necroptosis.8,20,29,32,33 For example, Qu et al found that the expression levels of RIP1 and RIP3 were significantly increased in developing brain after hypoxia-ischemia induction. Meanwhile, the interaction of RIP1–RIP3 complex with MLKL was also upregulated in such a condition.29 The activation of RIP1–RIP3–MLKL pathway has been considered as a step that leads to necroptosis.34 In line with these studies, our experimental data showed that these three critical necroptosis-related proteins, including RIP1, RIP3 and MLKL, were elevated in rats subjected to SAH, as compared to sham-operated rats, although the exact mechanisms that initiate necroptosis at an early stage after SAH remain unknown. Degterev et al proposed that pro-death signaling molecules, such as TNF-α and factor-related apoptosis ligand, triggered necroptosis through combining with their death receptors during ischemic brain injury.8 Consistently, the production of inflammatory cytokine TNF-α was also increased as a result of activation of NF-κB signaling after SAH induction in our study. Combined with the findings from our study, we believe that necroptosis is activated and closely related to the EBI after experimental SAH.
As a potent inhibitor of RIP1, Nec-1 inhibits the activation of RIP1 kinase and subsequently blocks the formation of necrosome. In our study, we further investigated the protective effects of a single dose of Nec-1 on SAH-induced EBI. Results showed that the necroptotic factors RIP1, RIP3 and MLKL decreased after Nec-1 treatment. Additionally, a series of cascade reactions that contributed to EBI after experimental SAH, including the breakdown of BBB, formation of brain edema, neural cell death and inflammatory reaction, were also dramatically ameliorated after Nec-1 treatment. Our results are consistent with findings from previous studies which showed the expression levels of necroptosis-related proteins were decreased in brain after Nec-1 treatment, accompanied by neurologic improvement.12,32 These results together support a neuroprotective role of Nec-1 on EBI following SAH. However, it is worth noting that inhibition of RIP1 kinase activation by Nec-1 cannot completely decrease necroptosis-related proteins to the baseline level in the sham-operated group, which suggests that other molecular mechanisms may also be involved in the process of necroptosis. One possible explanation for this result is that necroptosis can be initiated via RIP1-independent signaling pathways, which has been reported in recent studies. In these studies, the activation of necroptosis was mediated by Toll-like receptor signaling (TLR3/TLR4) without participation of RIP1.35,36 On the other hand, the RIP3-associated death complex assembly is necessary and sufficient to initiate necroptosis.37
Inflammatory response has been accepted as a dominant mechanism implicated in the pathogenesis of EBI after experimental SAH.38 Proinflammatory transcription factor NF-κB, a core component of proinflammatory signaling, has also been clarified clearly in this field.39,40 Here, we also determined the expressions of NF-κB and its downstream effector molecules, TNF-α and IL-1β. Our results showed that activation of necroptosis was accompanied by an increasing NF-κB translocation to the nucleus and release of inflammatory cytokines at 48 hours after SAH, while neuroinflammation was alleviated after Nec-1 treatment. Some explanations may exist. Firstly, similar to necrotic cell death, necroptosis presents cell death with organelle swelling, plasma membrane rupture and inflammatory cell infiltration, all of which facilitate inflammatory response. The RIP1/RIP3/MLKL signaling cascade can be blocked to some extent on treatment with Nec-1, which results in inhibition of necroptosis. As shown in our results, the number of PI-positive cells reduced greatly after Nec-1 treatment. That is, Nec-1 reduces neuroinflammation by inhibiting necroptosis signaling. Secondly, the mechanisms of various agents effective for EBI were associated with its anti-inflammatory effect. Moreover, Su et al also revealed that Nec-1 directly inhibited microglial activation and reduced inflammatory response.11 In this study, we cannot completely eliminate the anti-inflammatory effect of Nec-1 itself. Therefore, we believe that Nec-1 ameliorates EBI after experimental SAH, mainly by inhibiting necroptosis.
Meanwhile, there are several limitations in our study. First, the distribution of necroptosis besides neurons has not been illustrated in this study. Double immunofluorescence would be conducted to identify the specific cell types that express the abovementioned necroptosis-related proteins. Second, only Nec-1 was used to determine the neuroprotective role against necroptosis, while the inhibitors of RIP3 and MLKL should also be explored in further study. Third, only a single dose of Nec-1 was administered because of its half-life period; therefore, multiple administrations should also be considered in the follow-up study.
Conclusion
In summary, we first showed the role of necroptosis on EBI following SAH, and found that blockade of necroptosis was effective in alleviating tissue injury and then promoting functional recovery in our model. Our results indicate that neuronal necroptosis may be a crucial pathogenic mechanism involved in the EBI after SAH. Moreover, we believe that Nec-1 may serve as a novel therapeutic strategy for alleviating EBI from SAH.
Acknowledgments
This work was supported by grants from the major project of Fujian Provincial Department of Science And Technology (No 2014YZ0003), the National Natural Science Foundation of China (No 81501012), the Youth Project of Fujian Provincial Department of Health (No 2014-1-59), the Fujian Province Natural Science Foundation (No 2015J05149) and the key clinical specialty discipline construction program of Fujian, China. The authors would like to thank Linying Zhou from the Lab of Electron Microscopy, School of Basic Medical Science, Fujian Medical University for technical assistance.
Disclosure
The authors report no conflicts of interest in this work.
References
Cahill J, Calvert JH, Zhang JH. Mechanisms of early brain injury after subarachnoid hemorrhage. J Cereb Blood Flow Metab. 2006;26(11):1341–1353. | ||
Cahill J, Zhang JH. Subarachnoid hemorrhage: is it time for a new direction? Stroke. 2009;40(Suppl 3):S86–S87. | ||
Ostrowski RP, Colohan AR, Zhang JH. Molecular mechanisms of early brain injury after subarachnoid hemorrhage. Neurol Res. 2006;28(4):399–414. | ||
Li J, Chen J, Mo H, et al. Minocycline protects against NLRP3 inflammasome-induced inflammation and P53-associated apoptosis in early brain injury after subarachnoid hemorrhage. Mol Neurobiol. 2016;53(4):2668–2678. | ||
Park S, Yamaguchi M, Zhou C, Calvert JW, Tang J, Zhang JH. Neurovascular protection reduces early brain injury after subarachnoid hemorrhage. Stroke. 2004;35(10):2412–2417. | ||
Cahill J, Calvert JW, Solaroglu I, Zhang JH. Vasospasm and p53-induced apoptosis in an experimental model of subarachnoid hemorrhage. Stroke. 2006;37(7):1868–1874. | ||
Connolly ES Jr, Rabinstein AA, Carhuapoma JR, et al; American Heart Association Stroke Council; Council on Cardiovascular Radiology and Intervention; Council on Cardiovascular Nursing; Council on Cardiovascular Surgery and Anesthesia; Council on Clinical Cardiology. Guidelines for the management of aneurysmal subarachnoid hemorrhage: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2012;43(6):1711–1737. | ||
Degterev A, Huang Z, Boyce M, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1(2):112–119. | ||
Cho YS, Challa S, Moquin D, et al. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. 2009;137(6):1112–1123. | ||
Wang H, Sun L, Su L, et al. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell. 2014;54(1):133–146. | ||
Su X, Wang H, Kang D, et al. Necrostatin-1 ameliorates intracerebral hemorrhage-induced brain injury in mice through inhibiting RIP1/RIP3 pathway. Neurochem Res. 2015;40(4):643–650. | ||
Ofengeim D, Ito Y, Najafov A, et al. Activation of necroptosis in multiple sclerosis. Cell Rep. 2015;10(11):1836–1849. | ||
Fan H, Zhang K, Shan L, et al. Reactive astrocytes undergo M1 microglia/macrohpages-induced necroptosis in spinal cord injury. Mol Neurodegener. 2016;11:14. | ||
Kanno H, Ozawa H, Tateda S, Yahata K, Itoi E. Upregulation of the receptor-interacting protein 3 expression and involvement in neural tissue damage after spinal cord injury in mice. BMC Neuroscience. 2015;16:62. | ||
Zhang J, Yang Y, He W, Sun L. Necrosome core machinery: MLKL. Cell Mol Life Sci. 2016;73(11–12):2153–2163. | ||
Dillon Christopher P, Oberst A, Weinlich R, et al. Survival Function of the FADD-CASPASE-8-cFLIPL Complex. Cell Reports. 2012;1(5):401–407. | ||
Lin Y, Choksi S, Shen HM, et al. Tumor necrosis factor-induced nonapoptotic cell death requires receptor-interacting protein-mediated cellular reactive oxygen species accumulation. J Biol Chem. 2004;279(11):10822–10828. | ||
Matsumura H, Shimizu Y, Ohsawa Y, Kawahara A, Uchiyama Y, Nagata S. Necrotic death pathway in Fas receptor signaling. J Cell Biol. 2000;151(6):1247–1256. | ||
Kusaka G, Ishikawa M, Nanda A, Granger DN, Zhang JH. Signaling pathways for early brain injury after subarachnoid hemorrhage. J Cereb Blood Flow Metab. 2004;24(8):916–925. | ||
Northington FJ, Chavez-Valdez R, Graham EM, Razdan S, Gauda EB, Martin LJ. Necrostatin decreases oxidative damage, inflammation, and injury after neonatal HI. J Cereb Blood Flow Metab. 2011;31(1):178–189. | ||
Yan F, Cao S, Li J, et al. Pharmacological Inhibition of PERK Attenuates Early Brain Injury After Subarachnoid Hemorrhage in Rats Through the Activation of Akt. Mol Neurobiol. 2017;54(3):1808–1817. | ||
Garcia JH, Wagner S, Liu KF, Hu XJ. Neurological deficit and extent of neuronal necrosis attributable to middle cerebral artery occlusion in rats. Statistical validation. Stroke. 1995;26(4):627–634; discussion 635. | ||
Chen J, Li Y, Wang L, et al. Therapeutic benefit of intravenous administration of bone marrow stromal cells after cerebral ischemia in rats. Stroke. 2001;32(4):1005–1011. | ||
Kuan YH, Shih HC, Tang SC, Jeng JS, Shyu BC. Targeting P(2)X(7) receptor for the treatment of central post-stroke pain in a rodent model. Neurobiol Dis. 2015;78:134–145. | ||
Tang J, Zhu C, Li ZH, et al. Inhibition of the spinal astrocytic JNK/MCP-1 pathway activation correlates with the analgesic effects of tanshinone IIA sulfonate in neuropathic pain. J Neuroinflammation. 2015;12:57. | ||
Sugawara T, Ayer R, Jadhav V, Zhang JH. A new grading system evaluating bleeding scale in filament perforation subarachnoid hemorrhage rat model. J Neurosci Methods. 2008;167(2):327–334. | ||
Suzuki H, Hasegawa Y, Kanamaru K, Zhang JH. Mechanisms of osteopontin-induced stabilization of blood-brain barrier disruption after subarachnoid hemorrhage in rats. Stroke. 2010;41(8):1783–1790. | ||
Zhu Y, Cui H, Gan H, et al. Necroptosis mediated by receptor interaction protein kinase 1 and 3 aggravates chronic kidney injury of subtotal nephrectomised rats. Biochem Biophys Res Commun. 2015;461(4):575–581. | ||
Qu Y, Shi J, Tang Y, et al. MLKL inhibition attenuates hypoxia-ischemia induced neuronal damage in developing brain. Exp Neurol. 2016;279:223–231. | ||
Zhang D, Yan H, Li H, et al. TGFbeta-activated Kinase 1 (TAK1) Inhibition by 5Z-7-Oxozeaenol Attenuates Early Brain Injury after Experimental Subarachnoid Hemorrhage. J Biol Chem. 2015;290(32):19900–19909. | ||
Feng L, Chen Y, Ding R, et al. P2X7R blockade prevents NLRP3 inflammasome activation and brain injury in a rat model of intracerebral hemorrhage: involvement of peroxynitrite. J Neuroinflammation. 2015;12:190. | ||
You Z, Savitz SI, Yang J, et al. Necrostatin-1 reduces histopathology and improves functional outcome after controlled cortical impact in mice. J Cereb Blood Flow Metab. 2008;28(9):1564–1573. | ||
Vieira M, Fernandes J, Carreto L, et al. Ischemic insults induce necroptotic cell death in hippocampal neurons through the up-regulation of endogenous RIP3. Neurobiol Dis. 2014;68:26–36. | ||
Wang Z, Jiang H, Chen S, Du F, Wang X. The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways. Cell. 2012;148(1–2):228–243. | ||
Kaiser WJ, Sridharan H, Huang C, et al. Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J Biol Chem. 2013;288(43):31268–31279. | ||
He SD, Liang YQ, Shao F, Wang X. Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3-mediated pathway. Proc Natl Acad Sci U S A. 2011;108(50):20054–20059. | ||
Wu XN, Yang ZH, Wang XK, et al. Distinct roles of RIP1-RIP3 hetero- and RIP3-RIP3 homo-interaction in mediating necroptosis. Cell Death Differ. 2014;21(11):1709–1720. | ||
Dumont AS, Dumont RJ, Chow MM, et al. Cerebral vasospasm after subarachnoid hemorrhage: putative role of inflammation. Neurosurgery. 2003;53(1):123–133; discussion 133–125. | ||
Suzuki H, Ayer R, Sugawara T, et al. Protective effects of recombinant osteopontin on early brain injury after subarachnoid hemorrhage in rats. Crit Care Med. 2010;38(2):612–618. | ||
Echigo R, Shimohata N, Karatsu K, et al. Trehalose treatment suppresses inflammation, oxidative stress, and vasospasm induced by experimental subarachnoid hemorrhage. J Transl Med. 2012;10:80. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.