")
Back to Journals » OncoTargets and Therapy » Volume 11
Natural product pectolinarigenin inhibits proliferation, induces apoptosis, and causes G2/M phase arrest of HCC via PI3K/AKT/mTOR/ERK signaling pathway
Authors Wu T , Dong X, Yu D, Shen Z, Yu J, Yan S
Received 2 September 2018
Accepted for publication 6 November 2018
Published 3 December 2018 Volume 2018:11 Pages 8633—8642
DOI https://doi.org/10.2147/OTT.S186186
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Carlos E Vigil
Tianchun Wu,1–3 Xiaogang Dong,4 Dongdong Yu,1–3 Zhenhua Shen,1–3 Jinbei Yu,5 Sheng Yan1–3
1Division of Hepatobiliary and Pancreatic Surgery, Department of Surgery, First Affiliated Hospital, School of Medicine, Zhejiang University, Hangzhou, People’s Republic of China; 2Key Laboratory of Precision Diagnosis and Treatment for Hepatobiliary and Pancreatic Tumor of Zhejiang Province, Hangzhou, People’s Republic of China; 3State Key Laboratory & Collaborative Innovation Center for Diagnosis and Treatment of Infectious Diseases, First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, People’s Republic of China; 4Division of Hepatobiliary and Pancreatic Surgery, Department of Surgery, Affiliated Tumor Hospital of Xinjiang Medical University, Urumqi, Xinjiang Uygur Autonomous Region, People’s Republic of China; 5Department of Neurology, the First Affiliated Hospital of Zhengzhou University, Zhengzhou, People’s Republic of China
Background: Hepatocellular carcinoma (HCC) is characterized by considerable phenotypic and molecular heterogeneity, but the overall survival of HCC patients remains extremely poor. Thus, novel and efficient alternatives to antitumor agents are urgently needed. Pectolinarigenin, a flavonoid compound extract, has been previously reported for the treatment of nasopharyngeal cancer. However, the potential antitumor roles of pectolinarigenin in HCC have not been clearly elaborated. In the present study, we investigated its role in HCC treatment and explored the potential molecular mechanism(s).
Materials and methods: HCC cell lines SMMC7721 and PLC5 were cultured and treated with indicated concentrations of pectolinarigenin. For the HCC cell proliferation, after HCC cells were stimulated with indicated concentrations of pectolinarigenin, the cell viability was detected in CCK-8 and colony-forming assays. HCC cell invasion/migration assay was performed by Transwell and wound scratch methods. Additionally, cellular apoptosis and cell cycle arrest analysis was performed with flow cytometric analysis. Finally, the involved underlying signaling pathway, the PI3K/AKT/mTOR/ERK signaling-related molecular markers were detected through Western blot methods with indicated antibodies. Meanwhile, antitumor activity of pectolinarigenin was also assessed in tumor-bearing mice.
Results: The results indicated that the treatment with pectolinarigenin significantly inhibited cell proliferation and migratory and invasive abilities of SMMC7721 and PLC5 cells in concentration- and time-dependent manner. Meanwhile, pectolinarigenin markedly induced cell apoptosis and G2/M phase arrest in SMMC7721 and PLC5 cells, which was associated with apoptosis- and cell cycle-related protein levels, respectively. Furthermore, pectolinarigenin inhibited PI3K/AKT/mTOR/ERK signaling pathway. It also significantly suppressed HCC tumor growth in vivo.
Conclusion: Pectolinarigenin could suppress the viability and motility and cause apoptosis and G2/M phase arrest in HCC cell lines by inhibiting the PI3K/AKT/mTOR/ERK signaling pathway. This might be an appealing potential therapeutic agent for HCC treatment.
Keywords: hepatocellular carcinoma, pectolinarigenin, apoptosis, cell cycle arrest, antitumor
Introduction
Hepatocellular carcinoma (HCC) is ranked the third most frequent cancer-related death and the sixth most common neoplasm.1 Patients with HCC often embraced the basis of virus infectious, chronic alcohol consumption, and metabolic syndrome, all of which contributed to liver dysfunction and an extremely poor prognosis. Unfortunately, increasing incidences of HCC are forecasted in the future.2 Currently, more than two thirds of patients are in the advanced stage at the time of diagnosis when curative surgical therapies are contraindicated.3,4 In addition, the clinical therapeutic outcomes of sorafenib or lenvatinib to 2–3 months survival advantages are of great limit. Therefore, it is of great necessity to develop and explore a new therapeutic strategy on the basis of understanding of the biological characteristics of HCC.
Pectolinarigenin, a component extract of the Chinese herbal plant, was isolated from Chromolaena odorata, which has shown multifunctional bioactivities, including cytotoxic activity by inducing cell apoptosis,5,6 anti-inflammation,7 anti-allergy, and antitumor effect.8 It has drawn mounting attention as a potential candidate for the treatment of human malignancies. However, the exact biological role(s) and regulating mechanism(s) of pectolinarigenin in HCC has not yet been reported in detail.
In the present study, we evaluated the viability and effectiveness of the antitumor activity of pectolinarigenin and elucidated its potential mechanism in HCC. We used the functional assays and Western blot to elaborate that pectolinarigenin inhibited the viability and motility of HCC cell lines, induced apoptosis, and caused G2/M phase arrest. Moreover, further studies demonstrated that pectolinarigenin directly blocked the PI3K/AKT/mTOR/ERK signaling pathway.
Materials and methods
Reagents and cell culture
Pectolinarigenin was purchased from Abmole Bioscience Inc., Houston, TX, USA (M4748) and was dissolved in DMSO and stored at −20°C. Human HCC cell lines SMMC7721 and PLC5 were obtained from the Chinese Academy of Sciences (Beijing, China) and maintained in Roswell Park Memorial Institute 1640 medium (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% FBS (Thermo Fisher Scientific), 100 U/mL penicillin, and 100 μg/mL streptomycin (Thermo Fisher Scientific) and maintained at 37°C in a humidified incubator with 5% CO2.
Cell viability assay
The cell viability was quantified by the Cell Counting Kit-8 (CCK-8) assay (Dojindo Molecular Technologies, Inc., Kumamoto, Japan). In general, about 4×104 cells of SMMC7721 or PLC5 were plated into 96-well plates for 24 hours and were then treated with the indicated concentrations of pectolinarigenin (0, 5, 10, 25, 50, and 100 μM) for the indicated times. A total of 0.1% DMSO was used in the control group. After incubation at 37°C for various periods of time (24, 36, 48, and 72 hours), the 450-nm absorbance wavelength was measured using a microplate reader. Cell viability was determined when compared to DMSO-treated group. All experiments were independently repeated thrice.
Cell colony-forming assay
In the cell colony-forming assay, 1×103 cells of SMMC7721 or PLC5 were seeded in six-well plates for 24 hours and then maintained with or without the indicated concentrations of pectolinarigenin for 10 days. During the period, the culture medium with different concentrations of pectolinarigenin was replaced every 2 days. At the end, the colonies were fixed in 4% paraformaldehyde for 30 minutes and then stained with 0.3% crystal violet for 20 minutes at room temperature. Then, the colonies were photographed and counted.
Migration and invasion assays
For the HCC cell migration and invasion assays, the Transwell chamber assays were performed with the 24-well Transwell plate (Corning Incorporated, Corning, NY, USA). About 2×105 cells of SMMC7721 or PLC5 stimulated with indicated concentrations of pectolinarigenin in 200 μL culture medium without FBS were added to the upper chamber, and 500 μL of culture medium containing 20% FBS was added to the lower chamber. The cells were maintained in CO2 incubator for 48 hours. Then invasive cells in the lower chamber were fixed with 4% paraformaldehyde for 30 minutes and then stained with 0.3% crystal violet for 20 minutes at room temperature. Finally, the invasive cell numbers were photographed and counted using an optical microscope (BX53; Olympus Corporation, Tokyo, Japan).
Further, for the cell wound scratching assay, about 5×105 cells of SMMC7721 or PLC5 were plated into six-well plates and grown to 90% confluence. Then, the cell layer was scratched by using a 200 μL pipette tip, followed by replacing the medium with or without 10 μM pectolinarigenin. After incubation for 48 hours, images of the cell-free areas were photographed, and the percentage of the scratch spaces was measured using ImageJ software (National Institutes of Health, Bethesda, MA, USA).
Cell cycle arrest analysis
Cell cycle distribution and DNA content were determined using a FACSCalibur flow cytometer (Beckman Coulter, Brea, CA, USA). Generally, SMMC7721 and PLC5 cells were seeded in a six-well plate at a density of 5×105 cells/well for 24 hours, and then were treated with or without pectolinarigenin (10 μM) for 48 hours. Then the cells were fixed in 75% ethanol overnight at 4°C, followed by washing twice with PBS, and the fixed cells were then incubated with 300 μL propidium iodide (PI) working solution for 15–30 minutes, shielded from light at room temperature before flow cytometric analysis.
Apoptosis analysis
Annexin V/PI apoptosis detection kit (556420; BD Biosciences, San Jose, CA, USA) was used to detect cell apoptosis according to the manufacturer’s instructions. Briefly, cells were harvested and washed twice with PBS after treatment for 48 hours. Then the cells were suspended in 200 μL of binding buffer, 5 μL of Annexin V antibody conjugated with fluorescein isothiocyanate (FITC), and 5 μL of PI solution. After incubation in the mixture for 15 minutes at room temperature, shielded from light, the percentage of apoptotic cells was determined by flow cytometry (Beckman Coulter).
Western blotting
The total protein was extracted in ice-cold radioimmunoprecipitation assay lysis buffer (Beyotime, Shanghai, China), and the concentration of protein was quantified using a bicinchoninic acid protein assay kit (Beyotime). Equal amounts (20 μg/lane) of protein were fractionated using SDS-PAGE and transferred to polyvinylidene difluoride membranes. These membranes were then incubated with anti-Bax, anti-Caspase-3, anti-Cleaved Caspase-3, anti-Bcl-2, anti-cyclin B1, anti-cyclin D1, anti-P21, anti-CDC21, anti-PI3K, anti-p-mTOR, anti-mTOR, anti-p-EKR1/2, anti-EKR1/2, anti-pAKT, anti-AKT, and anti-GAPDH (Cell Signaling Technology, Danvers, MA, USA). Next the membranes were incubated with peroxidase-conjugated secondary antibodies. The Western blot bands were visualized using electrochemiluminescence kits in accordance with the manufacturer’s instructions (Abcam, Cambridge, UK). GAPDH was used for normalization of protein loading.
Animal and tumor xenograft studies
All animals were maintained under specific pathogen-free conditions. SMMC7721 cells (5×106 cells) were injected subcutaneously into the right side of each 5- to 6-week-old BALB/c male nude mice (Animal Center of the Chinese Academy of Science, Shanghai, China). The treatment was initiated when tumors reached a volume of 50–150 mm3. Tumor volume was calculated as the largest length×width2×0.5, and tumor diameter was measured thrice every week. Mice were divided into control group and pectolinarigenin group. Each group contained five animals. All animals were cared for, and the experiments were reviewed and approved by the Zhejiang University Animal Care Committee.
Statistical analysis
The statistical analysis was conducted using GraphPad Prism 7 software (GraphPad Software, La Jolla, CA, USA), two-tailed P<0.05 was considered statistically significant. The quantitative variables were summarized as mean ± SD. The data between groups were compared using the two-tailed Student’s t-test.
Results
Retardation of cell viability, proliferation, and invasive ability of HCC cells by pectolinarigenin
The cytotoxicity of pectolinarigenin on SMMC7721 and PLC5 cells was assessed by CCK-8 assay. Pectolinarigenin substantially retarded the viability of SMMC7721 and PLC5 cells in a time- and dose-dependent manner (Figure 1A and B). IC50 values of 24, 48, and 72 hours stimulation were 29.26, 19.23, and 11.59 μM, respectively, for SMMC7721, and 32.67, 20.9, and 11.97 μM, respectively, for PLC5. Colony formation assays also showed that when treated with 10 μM pectolinarigenin, both SMMC7721 and PLC5 cells showed fewer and smaller colonies than the control group (Figure 1C). Hence, 10 μM pectolinarigenin was used in the following assay to examine its effect on HCC cell lines to avoid the interference from cell death effect. The results of Transwell (Figure 1D) and scratch analysis (Figure 1E) showed that pectolinarigenin significantly suppressed invasion and migration. Taken together, pectolinarigenin could inhibit SMMC7721 and PLC5 cell invasion and migration.
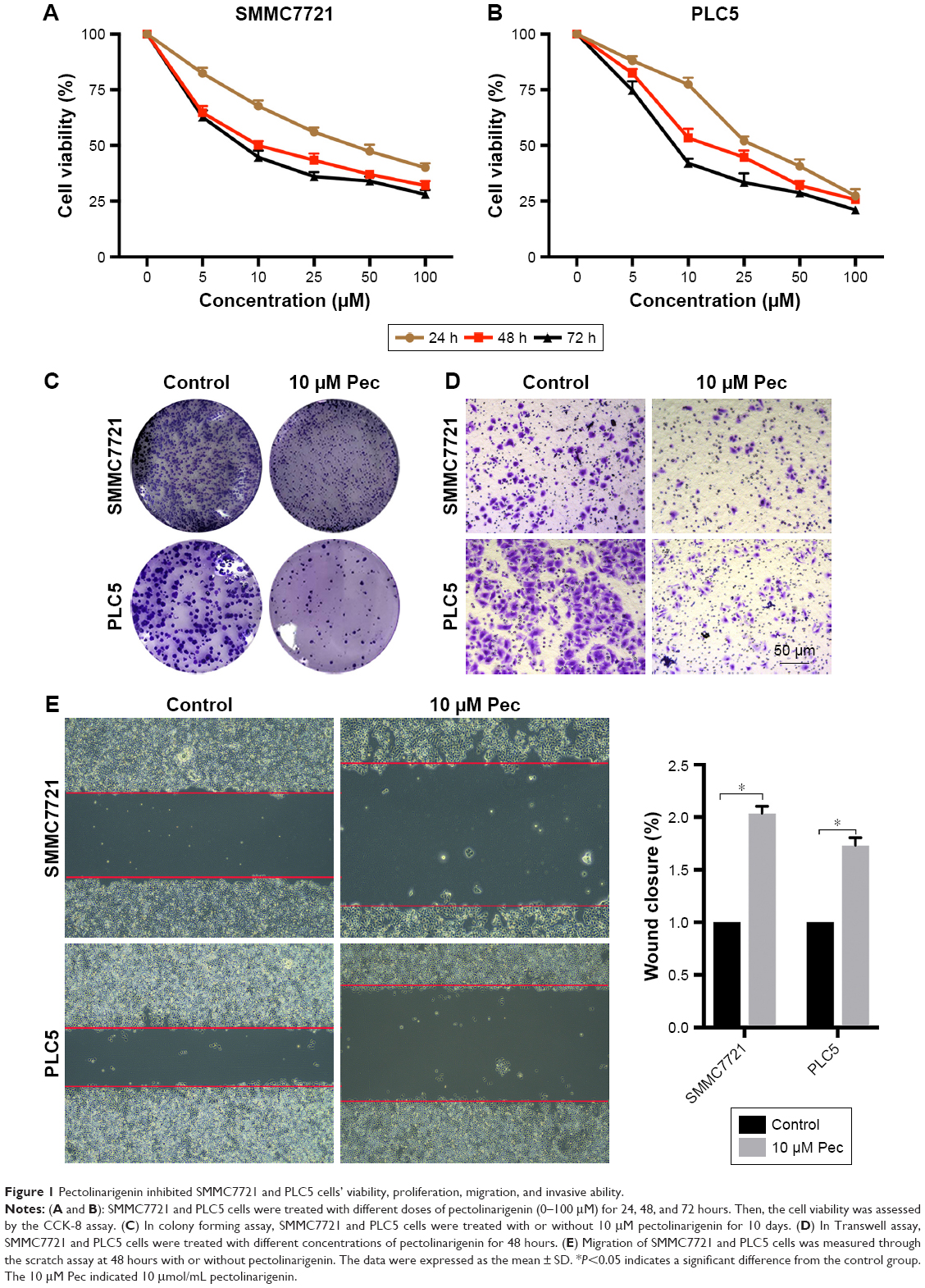 | Figure 1 Pectolinarigenin inhibited SMMC7721 and PLC5 cells’ viability, proliferation, migration, and invasive ability. |
Pectolinarigenin induced apoptosis in HCC cells
Apoptosis process is one of the proven antitumor pathways.9 Thus, to further confirm the effect of pectolinarigenin on HCC cell apoptosis process, Annexin V and PI staining was used to analyze whether pectolinarigenin caused apoptosis in SMMC7721 and PLC5 cells. As shown in Figure 2A, a significant increase in the ratio of apoptotic cells was observed in pectolinarigenin-treated SMMC7721 and PLC5 cells, ranging from 1.7% to 16.0% and from 5.3% to 38.5%, respectively. The difference was statistically significant (Figure 2B). Meanwhile, we further verified the protein expression levels of apoptosis-related proteins Bcl-2, Bax, and Caspase-3. Similar to flow cytometric results, exposure to pectolinarigenin distinctly downregulated the level of antiapoptotic Bcl-2 and upregulated the expression of the proapoptotic regulatory factor Bax (Figure 2C). Besides, the cleavage form of Caspase-3, as the executioner of apoptosis, was also upregulated significantly. Taken together, pectolinarigenin promotes apoptosis in HCC cells through the Bcl-2/Bax/Caspase-3 pathway.
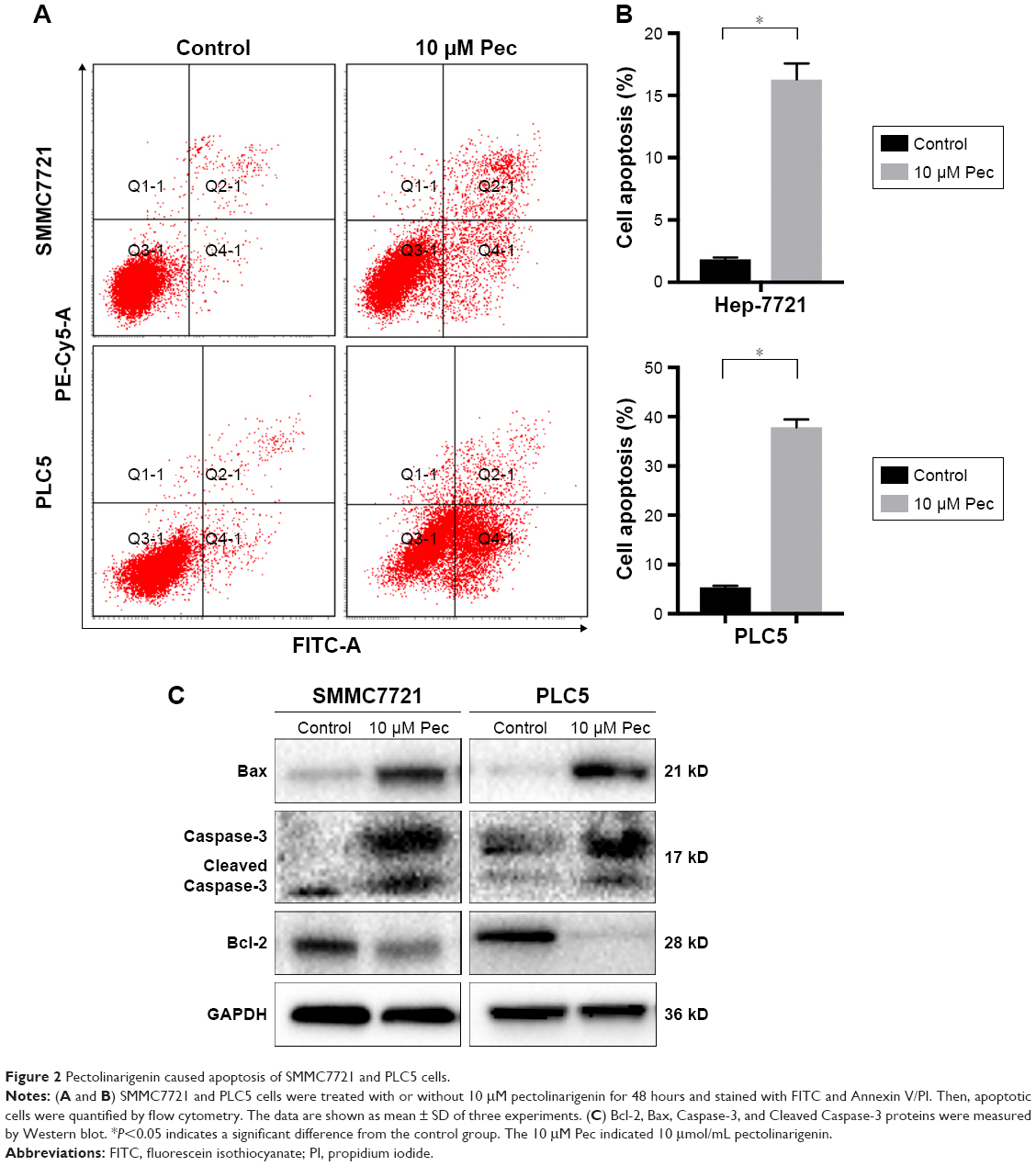 | Figure 2 Pectolinarigenin caused apoptosis of SMMC7721 and PLC5 cells. |
Pectolinarigenin triggered G2/M phase cell-cycle arrest in HCC cells
The effect of pectolinarigenin on cell-cycle distribution of HCC cells was examined by flow cytometric analysis. The results indicated that typical G2/M cell-cycle phase arrest pattern was observed when treated with pectolinarigenin. The proportion of cells at G2/M phase significantly increased in both SMMC7721 and PLC5 cells, ranging from 8.9% to 45.6% and from 14.4% to 42.0%, respectively (Figure 3A and B). To further clarify the underlying mechanism of molecules likely to be involved in cell-cycle progression, we examined the protein level by Western bolt for several cell-cycle regulatory factors in SMMC7721 and PLC5 cells. The expressions of G2/M cell cycle-regulatory proteins cyclin B1, cyclin D1, and CDC2 were downregulated and p21 expression was upregulated significantly (Figure 3C).
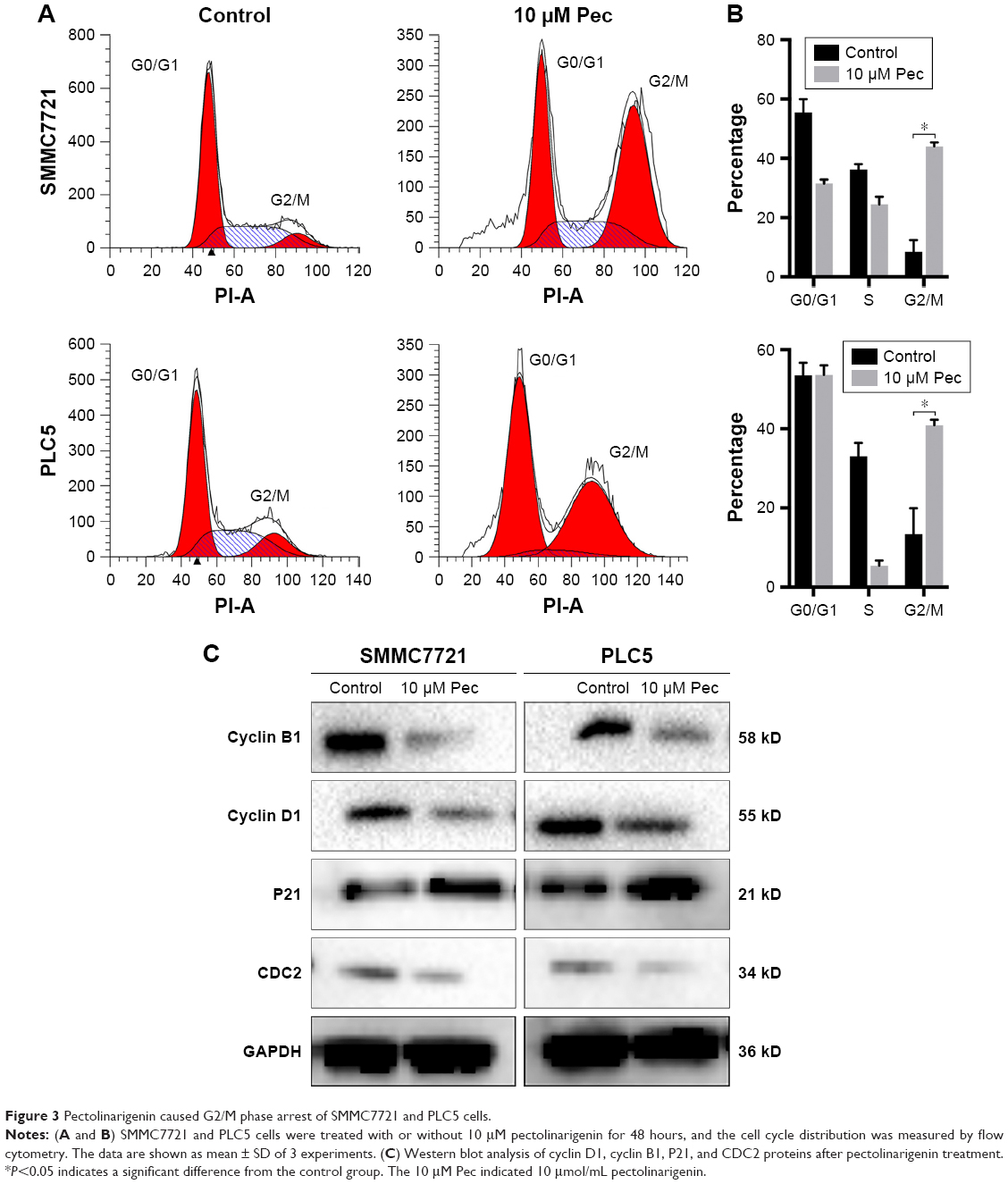 | Figure 3 Pectolinarigenin caused G2/M phase arrest of SMMC7721 and PLC5 cells. |
Pectolinarigenin suppressed PI3K/AKT/mTOR/ERK signaling pathway
To explore the potential mechanism by which pectolinarigenin induced HCC cell proliferation, we conducted Western blot to investigate the potential molecular targets of pectolinarigenin. The results indicated that pectolinarigenin effectively reduced PI3K, p-AKT, p-mTOR, and p-ERK1/2 protein levels (Figure 4) in both SMMC7721 and PLC5 cells. The present outcomes suggested that pectolinarigenin may cause proliferation retardation in HCC cells through PI3K/AKT/mTOR/ERK signaling pathway and suggested that it may be an alternative therapeutic strategy for HCC treatment.
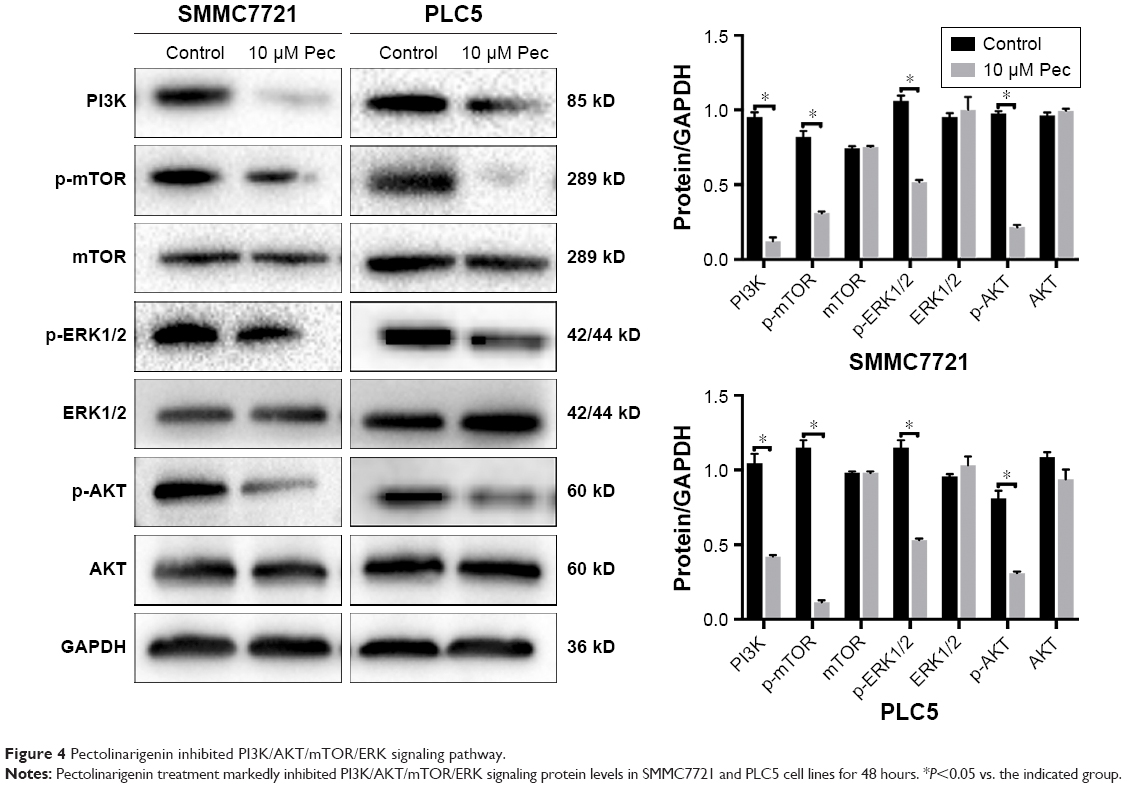 | Figure 4 Pectolinarigenin inhibited PI3K/AKT/mTOR/ERK signaling pathway. |
Pectolinarigenin suppressed HCC tumor growth in mouse xenograft models
The presented outcomes have suggested that pectolinarigenin significantly suppressed HCC cell proliferation in vitro and whether this phenomenon could be replicated similarly in vivo. After four cycles of pectolinarigenin treatment, the tumors of pectolinarigenin-treated mice were significantly smaller than those of the control group, as shown in Figure 5A, and the mean tumor weights were 0.44±0.04 and 0.14±0.04 g, respectively (n=5, P<0.001, Figure 5B). The mean tumor volumes in pectolinarigenin group were markedly smaller than those in the control group, 492.7±20.8 and 154.9±43.9 mm3, respectively (n=5, P<0.001, Figure 5C). Furthermore, immunohistochemistry staining was used to test the Ki-67 expression level. As shown in Figure 5D, Ki-67-positive cells reduced significantly in the pectolinarigenin group compared to that in the control group. Taken together, these results indicated that pectolinarigenin treatment could significantly inhibit HCC tumor proliferation in vivo.
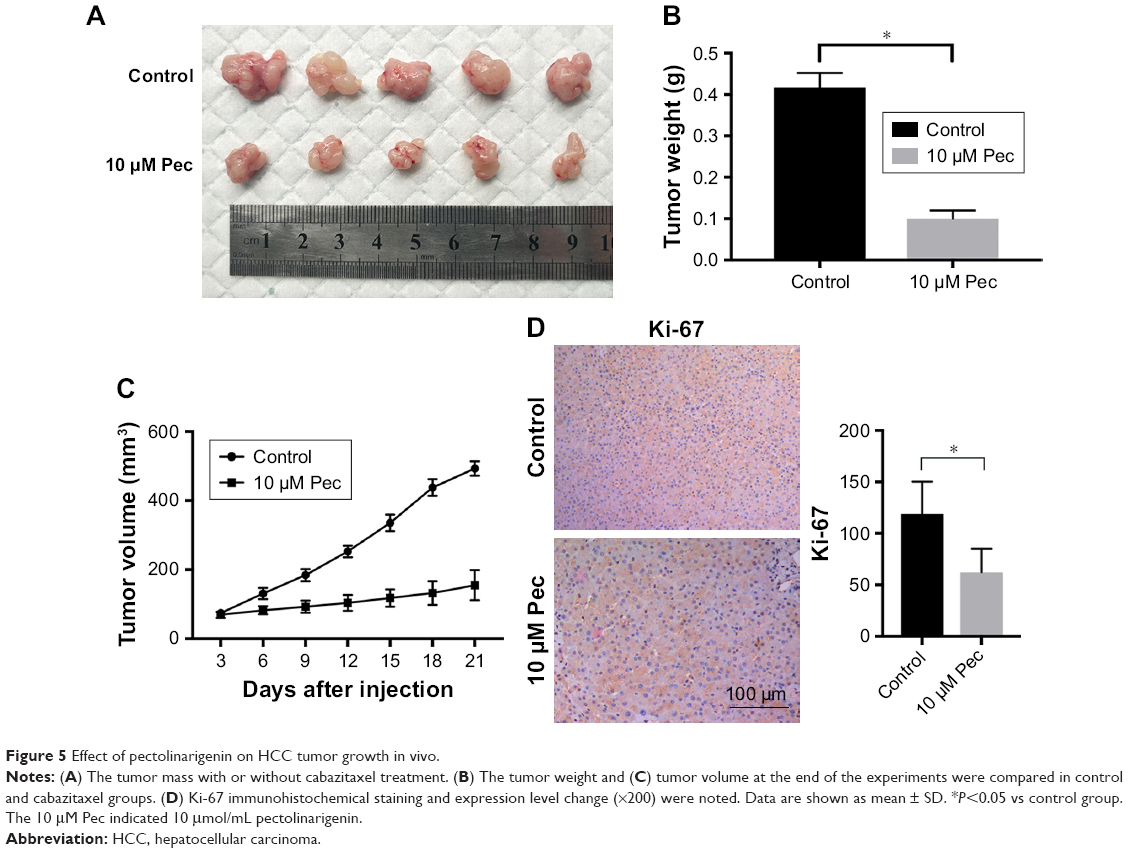 | Figure 5 Effect of pectolinarigenin on HCC tumor growth in vivo. |
Discussion
Currently, in Asia and Africa, the prevalence of liver cancer remains at the highest level than in other regions,10 and compared to other types of liver cancer, HCC accounts for 90% of primary liver cancers.11,12 The prognosis and 5-year survival of HCC treatment to some extent has been improved; however, the available systemic treatment options are finite due to phenotypic and molecular heterogeneity.13,14 Several progresses in the treatment of HCC have been achieved through advanced techniques and clinical targeted agents,15–18 but the clinical therapeutic outcomes of HCC patients remain unsatisfying, of which the clinical drug-resistance contributes as the greatest challenge. Therefore, an effective alternative to antitumor agents for the treatment of HCC is urgently required. In the present study, we offered several lines of evidence that pectolinarigenin, a natural Chinese herbal extract, could provide as a potential alternative in HCC treatment.
Recently, several studies have reported that pectolinarigenin possess anti-inflammation activities, leading to reduced eicosanoid production in inflammatory lesions, and antiviral and immunomodulatory potentials.7,19 In addition, a previous study reported that pectolinarigenin exhibited cytotoxic activity on human malignancies, including human small lung carcinoma and colorectal cancer.20 Meanwhile, in human large cell lung carcinoma, pectolinarigenin also exhibited strong cytotoxic activity against cell proliferation with an IC50 value of 4.07 μM.8 Based on the abovementioned antitumor effect of pectolinarigenin on several cancer types, we propose that it has an extent antitumor effect; however, the biological functions of pectolinarigenin in HCC remain unknown. To the best of our knowledge, this study elaborates the cytotoxic effect of pectolinarigenin on HCC cells. First, we identified that pectolinarigenin suppressed proliferation, promoted apoptosis, and caused G2/M arrest in HCC cancer cells. Second, we demonstrated that pectolinarigenin might play an antitumor role through PI3K/AKT/mTOR/ERK signaling pathway.
Resistance to cell death is represented as one of the hallmarks of cancer proliferation. Apoptosis has been regarded as a prevalent mode of “programmed” cell death.9 Gene-encoding21 or drugs22 has been used for cancer chemotherapy which could lead to apoptotic death. BCL-2, an antiapoptotic gene, was identified as a part of (14;18) chromosomal translocation in B-cell lymphomas.23,24 Previous studies demonstrated that overexpression or reduction of Bcl-2 protein was associated with inverse response of chemotherapy.25,26 Moreover, Bax played a role as a pro-apoptosis regulator through sequestering multi-domain pro-death BCL-2 proteins. Hence, it was reasoned that chemicals that disrupted the protein–protein interaction could offer a means to overcome blocks to cell death in cancer.27 In the present study, the percentage of early apoptosis cells was significantly increased when HCC cells were treated with pectolinarigenin as determined by flow cytometric analysis. The mechanism was, pectolinarigenin distinctly downregulated the protein levels of antiapoptotic Bcl-2 and upregulated the expression of the pro-apoptotic Bax. The cleavage form of Caspase-3 was the executioner of apoptosis that enhanced the apoptotic signaling.28,29 In our work, we also found that pectolinarigenin activated Caspase-3 in vitro. Therefore, pectolinarigenin may induce HCC cell apoptosis through the Bcl-2/Bax/Caspase-3 pathway.
Cell cycle arrest has been confirmed to be an effective approach in controlling tumor growth.30,31 In our study, we have found that pectolinarigenin markedly regulated the cell cycle in G2/M arrest. Previous works have suggested that p21 was a key regulator of the G1/S transition,32 which was consistent with our study that pectolinarigenin highly increased the p21 protein level. Besides, we also demonstrated that the G1/S phase checkpoint protein, cyclin D1, was significantly altered when HCC cells were treated with pectolinarigenin. The highly decreased cyclin B1 level observed in pectolinarigenin-treated cells may also impede G2/M transition and thus arrest the cells at the G2/M phase. These data suggested that pectolinarigenin may lead to cell growth via changes in cell cycle progression.
Accumulating evidence has indicated that the PI3K/AKT/mTOR/ERK signaling pathway was associated with aggressive phenotype and poor prognosis and was dysregulated in human HCC.33,34 One recent study has uncovered that KRas-driven cancer cells S-phase arrest was glutamine-dependent metabolic checkpoints,35 and another study reported that 5-aminoimidazole-4-carboxamide-1-β-4-ribofuranoside could induce breast cancer cells’ S-phase cell cycle arrest through the PI3K/mTOR/Akt signaling pathway.36 To better understand the mechanism by which pectolinarigenin caused HCC tumor cell proliferation, we focused on the PI3K/AKT/mTOR/ERK signaling pathway which was tightly involved in regulating HCC cell life development.34,37,38 In our research, we demonstrated that pectolinarigenin effectively suppressed the PI3K/AKT/mTOR/ERK signaling pathway by highly decreasing the phosphorylation level S of AKT, ERK1/2, and mTOR in human HCC SMMC7721 and PLC5 cell lines. These data suggested that pectolinarigenin significantly inhibited the PI3K/AKT/mTOR/ERK signaling pathway, which may induce apoptosis, cell cycle arrest, and migration/invasion delay in HCC cells.
Conclusion
In conclusion, our research indicated that pectolinarigenin could suppress HCC cell proliferation by inducing cell apoptosis and cell cycle arrest. Besides, pectolinarigenin also decreased HCC cell migration and invasion. Finally, we also proved that it could deactivate the PI3K/AKT/mTOR/ERK signaling pathway. The mechanisms underlying the anticancer effects by pectolinarigenin need to be further elucidated, but our findings suggest that it may be an alternative therapeutic strategy for HCC treatment.
Acknowledgments
We would like to acknowledge the National Natural Science Foundation of China (No. 81372626, No 81572975); Key Research and Development Project of Science and Technology Department of Zhejiang, China (No. 2015C03053); and Zhejiang Provincial Program for the Cultivation of High-level Innovative Health Talents (2016) for their help with the study.
Author contributions
SY designed the study. TCW and XGD managed the data collection of the study. TCW and XGD performed the data analysis. TCW wrote the final report. All authors contributed to critical revision of the final report. DDY, ZHS, and JBY are principal investigators of the study. All authors contributed to data analysis, drafting and revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Forner A, Reig M, Bruix J. Hepatocellular carcinoma. Lancet. 2018;391(10127):1301–1314. | ||
Forner A, Llovet JM, Bruix J. Hepatocellular carcinoma. Lancet. 2012;379(9822):1245–1255. | ||
Bruix J, Sherman M. American Association for the Study of Liver D. Management of hepatocellular carcinoma: an update. Hepatology. 2011;53(3):1020–1022. | ||
European Association For The Study Of The Liver, European Organisation For Research And Treatment Of Cancer. EASL-EORTC clinical practice guidelines: management of hepatocellular carcinoma. J Hepatol. 2012;56(4):908–943. | ||
Muthu C, Reegan AD, Kingsley S, Ignacimuthu S. Larvicidal activity of pectolinaringenin from Clerodendrum phlomidis L. against Culex quinquefasciatus Say and Aedes aegypti L. (Diptera: Culicidae). Parasitol Res. 2012;111(3):1059–1065. | ||
Sujatha P, Sreekanth G, Khasim S, Adavi Rao BV, Ravi Kumar B, Appa Rao AV. Flavonoids of dikamali: a phytochemical reinvestigation. Nat Prod Res. 2013;27(20):1930–1932. | ||
Lim H, Son KH, Chang HW, Bae K, Kang SS, Kim HP. Anti-inflammatory activity of pectolinarigenin and pectolinarin isolated from Cirsium chanroenicum. Biol Pharm Bull. 2008;31(11):2063–2067. | ||
Bonesi M, Tundis R, Deguin B, et al. In vitro biological evaluation of novel 7-O-dialkylaminoalkyl cytotoxic pectolinarigenin derivatives against a panel of human cancer cell lines. Bioorg Med Chem Lett. 2008;18(20):5431–5434. | ||
Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35(4):495–516. | ||
Bray F, Ferlay J, Laversanne M, et al. Cancer Incidence in Five Continents: inclusion criteria, highlights from Volume X and the global status of cancer registration. Int J Cancer. 2015;137(9):2060–2071. | ||
Llovet JM, Zucman-Rossi J, Pikarsky E, et al. Hepatocellular carcinoma. Nat Rev Dis Primers. 2016;2:16018. | ||
Zucman-Rossi J, Villanueva A, Nault JC, Llovet JM. Genetic landscape and biomarkers of hepatocellular carcinoma. Gastroenterology. 2015;149(5):1226–1239. | ||
Llovet JM, Lencioni R, Di Bisceglie AM, et al. EASL-EORTC Clinical practice guidelines: management of hepatocellular carcinoma. J Hepatol. 2012;56(4):908–943. | ||
Carlson RW, Larsen JK, Mcclure J, et al. International adaptations of NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw. 2014;12(5):643–648. | ||
Llovet JM, Ricci S, Mazzaferro V, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359(4):378–390. | ||
Bruix J, Qin S, Merle P, et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;389(10064):56–66. | ||
Cheng AL, Kang YK, Chen Z, et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: a phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009;10(1):25–34. | ||
El-Khoueiry AB, Sangro B, Yau T, et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet. 2017;389(10088):2492–2502. | ||
Lu M, Kong Q, Xu X, et al. Pectolinarigenin – A flavonoid compound from cirsium japonicum with potential anti-proliferation activity in MCF-7 breast cancer cell. Trop J Pharm Res. 2014;13(2):225–228. | ||
Woerdenbag HJ, Merfort I, Passreiter CM, et al. Cytotoxicity of flavonoids and sesquiterpene lactones from Arnica species against the GLC4 and the COLO 320 cell lines. Planta Med. 1994;60(5):434–437. | ||
Jia LT, Chen SY, Yang AG. Cancer gene therapy targeting cellular apoptosis machinery. Cancer Treat Rev. 2012;38(7):868–876. | ||
Gao X, Han L, Ding N, et al. Bafilomycin C1 induces G0/G1 cell-cycle arrest and mitochondrial-mediated apoptosis in human hepatocellular cancer SMMC7721 cells. J Antibiot. 2018;71(9):808–817. | ||
Tsujimoto Y, Finger LR, Yunis J, Nowell PC, Croce CM. Cloning of the chromosome breakpoint of neoplastic B cells with the t(14;18) chromosome translocation. Science. 1984;226(4678):1097–1099. | ||
Tsujimoto Y, Cossman J, Jaffe E, Croce CM. Involvement of the bcl-2 gene in human follicular lymphoma. Science. 1985;228(4706):1440–1443. | ||
Kitada S, Takayama S, de Riel K, Tanaka S, Reed JC. Reversal of chemoresistance of lymphoma cells by antisense-mediated reduction of bcl-2 gene expression. Antisense Res Dev. 1994;4(2):71–79. | ||
Tóthová E, Fricova M, Stecová N, Kafková A, Elbertová A. High expression of Bcl-2 protein in acute myeloid leukemia cells is associated with poor response to chemotherapy. Neoplasma. 2002;49(3):141–144. | ||
Croce CM, Reed JC. Finally, an apoptosis-targeting therapeutic for cancer. Cancer Res. 2016;76(20):5914–5920. | ||
Rai NK, Tripathi K, Sharma D, Shukla VK. Apoptosis: a basic physiologic process in wound healing. Int J Low Extrem Wounds. 2005;4(3):138–144. | ||
Cohen GM. Caspases: the executioners of apoptosis. Biochem J. 1997;326(1):1–16. | ||
Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer. 2009;9(3):153–166. | ||
Williams GH, Stoeber K. The cell cycle and cancer. J Pathol. 2012;226(2):352–364. | ||
Xiong Y, Hannon GJ, Zhang H, Casso D, Kobayashi R, Beach D. p21 is a universal inhibitor of cyclin kinases. Nature. 1993;366(6456):701–704. | ||
Matter MS, Decaens T, Andersen JB, Thorgeirsson SS. Targeting the mTOR pathway in hepatocellular carcinoma: current state and future trends. J Hepatol. 2014;60(4):855–865. | ||
Yang S, Liu G. Targeting the Ras/Raf/MEK/ERK pathway in hepatocellular carcinoma. Oncol Lett. 2017;13(3):1041–1047. | ||
Mukhopadhyay S, Saqcena M, Foster DA. Synthetic lethality in KRas-driven cancer cells created by glutamine deprivation. Oncoscience. 2015;2(10):807–808. | ||
Mukhopadhyay S, Chatterjee A, Kogan D, Patel D, Foster DA. 5-Aminoimidazole-4-carboxamide-1-β-4-ribofuranoside (AICAR) enhances the efficacy of rapamycin in human cancer cells. Cell Cycle. 2015;14(20):3331–3339. | ||
Kirstein MM, Boukouris AE, Pothiraju D, et al. Activity of the mTOR inhibitor RAD001, the dual mTOR and PI3-kinase inhibitor BEZ235 and the PI3-kinase inhibitor BKM120 in hepatocellular carcinoma. Liver Int. 2013;33(5):780–793. | ||
Ng KY, Chan LH, Chai S, et al. TP53INP1 downregulation activates a p73-Dependent DUSP10/ERK signaling pathway to promote metastasis of hepatocellular carcinoma. Cancer Res. 2017;77(17):4602–4612. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.