")
Back to Journals » Journal of Blood Medicine » Volume 5
Hemophagocytic lymphohistiocytosis: review of etiologies and management
Authors George M
Received 27 December 2013
Accepted for publication 8 March 2014
Published 12 June 2014 Volume 2014:5 Pages 69—86
DOI https://doi.org/10.2147/JBM.S46255
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Melissa R George
Department of Pathology, Penn State Milton S Hershey Medical Center, Hershey, PA, USA
Abstract: Hemophagocytic lymphohistiocytosis (HLH) covers a wide array of related life-threatening conditions featuring ineffective immunity characterized by an uncontrolled hyperinflammatory response. HLH is often triggered by infection. Familial forms result from genetic defects in natural killer cells and cytotoxic T-cells, typically affecting perforin and intracellular vesicles. HLH is likely under-recognized, which contributes to its high morbidity and mortality. Early recognition is crucial for any reasonable attempt at curative therapy to be made. Current treatment regimens include immunosuppression, immune modulation, chemotherapy, and biological response modification, followed by hematopoietic stem-cell transplant (bone marrow transplant). A number of recent studies have contributed to the understanding of HLH pathophysiology, leading to alternate treatment options; however, much work remains to raise awareness and improve the high morbidity and mortality of these complex conditions.
Keywords: macrophage activation syndrome, hyperinflammatory response
Introduction
Hemophagocytosis is the engulfment of hematopoietic cells by activated macrophages acting outside of usual immune system regulations. Hemophagocytic lymphohistiocytosis (HLH) covers a wide array of related diseases including HLH, autosomal recessive familial HLH (FHL), familial erythrophagocytic lymphohistiocytosis, viral-associated hemophagocytic syndrome, and autoimmune-associated macrophage activation syndrome (MAS). These disorders feature severe cytopenias due to this uncontrolled hemophagocytosis. Other laboratory signs and clinical symptoms result from disordered immune regulation and cytokine storm. The term primary HLH refers to an underlying genetic abnormality causing the disorder, whereas secondary HLH indicates that the disorder is secondary to underlying conditions such as infection, autoimmune/rheumatologic, malignant, or metabolic conditions. For the purposes of this review, FHL will indicate cases with a primary genetic cause, secondary HLH will refer to cases secondary to infection, malignancy, or metabolic disorders, and MAS will refer to cases associated with autoimmune diseases.
Much has been learned about HLH in the 75 years since it was first discovered. One of the earliest descriptions of the disease was in 1939 when Scott and Robb-Smith1 described a disorder featuring erythrophagocytosis by proliferating histiocytes in the lymphoreticular system and called it “histiocytic medullary reticulosis” or HMR. It was later classified among malignant histiocytosis. Later in 1952, the familial form of HLH, FHL, was more fully described by Farquhar and Claireaux2 with the cases of two siblings who succumbed to HLH, and later in 1958, another sibling from this same family presented in the same manner.3 Risdall was among the first to describe a viral association with HLH and proposed that the condition be called virus-associated HLH, distinct from malignant histiocytosis.4 In the years since, researchers have recognized the wide scope of this disease and the fact that infection often triggers both primary and secondary HLH. Regardless of cause, physiologically, HLH is characterized by defective cytotoxic cell function coupled with unbridled macrophage activity, leading to excessive cytokine production, subsequent immune dysregulation, and tissue damage. Left untreated, the dysregulated inflammatory response causes severe neutropenia, and patients often die from bacterial or fungal infections. The condition carries high morbidity and mortality.5 Long-term survival in 1983 was estimated to be as low as 4%.6 The median survival without treatment is estimated at <2 months.6
Diagnosis
The diagnosis of FHL or secondary HLH is based on a number of clinical signs and laboratory findings. Due to the relatively nonspecific nature of the clinical signs and symptoms, and significant overlap with other illnesses, diagnosis is often delayed. The official diagnosis of HLH, established by the Histiocyte Society, is based on fulfilling one or both of the following criteria:
- A molecular diagnosis consistent with HLH
- Five out of the following nine diagnostic criteria for HLH: fever, splenomegaly, cytopenias (affecting two or more of three lineages in the peripheral blood), hypertriglyceridemia, hypofibrinogenemia, elevated ferritin, hemophagocytosis in bone marrow/spleen/lymph nodes, low or absent natural killer (NK)-cell activity, or elevated soluble CD25 (interleukin [IL]-2 receptor).7,8
All of the clinical and laboratory findings are readily linked to the pathophysiology of HLH. Fever is the result of high IL levels. Splenomegaly is the direct result of infiltration by lymphocytes and macrophages. Cytopenias can be explained by high concentrations of tumor necrosis factor (TNF)-α and interferon (IFN)-γ, as well as direct hemophagocytosis. High triglycerides are secondary to decreased lipoprotein lipase activity initiated by increased TNF-α levels. Elevated ferritin >10,000 μg/L has been demonstrated to be 90% sensitive and 96% specific for HLH.9–12 Ferritin is believed to accumulate during the anti-inflammatory process of macrophage scavenging of heme via the CD163 receptor. High concentrations of soluble IL-2 receptor are produced by activated lymphocytes.13 A summary of clinical and laboratory findings is provided in Table 1.
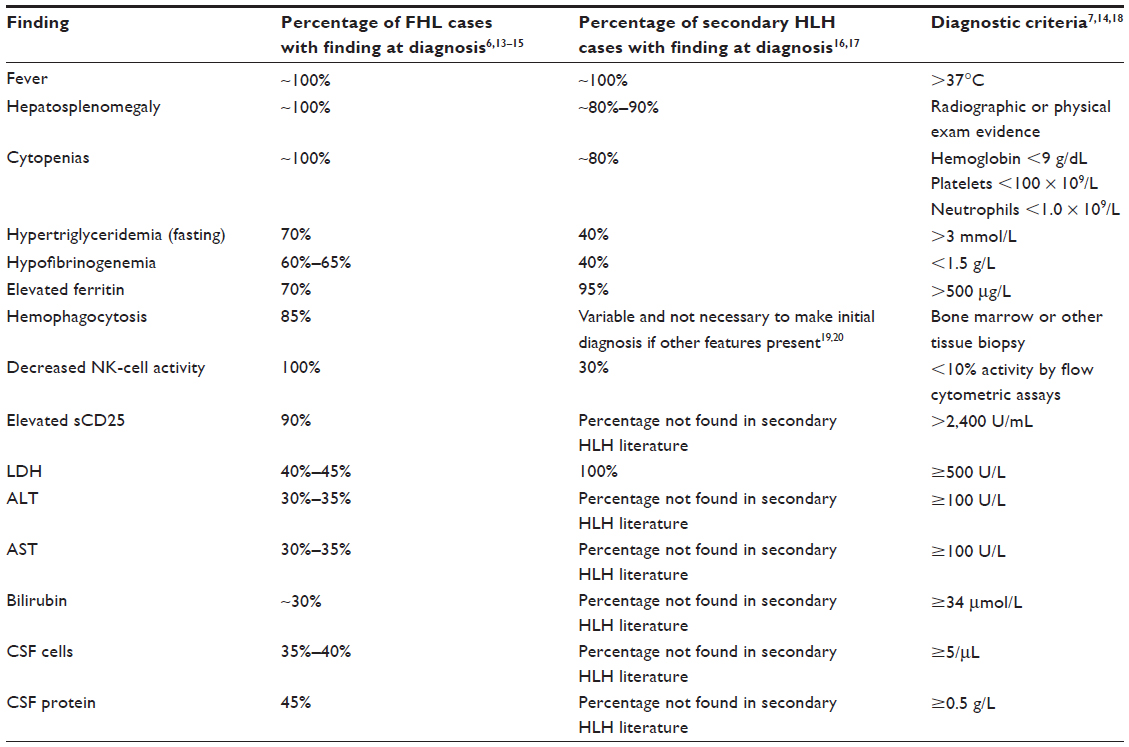 | Table 1 Incidences of clinical and laboratory findings in FHL and secondary HLH |
Coagulopathy is a prominent feature of HLH, as low fibrinogen is found in the majority of patients. Further coagulation studies have demonstrated normal factor V and VIII levels and an absence of fibrin split products. These findings provide evidence against disseminated intravascular coagulation, a diagnosis that may overlap with HLH due to the shared findings of thrombocytopenia and hypofibrinogenemia.21 It is believed that macrophages may secrete plasminogen activators which accelerate the conversion of plasminogen to plasmin, subsequently degrading fibrinogen. Fibrin split products may be phagocytized by macrophages in the reticuloendothelial system. Given the normal coagulation factors measured in HLH, liver failure, while often present, is not related to the coagulopathy. The hepatomegaly, elevated transaminases, and bilirubin are believed to be the direct result of organ infiltration by lymphocytes and histiocytes.18
Ironically, despite the fact that hemophagocytosis is prominently featured in the name of this disease, it is rarely found at presentation in secondary cases and may not be visible until late in disease progression. Bone marrow biopsies performed early in the course of secondary disease may be normal or demonstrate very nonspecific findings such as increased or decreased unilineage or multilineage hematopoiesis. Repeat studies may be necessary to show these findings. FHL, on the other hand, may demonstrate prominent hemophagocytosis from the start. An immunohistochemical stain for CD163 may be useful, as upregulation of this receptor facilitates hemophagocytosis.22 In cases in which cerebrospinal fluid is obtained, pleocytosis is sometimes noted with lymphocytes, histiocytes, and an increased protein level. Microscopic review of spun cerebrospinal fluid may demonstrate hemophagocytosis.6 While many of the laboratory tests are readily available, evaluation of IL-2 receptor and NK-cell activity may require sending specimens out to specialized reference laboratories and may not be a timely option for clinical diagnosis. When available, comparison of IFN-γ, IL-10, and IL-6 may be useful for distinguishing between bacterial sepsis, viral infections, and HLH. Using the criteria IFN-γ >75 pg/mL, and IL-10 60 pg/mL, sensitivity and specificity of diagnosing HLH is 98.9% and 93.0%, respectively.23 Additionally, measuring plasma levels of CD163, a receptor for hemoglobin-haptoglobin complexes, may also be helpful in distinguishing HLH from other purely infectious diseases.24
Flow cytometry may be used to identify and quantify levels of several useful markers involved in the pathophysiology of HLH. A rapid flow cytometric analysis of intracellular X-linked inhibitor of apoptosis protein (XIAP) has been developed for detection for X-linked lymphoproliferative disease and carrier state, which has also proven useful following bone marrow transplant to monitor reconstitution.25,26 A prospective evaluation of degranulation assays was found to be useful in the differential diagnosis of FHL. CD107 may serve as a useful surrogate marker for reduced or absent NK-cell and cytotoxic T-cell activity. Using an assay for surface upregulation of CD107a on NK-cells and cytotoxic T-lymphocytes in a large cohort of patients under evaluation for HLH, the vast majority of patients with FHL subtypes 3–5 and Griscelli syndrome (GS) type 2 or Chediak–Higashi syndrome (CHS) had abnormal resting NK-cell degranulation. NK-cell degranulation was found to be normal in the majority of patients with FHL type 2 and X-linked lymphoproliferative disease. Instead these patients were found to have diminished intracellular SAP (SLAM [signaling lymphocytic activation molecule]-associated protein), XIAP, and perforin expression. Most patients with secondary HLH did not have abnormalities of NK-cell degranulation. Thus, degranulation assays may help speed the diagnosis of HLH and allow treatment to begin more rapidly.27 Degranulation of CD107 may be associated with a specific type of FHL (FHL-5) in which missense mutations lead to decreased lymphocyte stability of syntaxin binding protein 2 (Munc18-2) and syntaxin 11. These proteins would normally be involved in regulating vesicle transport to the plasma membrane, which is key to the pathophysiology of HLH.28
Flow cytometry for perforin staining in cytotoxic lymphocytes, including NK-cells, CD8+ T-cells, and CD56+ T-cells, seems to be useful as a quick and reliable marker for perforin gene mutations seen in HLH. In a study of eleven unrelated HLH patients and 19 family members, four of seven patients with FHL showed lack of intracellular perforin in all cytotoxic cell types, which corresponded to mutations in the perforin gene. The parents of these patients also had abnormal perforin staining, indicative of their carrier state for perforin mutations. Evaluation of cytotoxic T-cells from the other three patients with FHL demonstrated normal percentages of perforin staining cytotoxic T-cells. The four patients with Epstein–Barr virus (EBV)-associated secondary HLH had depressed numbers of NK-cells but increased CD8+ T-cells with perforin expression.29 Flow cytometry studies have proven to be a rapid and useful modality to reliably diagnose HLH and shorten the time to treatment.29
Epidemiology
It is difficult to assess the true epidemiology of HLH. The disease has been studied extensively in Sweden, where the incidence of FHL is estimated at 1 in 50,000 live births.14,30 Overall, an estimate of HLH in children <18 years old across ethnicities and races is approximately 1 in 100,000.31 Other large series have been described in Hong Kong and Taiwan; however, worldwide, the incidence of FHL is unknown, and even less epidemiologic data are available regarding acquired HLH cases.15,32–34 It is largely believed that the condition is underrecognized, as hemophagocytosis is often not pathologically evident until autopsy. The familial types are usually diagnosed in childhood, while secondary HLH can occur at any age.
Associated illnesses
A number of conditions are associated with secondary HLH. By prevalence, these include viral infections (29%), other infections (20%), malignancies (27%), rheumatologic disorders (7%), and immune deficiency syndromes (6%).35 These inciting conditions are addressed below.
Infections
Both sporadic and familial cases of HLH can be initiated by infectious causes; however, it is important to distinguish between primary, genetic cases versus secondary, as treatment of the underlying condition may be attempted in the latter. All patients who meet the criteria should be tested for a precipitating infection, including culture of blood and urine, chest radiography, and screening for EBV, cytomegalovirus, parvovirus B19, human immunodeficiency virus (HIV), and human herpes virus-6. Throat and rectal swabs for viral culture may also prove useful. HLH has also been linked to HIV-associated infections such as pneumocystosis and histoplasmosis, so appropriate screening of infected patients should be considered. It is also recommended that patients with a travel history be screened for relevant infections.36
The possible role of infectious diseases in causing HLH was first elucidated in 1979 in a case series describing renal transplant patients with a viral-associated HLH (viral-associated hemophagocytic syndrome).4 EBV, a DNA (deoxyribonucleic acid) virus and member of the Herpesviridae family has been the most consistently reported virus associated with HLH.36–52 The majority of EBV-associated HLH cases have been reported in Asia, with limited information on the incidence elsewhere in the world. A Japanese study estimated the annual incidence of HLH at 1 in 800,000 persons per year, with 90% of these cases being secondary and one-third of these secondary cases being related to EBV. This leads to an estimated annual incidence of 0.4 cases of EBV-associated HLH per million persons. Some have theorized that the higher rates of EBV-associated HLH in Asian countries may be due to a more pathogenic strain of EBV.53 Several studies have performed sequence analysis of the EBV nuclear antigen 2 (EBNA-2) gene and latent membrane protein-1 (lmp-1). These studies have demonstrated that no single sub-strain of EBV has been linked to HLH as of yet.54–56 It appears that new infection as well as reactivation of latent EBV infection may predispose individuals to HLH. Quantitative determination of EBV genome copy numbers in peripheral blood may be useful in predicting prognosis and effectiveness of therapy.57,58 Interestingly, male patients with EBV-associated HLH may have mutations in the SH2D1A gene, which is traditionally associated with X-linked lymphoproliferative syndrome (XLPS). XLPS is characterized by immunodeficiency to EBV. A Japanese study performed genetic analysis of the SH2D1A gene in 40 male patients presenting with severe EBV-associated illnesses, including fulminant infectious mononucleosis, EBV-positive lymphoma, and chronic EBV infection, finding mutations in a quarter of these patients. Given this strong association, it is recommended that male patients with EBV-associated HLH be screened for XLPS.59 While fulminant infectious mononucleosis may overlap with EBV-associated HLH, higher viral loads are seen in EBV-associated HLH.54
While not as commonly reported nor as well defined as EBV, many other viruses may be associated with HLH. These can be best organized by DNA or RNA (ribonucleic acid) and, subsequently, by virus family, as listed in Table 2.
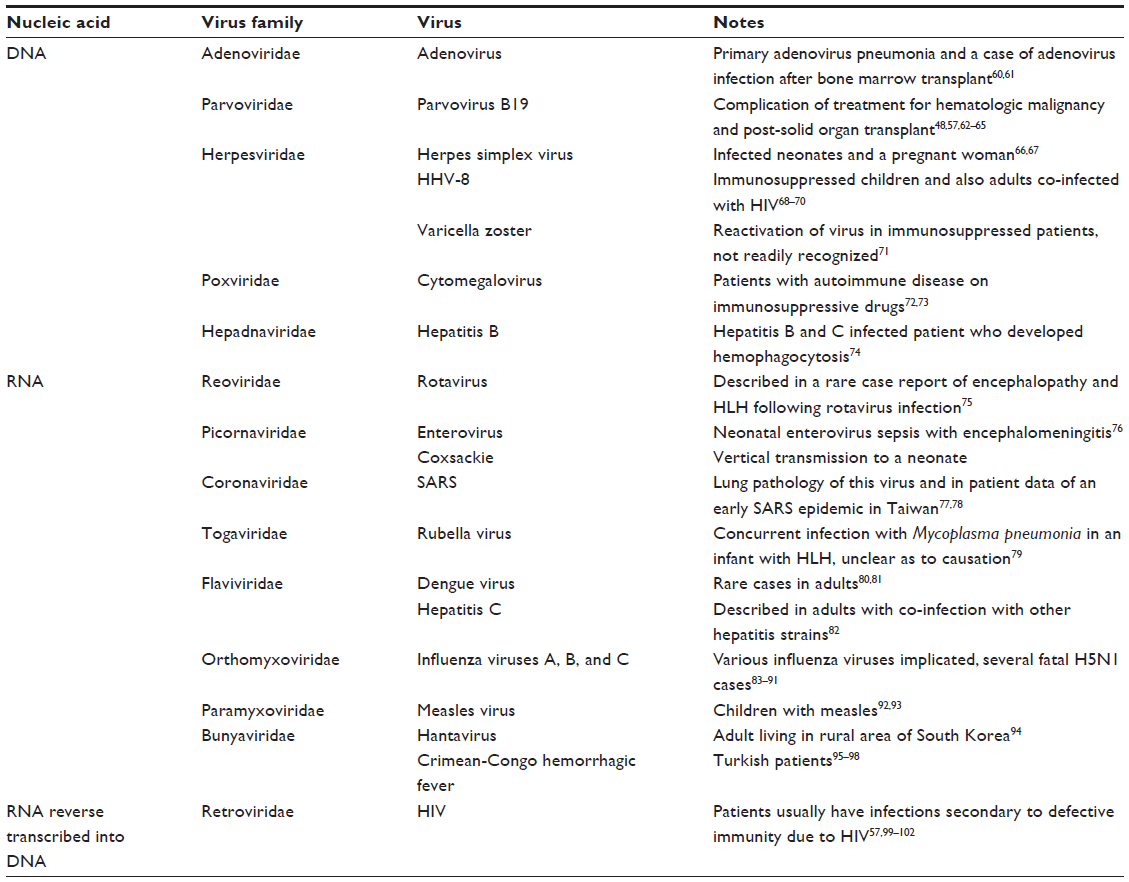 | Table 2 Viruses associated with secondary HLH |
A few years after Risdall et al103 described a possible viral etiology for HLH, this group discovered links to bacteria as well. The most common bacterial infections associated with HLH are given in Table 3.
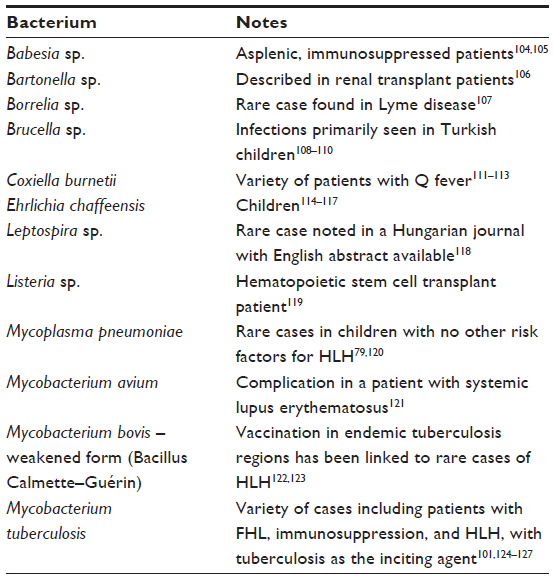 | Table 3 Bacteria associated with secondary HLH |
After the first bacteria-associated HLH cases were identified, a connection to protozoan, parasitic, and fungal infections was not far behind. Numerous reports of parasite-associated HLH have also been reported, with leishmaniasis83,128–140 and malaria141,142 being the most common. A more comprehensive discussion of zoonoses associated with HLH is presented in Cascio et al.83 The most common protozoans associated with HLH are presented in Table 4.
 | Table 4 Protozoa associated with secondary HLH |
Fungal organisms may cause HLH, either as a primary cause or in association with immunosuppression, as in HIV infection. Common fungal organisms associated with HLH are listed in Table 5.
 | Table 5 Fungi associated with secondary HLH |
Malignancy-related
Malignancies are associated with a significant percentage of secondary HLH cases (up to 27%).35 Among these cases, hematologic malignancies are most prevalent. These hematologic disorders can be best classified according to cell type. In children, HLH is most commonly associated with acute B-lymphoblastic leukemia.154 The cytokines released in malignancy, mainly IL-2, IL-6, IL-10, IL-12, IFN-γ, and TNF-α are believed to be the inciting factor for malignancy-associated HLH. An overview of malignancies associated with HLH is given in Table 6.
 | Table 6 Hematologic malignancies associated with secondary HLH |
An added complication to the understanding of HLH is the observation that HLH has been seen in the first 4 weeks following hematopoietic stem-cell transplant (HSCT) for malignant disease.61,63,166–168 Post-transplant HLH can be particularly difficult to diagnose, as clinical and laboratory signs overlap with recovering bone marrow.
Autoimmune disease
HLH is well known to occur in the setting of various autoimmune disorders. Lupus erythematosus is one of the more common conditions reported.35,169–178 MAS is a serious and often fatal complication in children with systemic juvenile idiopathic arthritis (sJIA)175,179–186 and in adults with Still’s disease.35,181,187–189 While MAS is generally considered a type of secondary HLH, some children with sJIA may have heterozygous expression of some genes associated with HLH.182 MAS often presents in sJIA children shortly after initiation of non-steroidal anti-inflammatory drugs. Other rheumatologic conditions have been associated with HLH, including polyarteritis nodosa, mixed connective disease, pulmonary sarcoidosis, systemic sclerosis, and Sjögren’s syndrome.35 MAS is considered a unique type of HLH and is detailed in the literature elsewhere.
Other conditions
The genetic causes of HLH can be divided into two groups: 1) FHL and 2) immune deficiencies such as CHS, GS, and XLPS. FHL was first described in 1952 by Farquhar and Claireaux,2 with HLH as the only manifestation.3 They described the tragic case of two siblings who developed a rapidly fatal illness, with fever, diarrhea, and vomiting, at 9 weeks of age and termed the illness familial hemophagocytic reticulosis. The children had hepatosplenomegaly without lymphadenopathy. They also had pancytopenia. Both infants died, and at autopsy, histiocyte proliferation with active hemophagocytosis was readily apparent in the lymph nodes, spleen, liver, and kidney. A third child from the same family was unaffected, but the fourth was affected, as reported by Farquhar in 1958.3 Numerous other cases under various other names have been reported in the medical literature, including familial erythrophagocytic lymphohistiocytosis, familial lymphohistiocytosis, and generalized lymphohistiocytic infiltration to name a few.190–196 A high percentage (up to 24%) of FHL cases are associated with parental consanguinity.8
As for immune deficiency syndromes associated with HLH, CHS, and GS, both have abnormalities of cellular granules. CHS typically features albinism and recurrent pyogenic infections due to deficient white blood cells with decreased chemotaxis and containing giant lysosomal inclusion bodies. GS also features albinism/hypopigmentation and neutrophil dysfunction. XLPS is associated with a marked vulnerability to EBV infection and subsequent viral-associated HLH and an immune system predisposed to lymphomas and dysgammaglobulinemia.18 Up to 60% of patients may develop EBV-HLH in this rare condition. Other conditions associated with a predisposition to HLH and reported in the literature include common variable immunodeficiency,197,198 renal transplant patients,144,199 Hermansky–Pudlak syndrome,200,201 to name a few.
Pathophysiology
An understanding of the defective function of several types of immune cells in HLH has greatly enhanced our knowledge of normal physiology of cytotoxic cells. Several cell types are involved in the pathophysiology of HLH, including macrophages, NK-cells, and cytotoxic T-lymphocytes. Macrophages typically serve as antigen presenting cells to present foreign antigens to lymphocytes for either direct destruction or antibody development. In various forms of HLH, macrophages become activated and secrete cytokines. Cytokines, in turn, can cause organ damage when excreted in excessive amounts. NK-cells directly destroy damaged or infected cells, independent of the major histocompatibility complex (MHC). Cytotoxic T-lymphocytes, while similar to NK-cells, kill autologous cells carrying foreign antigens associated with MHC Class I. An alternate theory proposes ineffective antigen removal, which results in ongoing immune stimulation and inappropriate hemophagocytosis.202 Defects in NK-cell function may vary within the various types of HLH, indicating that several aspects of cell signaling are likely involved in NK-cell dysfunction seen in the disease.203–205 In particular, patients with type 3 NK-cell deficiency with completely absent NK-cell function are likely to need hematopoietic cell transplant.203,204 Patients with CHS, which is associated with a high frequency of HLH, often have defects in CTLA-4 (cytotoxic T-lymphocyte-associated antigen 4), such that secretory lysosomes cannot move to the cell membrane.206 Deficient apoptosis appears to be the underlying mechanism behind at least some forms of FHL, a theory further supported by the success of etoposide, a drug used to trigger apoptosis.207
Genetics
FHL syndromes are sub-classified into FHL-1 through FHL-5, based upon functional protein anomalies and the prerequisite gene mutations responsible. The genetic abnormalities identified in these syndromes are outlined in Table 7.
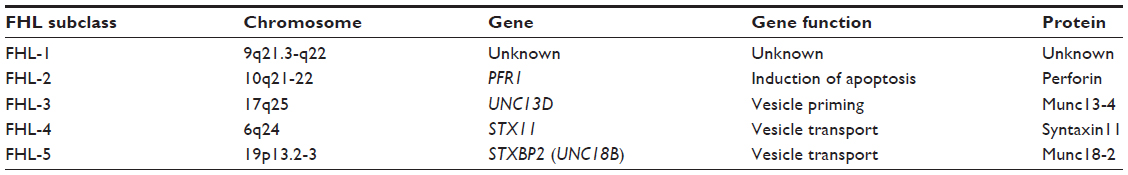 | Table 7 Genetic defects associated with FHL |
Generally, the forms of HLH associated with infants and young children are caused by defects in immune regulation, such as mutations in genes controlling the function of cytotoxic T-lymphocytes and NK-cells. With older children and adults, HLH is more likely to be secondary to infection, malignancy, or autoimmune disease. While rare, some familial cases have been undetected until adulthood. Even children with a defined genetic cause of HLH often have a secondary assault such as infection that triggers HLH, in keeping with the two-hit hypothesis required for the development of many diseases.
The first genetic defect described in FHL was a mutation in the perforin gene (PRF1) in 1999 by Stepp et al.208 Perforin is found in secretory granules of cytotoxic cells and plays an important role in apoptosis and immune modulation. Perforin mutations are causative in the majority of FHL cases, accounting for up to 58%, and are considered a defining feature of FHL-2.31,125,183,186,209–221 Patients presenting later in life may have residual perforin expression, but patients presenting in early childhood usually have no perforin in their NK- or CD8+ cells. The HLH Study Group of the Histiocyte Society identified 63 specific mutations within the perforin gene that demonstrate varying prevalence among ethnic groups. Three of the more common mutations were specifically associated with Turkish, African/African American, and Japanese origins, respectively. A common perforin polymorphism C272T (A91V) seen in late-onset FHL has been demonstrated to lead to a dysfunctional perforin.212,222 More detailed discoveries about the role of perforin in HLH, lymphoma, and other immune-mediated diseases have been recently described.31,210,212,213,216,218,222–229
Genes involved in cytotoxic granule exocytosis have been demonstrated to bear mutations in FHL-3, FHL-4, and FHL-5. FHL-3 cases, which account for 10%–32% of genetic HLH feature UNC13D mutations.30 The essential role of the UNC13D gene in the fusion of cytolytic granules and involvement in FHL was first described in 2003 by Feldmann et al.230 Patients with disruptive mutations presented at a younger age than FHL-3 patients with missense mutations, but older than FHL-2 patients. FHL-3 is marked by more central nervous system involvement than the other subclasses.30,180,182,211,218,231–237 FHL-4 is characterized by SYNTAXIN11 (STX11) mutations and is found almost exclusively in patients of Turkish/Kurdish descent.31,211,238–240 The gene mutation causing FHL-5 was described as recently as 2009 by Zur Stadt et al and also by Côte et al.28,240–243 The FHL-1 locus on chromosome 9p21.3-q22 codes for a yet unknown gene and protein involved in the development of FHL-1 and accounts for fewer cases of FHL at approximately 10%.212
Aside from FHL, the immune deficiency syndromes associated with HLH, including CHS-1, GS, and XLPS can be grouped based on molecular features. Similar to FHL, from a molecular standpoint, CHS-1 and GS can be characterized by intracellular vesicle content, docking, and fusion defects. Both are characterized by albinism, neutrophil granule dysfunction, and recurrent infections. XLPS on the other hand is molecularly distinct in that XLPS lacks normal immune function of T-lymphocytes rather than having neutrophil granule defects. Genetic defects associated with these conditions are presented in Table 8.
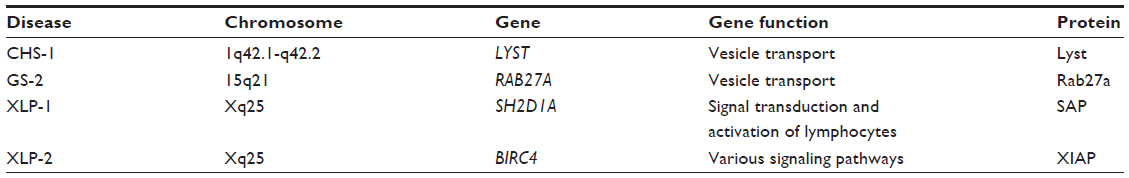 | Table 8 Immunodeficiency syndromes associated with HLH |
Gene expression analysis of mononuclear cells from patients with various genotypes of HLH demonstrates increased expression of IL-1b, TNF-α, IL-6, and IL-8. Along with the clinical diagnosis of HLH, gene expression profiling may be useful in predicting the risk of relapse and response to treatment.
Treatment
Prior to the use of modern treatment regimens, survival with HLH was close to 0%.202 Broadly, treatment of HLH involves immune-suppressive and modulatory agents, biological response modifiers, treatment of the inciting illness if secondary, and subsequent stem-cell transplantation. Therapy is aimed at suppressing the hyperinflammatory state and immune dysregulation that leads to life-threatening organ damage and susceptibility to deadly infections. It is also important to kill infected antigen-presenting cells to remove the stimulus for ongoing immune activation. Treatment of HLH may vary according to cause. Discussion of treatment is subdivided into FHL, infection-related, malignancy-associated, and autoimmune diseases.
FHL
Early attempts at treating HLH included vinblastine (a vinca alkaloid) and corticosteroids. The epipodophyllotoxins, etoposide (VP-16), and teniposide (VM-26), in combination with steroids showed some promise in achieving prolonged remissions. In 1994, the Histiocyte Society proposed the first protocol for the treatment of HLH (HLH-94). The protocol began in 1994, 5 years before genetic markers for FHL were found. At the time, FHL was defined by having an affected sibling. HLH-94 offered an established chemotherapy regimen (epidodophyllotoxin and corticosteroids) in conjunction with immunotherapy with cyclosporine A (CSA). The original eligibility requirements for the trial included only patients under the age of 16 years. HLH-94 included 8 weeks of initial chemotherapy and immunotherapy, attempting to achieve complete remission, followed by continuation therapy until an acceptable bone marrow donor could be found. The first 8 weeks consisted of dexamethasone at a starting dosage of 10 mg/m2 body surface area, then tapering down by half in 2-week increments for 6 weeks, then 1 week of 1.25 mg/m2, and a subsequent week of taper. Concurrently with the dexamethasone taper, VP-16 therapy was initiated twice weekly during the first 2 weeks, then weekly. After week 8, CSA therapy began and extended for the duration of therapy. Intrathecal methotrexate was included in selected patients with evidence of central nervous system (CNS) involvement. After 8 weeks, pulses of dexamethasone were given at regular intervals. HSCT was recommended for all children with a suitable allogeneic donor. Supportive care may have included an intensive care unit stay, broad-spectrum antibiotics until appropriate culture results were available, microbiological surveillance, HLA testing of patient and family in anticipation of HSCT, and prophylactic antifungals.7 Conditioning regimens prior to HSCT included busulfan, cyclophosphamide, etoposide, and if the donor was unrelated, antithymocyte globulin (ATG).244
Retrospective reviews of the HLH-94 treatment protocol morbidity and mortality for 249 patients with long-term follow-up revealed that overall survival and response to therapy did not differ in patients with versus without family history. Overall survival rates were 54%. In this study, 114 (46%) died, and 72 did not receive transplant. Of patient deaths, 64 (89%) occurred in the first year. Of the eight deaths occurring after 1 year of therapy, two had progressive disease without an available HSCT donor, one could not be transplanted due to severe neurological disease, and five relapsed. Overall, HLH-94 achieved complete remission or allowed survival to HSCT in 71% of patients.244
Given the success of the HLH-94 protocol, in 2004, a revised protocol called HLH-2004 was proposed, which aimed to 1) evaluate a revised initial and continuation therapy with an end goal of HSCT, 2) evaluate and improve results of HSCT with various types of donors, 3) evaluate the prognostic importance of state of remission at the time of HSCT, 4) evaluate long-term neurologic sequelae, and 5) improve understanding of the pathophysiology of HLH by including genotype–phenotype studies and evaluate the prognostic value of NK-cell activity subtyping. HLH-2004 essentially took HLH-94 and moved cyclosporine from later in the regimen to an initial therapy, concurrent with dexamethasone and VP-16. Dexamethasone and VP-16 dosing was the same as in HLH-94. Intrathecal methotrexate was still included for select patients with CNS involvement. In general, HLH-2004 approaches all patients with an initial 8 weeks of chemotherapy, and supports the patient until HSCT can be performed in the case of patients with genetically determined disease or persistent non-genetic disease. Cases which resolve or are non-genetic can cease therapy unless relapse occurs, in which case HSCT would be undertaken. Eligibility requirements include children <18 years old. Separate concurrent studies have been undertaken on patients >18 years old, and those with CHS, GS, or XLPS.7 The HLH-2004 protocol officially ceased enrolment at the end of 2011. Long-term outcomes are still under evaluation.
An alternative regimen for FHL was first described by Stephan et al245 in 1993 and in an expanded trial described by Mahlaoui et al246 in 2007. The development of these regimens was based on the recognition that T-lymphocytes play a role in the pathology of HLH as well as macrophages. This discovery was based on the detection of major histocompatibility complex class II positive T-cells, high levels of soluble serum IL-2, CD8, and IFN-γ in the serum of FHL patients. This regimen featured a combination of ATG with corticosteroids, CSA, and intrathecal methotrexate. The Stephan study evaluated six patients and was successful in achieving remission quickly, although two patients died from CNS disease. In the expanded study, the regimen was used to treat 38 consecutive patients with FHL over the course of 14 years. The patients received 45 courses of ATG, with a total dosage of 50 mg/kg or 25 mg/kg varying by severity of disease over the course of 5 days. Methylprednisolone at 4 mg/kg/day was administered with the ATG for 5 days then tapered. Intrathecal methotrexate and corticosteroids were given at various dosages determined by patient age and at intervals varying according to the severity of CNS involvement. Patients also received supportive care with fibrinogen infusions, irradiated packed red blood cells, and platelets. Broad spectrum antibiotics and intravenous immunoglobulins were also given. CSA was added to reach a plasma concentration of 150 ng/mL prior to HSCT. Immediate adverse effects of ATG were relatively minor and presented as fever and chills during infusion that rapidly resolved and did not preclude further treatment. After approximately 2 weeks of ATG treatment, bacterial, viral, or fungal infections were encountered. Infections were more common in patients receiving ATG as a secondary treatment for relapse than in first-line therapy. Once a complete response was achieved as evidenced by normalization of clinical and biological parameters, HSCT was performed for patients with an available HLA identical donor. Patients without a complete response or without a viable donor were given maintenance therapy. Overall, the efficacy of ATG therapy in achieving complete remission was 73%. Using ATG as a first-line treatment had a higher success rate of 82% of patients achieving complete remission versus only about 50% achieving complete remission with second-line ATG treatment. The best outcomes were seen with patients who received HSCT shortly after starting ATG therapy.246
Infection-associated HLH
While treating the inciting infectious agent is important in the treatment of infection-related HLH, treating the identified organism alone is not enough. Most cases of infection-related HLH should be treated aggressively with standard HLH protocols. The exception to this rule is in Leishmania-related HLH, which has been treated successfully with liposomal amphotericin alone.129–135,140,247 In particular, the prognosis for EBV-associated cases has improved dramatically with chemotherapy and immune modifying agents. In a multivariate analysis of patients on regimens consisting of corticosteroids alone, intravenous immunoglobulins alone, CSA alone, or a combination of treatments without etoposide versus another group of patients receiving etoposide, early introduction of etoposide was the only significant variable for improved survival.248 Etoposide appears to interfere with EBV-induced lymphocyte transformation and suppresses formation of EBV nuclear antigen. Despite its potential risks, the benefits of etoposide justify its early use in light of the fact that even seemingly mild cases may deteriorate quickly.
MAS
MAS is rather unique in that these HLH cases may respond quite well to high dose corticosteroids alone. One of the first reports of treating MAS associated HLH with corticosteroids, was a case series of sJIA in France which described seven children with hemorrhagic, hepatic, and neurologic features, later realized to be MAS. The children were treated with high-dose steroids, and five out of seven survived.249 Several other case series had similar outcomes.184 CSA therapy has also become a prominent therapy in addition to corticosteroids in MAS associated with sJIA, as CSA may preferentially inhibit lymphocytes by targeting transcription factors that activate various cytokine genes.250 CSA likely inhibits the cytokine storm of MAS. Cyclophosphamide has also been used to target lymphocytes in MAS.251 Etoposide-based regimens such as HLH-94 and HLH-2004 can be used in MAS, but the risks must be weighed carefully.
HSCT
Virtually all genetic cases of HLH and many secondary cases should be treated with HSCT. The first report of successful HSCT was reported in 1986.252 Several studies have demonstrated that HSCT is the only true hope for permanent control of the disease or essentially a cure.253–257 A study of 86 children treated with HLH-94 followed by HSCT demonstrated similar long-term disease-free survival (70% at 3 years) with matched unrelated donor transplants as with matched sibling transplants. Survival with family haploidentical donor transplants or mismatched unrelated transplants showed much less favorable results with long-term disease-free survival of only 50%.258 Cord blood transplant has been successful in some patients. However, overall transplant morbidity and mortality remains high. The same pediatric study showed a mortality rate of 26 out of 86 patients, with deaths resulting from pulmonary and liver complications.258 Patients responding well to pre-transplant induction therapy appear to respond best to HSCT. Pre-transplant conditioning regimens generally include busulfan, etoposide, and cyclophosphamide. Busulfan levels must be carefully monitored, and clonazepam or phenytoin may be useful as anticonvulsive prophylaxis. Dexamethasone may be used to prevent VP-16-induced anaphylactic-like symptoms. Mesna can be used for protection against cyclophosphamide-induced bladder injury. Trimethoprim/sulfamethoxazole may be used for pneumocystis prophylaxis, and acyclovir prophylaxis is recommended.7
Acute graft versus host disease (GVHD) appears to be the most common complication post-transplant, with rates as high as 32% and chronic GVHD rates at about 9%.259 Additionally, some patients may develop mixed chimerism necessitating regular donor lymphocyte infusions.30 With reduced intensity conditioning at an experienced transplant center, patients surviving to HSCT have an approximate survival rate of 92%.202 The unifying thread of all treatments is that the best success rates occur when complete remission is achieved rapidly and HSCT closely follows.
CNS manifestations
CNS manifestations are of particular concern, as long-term deficits are possible. Both primary and reactivation HLH protocols include systemic treatment with dexamethasone, which can cross the blood–brain barrier. For patients with persistent cerebrospinal fluid abnormalities, intrathecal therapy such as methotrexate is recommended, despite the fact that a study of 35 patients demonstrated that the probability of achieving normalization of CNS symptoms is roughly equal with or without intrathecal therapy.260 Given the devastating cognitive, neurological, and developmental defects that can result from HLH, many would argue that the benefits of intrathecal therapies justify potential risk.
Salvage therapy
Despite advances in treatment regimens, up to 25% of children with HLH cannot undergo HSCT due to advancing disease. Due to the rarity of HLH and short survival, salvage therapies have been described in various case reports but few large studies, leaving clinicians with few evidence-based options for refractory HLH. Removal of cytokines via plasmapheresis has been described to support patients until other therapies have reached therapeutic effect.90,261,262 Recombinant human thrombopoietin has been used as supportive therapy for thrombocytopenia in HLH.263 The use of monoclonal antibodies such as alemtuzumab, infliximab, and daclizumab has been described in various case reports. Alemtuzumab targets the CD-52 antigen, which is expressed on most lymphocytes, monocytes, macrophages, and dendritic cells.264–266 Infliximab targets TNF, and daclizumab targets CD-25. Both have been used with reported success.267–269 Additionally, etanercept, a TNF inhibitor, was used with success in a patient with acute lupus hemophagocytic syndrome.178 Various case reports have elaborated on treatment of refractory cases with splenectomy270 and even liver transplant for the damage caused by the unbridled macrophage activity.271 However, given the scarcity of literature on the subject, it is unclear what role these measures will play in the treatment of HLH.
Conclusion
HLH is a diverse condition with many causes and is likely under-recognized, which contributes to its high morbidity and mortality. Early recognition is crucial for any reasonable attempt at curative therapy to be made. HLH-94, HLH-2004, and ATG treatment regimens followed by HSCT have greatly increased survival in this devastating disease. A number of recent studies have contributed to the understanding of HLH pathophysiology, leading to alternate treatment options; however, much work remains to raise awareness and improve the effectiveness of treatment regimens.
Disclosure
The author has no disclosures to make in relation to this article.
References
Scott RB, Robb-Smith AHT. Histiocytic medullary reticulosis. Lancet. 1939;234:194–198. | |
Farquhar JW, Claireaux AE. Familial haemophagocytic reticulosis. Arch Dis Child. 1952;27(136):519–525. | |
Farquhar JW, Macgregor AR, Richmond J. Familial haemophagocytic reticulosis. BMJ. 1958;2(5112):1561–1564. | |
Risdall RJ, McKenna RW, Nesbit ME, et al. Virus-associated hemophagocytic syndrome: a benign histiocytic proliferation distinct from malignant histiocytosis. Cancer. 1979;44(3):993–1002. | |
Janka G. Hemophagocytic lymphohistiocytosis: when the immune system runs amok. Klinische Padiatrie. 2009;221(5):278–285. | |
Janka GE. Familial hemophagocytic lymphohistiocytosis. Eur J Pediatr. 1983;140(3):221–230. | |
Henter JI, Horne A, Arico M, et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48(2):124–131. | |
Arico M, Janka G, Fischer A, et al. Hemophagocytic lymphohistiocytosis. Report of 122 children from the International Registry. FHL Study Group of the Histiocyte Society. Leukemia. 1996;10(2):197–203. | |
Allen CE, Yu X, Kozinetz CA, McClain KL. Highly elevated ferritin levels and the diagnosis of hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2008;50(6):1227–1235. | |
Lin TF, Ferlic-Stark LL, Allen CE, Kozinetz CA, McClain KL. Rate of decline of ferritin in patients with hemophagocytic lymphohistiocytosis as a prognostic variable for mortality. Pediatr Blood Cancer. 2011;56(1):154–155. | |
Imashuku S. Hyperferritinemia in hemophagocytic lymphohistiocytosis and related diseases. Pediatr Blood Cancer. 2008;51(3):442. | |
Henter JI. Pronounced hyperferritinemia: expanding the field of hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2008;50(6):1127–1129. | |
Janka GE. Familial and acquired hemophagocytic lymphohistiocytosis. Eur J Pediatr. 2007;166(2):95–109. | |
Henter JI, Elinder G, Soder O, Ost A. Incidence in Sweden and clinical features of familial hemophagocytic lymphohistiocytosis. Acta Paediatr Scand. 1991;80(4):428–435. | |
Henter JI, Aricò M, Elinder G, Imashuku S, Janka G. Familial hemophagocytic lymphohistiocytosis. Primary hemophagocytic lymphohistiocytosis. Hematol Oncol Clin North Am. 1998;12(2):417–433. | |
Han AR, Lee HR, Park BB, et al. Lymphoma-associated hemophagocytic syndrome: clinical features and treatment outcome. Ann Hematol. 2007;86(7):493–498. | |
Park HS, Kim DY, Lee JH, et al. Clinical features of adult patients with secondary hemophagocytic lymphohistiocytosis from causes other than lymphoma: an analysis of treatment outcome and prognostic factors. Ann Hematol. 2012;91(6):897–904. | |
Janka G, zur Stadt U. Familial and acquired hemophagocytic lymphohistiocytosis. Hematology Am Soc Hematol Educ Program. 2005:82–88. | |
Gupta A, Tyrrell P, Valani R, Benseler S, Weitzman S, Abdelhaleem M. The role of the initial bone marrow aspirate in the diagnosis of hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2008;51(3):402–404. | |
Gupta A, Weitzman S, Abdelhaleem M. The role of hemophagocytosis in bone marrow aspirates in the diagnosis of hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2008;50(2):192–194. | |
McClure PD, Strachan P, Saunders EF. Hypofibrinogenemia and thrombocytopenia in familial hemophagocytic reticulosis. J Pediatr. 1974;85(1):67–70. | |
Filipovich AH. Hemophagocytic lymphohistiocytosis and related disorders. Curr Opin Allergy Clin Immunol. 2006;6(6):410–415. | |
Xu XJ, Tang YM, Song H, et al. Diagnostic accuracy of a specific cytokine pattern in hemophagocytic lymphohistiocytosis in children. J Pediatr. 2012;160(6):984–990. e1. | |
Filipovich AH. Hemophagocytic lymphohistiocytosis (HLH) and related disorders. Hematology Am Soc Hematol Educ Program. 2009:127–131. | |
Marsh RA, Villanueva J, Zhang K, et al. A rapid flow cytometric screening test for X-linked lymphoproliferative disease due to XIAP deficiency. Cytometry B Clin Cytom. 2009;76(5):334–344. | |
Marsh RA, Bleesing JJ, Filipovich AH. Using flow cytometry to screen patients for X-linked lymphoproliferative disease due to SAP deficiency and XIAP deficiency. J Immunol Methods. 2010;362(1–2):1–9. | |
Bryceson YT, Pende D, Maul-Pavicic A, et al. A prospective evaluation of degranulation assays in the rapid diagnosis of familial hemophagocytic syndromes. Blood. 2012;119(12):2754–2763. | |
zur Stadt U, Rohr J, Seifert W, et al. Familial hemophagocytic lymphohistiocytosis type 5 (FHL-5) is caused by mutations in Munc18-2 and impaired binding to syntaxin 11. Am J Hum Genet. 2009;85(4):482–492. | |
Kogawa K, Lee SM, Villanueva J, Marmer D, Sumegi J, Filipovich AH. Perforin expression in cytotoxic lymphocytes from patients with hemophagocytic lymphohistiocytosis and their family members. Blood. 2002;99(1):61–66. | |
Janka GE. Familial and acquired hemophagocytic lymphohistiocytosis. Annu Rev Med. 2012;63:233–246. | |
Niece JA, Rogers ZR, Ahmad N, Langevin AM, McClain KL. Hemophagocytic lymphohistiocytosis in Texas: observations on ethnicity and race. Pediatr Blood Cancer. 2010;54(3):424–428. | |
Wong KF, Chan JK. Reactive hemophagocytic syndrome – a clinicopathologic study of 40 patients in an Oriental population. Am J Med. 1992;93(2):177–180. | |
Wong KF, Chan JK, Lo ES, Wong CS. A study of the possible etiologic association of Epstein-Barr virus with reactive hemophagocytic syndrome in Hong Kong Chinese. Hum Pathol. 1996;27(11):1239–1242. | |
Janka GE. Hemophagocytic lymphohistiocytosis. Hematology. 2005; 10 Suppl 1:104–107. | |
Dhote R, Simon J, Papo T, et al. Reactive hemophagocytic syndrome in adult systemic disease: report of twenty-six cases and literature review. Arthritis Rheum. 2003;49(5):633–639. | |
Fisman DN. Hemophagocytic syndromes and infection. Emerging Infectious Diseases. 2000;6(6):601–608. | |
Belyea B, Hinson A, Moran C, Hwang E, Heath J, Barfield R. Spontaneous resolution of Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2010;55(4):754–746. | |
Beutel K, Gross-Wieltsch U, Wiesel T, Stadt UZ, Janka G, Wagner HJ. Infection of T lymphocytes in Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis in children of non-Asian origin. Pediatr Blood Cancer. 2009;53(2):184–190. | |
Buza N, Balint I, Schneider T, Koltai L, Orosz Z. Unusual clinical manifestation of virus-associated hemophagocytic syndrome. Pathol Res Pract. 2003;199(11):755–759. | |
Chellapandian D, Das R, Zelley K, et al. Treatment of Epstein Barr virus-induced haemophagocytic lymphohistiocytosis with rituximab-containing chemo-immunotherapeutic regimens. Br J Haematol. 2013;162(3):376–382. | |
Chuang HC, Lay JD, Hsieh WC, et al. Epstein-Barr virus LMP1 inhibits the expression of SAP gene and upregulates Th1 cytokines in the pathogenesis of hemophagocytic syndrome. Blood. 2005;106(9):3090–3096. | |
Fox CP, Shannon-Lowe C, Gothard P, et al. Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis in adults characterized by high viral genome load within circulating natural killer cells. Clin Infect Dis. 2010;51(1):66–69. | |
George MR, Herman JH, Holdbrook T, Cui C, Vardhana HG, Behling EM. Platelet refractoriness in acquired hemophagocytic syndrome. Transfusion. 2011;51(11):2319–2326. | |
George TI, Jeng M, Berquist W, Cherry AM, Link MP, Arber DA. Epstein-Barr virus-associated peripheral T-cell lymphoma and hemophagocytic syndrome arising after liver transplantation: case report and review of the literature. Pediatr Blood Cancer. 2005;44(3):270–276. | |
Gonzalo DH, Rodriguez G, Marcilla D. Diagnostic difficulties of the hemophagocytic lymphohistiocytosis (HLH) associated with the Epstein-Barr virus. J Pediatr Hematol Oncol. 2007;29(3):206–207. | |
Halasa NB, Whitlock JA, McCurley TL, et al. Fatal hemophagocytic lymphohistiocytosis associated with Epstein-Barr virus infection in a patient with a novel mutation in the signaling lymphocytic activation molecule-associated protein. Clin Infect Dis. 2003;37(10):e136–e141. | |
Han K, Kim Y, Kahng J, et al. In situ hybridization studies of cytomegalovirus and Epstein-Barr virus in reactive histiocytic hyperplasia with hemophagocytosis. Acta Haematol. 1996;96(3):140–145. | |
Henter JI, Ehrnst A, Andersson J, Elinder G. Familial hemophagocytic lymphohistiocytosis and viral infections. Acta Paediatr. 1993;82(4):369–372. | |
Imashuku S. Clinical features and treatment strategies of Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis. Crit Rev Oncol Hematol. 2002;44(3):259–272. | |
Imashuku S. Treatment of Epstein-Barr virus-related hemophagocytic lymphohistiocytosis (EBV-HLH); update 2010. J Pediatr Hematol Oncol. 2011;33(1):35–39. | |
Imashuku S, Hibi S, Kuriyama K, et al. Management of severe neutropenia with cyclosporin during initial treatment of Epstein-Barr virus-related hemophagocytic lymphohistiocytosis. Leuk Lymphoma. 2000;36(3–4):339–346. | |
Imashuku S, Hibi S, Ohara T, et al. Effective control of Epstein-Barr virus-related hemophagocytic lymphohistiocytosis with immunochemotherapy. Histiocyte Society. Blood. 1999;93(6):1869–1874. | |
Tabata YS, Teramura T, Kuriyama K, et al. Molecular analysis of latent membrane protein 1 in patients with Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis in Japan. Leuk Lymphoma. 2000;38(3–4):373–380. | |
Kelesidis T, Humphries R, Terashita D, et al. Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis in Los Angeles County. J Med Virol. 2012;84(5):777–785. | |
Aitken C, Sengupta SK, Aedes C, Moss DJ, Sculley TB. Heterogeneity within the Epstein-Barr virus nuclear antigen 2 gene in different strains of Epstein-Barr virus. J Gen Virol. 1994;75(Pt 1):95–100. | |
Schuster V, Seidenspinner S, Kreth HW. Detection of a nuclear antigen 2 (EBNA2)-variant Epstein-Barr virus strain in two siblings with fatal lymphoproliferative disease. J Med Virol. 1996;48(1):114–120. | |
Rouphael NG, Talati NJ, Vaughan C, Cunningham K, Moreira R, Gould C. Infections associated with haemophagocytic syndrome. Lancet Infect Dis. 2007;7(12):814–822. | |
Sonke GS, Ludwig I, van Oosten H, et al. Poor outcomes of chronic active Epstein-Barr virus infection and hemophagocytic lymphohistiocytosis in non-Japanese adult patients. Clin Infect Dis. 2008;47(1):105–108. | |
Sumazaki R, Kanegane H, Osaki M, et al. SH2D1A mutations in Japanese males with severe Epstein-Barr virus-associated illnesses. Blood. 2001;98(4):1268–1270. | |
Morimoto A, Teramura T, Asazuma Y, Mukoyama A, Imashuku S. Hemophagocytic syndrome associated with severe adenoviral pneumonia: usefulness of real-time polymerase chain reaction for diagnosis. Int J Hematol. 2003;77(3):295–298. | |
Levy J, Wodell RA, August CS, Bayever E. Adenovirus-related hemophagocytic syndrome after bone marrow transplantation. Bone Marrow Transplant. 1990;6(5):349–352. | |
Ardalan MR, Shoja MM, Tubbs RS, Esmaili H, Keyvani H. Postrenal transplant hemophagocytic lymphohistiocytosis and thrombotic microangiopathy associated with parvovirus b19 infection. Am J Transplant. 2008;8(6):1340–1344. | |
Ishikawa J, Maeda T, Miyazaki T, et al. Early onset of hemophagocytic syndrome following allogeneic bone marrow transplantation. Int J Hematol. 2000;72(2):243–246. | |
Matsubara K, Uchida Y, Wada T, et al. Parvovirus B19-associated hemophagocytic lymphohistiocytosis in a child with precursor B-cell acute lymphoblastic leukemia under maintenance chemotherapy. J Pediatr Hematol Oncol. 2011;33(7):565–569. | |
Yilmaz S, Oren H, Demircioglu F, Firinci F, Korkmaz A, Irken G. Parvovirus B19: a cause for aplastic crisis and hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2006;47(6):861. | |
Wada Y, Kai M, Tanaka H, Shimizu N, Shimatani M, Oshima T. Computed tomography findings of the liver in a neonate with Herpes simplex virus-associated hemophagocytic lymphohistiocytosis. Pediatr Int. 2011;53(5):773–776. | |
Yamaguchi K, Yamamoto A, Hisano M, Natori M, Murashima A. Herpes simplex virus 2-associated hemophagocytic lymphohistiocytosis in a pregnant patient. Obstetr Gynecol. 2005;105(5 Pt 2):1241–1244. | |
Fardet L, Blum L, Kerob D, et al. Human herpesvirus 8-associated hemophagocytic lymphohistiocytosis in human immunodeficiency virus-infected patients. Clin Infect Dis. 2003;37(2):285–291. | |
Grossman WJ, Radhi M, Schauer D, Gerday E, Grose C, Goldman FD. Development of hemophagocytic lymphohistiocytosis in triplets infected with HHV-8. Blood. 2005;106(4):1203–1206. | |
Pasic S, Cupic M, Lazarevic I. HHV-8-related hemophagocytic lymphohistiocytosis in a boy with XLP phenotype. J Pediatr Hematol Oncol. 2012;34(6):467–471. | |
van der Werff ten Bosch JE, Kollen WJ, Ball LM, et al. Atypical varicella zoster infection associated with hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2009;53(2):226–228. | |
Maruyama K, Koizumi T, Hirato J. Cytomegalovirus infections associated hemophagocytic lymphohistiocytosis in a premature infant. Pediatr Int. 2006;48(6):648–650. | |
N’Guyen Y, Baumard S, Salmon JH, et al. Cytomegalovirus associated hemophagocytic lymphohistiocytosis in patients suffering from Crohn’s disease treated by azathioprine: a series of four cases. Inflammatory Bowel Diseases. 2011;17(9):E116–E118. | |
Faurschou M, Nielsen OJ, Hansen PB, Juhl BR, Hasselbalch H. Fatal virus-associated hemophagocytic syndrome associated with coexistent chronic active hepatitis B and acute hepatitis C virus infection. Am J Hematol. 1999;61(2):135–138. | |
Takahashi S, Oki J, Miyamoto A, et al. Encephalopathy associated with haemophagocytic lymphohistiocytosis following rotavirus infection. Eur J Pediatr. 1999;158(2):133–137. | |
Lindamood KE, Fleck P, Narla A, et al. Neonatal enteroviral sepsis/meningoencephalitis and hemophagocytic lymphohistiocytosis: diagnostic challenges. Am J Perinatol. 2011;28(5):337–346. | |
Nicholls JM, Poon LL, Lee KC, et al. Lung pathology of fatal severe acute respiratory syndrome. Lancet. 2003;361(9371):1773–1778. | |
Hsueh PR, Chen PJ, Hsiao CH, et al. Patient data, early SARS epidemic, Taiwan. Emerg Infect Dis. 2004;10(3):489–493. | |
Yoshiyama M, Kounami S, Nakayama K, Aoyagi N, Yoshikawa N. Clinical assessment of Mycoplasma pneumoniae-associated hemophagocytic lymphohistiocytosis. Pediatr Int. 2008;50(4):432–435. | |
Wong KF, Chan JK, Chan JC, Lim WW, Wong WK. Dengue virus infection-associated hemophagocytic syndrome. Am J Hematol. 1991;38(4):339–340. | |
Jain D, Singh T. Dengue virus related hemophagocytosis: a rare case report. Hematology. 2008;13(5):286–288. | |
Wu CS, Chang KY, Dunn P, Lo TH. Acute hepatitis A with coexistent hepatitis C virus infection presenting as a virus-associated hemophagocytic syndrome: a case report. Am J Gastroenterol. 1995; 90(6):1002–1005. | |
Cascio A, Pernice LM, Barberi G, et al. Secondary hemophagocytic lymphohistiocytosis in zoonoses. A systematic review. Eur Rev Med Pharmacol Sci. 2012;16(10):1324–1337. | |
Harms PW, Schmidt LA, Smith LB, et al. Autopsy findings in eight patients with fatal H1N1 influenza. Am J Clin Pathol. 2010;134(1):27–35. | |
Lai S, Merritt BY, Chen L, Zhou X, Green LK. Hemophagocytic lymphohistiocytosis associated with influenza A (H1N1) infection in a patient with chronic lymphocytic leukemia: an autopsy case report and review of the literature. Ann Diagn Pathol. 2012;16(6):477–484. | |
Mou SS, Nakagawa TA, Riemer EC, McLean TW, Hines MH, Shetty AK. Hemophagocytic lymphohistiocytosis complicating influenza A infection. Pediatrics. 2006;118(1):e216–e219. | |
Ozdemir H, Ciftci E, Ince EU, Ertem M, Ince E, Dogru U. Hemophagocytic lymphohistiocytosis associated with 2009 pandemic influenza A (H1N1) virus infection. J Pediatr Hematol Oncol. 2011;33(2):135–137. | |
Shrestha B, Omran A, Rong P, Wang W. Report of a fatal pediatric case of hemophagocytic lymphohistiocytosis associated with pandemic influenza A (H1N1) infection in 2009. Pediatr Neonatol. Epub April 18, 2013. | |
Willekens C, Cornelius A, Guerry MJ, Wacrenier A, Fourrier F. Fulminant hemophagocytic lymphohistiocytosis induced by pandemic A (H1N1) influenza: a case report. J Med Case Rep. 2011;5:280. | |
Zhang XY, Ye XW, Feng DX, Han J, Li D, Zhang C. Hemophagocytic lymphohistiocytosis induced by severe pandemic influenza A (H1N1) 2009 virus infection: a case report. Case Rep Med. 2011;2011:951910. | |
Zheng Y, Yang Y, Zhao W, Wang H. Novel swine-origin influenza A (H1N1) virus-associated hemophagocytic syndrome – a first case report. Am J Trop Med Hyg. 2010;82(4):743–745. | |
Iaria C, Leonardi MS, Buda A, Toro ML, Cascio A. Measles and secondary hemophagocytic lymphohistiocytosis. Emerg Infect Dis. 2012;18(9):1529; author reply 1529–1530. | |
Howells DW, Strobel S, Smith I, Levinsky RJ, Hyland K. Central nervous system involvement in the erythrophagocytic disorders of infancy: the role of cerebrospinal fluid neopterins in their differential diagnosis and clinical management. Pediatr Res. 1990;28(2):116–119. | |
Lee JJ, Chung IJ, Shin DH, et al. Hemorrhagic fever with renal syndrome presenting with hemophagocytic lymphohistiocytosis. Emerg Infect Dis. 2002;8(2):209–210. | |
Erduran E, Cakir M. Reactive hemophagocytic lymphohistiocytosis and Crimean-Congo hemorrhagic fever. Int J Infect Dis. 2010;14 Suppl 3:e349; author reply e350. | |
Barut S, Dincer F, Sahin I, Ozyurt H, Akkus M, Erkorkmaz U. Increased serum ferritin levels in patients with Crimean-Congo hemorrhagic fever: can it be a new severity criterion? Int J Infect Dis. 2010;14(1):e50–e54. | |
Cagatay A, Kapmaz M, Karadeniz A, et al. Haemophagocytosis in a patient with Crimean Congo haemorrhagic fever. J Med Microbiol. 2007;56(Pt 8):1126–1128. | |
Tasdelen Fisgin N, Fisgin T, Tanyel E, et al. Crimean-Congo hemorrhagic fever: five patients with hemophagocytic syndrome. Am J Hematol. 2008;83(1):73–76. | |
Cascio A, Todaro G, Bonina L, Iaria C. Please, do not forget secondary hemophagocytic lymphohistiocytosis in HIV-infected patients. Int J Infect Dis. 2011;15(12):e885–e886. | |
Chen TL, Wong WW, Chiou TJ. Hemophagocytic syndrome: an unusual manifestation of acute human immunodeficiency virus infection. Int J Hematol. 2003;78(5):450–452. | |
Wong CK, Wong BC, Chan KC, et al. Cytokine profile in fatal human immunodeficiency virus tuberculosis Epstein-Barr virus associated hemophagocytic syndrome. Arch Intern Med. 2007;167(17):1901–1903. | |
Sun HY, Chen MY, Fang CT, Hsieh SM, Hung CC, Chang SC. Hemophagocytic lymphohistiocytosis: an unusual initial presentation of acute HIV infection. J Acquir Immune Defic Syndr. 2004;37(4):1539–1540. | |
Risdall RJ, Brunning RD, Hernandez JI, Gordon DH. Bacteria-associated hemophagocytic syndrome. Cancer. 1984;54(12):2968–2972. | |
Gupta P, Hurley RW, Helseth PH, Goodman JL, Hammerschmidt DE. Pancytopenia due to hemophagocytic syndrome as the presenting manifestation of babesiosis. Am J Hematol. 1995;50(1):60–62. | |
Slovut DP, Benedetti E, Matas AJ. Babesiosis and hemophagocytic syndrome in an asplenic renal transplant recipient. Transplantation. 1996;62(4):537–539. | |
Karras A, Thervet E, Legendre C. Hemophagocytic syndrome in renal transplant recipients: report of 17 cases and review of literature. Transplantation. 2004;77(2):238–243. | |
Cantero-Hinojosa J, Diez-Ruiz A, Santos-Perez JL, Aguilar-Martinez JL, Ramos-Jimenez A. Lyme disease associated with hemophagocytic syndrome. Clin Investig. 1993;71(8):620. | |
Karakukcu M, Patiroglu T, Ozdemir MA, Gunes T, Gumus H, Karakukcu C. Pancytopenia, a rare hematologic manifestation of brucellosis in children. J Pediatr Hematol Oncol. 2004;26(12):803–806. | |
Martin-Moreno S, Soto-Guzman O, Bernaldo-de-Quiros J, Reverte- Cejudo D, Bascones-Casas C. Pancytopenia due to hemophagocytosis in patients with brucellosis: a report of four cases. J Infect Dis. 1983;147(3):445–449. | |
Ullrich CH, Fader R, Fahner JB, Barbour SD. Brucellosis presenting as prolonged fever and hemophagocytosis. Am J Dis Child. 1993;147(10):1037–1038. | |
Estrov Z, Bruck R, Shtalrid M, Berrebi A, Resnitzky P. Histiocytic hemophagocytosis in Q fever. Arch Pathol Lab Med. 1984; 108(1):7. | |
Harris P, Dixit R, Norton R. Coxiella burnetii causing haemophagocytic syndrome: a rare complication of an unusual pathogen. Infection. 2011;39(6):579–582. | |
Hufnagel M, Niemeyer C, Zimmerhackl LB, Tuchelmann T, Sauter S, Brandis M. Hemophagocytosis: a complication of acute Q fever in a child. Clin Infect Dis. 1995;21(4):1029–1031. | |
Abbott KC, Vukelja SJ, Smith CE, et al. Hemophagocytic syndrome: a cause of pancytopenia in human ehrlichiosis. Am J Hematol. 1991;38(3):230–234. | |
Burns S, Saylors R, Mian A. Hemophagocytic lymphohistiocytosis secondary to Ehrlichia chaffeensis infection: a case report. J Pediatr Hematol Oncol. 2010;32(4):e142–e143. | |
Hanson D, Walter AW, Powell J. Ehrlichia-induced hemophagocytic lymphohistiocytosis in two children. Pediatr Blood Cancer. 2011;56(4):661–663. | |
Hanson D, Walter AW, Powell J. Ehrlichia-induced hemophagocytic lymphohistiocytosis in two children. Pediatr Blood Cancer. 2011; 56(4):661–663. | |
Niller HH. Myelodysplastic syndrome (MDS) as a late stage of subclinical hemophagocytic lymphohistiocytosis (HLH): a putative role for Leptospira infection. A hypothesis. Acta Microbiol Immunol Hung. 2010;57(3):181–189. | |
Lambotte O, Fihman V, Poyart C, Buzyn A, Berche P, Soumelis V. Listeria monocytogenes skin infection with cerebritis and haemophagocytosis syndrome in a bone marrow transplant recipient. J Infect. 2005;50(4):356–358. | |
Ishida Y, Hiroi K, Tauchi H, Oto Y, Tokuda K, Kida K. Hemophagocytic lymphohistiocytosis secondary to Mycoplasma pneumoniae infection. Pediatr Int. 2004;46(2):174–177. | |
Yang WK, Fu LS, Lan JL, et al. Mycobacterium avium complex-associated hemophagocytic syndrome in systemic lupus erythematosus patient: report of one case. Lupus. 2003;12(4):312–316. | |
Wali Y, Beshlawi I. BCG lymphadenitis in neonates with familial hemophagocytic lymphohistiocytosis. Pediatr Infect Dis J. 2012;31(3):324. | |
González MJ, Franco AG, Alvaro CG. Hemophagocytic lymphohistiocytosis secondary to Calmette-Guèrin bacilli infection. Eur J Intern Med. 2008;19(2):150. | |
Maheshwari P, Chhabra R, Yadav P. Perinatal tuberculosis associated hemophagocytic lymphohistiocytosis. Indian J Pediatr. 2012;79(9):1228–1229. | |
Mancebo E, Allende LM, Guzman M, et al. Familial hemophagocytic lymphohistiocytosis in an adult patient homozygous for A91V in the perforin gene, with tuberculosis infection. Haematologica. 2006;91(9):1257–1260. | |
Su NW, Chen CK, Chen GS, Hsieh RK, Chang MC. A case of tuberculosis-induced hemophagocytic lymphohistiocytosis in a patient under hemodialysis. Int J Hematol. 2009;89(3):298–301. | |
Brastianos PK, Swanson JW, Torbenson M, Sperati J, Karakousis PC. Tuberculosis-associated haemophagocytic syndrome. Lancet Infect Dis. 2006;6(7):447–454. | |
Gurgey A, Secmeer G, Tavil B, et al. Secondary hemophagocytic lymphohistiocytosis in Turkish children. Pediatr Infect Dis J. 2005;24(12):1116–1117. | |
Koliou MG, Soteriades ES, Ephros M, et al. Hemophagocytic lymphohistiocytosis associated with Epstein Barr virus and Leishmania donovani coinfection in a child from Cyprus. J Pediatr Hematol Oncol. 2008;30(9):704–707. | |
Marom D, Offer I, Tamary H, Jaffe CL, Garty BZ. Hemophagocytic lymphohistiocytosis associated with visceral leishmaniasis. Pediatr Hematol Oncol. 2001;18(1):65–70. | |
Ozyurek E, Ozcay F, Yilmaz B, Ozbek N. Hemophagocytic lymphohistiocytosis associated with visceral leishmaniasis: a case report. Pediatr Hematol Oncol. 2005;22(5):409–414. | |
Rajagopala S, Dutta U, Chandra KS, Bhatia P, Varma N, Kochhar R. Visceral leishmaniasis associated hemophagocytic lymphohistiocytosis – case report and systematic review. J Infect. 2008;56(5):381–388. | |
Tapisiz A, Belet N, Ciftci E, Ince E, Dogru U. Hemophagocytic lymphohistiocytosis associated with visceral leishmaniasis. J Trop Pediatr. 2007;53(5):359–361. | |
Ay Y, Yildiz B, Unver H, Karapinar DY, Vardar F. Hemophagocytic lymphohistiocytosis associated with H1N1 virus infection and visceral leishmaniasis in a 4.5-month-old infant. Rev Soc Bras Med Trop. 2012;45(3):407–409. | |
Cancado GG, Freitas GG, Faria FH, de Macedo AV, Nobre V. Hemophagocytic lymphohistiocytosis associated with visceral leishmaniasis in late adulthood. Am J Trop Med Hyg. 2013;88(3):575–577. | |
Cerdan Vera MT, Bernal Ferrer AM, Sequi Canet JM, Sifre Aranda M. [Pericardial effusion in a case of hemophagocytic lymphohistiocytosis secondary to leishmaniasis]. An Pediatr (Barc). 2012;77(6):422–423. Spanish. | |
Fathalla M, Hashim J, Alkindy H, Wali Y. Cerebrospinal fluid involvement in a case of visceral leishmaniasis associated with hemophagocytic lymphohistiocytosis. Sultan Qaboos Univ Med J. 2007;7(3):253–256. | |
Kilani B, Ammari L, Kanoun F, Ben Chaabane T, Abdellatif S, Chaker E. Hemophagocytic syndrome associated with visceral leishmaniasis. Int J Infect Dis. 2006;10(1):85–86. | |
Koubaa M, Maaloul I, Marrakchi C, et al. Hemophagocytic syndrome associated with visceral leishmaniasis in an immunocompetent adult-case report and review of the literature. Ann Hematol. 2012;91(7):1143–1145. | |
Martin A, Marques L, Soler-Palacin P, et al. Visceral leishmaniasis associated hemophagocytic syndrome in patients with chronic granulomatous disease. Pediatr Infect Dis J. 2009;28(8):753–754. | |
Bae E, Jang S, Park CJ, Chi HS. Plasmodium vivax malaria-associated hemophagocytic lymphohistiocytosis in a young man with pancytopenia and fever. Ann Hematol. 2011;90(4):491–492. | |
Saribeyoglu ET, Anak S, Agaoglu L, Boral O, Unuvar A, Devecioglu O. Secondary hemophagocytic lymphohistiocytosis induced by malaria infection in a child with Langerhans cell histiocytosis. Pediatr Hematol Oncol. 2004;21(3):267–272. | |
Kumar N, Jain S, Singh ZN. Disseminated histoplasmosis with reactive hemophagocytosis: aspiration cytology findings in two cases. Diagn Cytopathol. 2000;23(6):422–424. | |
Lo MM, Mo JQ, Dixon BP, Czech KA. Disseminated histoplasmosis associated with hemophagocytic lymphohistiocytosis in kidney transplant recipients. Am J Transplant. 2010;10(3):687–691. | |
Phillips J, Staszewski H, Garrison M. Successful treatment of secondary hemophagocytic lymphohistiocytosis in a patient with disseminated histoplasmosis. Hematology. 2008;13(5):282–285. | |
Wang Z, Duarte AG, Schnadig VJ. Fatal reactive hemophagocytosis related to disseminated histoplasmosis with endocarditis: an unusual case diagnosed at autopsy. South Med J. 2007;100(2):208–211. | |
Sung PS, Kim IH, Lee JH, Park JW. Hemophagocytic lymphohistiocytosis (HLH) associated with plasmodium vivax infection: case report and review of the literature. Chonnam Med J. 2011;47(3):173–176. | |
Duband S, Cornillon J, Tavernier E, Dumollard JM, Guyotat D, Peoc’h M. Toxoplasmosis with hemophagocytic syndrome after bone marrow transplantation: diagnosis at autopsy. Transpl Infect Dis. 2008;10(5):372–374. | |
Segall L, Moal MC, Doucet L, Kergoat N, Bourbigot B. Toxoplasmosis-associated hemophagocytic syndrome in renal transplantation. Transpl Int. 2006;19(1):78–80. | |
Bhatia S, Bauer F, Bilgrami SA. Candidiasis-associated hemophagocytic lymphohistiocytosis in a patient infected with human immunodeficiency virus. Clin Infect Dis. 2003;37(11):e161–e166. | |
Numata K, Tsutsumi H, Wakai S, Tachi N, Chiba S. A child case of haemophagocytic syndrome associated with cryptococcal meningoencephalitis. J Infect. 1998;36(1):118–119. | |
Chemlal K, Andrieu-Bautru V, Couvelard A. Hemophagocytic syndrome during Histoplasma capsulatum infection. Haematologica. 1997;82(6):726. | |
Chim CS, Fong CY, Ma SK, Wong SS, Yuen KY. Reactive hemophagocytic syndrome associated with Penicillium marneffei infection. Am J Med. 1998;104(2):196–197. | |
Celkan T, Berrak S, Kazanci E, et al. Malignancy-associated hemophagocytic lymphohistiocytosis in pediatric cases: a multicenter study from Turkey. Turk J Pediatr. 2009;51(3):207–213. | |
Vella JE, El-Daly H. Hemophagocytic lymphohistiocytosis in a patient with angioimmunoblastic lymphoma: a case report and review of the literature. International Journal of Surgical Pathology. 2012;20(6):606–609. | |
Varghese D, Haseer Koya H, Cherian SV, et al. Hemophagocytic lymphohistiocytosis: an uncommon presentation of enteropathy-associated T-cell lymphoma. J Clin Oncol. 2013;31(13):e226–e230. | |
Alomari A, Hui P, Xu M. Composite peripheral T-cell lymphoma not otherwise specified, and B-cell small lymphocytic lymphoma presenting with hemophagocytic lymphohistiocytosis. Int J Surg Pathol. 2013;21(3):303–308. | |
Allory Y, Challine D, Haioun C, et al. Bone marrow involvement in lymphomas with hemophagocytic syndrome at presentation: a clinicopathologic study of 11 patients in a Western institution. Am J Surg Pathol. 2001;25(7):865–874. | |
Krenova Z, Sterba J, Blatny J, Kren L, Slany J. A case of anaplastic large cell lymphoma-induced hemophagocytic lymphohistiocytosis in an adolescent female. Pediatr Blood Cancer. 2007;49(7):1056. | |
Takahashi N, Miura I, Chubachi A, Miura AB, Nakamura S. A clinicopathological study of 20 patients with T/natural killer (NK)-cell lymphoma-associated hemophagocytic syndrome with special reference to nasal and nasal-type NK/T-cell lymphoma. Int J Hematol. 2001;74(3):303–308. | |
Shimazaki C, Inaba T, Shimura K, et al. B-cell lymphoma associated with haemophagocytic syndrome: a clinical, immunological and cytogenetic study. Br J Haematol. 1999;104(4):672–679. | |
Murase T, Nakamura S, Kawauchi K, et al. An Asian variant of intravascular large B-cell lymphoma: clinical, pathological and cytogenetic approaches to diffuse large B-cell lymphoma associated with haemophagocytic syndrome. Br J Haematol. 2000;111(3):826–834. | |
Machaczka M, Vaktnas J, Klimkowska M, Hagglund H. Malignancy-associated hemophagocytic lymphohistiocytosis in adults: a retrospective population-based analysis from a single center. Leuk Lymphoma. 2011;52(4):613–619. | |
Chan K, Behling E, Strayer DS, Kocher WS, Dessain SK. Prolonged hemophagocytic lymphohistiocytosis syndrome as an initial presentation of Hodgkin lymphoma: a case report. J Med Case Rep. 2008;2:367. | |
Hagihara M, Inoue M, Hua J, Iwaki Y. Lymphocyte-depleted Hodgkin lymphoma complicating hemophagocytic lymphohistiocytosis as an initial manifestation: a case report and review of the literature. Intern Med. 2012;51(21):3067–3072. | |
Kounami S, Nakayama K, Yoshiyama M, et al. Early-onset hemophagocytic lymphohistiocytosis after the start of chemotherapy for advanced neuroblastoma. Pediatric Hematology and Oncology. 2012;29(1):99–103. | |
Aung W, Koslosky W, Chin NW, Pincus MR. Diffuse large cell lymphoma of B-cell type associated with reactive hemophagocytosis. Annals of Clinical and Laboratory Science. 1996;26(5):433–436. | |
Reardon DA, Roskos R, Hanson CA, Castle V. Virus-associated hemophagocytic syndrome following bone marrow transplantation. Am J Pediatr Hematol Oncol. 1991;13(3):305–309. | |
Furukawa H, Kawasaki A, Oka S, et al. Association of a single nucleotide polymorphism in the SH2D1A intronic region with systemic lupus erythematosus. Lupus. 2013;22(5):497–503. | |
Wong KF, Hui PK, Chan JK, Chan YW, Ha SY. The acute lupus hemophagocytic syndrome. Ann Intern Med. 1991;114(5):387–390. | |
Kaneko K, Matsuda M, Sekijima Y, et al. Acute respiratory distress syndrome due to systemic lupus erythematosus with hemophagocytic syndrome: an autopsy report. Clin Rheumatol. 2005;24(2):158–161. | |
Keith MP, Pitchford C, Bernstein WB. Treatment of hemophagocytic lymphohistiocytosis with alemtuzumab in systemic lupus erythematosus. J Clin Rheumatol. 2012;18(3):134–137. | |
Kwon CM, Jung YW, Yun DY, et al. A case of acute pericarditis with hemophagocytic syndrome, cytomegalovirus infection and systemic lupus erythematosus. Rheumatol Int. 2008;28(3):271–273. | |
Parodi A, Davi S, Pringe AB, et al. Macrophage activation syndrome in juvenile systemic lupus erythematosus: a multinational multicenter study of thirty-eight patients. Arthritis Rheum. 2009;60(11):3388–3399. | |
Pringe A, Trail L, Ruperto N, et al. Macrophage activation syndrome in juvenile systemic lupus erythematosus: an under-recognized complication? Lupus. 2007;16(8):587–592. | |
Ruppert P, Edmonds EC, Brook M, Musil S, Han SD. Neuropsychological assessment in a case of adult-onset hemophagocytic lymphohistiocytosis (HLH). Clin Neuropsychol. 2012;26(6):1038–1052. | |
Takahashi K, Kumakura S, Ishikura H, Murakawa Y, Yamauchi Y, Kobayashi S. Reactive hemophagocytosis in systemic lupus erythematosus. Intern Med. 1998;37(6):550–553. | |
Takahashi N, Naniwa T, Banno S. Successful use of etanercept in the treatment of acute lupus hemophagocytic syndrome. Mod Rheumatol. 2008;18(1):72–75. | |
Davi S, Consolaro A, Guseinova D, et al. An international consensus survey of diagnostic criteria for macrophage activation syndrome in systemic juvenile idiopathic arthritis. J Rheumatol. 2011;38(4):764–768. | |
Donn R, Ellison S, Lamb R, Day T, Baildam E, Ramanan AV. Genetic loci contributing to hemophagocytic lymphohistiocytosis do not confer susceptibility to systemic-onset juvenile idiopathic arthritis. Arthritis Rheum. 2008;58(3):869–874. | |
Felix FH, Leal LK, Fontenele JB. Cloak and dagger: the case for adult onset still disease and hemophagocytic lymphohistiocytosis. Rheumatol Int. 2009;29(8):973–974. | |
Hazen MM, Woodward AL, Hofmann I, et al. Mutations of the hemophagocytic lymphohistiocytosis-associated gene UNC13D in a patient with systemic juvenile idiopathic arthritis. Arthritis Rheum. 2008;58(2):567–570. | |
Lee WI, Chen SH, Hung IJ, et al. Clinical aspects, immunologic assessment, and genetic analysis in Taiwanese children with hemophagocytic lymphohistiocytosis. Pediatr Infect Dis J. 2009;28(1):30–34. | |
Ravelli A, Grom AA, Behrens EM, Cron RQ. Macrophage activation syndrome as part of systemic juvenile idiopathic arthritis: diagnosis, genetics, pathophysiology and treatment. Genes Immun. 2012;13(4):289–298. | |
Ravelli A, Magni-Manzoni S, Pistorio A, et al. Preliminary diagnostic guidelines for macrophage activation syndrome complicating systemic juvenile idiopathic arthritis. J Pediatr. 2005;146(5):598–604. | |
Vastert SJ, van Wijk R, D’Urbano LE, et al. Mutations in the perforin gene can be linked to macrophage activation syndrome in patients with systemic onset juvenile idiopathic arthritis. Rheumatol (Oxford). 2010;49(3):441–449. | |
Kirino Y, Takeno M, Iwasaki M, et al. Increased serum HO-1 in hemophagocytic syndrome and adult-onset Still’s disease: use in the differential diagnosis of hyperferritinemia. Arthritis Res Ther. 2005;7(3):R616–R624. | |
Morris JA, Adamson AR, Holt PJ, Davson J. Still’s disease and the virus-associated haemophagocytic syndrome. Ann Rheum Dis. 1985;44(5):349–353. | |
Wang Z, Wang Y, Wang J, Feng C, Tian L, Wu L. Early diagnostic value of low percentage of glycosylated ferritin in secondary hemophagocytic lymphohistiocytosis. Int J Hematol. 2009;90(4):501–505. | |
Bell RJ, Brafield AJ, Barnes ND, France NE. Familial haemophagocytic reticulosis. Arch Dis Child. 1968;43(231):601–606. | |
Blennow G, Berg B, Brandt L, Messeter L, Low B, Soderstrom N. Haemophagocytic reticulosis. A state of chimerism? Arch Dis Child. 1974;49(12):960–962. | |
Buist NR, Jones RN, Cavens TR. Familial haemophagocytic reticulosis in first cousins. Arch Dis Child. 1971;46(249):728–729. | |
Fullerton P, Ekert H, Hosking C, Tauro GP. Hemophagocytic reticulosis. A case report with investigations of immune and white cell function. Cancer. 1975;36(2):441–445. | |
Goodall HB, Guthrie W, Buist NR. Familial haemophagocytic reticulosis. Scott Med J. 1965;10(11):425–438. | |
Marrian VJ, Sanerkin NG. Familial histiocytic reticulosis (familial haemophagocytic reticulosis). J Clin Pathol. 1963;16(1):65–69. | |
Macmahon HE, Bedizel M, Ellis CA. Familial erythrophagocytic lymphohistiocytosis. Pediatrics. 1963;32:868–879. | |
Asano T, Kogawa K, Morimoto A, et al. Hemophagocytic lymphohistiocytosis after hematopoietic stem cell transplantation in children: a nationwide survey in Japan. Pediatr Blood Cancer. 2012;59(1):110–114. | |
Filipovich AH. Hemophagocytic lymphohistiocytosis and other hemophagocytic disorders. Immunol Allergy Clin North Am. 2008; 28(2):293–313, viii. | |
Rostaing L, Fillola G, Baron E, Cisterne JM, Durand D. Course of hemophagocytic histiocytic syndrome in renal transplant patients. Transplantation. 1995;60(5):506–509. | |
Enders A, Zieger B, Schwarz K, et al. Lethal hemophagocytic lymphohistiocytosis in Hermansky-Pudlak syndrome type II. Blood. 2006;108(1):81–87. | |
Jessen B, Bode SF, Ammann S, et al. The risk of hemophagocytic lymphohistiocytosis in Hermansky-Pudlak syndrome type 2. Blood. 2013;121(15):2943–2951. | |
Weitzman S. Approach to hemophagocytic syndromes. Hematology Am Soc Hematol Educ Program. 2011;2011:178–183. | |
Schneider EM, Lorenz I, Muller-Rosenberger M, Steinbach G, Kron M, Janka-Schaub GE. Hemophagocytic lymphohistiocytosis is associated with deficiencies of cellular cytolysis but normal expression of transcripts relevant to killer-cell-induced apoptosis. Blood. 2002;100(8):2891–2898. | |
Schneider EM, Lorenz I, Walther P, Janka-Schaub GE. Natural killer deficiency: a minor or major factor in the manifestation of hemophagocytic lymphohistiocytosis? J Pediatr Hematol Oncol. 2003;25(9):680–683. | |
Marcenaro S, Gallo F, Martini S, et al. Analysis of natural killer-cell function in familial hemophagocytic lymphohistiocytosis (FHL): defective CD107a surface expression heralds Munc13-4 defect and discriminates between genetic subtypes of the disease. Blood. 2006;108(7):2316–2323. | |
Yoshiyama M, Kounami S, Nakayama K, Okutani T, Aoyagi N, Yoshikawa N. Cytotoxic T-lymphocyte-associated antigen 4 gene polymorphisms in Japanese children with infection-associated hemophagocytic lymphohistiocytosis. Acta Haematol. 2010;123(3):186–190. | |
Henter JI. Biology and treatment of familial hemophagocytic lymphohistiocytosis: importance of perforin in lymphocyte-mediated cytotoxicity and triggering of apoptosis. Med Pediatr Oncol. 2002;38(5):305–309. | |
Stepp SE, Dufourcq-Lagelouse R, Le Deist F, et al. Perforin gene defects in familial hemophagocytic lymphohistiocytosis. Science. 1999;286(5446):1957–1959. | |
Molleran Lee S, Villanueva J, Sumegi J, et al. Characterisation of diverse PRF1 mutations leading to decreased natural killer cell activity in North American families with haemophagocytic lymphohistiocytosis. J Med Genet. 2004;41(2):137–144. | |
Gerard LM, Xing K, Sherifi I, et al. Adult hemophagocytic lymphohistiocytosis with severe pulmonary hypertension and a novel perforin gene mutation. Int J Hematol. 2012;95(4):445–450. | |
Gholam C, Grigoriadou S, Gilmour KC, Gaspar HB. Familial haemophagocytic lymphohistiocytosis: advances in the genetic basis, diagnosis and management. Clin Exp Immunol. 2011;163(3):271–283. | |
Göransdotter Ericson K, Fadeel B, Nilsson-Ardnor S, et al. Spectrum of perforin gene mutations in familial hemophagocytic lymphohistiocytosis. Am J Hum Genet. 2001;68(3):590–597. | |
Gupta S, Weitzman S. Primary and secondary hemophagocytic lymphohistiocytosis: clinical features, pathogenesis and therapy. Expert Rev Clin Immunol. 2010;6(1):137–154. | |
Gurgey A, Gogus S, Ozyurek E, et al. Primary hemophagocytic lymphohistiocytosis in Turkish children. Pediatr Hematol Oncol. 2003;20(5):367–371. | |
Lee SM, Sumegi J, Villanueva J, et al. Patients of African ancestry with hemophagocytic lymphohistiocytosis share a common haplotype of PRF1 with a 50delT mutation. J Pediatr. 2006;149(1):134–137. | |
Nagafuji K, Nonami A, Kumano T, et al. Perforin gene mutations in adult-onset hemophagocytic lymphohistiocytosis. Haematologica. 2007;92(7):978–981. | |
Zipursky A. Perforin deficiency and familial hemophagocytic lymphohistiocytosis. Pediatr Res. 2001;49(1):3. | |
Zur Stadt U, Beutel K, Kolberg S, et al. Mutation spectrum in children with primary hemophagocytic lymphohistiocytosis: molecular and functional analyses of PRF1, UNC13D, STX11, and RAB27A. Hum Mutat. 2006;27(1):62–68. | |
zur Stadt U, Pruggmayer M, Jung H, et al. Prenatal diagnosis of perforin gene mutations in familial hemophagocytic lymphohistiocytosis (FHLH). Prenat Diagn. 2002;22(1):80–81. | |
van Montfrans JM, Rudd E, van de Corput L, et al. Fatal hemophagocytic lymphohistiocytosis in X-linked chronic granulomatous disease associated with a perforin gene variant. Pediatr Blood Cancer. 2009;52(4):527–529. | |
Verbsky JW, Grossman WJ. Hemophagocytic lymphohistiocytosis: diagnosis, pathophysiology, treatment, and future perspectives. Ann Med. 2006;38(1):20–31. | |
Clementi R, Emmi L, Maccario R, et al. Adult onset and atypical presentation of hemophagocytic lymphohistiocytosis in siblings carrying PRF1 mutations. Blood. 2002;100(6):2266–2267. | |
Galehdari H, Mohammadi E, Andashti B, Naderi A, Molavi MA. Perforin gene analaysis in an Iranian family with familial hemophagocytic lymphohistiocytosis. Iran J Immunol. 2007;4(2):122–126. | |
Grunebaum E, Roifman CM. Gene abnormalities in patients with hemophagocytic lymphohistiocytosis. Isr Med Assoc J. 2002;4(5):366–369. | |
Muralitharan S, Al Lamki Z, Dennison D, et al. An inframe perforin gene deletion in familial hemophagocytic lymphohistiocytosis is associated with perforin expression. Am J Hematol. 2005;78(1):59–63. | |
Muralitharan S, Wali YA, Dennison D, et al. Novel spectrum of perforin gene mutations in familial hemophagocytic lymphohistiocytosis in ethnic Omani patients. Am J Hematol. 2007;82(12):1099–1102. | |
Okur H, Balta G, Akarsu N, et al. Clinical and molecular aspects of Turkish familial hemophagocytic lymphohistiocytosis patients with perforin mutations. Leuk Res. 2008;32(6):972–975. | |
Risma K, Jordan MB. Hemophagocytic lymphohistiocytosis: updates and evolving concepts. Curr Opin Pediatr. 2012;24(1):9–15. | |
Risma KA, Frayer RW, Filipovich AH, Sumegi J. Aberrant maturation of mutant perforin underlies the clinical diversity of hemophagocytic lymphohistiocytosis. J Clin Invest. 2006;116(1):182–192. | |
Feldmann J, Callebaut I, Raposo G, et al. Munc13-4 is essential for cytolytic granules fusion and is mutated in a form of familial hemophagocytic lymphohistiocytosis (FHL3). Cell. 2003;115(4):461–473. | |
Horne A, Ramme KG, Rudd E, et al. Characterization of PRF1, STX11 and UNC13D genotype-phenotype correlations in familial hemophagocytic lymphohistiocytosis. Br J Haematol. 2008;143(1):75–83. | |
Meeths M, Chiang SC, Wood SM, et al. Familial hemophagocytic lymphohistiocytosis type 3 (FHL3) caused by deep intronic mutation and inversion in UNC13D. Blood. 2011;118(22):5783–5793. | |
Rohr J, Beutel K, Maul-Pavicic A, et al. Atypical familial hemophagocytic lymphohistiocytosis due to mutations in UNC13D and STXBP2 overlaps with primary immunodeficiency diseases. Haematologica. 2010;95(12):2080–2087. | |
Santoro A, Cannella S, Trizzino A, et al. Mutations affecting mRNA splicing are the most common molecular defect in patients with familial hemophagocytic lymphohistiocytosis type 3. Haematologica. 2008;93(7):1086–1090. | |
Yoon HS, Kim HJ, Yoo KH, et al. UNC13D is the predominant causative gene with recurrent splicing mutations in Korean patients with familial hemophagocytic lymphohistiocytosis. Haematologica. 2010;95(4):622–626. | |
Zhang K, Filipovich AH, Johnson J, Marsh RA, Villanueva J. Hemophagocytic lymphohistiocytosis, Familial. In: Pagon RA, Adam MP, Bird TD, Dolan CR, Fong CT, Stephens K, editors. Gene Reviews. Seattle, WA: University of Washington, Seattle; 1993. | |
Zhizhuo H, Junmei X, Yuelin S, et al. Screening the PRF1, UNC13D, STX11, SH2D1A, XIAP, and ITK gene mutations in Chinese children with Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2012;58(3):410–414. | |
Bryceson YT, Rudd E, Zheng C, et al. Defective cytotoxic lymphocyte degranulation in syntaxin-11 deficient familial hemophagocytic lymphohistiocytosis 4 (FHL4) patients. Blood. 2007;110(6):1906–1915. | |
Danielian S, Basile N, Rocco C, et al. Novel syntaxin 11 gene (STX11) mutation in three Argentinean patients with hemophagocytic lymphohistiocytosis. J Clin Immunol. 2010;30(2):330–337. | |
Filipovich AH. The expanding spectrum of hemophagocytic lymphohistiocytosis. Curr Opin Allergy Clin Immunol. 2011;11(6):512–516. | |
Côte M, Ménager MM, Burgess A, et al. Munc18-2 deficiency causes familial hemophagocytic lymphohistiocytosis type 5 and impairs cytotoxic granule exocytosis in patient NK cells. J Clin Invest. 2009;119(12):3765–3773. | |
Pagel J, Beutel K, Lehmberg K, et al. Distinct mutations in STXBP2 are associated with variable clinical presentations in patients with familial hemophagocytic lymphohistiocytosis type 5 (FHL5). Blood. 2012;119(25):6016–6024. | |
Sieni E, Cetica V, Mastrodicasa E, et al. Familial hemophagocytic lymphohistiocytosis: a model for understanding the human machinery of cellular cytotoxicity. Cell Mol Life Sci. 2012;69(1):29–40. | |
Trottestam H, Horne A, Arico M, et al. Chemoimmunotherapy for hemophagocytic lymphohistiocytosis: long-term results of the HLH-94 treatment protocol. Blood. 2011;118(17):4577–4584. | |
Stephan JL, Donadieu J, Ledeist F, Blanche S, Griscelli C, Fischer A. Treatment of familial hemophagocytic lymphohistiocytosis with antithymocyte globulins, steroids, and cyclosporin A. Blood. 1993;82(8):2319–2323. | |
Mahlaoui N, Ouachee-Chardin M, de Saint Basile G, et al. Immunotherapy of familial hemophagocytic lymphohistiocytosis with antithymocyte globulins: a single-center retrospective report of 38 patients. Pediatrics. 2007;120(3):e622–e628. | |
Sukova M, Stary J, Houskova J, Nohynkova E. [Hemophagocytic lymphohistiocytosis as a manifestation of visceral leishmaniasis]. Cas Lek Cesk. 2002;141(18):581–584. Czech. | |
Imashuku S, Kuriyama K, Teramura T, et al. Requirement for etoposide in the treatment of Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis. J Clin Oncol. 2001;19(10):2665–2673. | |
Hadchouel M, Prieur AM, Griscelli C. Acute hemorrhagic, hepatic, and neurologic manifestations in juvenile rheumatoid arthritis: possible relationship to drugs or infection. J Pediatr. 1985;106(4):561–566. | |
Rao A, Luo C, Hogan PG. Transcription factors of the NFAT family: regulation and function. Ann Rev Immunol. 1997;15:707–747. | |
Wallace CA, Sherry DD. Trial of intravenous pulse cyclophosphamide and methylprednisolone in the treatment of severe systemic-onset juvenile rheumatoid arthritis. Arthritis Rheum. 1997;40(10):1852–1855. | |
Fischer A, Cerf-Bensussan N, Blanche S, et al. Allogeneic bone marrow transplantation for erythrophagocytic lymphohistiocytosis. J Pediatr. 1986;108(2):267–270. | |
Baker KS, DeLaat CA, Steinbuch M, et al. Successful correction of hemophagocytic lymphohistiocytosis with related or unrelated bone marrow transplantation. Blood. 1997;89(10):3857–3863. | |
Baker KS, Filipovich AH, Gross TG, et al. Unrelated donor hematopoietic cell transplantation for hemophagocytic lymphohistiocytosis. Bone Marrow Transplant. 2008;42(3):175–180. | |
Blanche S, Caniglia M, Girault D, Landman J, Griscelli C, Fischer A. Treatment of hemophagocytic lymphohistiocytosis with chemotherapy and bone marrow transplantation: a single-center study of 22 cases. Blood. 1991;78(1):51–54. | |
Durken M, Finckenstein FG, Janka GE. Bone marrow transplantation in hemophagocytic lymphohistiocytosis. Leuk Lymphoma. 2001;41(1–2):89–95. | |
Durken M, Horstmann M, Bieling P, et al. Improved outcome in haemophagocytic lymphohistiocytosis after bone marrow transplantation from related and unrelated donors: a single-centre experience of 12 patients. Br J Haematol. 1999;106(4):1052–1058. | |
Horne A, Janka G, Maarten Egeler R, et al. Haematopoietic stem cell transplantation in haemophagocytic lymphohistiocytosis. Br J Haematol. 2005;129(5):622–630. | |
Horne A, Zheng C, Lorenz I, et al. Subtyping of natural killer cell cytotoxicity deficiencies in haemophagocytic lymphohistocytosis provides therapeutic guidance. Br J Haematol. 2005;129(5):658–666. | |
Henter JI, Samuelsson-Horne A, Arico M, et al. Treatment of hemophagocytic lymphohistiocytosis with HLH-94 immunochemotherapy and bone marrow transplantation. Blood. 2002;100(7):2367–2373. | |
Raschke RA, Garcia-Orr R. Hemophagocytic lymphohistiocytosis: a potentially underrecognized association with systemic inflammatory response syndrome, severe sepsis, and septic shock in adults. Chest. 2011;140(4):933–938. | |
Tong H, Ren Y, Liu H, et al. Clinical characteristics of T-cell lymphoma associated with hemophagocytic syndrome: comparison of T-cell lymphoma with and without hemophagocytic syndrome. Leuk Lymphoma. 2008;49(1):81–87. | |
Wang Y, Wang Z, Wu L, Zhang J, Wang J, Yan L. Recombinant human thrombopoietin is an effective treatment for thrombocytopenia in hemophagocytic lymphohistiocytosis. Ann Hematology. 2013;92(12):1695–1699. | |
Machaczka M, Vaktnas J, Chiang SC, Bryceson YT. Alemtuzumab treatment for hemophagocytic lymphohistiocytosis. Nat Rev Clin Oncol. 2010;7(10). | |
Marsh RA, Allen CE, McClain KL, et al. Salvage therapy of refractory hemophagocytic lymphohistiocytosis with alemtuzumab. Pediatr Blood Cancer. 2013;60(1):101–109. | |
Strout MP, Seropian S, Berliner N. Alemtuzumab as a bridge to allogeneic SCT in atypical hemophagocytic lymphohistiocytosis. Nat Rev Clin Oncol. 2010;7(7):415–420. | |
Henzan T, Nagafuji K, Tsukamoto H, et al. Success with infliximab in treating refractory hemophagocytic lymphohistiocytosis. Am J Hematology. 2006;81(1):59–61. | |
Oda Y, Urushidani Y, Ooi S, et al. Hemophagocytic lymphohistiocytosis in a rheumatoid arthritis patient treated with infliximab. Intern Med. 2012;51(6):655–657. | |
Olin RL, Nichols KE, Naghashpour M, et al. Successful use of the anti-CD25 antibody daclizumab in an adult patient with hemophagocytic lymphohistiocytosis. Am J Hematology. 2008;83(9):747–749. | |
Machaczka M. Splenectomy as a therapeutic approach in refractory hemophagocytic lymphohistiocytosis. Biomed Pharmacother. 2012;66(2):159–160. | |
Wright G, Wilmore S, Makanyanga J, et al. Liver transplant for adult hemophagocytic lymphohistiocytosis: case report and literature review. Exp Clin Transplant. 2012;10(5):508–512. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.