")
Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 15
Necroptosis Mediates Cigarette Smoke-Induced Inflammatory Responses in Macrophages
Authors Wang Y, Wang XK, Wu PP, Wang Y, Ren LY, Xu AH
Received 8 October 2019
Accepted for publication 12 March 2020
Published 18 May 2020 Volume 2020:15 Pages 1093—1101
DOI https://doi.org/10.2147/COPD.S233506
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Chunxue Bai
Yong Wang, Xiao-Ke Wang, Pei-Pei Wu, Yi Wang, Liang-Yu Ren, Ai-Hui Xu
Department of Respiratory and Critical Care Medicine, First Affiliated Hospital of Anhui Medical University, Hefei 230022, People’s Republic of China
Correspondence: Ai-Hui Xu
Department of Respiratory and Critical Care Medicine, First Affiliated Hospital of Anhui Medical University, 218 Jixi Road, Hefei 230022, People’s Republic of China
Email [email protected]
Introduction: Cigarette smoke (CS)-induced inflammation in macrophages is involved in the pathological process of chronic obstructive pulmonary disease (COPD). Necroptosis, which is a form of programmed necrosis, has a close relationship with robust inflammation, while its roles in COPD are unclear.
Materials and Methods: Necroptosis markers were measured in mouse alveolar macrophages and cultured bone marrow-derived macrophages (BMDMs). Necroptosis inhibitors were used to block necroptosis in BMDMs, and inflammatory cytokines were detected. We further explored the related signaling pathways.
Results: In this study, we demonstrated the way in which necroptosis, in addition to its upstream and downstream signals, regulates CS-induced inflammatory responses in macrophages. We observed that CS exposure caused a significant increase in the levels of necroptosis markers (receptor interacting kinases [RIPK] 1 and 3) in mouse alveolar macrophages and BMDMs. Pharmacological inhibition of RIPK1 or 3 caused a significant suppression in CS extract (CSE)-induced inflammatory cytokines, chemokine ligands (CXCL) 1 and 2, and interleukin (IL)-6 in BMDMs. CSE-induced necroptosis was regulated by mitochondrial reactive oxygen species (mitoROS), which also promoted inflammation in BMDMs. Furthermore, necroptosis regulated CSE-induced inflammatory responses in BMDMs, most likely through activation of the nuclear factor-κB pathway.
Conclusion: Taken together, our results demonstrate that mitoROS-dependent necroptosis is essential for CS-induced inflammation in BMDMs and suggest that inhibition of necroptosis in macrophages may represent effective therapeutic approaches for COPD patients.
Keywords: cigarette smoke, macrophage, necroptosis, inflammatory response, NF-κB pathway
Introduction
Chronic obstructive pulmonary disease (COPD) is characterized by irreversible airflow obstruction and abnormal lung inflammation. COPD was responsible for around 6% of all deaths worldwide in 2012, and it is the fourth leading cause of death globally.1,2 This disease encompasses two major clinical phenotypes: chronic bronchitis and emphysema.3 Although multiple factors increase the risk for COPD, tobacco smoke remains the main cause. However, the cellular and molecular mechanisms that mediate cigarette smoke (CS)-induced COPD pathogenesis remain unknown.
Macrophages serve as the first line of defense and act as immune effector cells in the lung, which are reactive and respond to endogenous and exogenous stimuli. Accumulating evidence has shown that macrophage numbers are elevated in the alveoli and bronchioles and induce sputum formation in smokers and COPD patients.4 Additional studies suggest that there is a positive association between macrophage numbers in the alveolar walls and COPD severity. Macrophages are the major inflammatory cells in COPD, and they generate a host of inflammatory mediators and matrix metalloproteinases (MMPs), which cause defective immune surveillance and tissue damage that lead to COPD progression.5 However, the potential effects and detailed mechanisms of macrophages in regulation of CS-induced inflammatory responses remain unclear.
Necroptosis is a regulated form of necrosis that is characterized by cellular organellar swelling, cell membrane rupture, and proinflammatory intracellular component release, which relies on the enzymatic activity of receptor-interacting proteins (RIP) 1 and 3 in various diseases.6 RIP1/3 kinases (RIPK), which form a multiprotein complex called the necrosome, are key regulators of necroptosis.7 Mizumura et al showed that necroptosis participates in the process of COPD,8 and we also found that necroptosis plays an important role in CS-induced airway injury.9 Airway epithelial necroptosis is closely related to COPD pathogenesis. However, the underlying mechanisms of necroptosis in COPD have yet to be elucidated. We speculate that necroptosis may be involved in CS-induced macrophage inflammatory responses.
The present study aimed to explore roles and detailed mechanisms of necroptosis in regulation of CS-induced inflammatory responses in macrophages using pharmacological approaches. We propose that CS-induced necroptosis in macrophages as a specific inflammatory response mechanism. Administration of necroptosis inhibitors may, thus, represent a potential therapy for COPD.
Materials and Methods
Cigarette Smoke Extract Preparation and Cell Viability
CS extract (CSE) was prepared and treated as described previously.10,11 Cell viability was determined using the CCK8 (cell counting kit 8) assay (Liankebio, Hangzhou, China), according to the manufacturer’s instructions.
Chemicals and Reagents
GSK’872, BAY 11–7082, and MITO-TEMPO were purchased from Medchem Express (USA). Necrostatin-1 (NEC-1) and RBC lysing buffer were from Sigma-Aldrich (USA). Antibodies against RIPK3, RIPK1, p-P65, and actin were from Cell Signaling Technology and Abcam. RIPK3, RIPK1 and p-P65 were diluted at 1:1000 and actin were at 1:2500. Goat anti-Mouse and Goat anti-Rabbit secondary antibodies (diluted at 1:2500) were from Erath. Recombinant mouse M-CSF were from R&D Systems.
Bone Marrow-Derived Macrophages
Bone marrow-derived macrophages (BMDMs) were isolated and cultured as described previously.12 Eight-week-old mice were sacrificed and each entire mouse body soaked in 75% ethanol for 10 minutes. Bone marrow cells were flushed out from the murine femurs and tibias, and then were centrifuged for 5 min at 400 ×g. RBC lysing buffer was then used to eliminate erythrocytes. The remaining cells were cultured in DMEM containing 10% fetal bovine serum at 37°C, 5% CO2, and 10 ng/mL M-CSF for 7 days.
RNA Isolation and Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
Total RNA from BMDMs was isolated using RNAiso Plus (Takara Biotechnology, Beijing, China). Reverse Transcription Reagents (Takara Biotechnology) were applied to reverse-transcribe RNA. qRT-PCR using SYBR Green system (Takara Biotechnology) was applied to quantify mouse chemokine ligand (CXCL)1, CXCL2, and interleukin (IL)-6 gene expression.
ELISA
Cellular supernatants were collected from treated cells. Mouse CXCL1, CXCL2, and IL-6 ELISA kits (R&D Systems) were used to measure CXCL1, CXCL2, and IL-6 levels in cellular supernatants, according to the manufacturer’s protocols.
Western Blotting
Total proteins from cells were extracted using an ice-cold radioimmunoprecipitation assay containing protease (Roche Diagnostics) and phosphatase inhibitors (Roche Diagnostics). Protein samples were separated and immunoblotted using relevant antibodies, according to standard methods. Band densities were quantified using the Image J software.
Mice
Wild-type C57BL/6 mice were from the SLACCAS, Shanghai Laboratory Animal Center, CAS. All the mice were exposed for 1 month to room air or whole-body CS (2 h per day, 5 days per week) that was generated from research-grade cigarettes (3R4F; University of Kentucky, Lexington, KY, USA) in 5-l smoking chambers, as previously described.13 All the animal experimental protocols were approved by the Ethics Committee for Animal Studies at Anhui Medical University and were in accordance with the “Guide for the care and use of laboratory animals” approved by the committee.
Statistical Analysis
Data are presented as the mean ± standard error of the mean (SEM) and were analyzed using GraphPad Prism 7.00 (GraphPad software, CA, USA). Differences between experimental and control groups were identified using an unpaired, two-tailed Student’s t-test. A p value < 0.05 was considered to be significant.
Results
Cigarette Smoke Exposure Induces Necroptosis in Macrophages
To determine whether necroptosis was activated in macrophages upon CS exposure, we first assessed the role of necroptosis in CSE-induced cell death. As expected, we observed that CSE-induced cell death was significantly decreased after addition of the necroptosis inhibitor NEC-1, which targets RIPK1 activity (Figure 1A). Next, we measured the expression of necroptosis-related molecules in vivo and in vitro. Wild-type mice were exposed to air or CS for 1 month. We found that CS exposure increased the total number of inflammatory cells and inflammatory cytokines CXCL1 in BALF (Figure 1B and C). Alveolar macrophages were then isolated. RIP3 levels showed a significant increase in alveolar macrophages in the CS-exposed mouse, as indicated by Western blotting analysis (Figure 1D and E). Additionally, Western blotting analysis demonstrated that CSE caused an elevation in RIP1 and 3 in BMDMs (Figure 1F and G), indicating an increase in CS-induced necroptotic cell death in macrophages. These results suggest that CSE induces necroptosis in macrophages.
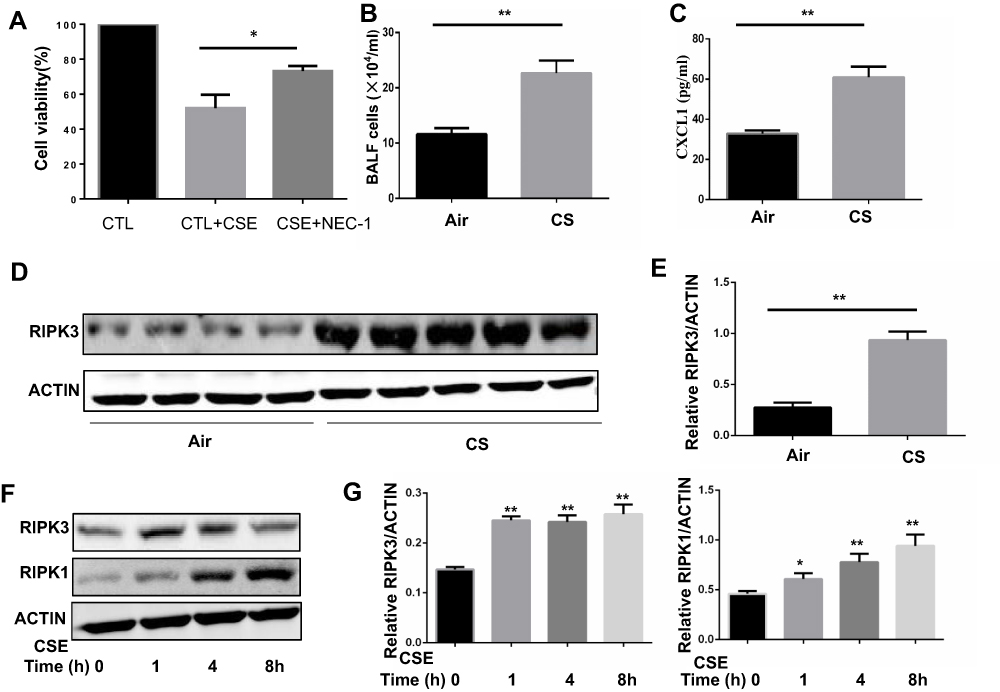 |
Figure 1 Cigarette smoke (CS) induces necroptosis in macrophages. (A) After pretreated with NEC-1 (50 μM) or vehicle, BMDMS were incubated with 2% CSE for 24h, and cell death was determined by CCK8 assay. (B and C) Mice were exposed to CS for 1 months, and the number of total inflammatory cells and CXCL1 in the bronchoalveolar lavage fluid (BALF) were measured.(D and E) Protein expression of RIPK3 in alveolar macrophages isolated from mice exposed to room air or whole-body CS is measured by Western blotting analysis. (F and G) Time-dependent expression of RIPK3 and RIPK1 in BMDMs treated with 1%CSE. Data are mean ± SEM of 3 independent experiments. Western blot data are representative of 3 independent experiments. *P < 0.05, **P < 0.01 (Student’s t-test). Abbreviations: CS, Cigarette smoke; BMDMs, bone marrow–derived macrophages; CSE, cigarette smoke extract; SEM, standard error of the mean. |
Necroptosis Regulates CSE-Induced Inflammatory Responses in BMDMs
Proinflammatory factors, such as CXCL1 and 2 and IL-6, were shown to be involved in COPD pathogenesis.14–16 We further explored the function of necroptosis in CS-induced inflammatory responses in BMDMs. Two commonly used pharmacological inhibitors, NEC-1 and GSK’872 that inhibit RIPK1 and 3, respectively, were used to block necroptosis. CSE exposure increased mRNA transcription and caused an increase in secreted CXCL1 and 2 and IL-6 protein levels in BMDMs, which was significantly reversed by NEC-1 and GSK’872 (Figures 2 and 3). Taken together, these data suggest that necroptosis promotes CSE-induced inflammation in macrophages.
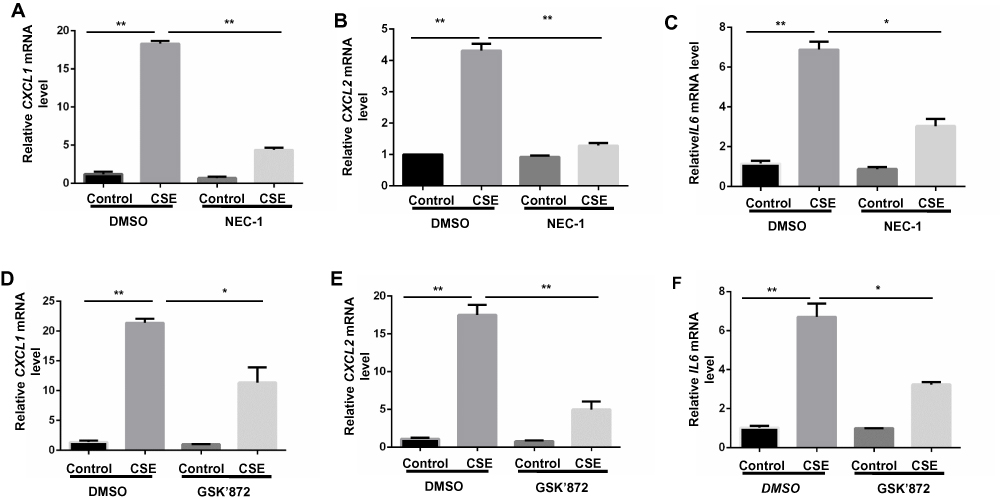 |
Figure 2 Inhibition of necroptosis alleviates CSE-induced mRNA transcripts of inflammatory cytokine in BMDMs. BMDMs were incubated with 1%CSE for 24h in the absence or presence of the indicated necroptosis inhibitor. Relative mRNA levels of CXCL1, CXCL2 and IL6 were detected by qRT-PCR (A–F). Data are mean ± SEM of 3 independent experiments. *P < 0.05, **P < 0.01 (Student’s t-test). Abbreviations: CSE, cigarette smoke extract; BMDMs, bone marrow–derived macrophages; NEC-1, necrostatin-1; qRT-PCR, quantitative real-time polymerase chain reaction; SEM, standard error of the mean. |
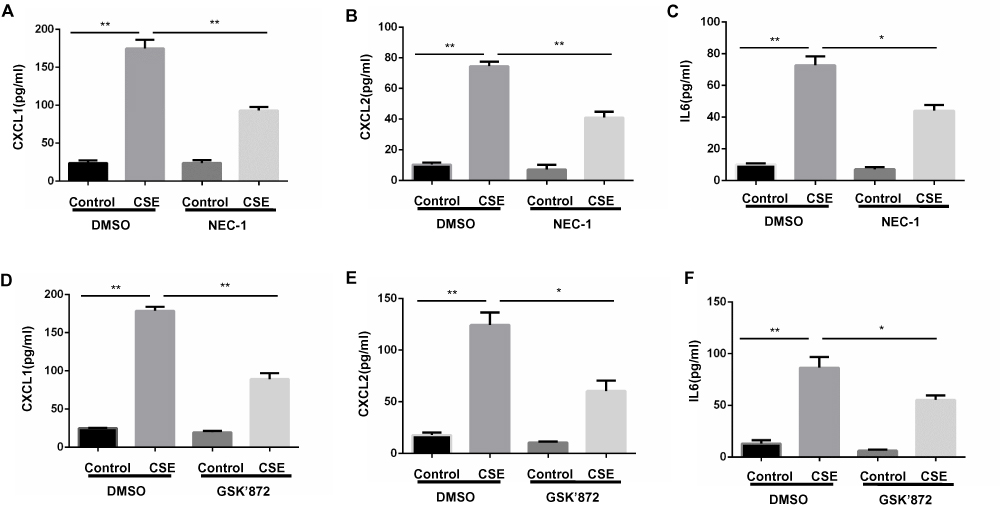 |
Figure 3 Inhibition of necroptosis alleviates CSE-induced protein levels of inflammatory cytokine in BMDMs. BMDMs were incubated with 1%CSE for 24h in the absence or presence of the indicated necroptosis inhibitor. Relative protein levels of CXCL1, CXCL2 and IL6 in the culture supernatants were detected by ELISA (A–F). Data are mean ± SEM of 3 independent experiments. *P < 0.05, **P < 0.01 (Student’s t-test). Abbreviations: CSE, cigarette smoke extract; BMDMs, bone marrow–derived macrophages; NEC-1, necrostatin-1; qRT-PCR, quantitative real-time polymerase chain reaction; SEM, standard error of the mean. |
mitoROS Mediates CSE-Induced Necroptosis and Inflammatory Responses in BMDMs
We next sought to examine the upstream pathways that mediate CSE-induced necroptosis and inflammatory responses. CS contains a high concentration of oxidants and reactive oxygen species (ROS), which are normally considered to be critical regulators of necroptotic signaling.17 CS-induced mitochondrial ROS (mitoROS), which can specifically promote necroptosis,18 appears to be a key contributor to COPD pathophysiology.19 Consequently, we intended to examine whether mitoROS is related to CSE-induced necroptosis and inflammatory responses in BMDMs. Specific inhibition of mitoROS by mitochondria-targeted antioxidant Mito-TEMPO effectively decreased CSE-induced cell death (Figure 4A) and RIP1 and 3 protein expression (Figure 4B and C). As expected, CXCL1 and 2 and IL-6 mRNA expression was significantly reduced by Mito-TEMPO (Figure 4D–F).
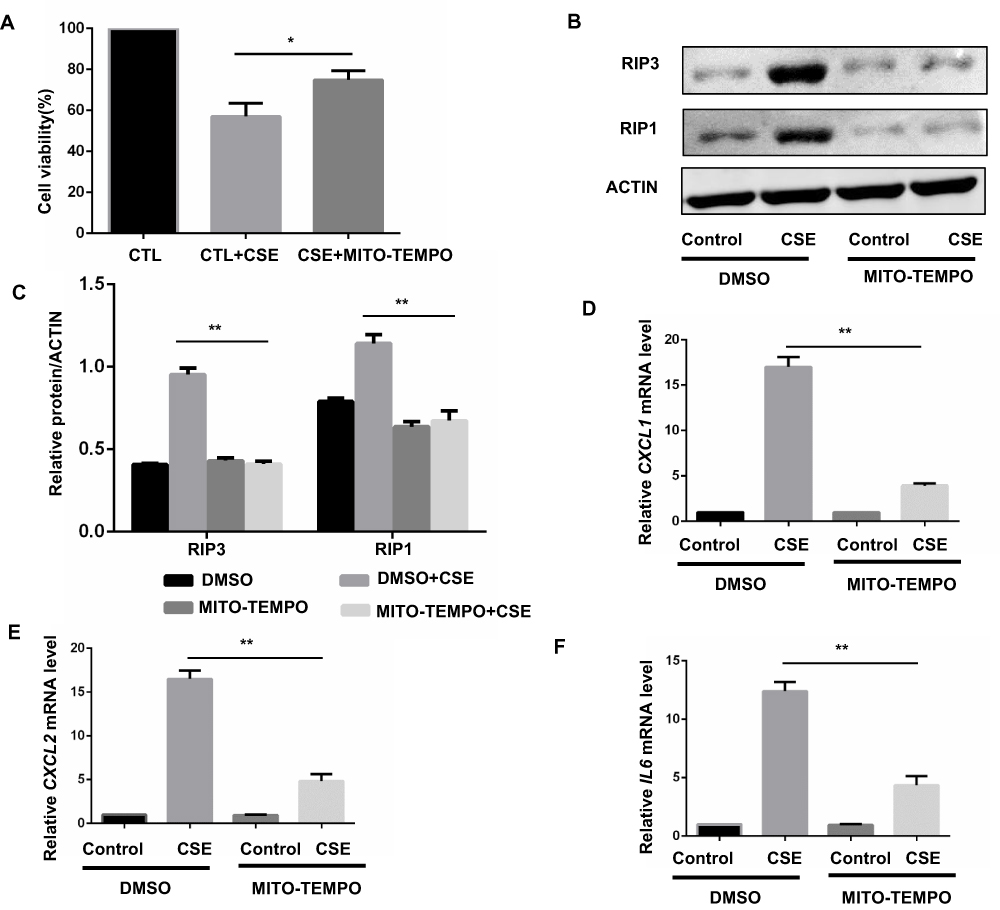 |
Figure 4 MitoROS mediate CSE-induced necroptosis and inflammatory responses in BMDMS. (A) After pretreated with MITO-TEMPO (10 μM) or vehicle, BMDMS were incubated with 2% CSE for 24h, and cell death was determined by CCK8 assay. (B and C) After pretreated with MITO-TEMPO or vehicle, BMDMS were incubated with 1% CSE for 8h and cell lysates were then subjected to Western blotting for RIPK3 and RIPK1. (D–F) BMDMs were incubated with 1%CSE for 24h in the absence or presence of the MITO-TEMPO. Relative mRNA levels of CXCL1, CXCL2 and IL6 were detected by qRT-PCR. Data are mean ± SEM of 3 independent experiments. Western blot data are representative of 3 independent experiments. *P < 0.05, **P < 0.01 (Student’s t-test). Abbreviations: CSE, cigarette smoke extract; BMDMs, bone marrow–derived macrophages; qRT-PCR, quantitative real-time polymerase chain reaction; SEM, standard error of the mean. |
Necroptosis Modulated Inflammatory Responses Through NF-κB Pathway in BMDMs
Nuclear factor (NF)-κB is closely associated with the regulation of COPD-related inflammatory cytokines,20 and we sought to clarify whether the NF-κB pathway acts as the downstream pathway that mediates the regulation of necroptosis in CS-induced inflammation. Previous studies revealed that the NF-κB pathway is required for the inflammation response in BMDMs.12 We also confirmed that inhibition of the NF-κB pathway by BAY 11–7082 suppressed CSE-induced CXCL1 and 2 and IL-6 expression in BMDMs (Figure 5A–C). Inhibition of necroptosis by NEC-1 or GSK’872 caused a significant decrease in p-P65 activation (Figure 5D and E). These data suggest that necroptosis upregulates CSE-induced inflammation through activation of the NF-κB pathway.
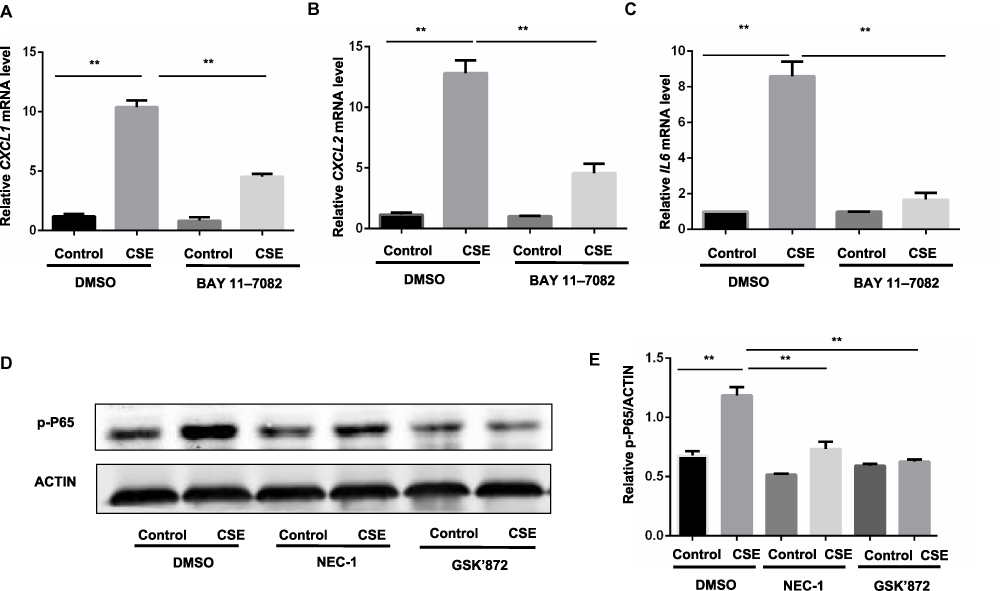 |
Figure 5 Necroptosis regulate CSE-induced inflammation through the activation of NF-κB pathway in BMDMs. (A–C) BMDMs were cultured with BAY 11–7082 (2.5μM) or vehicle together with 1% CSE for 24h, CXCL1, CXCL2 and IL6 mRNA transcripts were detected by qRT-PCR. (D and E) BMDMs were pretreated with the indicated necroptosis inhibitor, following treatment with 1% CSE for 12h, and the levels of p-P65 were assessed by Western blotting. **P < 0.01 (Student’s t-test). Abbreviations: CSE, cigarette smoke extract; BMDMs, bone marrow–derived macrophages; qRT-PCR, quantitative real-time polymerase chain reaction; SEM, standard error of the mean. |
Discussion
In this study, we demonstrated that CS elicits necroptosis through mitoROS production in macrophages, which is essential for CS-induced pulmonary inflammation in COPD pathogenesis. Necroptosis regulates CS-induced inflammatory responses, which may occur through the NF-κB pathway. Taken together, these data provide new insights into the deleterious role of necroptosis in COPD pathogenesis.
As a novel cell death mode, necroptosis has been shown to be involved in retinal degeneration, brain impact trauma, liver injury, and septic shock, and so on.21 Necroptosis execution depends on RIPK1 and 3 activation and it can be blocked by their inhibitors. Accompanied by robust inflammation, necroptotic cells can release damage-associated molecular patterns (DAMPs) that can initiate an inflammatory response; thus, necroptosis appears to be closely related to inflammation.22 A growing body of evidence has shown that necroptosis is elicited in idiopathic pulmonary fibrosis, acute lung injury, and environmental particle-induced pulmonary inflammation.23 In accordance with these studies, results from this research demonstrates that CS promotes necroptosis in alveolar macrophages and BMDMs. We also observed that inhibition of RIP1 or 3 caused a marked attenuation of CSE-induced inflammatory responses in BMDMs, indicating that necroptosis plays a deleterious role in COPD pathogenesis. In terms of mechanisms, we also provided strong evidence that CSE initially triggers mitoROS production and subsequently elicits necroptosis in BMDMs, which regulates inflammation that may occur through modulation of the NF-κB signaling. These data provide new insights in which blocking necroptosis in macrophages provides therapeutic benefits by alleviating CS-induced inflammation.
Previous study showed that CSE induced necroptosis in pulmonary epithelial cells8. We and others further found that necroptosis in bronchial epithelial cells play an important role in CS-induced airway inflammation.9,24 The present study indicated that necroptosis also regulated CSE-induced inflammatory responses in BMDMs. There is a dynamic interaction between macrophages and airway epithelium in the lung, and they may cooperatively regulated inflammatory responses in the COPD pathogenesis.
Oxidative stress occurs because of excessive free radical production and reduced antioxidant availability. Oxidative stress is integral to the general inflammatory responses, in addition to physiological signaling and activation of the host’s defenses.19 Excessive ROS accumulation, which could be from exogenous CS, leads to oxidative stress.25 In the respiratory system, ROS could damage lung tissue and act as a trigger for enzymatically-generated ROS that are released from respiratory and inflammatory cells, and this is essential for COPD development and progression.25 Accumulating evidence suggests that ROS is an important driving force in necroptosis.26,27 Recent evidence has suggested that mitoROS promotes RIP1 autophosphorylation on serine residue 161 and subsequently recruits RIP3 to form a functional necrosome, which controls necroptosis formation.18 Consistent with previous findings, the current study results showed that mitoROS mediates CS-induced necroptosis and inflammatory responses in macrophages, suggesting that CS elicits necroptosis that is most likely dependent on mitoROS production.
Inflammation is a critical feature in lung injury, and it causes the release of inflammatory mediators and substantial proteases, thereby ultimately contributing to COPD development.28 As the major inflammatory cells in the COPD lung, macrophages secrete inflammatory factors and enzymes that are involved in the process of airway injury and emphysematous lung destruction.29,30 Numerous studies have shown that inhibition of necroptosis can alleviate lung injury in several pulmonary diseases.31–33 However, a recent study has reported that induction of necroptosis caused a significant reduction in lung metastasis that was caused by an osteosarcoma.34 Our current study results are consistent with previous results that showed that necroptosis inhibitors greatly reduce the CS-induced inflammatory response in BMDMs, indicating that necroptosis plays a deleterious role in COPD development. Because pulmonary disease is complex, the function of necroptosis in the lung may be either protective or deleterious.
Generally, NF-κB signaling plays a large role in initiating inflammation. Substantial evidence has showed that CS induces the inflammatory response in the COPD lung through NF-κB signaling, the intensity of which is positively related to the severity of COPD.35,36
NF-κB typically consists of P50/P65 heterodimers, and P65 often controls the NF-κB transcriptional activation.37 Similarly, the present study provides strong evidence that the NF-κB pathway is essential for the regulating CS-induced inflammation in BMDMs. We further demonstrated that necroptosis is responsible for modulating the p65 phosphorylation status and, in turn, regulating inflammatory gene transcription in BMDMs; this suggests that necroptosis is a pivotal regulator in NF-κB signaling. Yatim et al38 reported that NF-κB activation is RIPK1-dependent, and RIPK3 was also shown to promote inflammatory responses through the NF-κB pathway.39 However, the specific necroptosis-related mechanisms that are involved in regulation of the NF-κB pathway remain unknown, and further studies are needed.
Conclusion
Thus, we demonstrated the manner in which CSE induces mitoROS and subsequently activates necroptosis in macrophages, which are essential events for inflammatory responses, and this activation may occur through modulation of the NF-κB pathway. Thus, our results suggest that inhibition of necroptosis and/or mitoROS in BMDMs could represent novel therapeutic strategies for CS-induced inflammation in COPD patients.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Pauwels RA, Rabe KF. Burden and clinical features of chronic obstructive pulmonary disease (COPD). Lancet. 2004;364(9434):613–620. doi:10.1016/S0140-6736(04)16855-4
2. Vestbo J, Hurd SS, Agusti AG, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am J Respir Crit Care Med. 2013;187(4):347–365. doi:10.1164/rccm.201204-0596PP
3. Decramer M, Janssens W, Miravitlles M. Chronic obstructive pulmonary disease. Lancet. 2012;379(9823):1341–1351. doi:10.1016/S0140-6736(11)60968-9
4. Wang Y, Xu J, Meng Y, Adcock IM, Yao X. Role of inflammatory cells in airway remodeling in COPD. Int J Chron Obstruct Pulmon Dis. 2018;13:3341–3348. doi:10.2147/COPD.S176122
5. Yamasaki K, Eeden SFV. Lung macrophage phenotypes and functional responses: role in the pathogenesis of COPD. Int J Mol Sci. 2018;19(2):E582. doi:10.3390/ijms19020582
6. Linkermann A, Green DR. Necroptosis. N Engl J Med. 2014;370(5):455–465. doi:10.1056/NEJMra1310050
7. Zhang D-W, Shao J, Lin J, et al. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science. 2009;325(5938):332–336. doi:10.1126/science.1172308
8. Mizumura K, Cloonan S, Nakahira K, et al. Mitophagy-dependent necroptosis contributes to the pathogenesis of COPD. J Clin Invest. 2014;124(9):3987–4003. doi:10.1172/JCI74985
9. Wang Y, Zhou JS, Xu XC, et al. Endoplasmic reticulum chaperone GRP78 mediates cigarette smoke-induced necroptosis and injury in bronchial epithelium. Int J Chron Obstruct Pulmon Dis. 2018;13:571–581. doi:10.2147/COPD.S150633
10. Chen ZH, Kim HP, Sciurba FC, et al. Egr-1 regulates autophagy in cigarette smoke-induced chronic obstructive pulmonary disease. PLoS One. 2008;3(10):e3316. doi:10.1371/journal.pone.0003316
11. Chen ZH, Lam HC, Jin Y, et al. Autophagy protein microtubule-associated protein 1 light chain-3B (LC3B) activates extrinsic apoptosis during cigarette smoke-induced emphysema. Proc Natl Acad Sci U S A. 2010;107(44):18880–18885. doi:10.1073/pnas.1005574107
12. Li Z, Wu Y, Chen HP, et al. MTOR suppresses environmental particle-induced inflammatory response in macrophages. J Immunol. 2018;200(8):2826–2834. doi:10.4049/jimmunol.1701471
13. Zhou J, Zhao Y, Zhou H, et al. Autophagy plays an essential role in cigarette smoke-induced expression of MUC5AC in airway epithelium. Am J Physiol Lung Cell Mol Physiol. 2016;310(11):L1042–L1052. doi:10.1152/ajplung.00418.2015
14. Su B, Liu T, Fan H, Chen F, Ding H. Inflammatory markers and the risk of chronic obstructive pulmonary disease: a systematic review and meta-analysis. PLoS One. 2016;11(4):e0150586. doi:10.1371/journal.pone.0150586
15. Lazar Z, Mullner N, Lucattelli M, et al. NTPDase1/CD39 and aberrant purinergic signalling in the pathogenesis of COPD. Eur Respir J. 2016;47(1):254–263. doi:10.1183/13993003.02144-2014
16. Baudiß K, Ayata CK, Lazar Z, et al. Ceramide-1-phosphate inhibits cigarette smoke-induced airway inflammation. Eur Respir J. 2015;45(6):1669–1680. doi:10.1183/09031936.00080014
17. Schenk B, Fulda S. Reactive oxygen species regulate Smac mimetic/TNFα-induced necroptotic signaling and cell death. Oncogene. 2015;34(47):5796. doi:10.1038/onc.2015.35
18. Zhang Y, Su SS, Zhao S, et al. RIP1 autophosphorylation is promoted by mitochondrial ROS and is essential for RIP3 recruitment into necrosome. Nat Commun. 2017;8(1):14329. doi:10.1038/ncomms14329
19. Boukhenouna S, Wilson MA, Bahmed K, Kosmider B. Reactive oxygen species in chronic obstructive pulmonary disease. Oxid Med Cell Longev. 2018;2018:5730395. doi:10.1155/2018/5730395
20. Lee IT, Yang CM. Inflammatory signalings involved in airway and pulmonary diseases. Mediators Inflamm. 2013;2013:791231. doi:10.1155/2013/791231
21. Weinlich R, Oberst A, Beere HM, Green DR. Necroptosis in development, inflammation and disease. Nat Rev Mol Cell Bio. 2016;18(2):127–136. doi:10.1038/nrm.2016.149
22. Fan EKY, Fan J. Regulation of alveolar macrophage death in acute lung inflammation. Resp Res. 2018;19(1):50. doi:10.1186/s12931-018-0756-5
23. Xu F, Luo M, He L, et al. Necroptosis contributes to urban particulate matter-induced airway epithelial injury. Cell Physio Biochem. 2018;46(2):699–712. doi:10.1159/000488726
24. Pouwels SD, Zijlstra GJ, Toorn M, et al. Cigarette smoke-induced necroptosis and DAMP release trigger neutrophilic airway inflammation in mice. Am J Physiol Lung Cell Mol Physiol. 2016;310(4):L377–L386. doi:10.1152/ajplung.00174.2015
25. Domej W, Oettl K, Renner W. Oxidative stress and free radicals in COPD–implications and relevance for treatment. Int J Chron Obstruct Pulmon Dis. 2014;9:1207–1224. doi:10.2147/COPD.S51226
26. Schulze-Osthoff K, Bakker AC, Vanhaesebroeck B, Beyaert R, Jacob WA, Fiers W. Cytotoxic activity of tumor necrosis factor is mediated by early damage of mitochondrial functions. Evidence for the involvement of mitochondrial radical generation. J Biol Chem. 1992;267(8):5317–5323.
27. Locatelli SL, Cleris L, Stirparo GG, et al. BIM upregulation and ROS-dependent necroptosis mediate the antitumor effects of the HDACi givinostat and sorafenib in hodgkin lymphoma cell line xenografts. Leukemia. 2014;28(9):1861–1871. doi:10.1038/leu.2014.81
28. Sharafkhaneh A, Hanania NA, Kim V. Pathogenesis of emphysema: from the bench to the bedside. Proc Am Thorac Soc. 2008;5(4):475–477. doi:10.1513/pats.200708-126ET
29. Arora S, Dev K, Agarwal B, Das P, Syed MA. Macrophages: their role, activation and polarization in pulmonary diseases. Immunobiology. 2017;223(4–5):S0171298517302073.
30. Barnes PJ. Cellular and molecular mechanisms of chronic obstructive pulmonary disease. Clin Chest Med. 2014;35(1):71–86. doi:10.1016/j.ccm.2013.10.004
31. Su HL, Ju HS, Song JH, et al. Inhibition of insulin-like growth factor receptor-1 reduces necroptosis-related markers and attenuates LPS-induced lung injury in mice. Biochem Biophys Res Commun. 2018;498(4):877–883. doi:10.1016/j.bbrc.2018.03.074
32. Bolognese AC, Yang WL, Hansen LW, et al. Inhibition of necroptosis attenuates lung injury and improves survival in neonatal sepsis. Surgery. 2018;S0039–6060(18):30096–30105.
33. Zhao H, Ning J, Lemaire A, et al. Necroptosis and parthanatos are involved in remote lung injury after receiving ischemic renal allografts in rats. Kidney Int. 2015;87(4):738–748. doi:10.1038/ki.2014.388
34. Fu Z, Deng B, Liao Y, et al. The anti-tumor effect of shikonin on osteosarcoma by inducing RIP1 and RIP3 dependent necroptosis. BMC Cancer. 2013;13(1):580. doi:10.1186/1471-2407-13-580
35. Di Stefano A, Caramori G, Oates T, et al. Increased expression of nuclear factor- B in bronchial biopsies from smokers and patients with COPD. Eur Respir J. 2002;20(3):556–563. doi:10.1183/09031936.02.00272002
36. Marwick JA, Kirkham PA, Stevenson CS, et al. Cigarette smoke alters chromatin remodeling and induces proinflammatory genes in rat lungs. Am J Respir Cell Mol Biol. 2004;31(6):633–642. doi:10.1165/rcmb.2004-0006OC
37. Campbell KJ, Perkins ND. Post-translational modification of RelA(p65) NF-kappaB. Biochem Soc Trans. 2004;32(6):1087–1089. doi:10.1042/BST0321087
38. Yatim N, Hélène JS, Susana O, et al. RIPK1 and NF-κB signaling in dying cells determines cross-priming of CD8 T cells. Science. 2015;350(6258):328. doi:10.1126/science.aad0395
39. Moriwaki K, Balaji S, Mcquade T, Malhotra N, Kang J, Chan FKM. The necroptosis adaptor RIPK3 promotes injury-induced cytokine expression and tissue repair. Immunity. 2014;41(4):567–578. doi:10.1016/j.immuni.2014.09.016
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.