")
Back to Journals » International Journal of Nanomedicine » Volume 13
Nanostructured lipid carriers co-delivering lapachone and doxorubicin for overcoming multidrug resistance in breast cancer therapy
Received 28 January 2018
Accepted for publication 5 March 2018
Published 12 July 2018 Volume 2018:13 Pages 4107—4119
DOI https://doi.org/10.2147/IJN.S163929
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Linlin Sun
Xin Li,1 Xiaoqian Jia,2 Hu Niu2
1Department of Breast and Thyroid Surgery, Heze Municipal Hospital, Heze, Shandong, China; 2Department of General Surgery 2, The Fourth People’s Hospital of Jinan, Jinan, Shandong, China
Background: Multidrug resistance is responsible for the poor outcome in breast cancer therapy. Lapa is a novel therapeutic agent that generates ROS through the catalysis of the NAD(P)H:quinone oxidoreductase-1 (NQO1) enzyme which significantly facilitate the intracellular accumulation of the co-delivered DOX to overcome MDR in cancer cells.
Purpose: Herein, in our study, nanostructured lipid carrier (NLC) co-delivering β-lapachone (Lapa) and doxorubicin (DOX) was developed (LDNLC) with the aim to overcome the multidrug resistance (MDR) in breast cancer therapy.
Patients and methods: Lapa and DOX were loaded into NLC to prepare LDNLC using melted ultrasonic dispersion method.
Results: The well designed LDNLC was nanoscaled particles that exhibited preferable stability in physiological environment. In vitro cell experiments on MCF-7 ADR cells showed increased DOX retention as compared to DOX mono-delivery NLC (DNLC). In vivo anti-cancer assays on MCF-7 ADR tumor bearing mice model also revealed significantly enhanced efficacy of LDNLC than mono-delivery NLCs (DNLC and LNLC).
Conclusion: LDNLC might be a promising platform for effective breast cancer therapy.
Keywords: β-lapachone, doxorubicin, nanostructured lipid carriers, multidrug resistance, breast cancer
Introduction
Breast cancer has been well recognized as one of the most fatal diseases that greatly threaten the quality of life of women across the world.1 Although chemotherapy is a mainstay in clinical breast cancer treatment, its outcome usually succumbs to multidrug resistance (MDR) which can be found in various cancer cells.2 As a result, many current studies have made great efforts to overcome MDR, for more effective cancer therapy.3–5
To date, much research has provided solid proof in order to demonstrate that MDR is greatly associated with P-gp.6,7 P-gp is generally known as MDR1, which is one of the most important members in the ABC subfamily.8 It is an important protein of the cell membrane that pumps various foreign substances out of cells. The expression of P-gp is abnormally high in cancer cells as compared to normal ones, due to their inherent genetic characteristics. As a result, cancer cells exert superior resistance to many chemotherapeutics.9,10
Like P-gp overexpression, the existence of NQO1 enzyme is also upregulated by up to 100-fold in different cancer cells compared to normal ones.11 It has been reported that β-lapachone (Lapa) is a novel therapeutic agent that generates ROS through the catalysis of NQO1, which is usually accompanied by consuming reduced NAD(P)H and ATP through NQO1-catalyzed futile redox cycling. As a result, Lapa exhibits an extremely high selectivity for ROS generation as well as NAD(P)H and ATP consumption in tumor cells compared to healthy ones.12,13 Thus, Lapa dramatically influences various P-gp-related pathways, such as NF-κB, HIF-1α, and caspase, that could downregulate P-gp expression.14 Because P-gp-mediated drug efflux is an ATP-dependent process, Lapa could significantly facilitate the intracellular accumulation of other chemotherapy agents in cancer cells to overcome MDR in cancer cells.15
Doxorubicin (DOX) is a chemotherapy medication used to treat various kinds of cancers, including breast cancer, bladder cancer.16 By intercalation and inhibition of macromolecular biosynthesis via interaction with intracellular DNAs, DOX can quickly exert a strong cytotoxic effect on rapidly dividing breast cancer cells.16 However, both clinical and fundamental research has proven that the performance of DOX could be greatly impaired by MDR, especially acquired MDR, after cells’ exposure to a certain dosage of DOX. Combination chemotherapy as cancer therapy is considered an important protocol for enhanced therapeutic effects and reduced systemic toxicity, by simultaneously modulating multiple cell-signaling pathways and overcoming MDR. As a result, DOX is often used together with other chemotherapy agents to further improve anticancer performance, as well as reducing the side effects.17,18
Nanostructured lipid carriers (NLCs) are the second-generation of lipid carriers based on solid-lipid based nanoparticles.19,20 They were developed as an alternative to overcome the disadvantages of other colloidal carriers, such as lipid nanoemulsions and liposomes, for increased stability, controlled drug release as well as large-scale production.21 The lipid matrix in NLCs, which is composed of optimized solid and liquid lipids, could result in an imperfect crystal structure and therefore provide more room for drug accommodation, especially for hydrophobic drugs.22
Here, in our study, we successfully developed a PEG-modified NLC co-delivering Lapa and DOX (LDNLC) for overcoming MDR in breast cancer therapy. The PEG modification could increase the stability and circulation time of the NLC which could contribute to its increased accumulation in the tumor tissue via the enhanced permeability and retention (EPR) effect. After endocytosis by cancer cells, the released Lapa would exert a strong influence on P-gp expression and P-gp-mediated drug efflux via NQO1 catalysis, which could dramatically reverse drug resistance in cancer cells and promote the nuclear transportation of DOX.
Materials and methods
Materials
Compritol® 888 ATO, PEG-SA, soybean phosphatidylcholine (S100), oleic acid, glycerin monostearate, Lapa, DOX, dicoumarol (Di), MTT, DAPI, and DiR were purchased from Sigma-Aldrich Co. (St. Louis, MO, USA). All other organic reagents were purchased from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China).
Preparation of drug-loaded formulations
The drug-loaded formulations were prepared by melted ultrasonic dispersion method, similar to a previous report, with some modifications.23 In brief, Lapa and DOX were dissolved in 1 mL of DMSO and then added to the hot lipid phase consisting of 20 mg Compritol® 888 ATO, 45 mg oleic acid, 45 mg glycerin monostearate, 90 mg S100, and 20 mg PEG-SA. The aqueous phase was prepared by dissolving 20 mg F68 in 10 mL of distilled water. Both phases were maintained for 15 min at 60°C in the oil bath under magnetic stirring. Then, the hot lipid phase was added slowly to the aqueous solution and dispersed using a high-speed homogenizer for 5 min at 10,000 rpm. The crude emulsion was additionally treated by a probe type ultrasonicator (VCX800; Sonics, CT, USA) for 5 min at 3 W. Then, the hot emulsion was cooled at 4°C in an ice bath, maintaining the mechanical stirring for another 30 min. The NLC suspension was dissolved with Milli-Q water and then dialyzed against Milli-Q water for 12 h to remove DMSO and impurities. Afterward, the LDNLC was subjected to lyophilization using the following protocol: 1) freezing right after production at −80°C; 2) thermal treatment at −22°C for 2 h; 3) cooling down to −40°C for another 2 h; 4) primary drying at 1.03 mbar (−30°C 7 h, −10°C 2 h, 20°C 12 h); 5) secondary drying at 30°C (0.001 mbar) for 3 h (the stability of particles post-lyophilization remained unchanged according to our tests). The obtained powder was stored at 4°C until further use. Drug-free NLC or Lapa mono-delivery NLC (LNLC) or DOX mono-delivery NLC (DNLC) was prepared using the same method accordingly.
Drug loading
The remaining drug loading content of the as-prepared NLC was determined. The drug-loaded NLC was dispersed in ethanol and sonicated for 30 min at 70°C to fully destroy the nanoparticles and release the encapsulated drugs. Drug content in the ethanol solution was determined using high- performance liquid chromatography (HPLC) as described in previous reports.39,40 In brief, Agilent 1260 Infinity II equipped with ZORBAX Eclipse XDB-C18 column (4.6 × 250 mm2, 5 μM) was used. For the determination of DOX, the mobile phase consisted of methanol, acetonitrile, and phosphate buffer (25 mmol/L Na2HPO4−, 30 mmol/L NaH2PO4, pH 5.0) with a volume ratio of 49:20:31; determined wavelength of 233 nm; flow rate of 1 mL/min, and temperature of 25°C.24 For Lapa, the mobile phase consisted of 7:2 acetonitrile:water, the total flow rate was 0.9 mL/min and the temperature was 25°C. Absorption at 260 nm was used to determine Lapa concentration.15
Encapsulation efficiency and loading efficiency were calculated by the following equations:
|

|
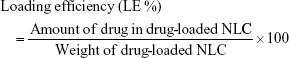
Particle size, zeta potential, and morphology measurements
The particle size and zeta potential of all nanoparticles were measured using a ZetaPlus Zeta Potential Analyzer (Brookhaven Instruments Corp., Holtsville, NY, USA). For structural observation, a single drop of each solution was deposited on a transmission electron microscopy (TEM) grid and allowed to air-dry. All silica nanoparticle samples and conjugates were imaged using a JEM-200CX transmission electron microscope (JEOL, Tokyo, Japan) with an acceleration voltage of 80 kV.25
Stability and hemolysis assay
For colloidal stability test, the freshly prepared LDNLC were diluted with PBS (pH 7.4) at the volume ratio of 1:10. The change in particle size was recorded at predetermined time intervals (3, 6, 9, 12, 24, 36, and 48 h) for up to 48 h. For hemolysis assay, red blood cells (RBCs) were first obtained from a New Zealand rabbit and diluted to 2% suspension with saline solution. LDNLC was added into 2% RBCs suspension with the same volume to achieve the designated concentrations (0.1, 0.25, 0.5, 0.75, and 1 mg/mL) and incubated at 37°C for 1 h. In addition, RBCs suspension was also incubated with saline and distilled water under the same conditions as negative (0% hemolysis) and positive controls (100% hemolysis), respectively. After that, all the samples were centrifuged at 3,000 rpm for 10 min, and the absorption values of the same volume of supernatants which represented the counts of released hemoglobin were measured by a UV spectrophotometer (SPECTRONIC 200; Thermo Fisher Scientific, Waltham, MA, USA).26
In vitro drug release behavior
The release behavior of Lapa and DOX from NLCs was assessed in PBS by the dialysis method.16 Briefly, the NLCs (containing 5 mg of Lapa and DOX) were suspended in 3 mL of the PBS release medium and transferred into a dialysis bag (MWCO 3,500 Da). The release experiments were started by placing the dialysis bag into 37 mL of PBS with 1% Tween 80 to afford the sink condition. The experiment proceeded with continuous shaking at 100 rpm at 37°C. At a predetermined time, a 100 μL aliquot of the solution outside the bag was withdrawn for HPLC analysis as described previously.
In vitro uptake
To evaluate the cellular uptake of LDNLC in an MCF-7 ADR cell line (Cell Bank of Shanghai Institute of Biochemistry and Cell Biology; Chinese Academy of Sciences, Shanghai, China), MCF-7 ADR cells were seeded in confocal dishes (Φ=15 mm; Corning Incorporated, Corning, NY, USA) at a density of 1 × 105 cells/dish and cultured overnight (humidified atmosphere containing 5% CO2 at 37°C). The dishes were then supplemented with 2 mL serum-free medium (DMEM containing 10% FBS; Thermo Fisher Scientific). The culture medium mentioned in the following section was DMEM (unless otherwise stated) containing DNLC or LDNLC. Additionally, cells were incubated with 60 μM of Di (an inhibitor of NQO1) solution at 37°C for 3 h prior to nanoparticles’ addition. Following 12 h of incubation with nanoparticles, cells were fixed with 4% formaldehyde for 15 min at 37°C. Cells were qualitatively assessed using a Leica confocal laser scanning microscope (LSM 710, ×600; Carl Zeiss Meditec AG, Jena, Germany).27 DOX fluorescence (excitation 510 nm, emission 550 nm) was expressed as red, Hoechst 33342 (excitation 405 nm, emission 460 nm) was expressed as blue.
Intracellular ATP level measurements and Western blot analysis
Intracellular ATP level was measured using a CellTiter-Glo® luminescent cell viability assay kit (Promega Corporation, Fitchburg, WI, USA). The assay was conducted according to the recommended procedure of the kit and a previous report.15 In brief, MCF-7 ADR cells were seeded in 96-well plates (Corning Incorporated) with 5,000 cells per well for 24 h before treatment. The cells were exposed to LNLC (10 μM Lapa), DNLC (10 μM DOX) or the equivalent dose of LDNLC with or without 60 μM Di for 4 h. Next, the medium was placed in 100 μL of fresh medium, and 100 μL of CellTiter-Glo reagent was added. The plate was shaken for 2 min and incubated at room temperature for 10 min, and then the luminescent signal, which is proportional to the amount of ATP, was measured on a Lumat LB9507 luminometer (Berthold, Bad Wildbad, Germany). The luminescent signal of the untreated cells was taken as the 100% value to determine the intracellular ATP level variations in response to different treatments.
The determination of the P-gp protein expression levels in cells was also in accordance with a previous report.15 In detail, MCF-7 ADR cells were seeded in 6-well plates at 1.5 × 105 cells/well as described previously, and treated with different drugs for 48 h. After being washed with PBS three times, the cells were harvested in lysis buffer (Beyotime, Shanghai, China) for 30 min and centrifuged at 13,000 rpm for 10 min, and the protein content in the supernatant was detected using a BCA protein assay (Beyotime). Equal amounts of the protein extracts (12 μg) were loaded and separated on an 8% SDS-PAGE gel and transferred onto a nitrocellulose membrane. The membrane was incubated, first with 5% nonfat milk powder in TBST buffer for 1 h to block nonspecific binding sites, and then with primary antibodies for P-gp (Abcam, Cambridge, MA, USA; 1:4,000) and β-tubulin (Abcam; 1:1,000) at 4°C overnight. After being washed with TBST, the membranes were further incubated with horseradish peroxidase-labeled goat anti-rabbit or mouse antibodies (1:1,000) at room temperature for 1 h. Finally, the membranes were rinsed and visualized with an electrochemiluminescence detection reagent (Beyotime). Protein expression was normalized to β-tubulin.
In vitro cytotoxicity studies
The cytotoxicity of LDNLC was evaluated using the standard MTT assay. MCF-7 ADR cells were seeded at 1.0 × 104 cells/well into 96-well plates and cultured until they reached 70%–80% confluence. The primary growth medium was replaced by 200 μL of fresh serum-free DMEM, to which drug-free NLC or DNLC, LNLC, LDNLC with or without 60 μM Di was added to achieve various concentrations (drug-free NLC ranging from 10 to 200 μg/mL; Lapa and DOX concentrations ranging from 0.5 to 100 μM; the weight/weight ratio of Lapa and DOX was 1). Plates were then returned to the incubator for up to 48 h of incubation, after which 20 μL of 5 mg/mL MTT solution in PBS was added to each well for an additional 4 h of incubation. Subsequently, the medium was carefully removed and replaced by 150 μL DMSO and measured at 570 nm using a microplate reader (EL800; BioTek Instruments Inc., Winooski, VT, USA). Untreated cells were used as a control with 100% viability.28 In addition, apoptosis assay using Annexin-V FITC (BD Biosciences, San Jose, CA, USA) was performed according to the manufacturer’s instructions.
In vivo distribution
Female BALB/c nude mice (20–22 g) aged 6 weeks were purchased from Shanghai Laboratory Animal Center (SLAC, China). All mice were housed in the SPF II lab and given free access to food and water. All procedures were conducted in compliance with NIH guidelines for care and use of research animals and approved by the ethics committee of The Fourth People’s Hospital of Jinan. The tumor-bearing mice were produced by inoculating a suspension of MCF-7 ADR cells (1 × 107 cells in 0.1 mL physiological saline) subcutaneously into the left flank. When the tumor volume reached 200 mm3, these mice were intravenously administered DiR-loaded LDNLC at 10 μg DiR per mouse. The in vivo tumor targeting efficacy and biodistribution of DiR-loaded LDNLC was evaluated using In Vivo Imaging System (FXPRO; Kodak, NY, USA) equipped with DiR filter sets (excitation/emission, 720/790 nm).29
The in vivo plasma pharmacokinetic and DOX biodistribution studies were performed according to a previous report.15 In brief, female BALB/c nude mice were randomly divided into two groups (n = 3), and intravenously injected with free DOX or LDNLC (DOX dosage: 5 mg/kg). At predetermined time intervals, blood samples were collected and centrifuged at 5,000 rpm and 4°C for 10 min. An amount of 100 μL of the supernatant plasma was mixed with another 900 μL of acetonitrile to precipitate the proteins and extract the DOX within. After centrifugation, the organic layer was collected, concentrated, and subjected to HPLC to determine the DOX level as described previously. In the drug biodistribution experiments, female BALB/c mice were inoculated with MCF-7 ADR tumors as mentioned previously. When the tumor volume reached 200 mm3, the mice were randomly divided and treated with free DOX or LDNLC as described previously. At 24 h after injection, the mice were sacrificed and their major organs (heart, liver, spleen, lung, kidneys) as well as tumors, were excised. The organs or tissues were cut into small pieces and homogenized, and DOX was extracted with 200 μL of ice-cold acetonitrile containing 0.5% acetic acid. The mixture was centrifuged at 13,000 rpm for 10 min, and 200 μL of the supernatant was concentrated and subjected to HPLC determination of DOX concentration.
In vivo anticancer efficiency
An MCF-7 ADR tumor-bearing nude mouse model was established as described previously. When the tumors reached a mean volume of approximately 100 mm3, mice were randomly assigned to four treatment groups (n = 6): saline, DNLC (DOX dosage: 5 mg/kg), LNLC (Lapa equivalent of 25 mg/kg), and LDNLC (DOX equivalent of 5 mg/kg, Lapa equivalent of 25 mg/kg) with reference to a previous report.15 The treatment was implemented by intravenous injection every 2 days (seven times). Tumor volume (mm3) was calculated using the formula: tumor volume = (shortest diameter)2 × (longest diameter) × 0.5. The tumor growth and body weight variation curves were plotted using the average tumor volume versus days after the first treatment. All the mice were sacrificed 14 days after the first treatment, and their tumors were resected. The excised tumors in each group were fixed using 4% paraformaldehyde buffer, embedded in paraffin and sectioned into 5 μM thick slices. The sections were stained with H and E (Beyotime) for histological examinations and imaged under an inverted microscope.
Results and discussion
Characterization of nanoparticles
The as-prepared NLC was characterized based on its particles’ size, zeta potential as well as morphology. According to results obtained by Zetasizer using dynamic light scattering method (Figure 1A), the size distribution of LDNLC was approximately 100 nm with narrow distribution range, which was reflected by its relatively small polydispersity index of 0.123 (<0.3). This was also confirmed by morphological observation using TEM (Figure 1C). It has been reported that nanoparticles with size under 200 nm could preferably take advantage of the high permeability of the tumor tissue to consistently accumulate to the targeted site, via the well-known EPR effect.30,31 In addition to particle size, the surface zeta potential also played an important role in determining the in vivo fate of the nanoparticles. It is well recognized that nanoparticles with negative surface charge could be more favorable than their positively charged counterparts in circulation, since the majority of plasma proteins in the blood stream are negatively charged.29 As a result, upon being introduced into the circulation, the positively charged nanoparticles would be recognized and retarded by the abundant plasma proteins due to electrostatic interaction between reverse-charged particles, which could have disappointing results due to the fact that these nanoparticles cannot travel far or long enough to satisfy the minimal requirement of drug delivery.31 In our study, the components of the as-prepared nanoparticles were mostly negatively charged lipids that finally resulted in the formation of negatively charged-NLC. As shown in Figure 1B, the zeta potential of LDNLC was −23.45 mV. Combined with PEG modification, the resultant LDNLC was expected to ensure the safe delivery of encapsulated drugs in vitro. DNLC and LNLC prepared using the same method, showed similar properties to that of LDNLC (Table 1). According to the HPLC results, the drug loading content reached up to 6.4% for Lapa and 5.1% for DOX with drug loading efficiency of 83.8% and 92.3%, respectively.
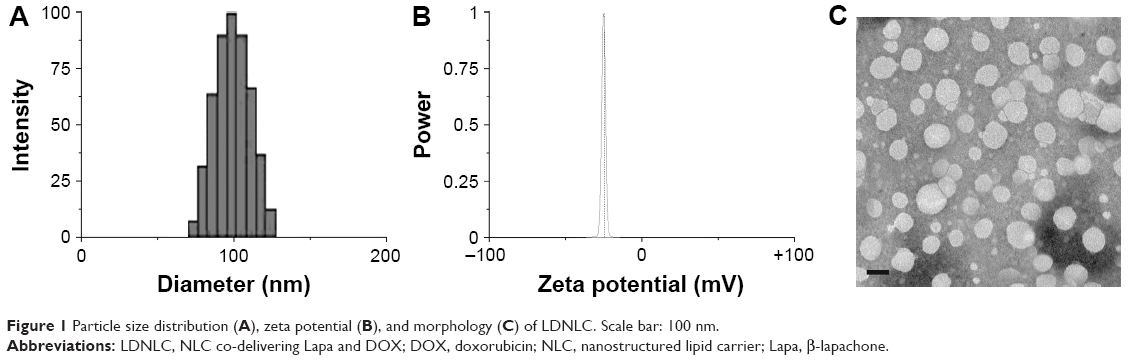 | Figure 1 Particle size distribution (A), zeta potential (B), and morphology (C) of LDNLC. Scale bar: 100 nm. |
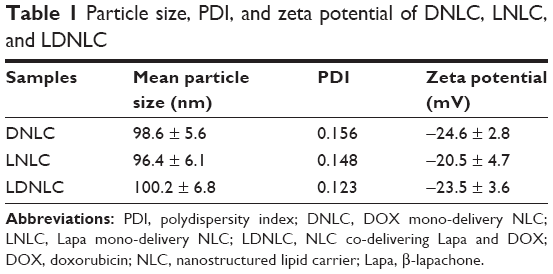 | Table 1 Particle size, PDI, and zeta potential of DNLC, LNLC, and LDNLC |
Stability and hemolysis assay
It has been generally recognized that several basic requirements are needed if a drug delivery system is intended to safely deliver the encapsulated drug molecules. Since particle size is a critical parameter in determining the in vivo fate of the drug delivery system, with the aim to bypass multiple extracellular barriers, the drug delivery system should be able to maintain its morphology in physiological conditions without significant size change until arriving at the targeted sites.32 Second, in order to reduce the systemic toxicity, the drug delivery system should not induce any adverse reactions during the whole process.26
As a result, LDNLC was studied regarding its stability as well as hemolysis as a function of time and concentration, respectively. To estimate the colloidal stability of the described NLC, the change in particle size of LDNLC, which serves as an indicator of potential instability under physiological conditions, was monitored in PBS (pH 7.4) for 48 h. As shown in Figure 2A, the size of LDNLC remained stable during the whole period with only slight fluctuation, which indicated that the as-prepared LDNLC was capable of maintaining its size without being significantly affected by the environment, for a relatively long time. Serum stability of LDNLC was further determined, which also confirmed the conclusion. The preferable stability of LDNLC offers the potential for controllable drug delivery for cancer therapy and is beneficial for its further development as a stable platform with predictable behavior to meet the demands of more extensive applications.
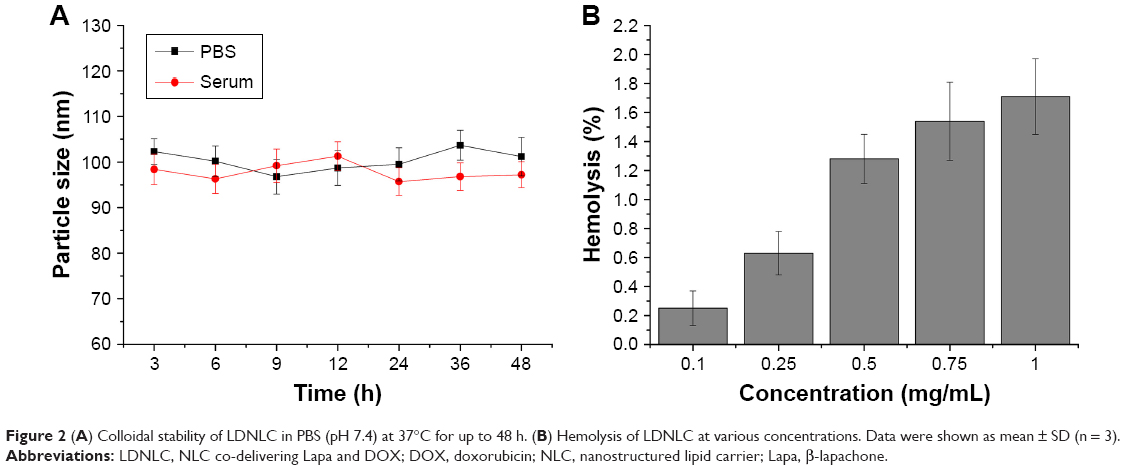 | Figure 2 (A) Colloidal stability of LDNLC in PBS (pH 7.4) at 37°C for up to 48 h. (B) Hemolysis of LDNLC at various concentrations. Data were shown as mean ± SD (n = 3). |
Apart from steady delivery and controlled release properties, hemolysis of the drug-loaded NLC was also investigated as a safety guide for intravenous administration and medicinal application. As shown in Figure 2B, LDNLC displayed no or negligible hemolysis (merely 1.71%) of RBCs at the highest concentration of 1 mg/mL. Considering that when being applied in vivo, the LDNLC concentration in the circulation would be diluted to a much lower one compared to those being tested in our study, it was suggested that the as-prepared LDNLC might be a relatively safe nanoparticle without inducing noticeable hemolysis. In all, it has been demonstrated that LDNLC exerted a stable profile with high biocompatibility which holds great promise for therapeutic efficacy in vivo.
Drug release
Lapa and DOX loaded into NLC is controllable and reproducible. The drug release profile of LDNLC was investigated. As shown in Figure 3, both the encapsulated drugs showed a sustained release behavior compared to free drugs since the initiation of the experiment. However, almost 90% of the drugs in LDNLC was released after 24 h of incubation, which indicated that drug loading and release in LDNLC was a reversible process that provided preliminary insurance to sufficiently release the majority of the encapsulated drugs. In addition, unlike free drugs that could be quickly diffused in a short time (50% in 4 h and over 90% in 12 h), it was also noted that the drug release rate of both drugs was maintained at a relatively low and stable speed before most of the drugs were released. At the same time, the release of Lapa and DOX showed a similar profile with synchronized release behaviors throughout the whole experiment and was near linear before reaching the balance. These observations indicated that Lapa and DOX were not only successfully loaded into the NLC but were also evenly distributed in the matrix of the nanoparticles. The successful loading of both drugs might be able to change their in vitro and in vivo behaviors to achieve a more sustainable and targeted profile, which makes LDNLC beneficial as a preferable drug delivery system.30 Drug release in acidic acetate buffer (pH 5.0) showed a similar profile to that in PBS (data not shown).
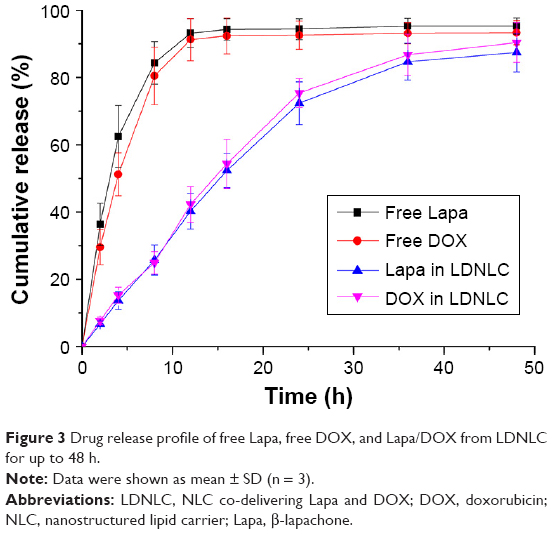 | Figure 3 Drug release profile of free Lapa, free DOX, and Lapa/DOX from LDNLC for up to 48 h. |
In vitro uptake
The in vitro drug uptake was studied using confocal laser scanning microscopy to observe the subcellular drug distribution and release in MCF-7 ADR cells treated with DNLC and LDNLC. It has been reported that DOX acts on topoisomerase II and DNA located in the nucleus. As a result, DOX accumulation in the nucleus is crucial to apoptosis induction and therapeutic effectiveness.16 As shown in Figure 4, only weak fluorescence could be observed in the cytoplasm of the cells, which indicated that only a small proportion of DOX was internalized into the cells, possibly due to the strong MDR effect of MCF-7 ADR cells. Under such conditions, almost no DOX signal was observed in the nucleus of cells, suggesting that DNLC might not effectively deliver the encapsulated DOX into the cells and the targeted site (nucleus) to fully exert the cytotoxicity of DOX. In contrast, cells treated with LDNLC showed much more intense fluorescence, mostly accumulated in the nucleus. It was inferred that upon entering the cells, the released Lapa in LDNLC could significantly inhibit the MDR process of cells to facilitate the accumulation as well as nucleus entering of DOX, which showed superior benefit compared to DNLC. In order to further confirm our assumption, we co-incubated LDNLC with the NQO1 inhibitor Di. As expected, in MCF-7 ADR cells, the whole intracellular fluorescence as well as DOX content in nucleus for this group decreased to the same level as that for the DNLC group, indicating that Di strongly blocked the anti-MDR to Lapa and impaired nuclear transport processes in these cells. It was inferred that the strong DOX resistance capability of MCF-7 ADR cells could quickly pump the released DOX outside the cells. It was noted that only free DOX can enter the nuclei, as a result, it was observed that DOX accumulated in nuclei for LDNLC, but in cytoplasm for DNLC and LDNLC + Di.15 Remarkably, these results indicated that LDNLC could overcome MDR and that NQO1-induced reduction of Lapa played a crucial role in this process.
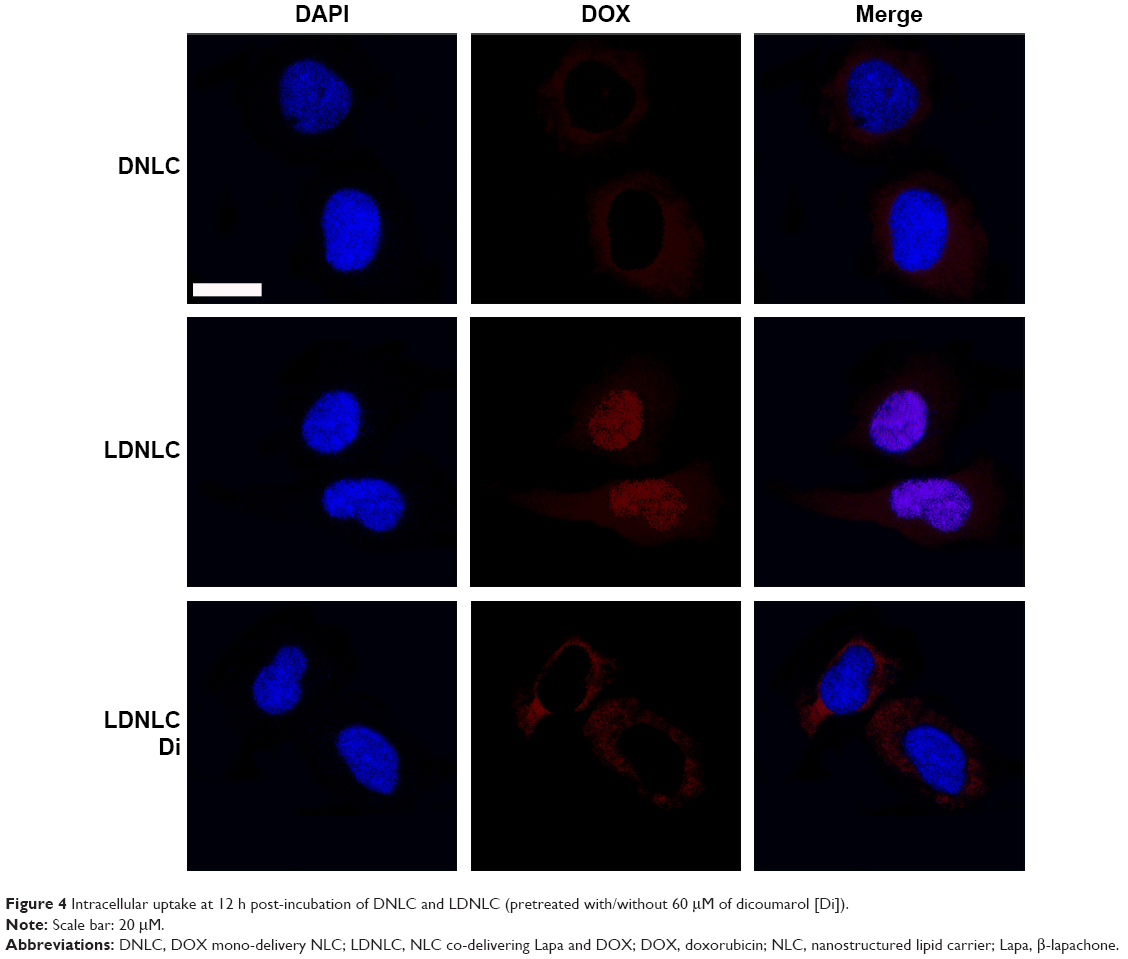 | Figure 4 Intracellular uptake at 12 h post-incubation of DNLC and LDNLC (pretreated with/without 60 μM of dicoumarol [Di]). |
Intracellular ATP level measurements and Western blot analysis
It has been generally recognized that P-gp-induced drug efflux is the principal mechanism leading to MDR in MCF-7 ADR cells. As a result, many previous articles have demonstrated that blocking the energy supply, as well as downregulating P-gp expression could be the main methods to inhibit the efflux.33,34 We therefore suggested that Lapa in LDNLC might also achieve anti-MDR through these approaches. In order to investigate the mechanism by which LDNLC could overcome the MDR of MCF-7 ADR cells, the intracellular ATP as well P-gp levels were determined. As shown in Figure 5A, the intracellular ATP variations influenced by different formulations in MCF-7 ADR cells were studied. In contrast to the DNLC group that showed an almost similar ATP level to the blank group, both the LNLC and LDNLC groups had ATP levels decreased by over 50%. Furthermore, the NQO1 inhibitor Di remarkably increased the ATP level back to 86% of the control group, indicating that NQO1-induced Lapa futile redox cycling led to NAD(P)H consumption along with ATP depletion, which would cut off the energy supply for P-gp.15 In addition, the P-gp level in cells determined by Western blot assay, also showed similar results. As shown in Figure 5B and C, DNLC treatment could elevate the P-gp expression in MCF-7 ADR cells compared with the blank group, indicating that acquired MDR was involved in this process. However, like LNLC, LDNLC co-delivering DOX and Lapa could also significantly downregulate P-gp expression, suggesting that Lapa could effectively reverse acquired MDR through multiple pathways, which include activating the MAPK pathways and downregulating NF-kB and HIF-1a expression, according to previous reports.13,35
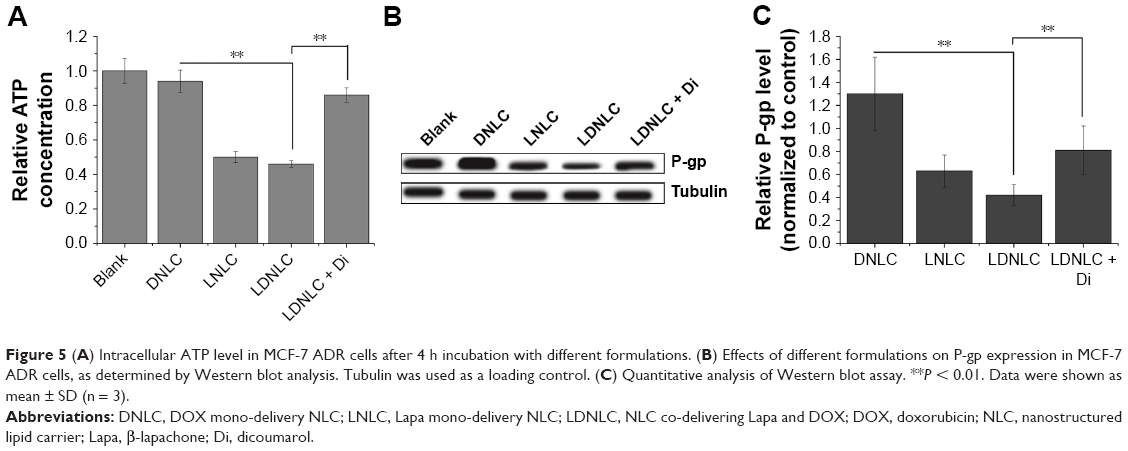 | Figure 5 (A) Intracellular ATP level in MCF-7 ADR cells after 4 h incubation with different formulations. (B) Effects of different formulations on P-gp expression in MCF-7 ADR cells, as determined by Western blot analysis. Tubulin was used as a loading control. (C) Quantitative analysis of Western blot assay. **P < 0.01. Data were shown as mean ± SD (n = 3). |
In vitro cytotoxicity
The in vitro cytotoxicity of LDNLC was investigated. The cytotoxic effect of drug-free NLC was first determined to verify its safety profile. As shown in Figure 6A, drug-free NLC showed relatively low cytotoxicity on MCF-7 ADR cells at all tested concentrations and time intervals. Even at the highest concentration of 200 μg/mL for 48 h, the cell viability of NLC-treated cells still had no significant differences to that of blank ones. This is higher than that adopted in the following cytotoxicity assay. As a result, it was inferred that the following cytotoxicity was all attributable to the loaded drugs but not to the carrier.
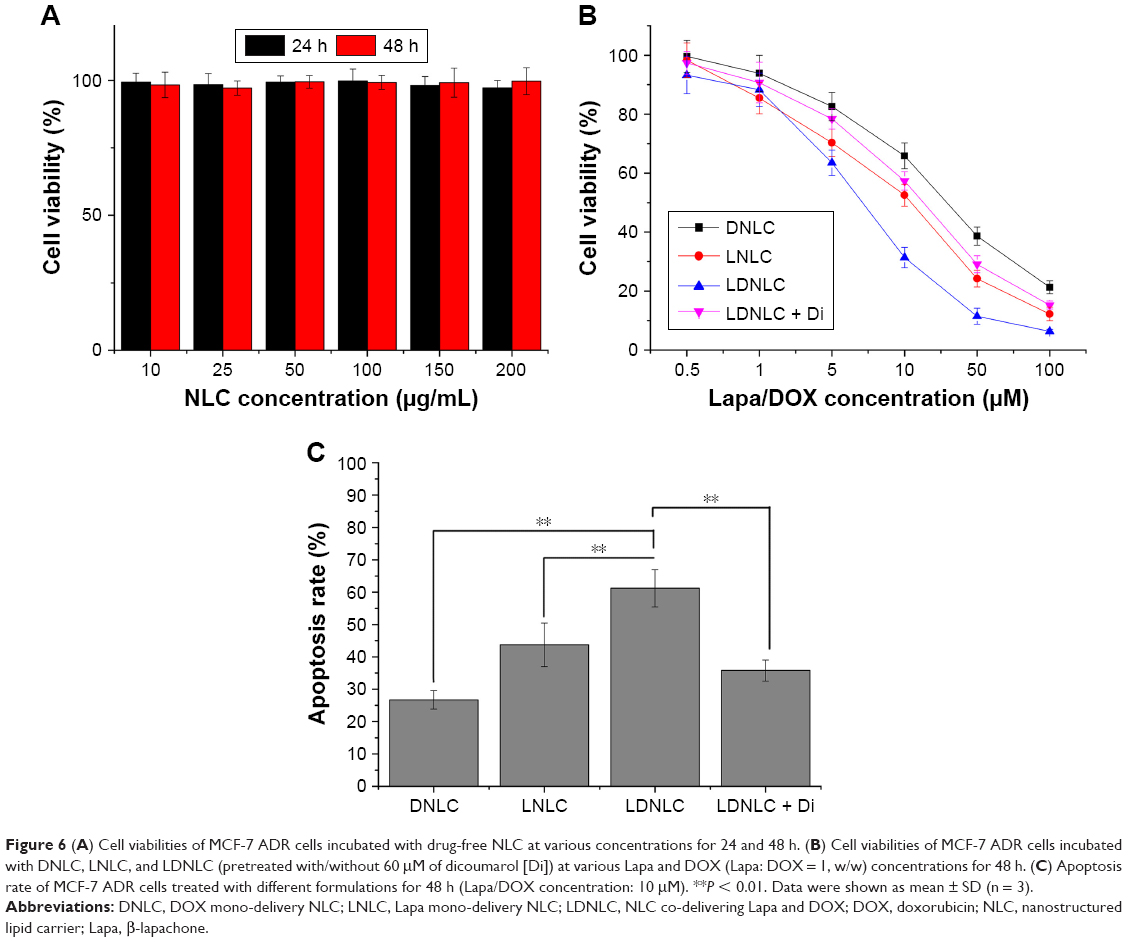 | Figure 6 (A) Cell viabilities of MCF-7 ADR cells incubated with drug-free NLC at various concentrations for 24 and 48 h. (B) Cell viabilities of MCF-7 ADR cells incubated with DNLC, LNLC, and LDNLC (pretreated with/without 60 μM of dicoumarol [Di]) at various Lapa and DOX (Lapa: DOX = 1, w/w) concentrations for 48 h. (C) Apoptosis rate of MCF-7 ADR cells treated with different formulations for 48 h (Lapa/DOX concentration: 10 μM). **P < 0.01. Data were shown as mean ± SD (n = 3). |
To observe the synergistic effect of Lapa and DOX, the molar ratio of the two drugs in the LDNLC was established as 1:1. As displayed in Figure 6B, DNLC with only DOX showed the most inferior cytotoxicity in MCF-7 ADR cells, which might be due to the acquired MDR effect of DOX upon treatment with DNLC. In contrast, LNLC showed increased cytotoxicity compared to DNLC, which might be due to the Lapa-related NAD(P)H consumption and ROS surge that significantly triggered apoptosis within cells. As expected, those treated with LDNLC showed the most effective cytotoxicity in MCF-7 ADR cells due to the synergistic effect of Lapa and DOX, as mentioned previously. Further confirmation using Di as NQO1 inhibitor also revealed a moderate cytotoxic difference between DNLC and LNLC, which is in accordance with our prediction. This conclusion was also supported by Annexin-V FITC assay (Figure 6C).
To comprehensively analyze the synergy between Lapa and DOX, the combination index (CI) of their IC50 values was calculated according to previous reports,15,36 applying the following formula: CI = IC50 (DOX in LDNLC)/IC50 (DOX) + IC50 (Lapa in LDNLC)/IC50 (Lapa). When the ratio (CI) is 1 the effect is considered additive; when the CI is significantly greater than or less than 1, the effect is considered subadditive (negative synergism) or supra-additive (positive synergism). The IC50 for MCF-7 ADR cells for LDNLC was calculated as 7.26 μM with a CI of 0.31, thus indicating a remarkable synergism in the drug-resistant strain. The synergy between the two chemotherapy drugs in MCF-7 ADR cells suggested that Lapa might minimize drug resistance in cancer cells.
In vivo distribution
The in vivo distribution of DiR-loaded LDNLC was investigated using a noninvasive real-time fluorescence imaging machine. As displayed in Figure 7A, free DiR showed quick accumulation at the tumor tissue but was also rapidly eliminated as a function of time. In contrast, the LDNLC showed visible fluorescence at the tumor site when intravenously administered for merely 2 h. On the other hand, it was observed that the fluorescence signal at the tumor site increased as a function of time, which indicated that the PEG-modified NLC could preferably take advantage of the EPR effect of the tumor tissue to passively target the tumor tissue.
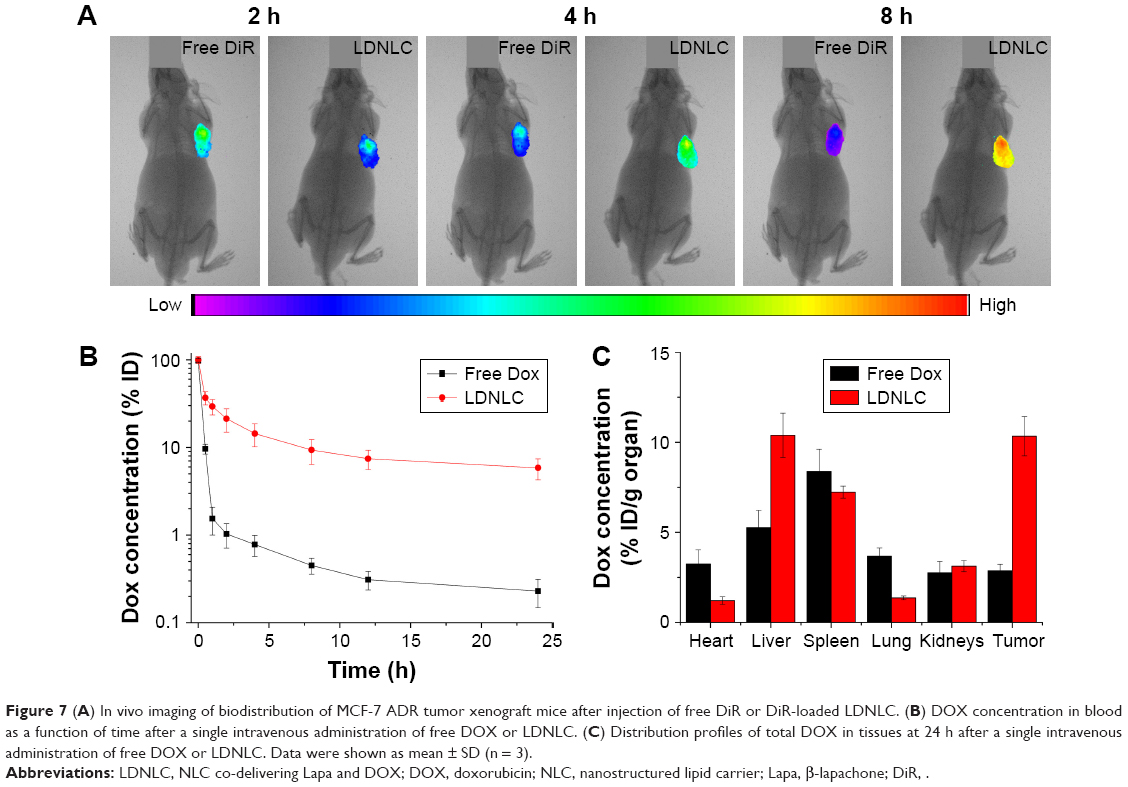 | Figure 7 (A) In vivo imaging of biodistribution of MCF-7 ADR tumor xenograft mice after injection of free DiR or DiR-loaded LDNLC. (B) DOX concentration in blood as a function of time after a single intravenous administration of free DOX or LDNLC. (C) Distribution profiles of total DOX in tissues at 24 h after a single intravenous administration of free DOX or LDNLC. Data were shown as mean ± SD (n = 3). |
Subsequently, we compared the in vivo pharmacokinetics of the LDNLC to those of free DOX. After a single intravenous injection of both formulations (DOX concentration: 5 mg/kg, n = 3), blood samples were withdrawn at different intervals and DOX concentrations in plasma were determined using HPLC as described previously. As illustrated in Figure 7B, due to PEG modification, LDNLC displayed a much longer blood circulation half-life than free DOX, with a 5.3 times increase in area under the concentration curve. At 8 h after injection, nearly 9.34% of the injected LDNLC remained in the plasma compared with only 0.45% of the free DOX. In addition to this, the drug distribution in main organs and tumors was also determined. As shown in Figure 7C, free DOX showed more accumulation in the heart than LDNLC, which raised the possibility of inducing severe cardiotoxicity, as reported by previous reports.37,38 In addition, LDNLC showed much more preferable accumulation in the tumor tissue compared to free DOX, which was almost fivefold of that in free DOX. This was beneficial for enhancing its anticancer effect in vivo.
In vivo anticancer efficiency
The in vivo therapeutic efficacy of the LDNLC with a DOX equivalent dose of 5 mg/kg and Lapa dose of 25 mg/kg was compared with that of DNLC, LNLC, and saline in an MCF-7 ADR tumor xenograft model. Once tumor volume reached 100 mm3, mice were randomly divided into four groups with six mice included in each one. The treatments were administered by intravenous injection every 2 days for a period of 14 days (total of seven treatments). Compared with the saline group, growth inhibition was observed in all drug-treated groups. However, due to the MDR in cancer cells, the anticancer efficacy of DNLC group was very limited. Unfortunately, LNLC showed similar efficacy to that of DNLC group due to the lack of effective anticancer drug. In contrast, the LDNLC group resulted in much better therapeutic efficacy (Figure 8A and B) than the other groups. An additional histological study was conducted by staining tumor tissue sections with H and E (Figure 8C). Compared with the tightly and regularly packed spherical cells in the saline (control) group, the cells treated with LDNLC swelled and exhibited severe vacuolization, significantly decreased cellularity, and shrinking nucleus, which were typical apoptotic characteristics. Vacuolization was also found in the DNLC and LNLC groups, but to a much lesser extent than the LDNLC group. The excellent in vivo therapeutic effect of the LDNLC against MDR tumors was well correlated with the in vitro cytotoxicity data and proved that the cascade drug release process and synergy of Lapa and DOX could overcome MDR.
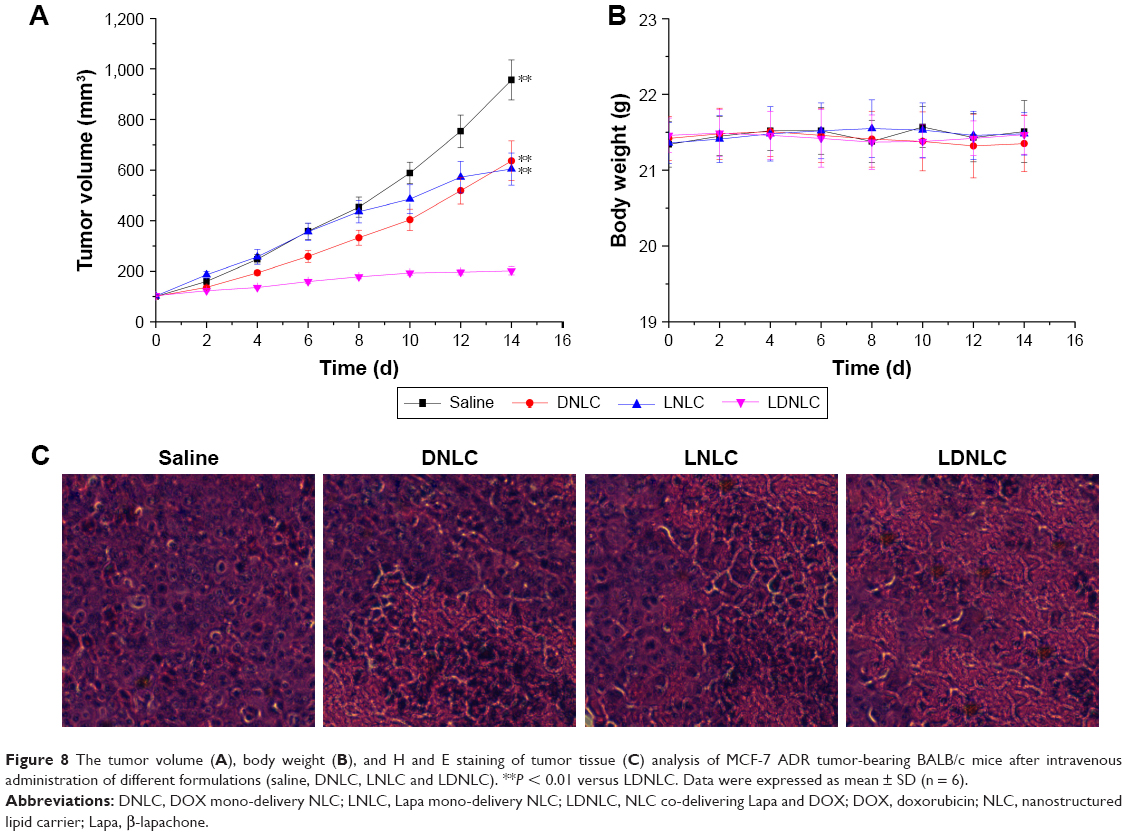 | Figure 8 The tumor volume (A), body weight (B), and H and E staining of tumor tissue (C) analysis of MCF-7 ADR tumor-bearing BALB/c mice after intravenous administration of different formulations (saline, DNLC, LNLC and LDNLC). **P < 0.01 versus LDNLC. Data were expressed as mean ± SD (n = 6). |
Conclusion
We successfully prepared NLCs co-delivering Lapa and DOX (LDNLC) with the aim to overcome MDR in breast cancer therapy. The well-designed LDNLC showed increased DOX retention as compared to DNLCs in MCF-7 ADR cells. In vivo anticancer assays on MCF-7 ADR tumor-bearing mouse model also revealed significantly enhanced efficacy of LDNLC. The LDNLC might be a promising platform toward overcoming MDR for enhanced breast cancer therapy.
Disclosure
The authors report no conflicts of interest in this work.
References
Verma S, Miles D, Gianni L, et al. Trastuzumab emtansine for HER2-positive advanced breast cancer. N Engl J Med. 2012;368(19):1783–1791. | ||
Liu F, Liu S, He S, et al. Survivin transcription is associated with P-glycoprotein/MDR1 overexpression in the multidrug resistance of MCF-7 breast cancer cells. Oncol Rep. 2010;23(5):1469–1475. | ||
Chen H, Tian J, Liu D, He W, Guo Z. Dual aptamer modified dendrigraft poly-l-lysine nanoparticles for overcoming multi-drug resistance through mitochondrial targeting. J Mater Chem B. 2017;5(5):972–979. | ||
Kankala RK, Tsai PY, Kuthati Y, et al. Overcoming multidrug resistance through co-delivery of ROS-generating nano-machinery in cancer therapeutics. J Mater Chem B. 2017;5(7):1507–1517. | ||
Zhang M, Jing S, Zhang J, et al. Intracellular release of PluronicL64 unimers into MCF-7/ADR cells to overcome multidrug resistance by surface-modified PAMAM. J Mater Chem B. 2017;5(21):3970–3981. | ||
Abraham I, Jain S, Wu CP, et al. Marine sponge-derived sipholane triterpenoids reverse P-glycoprotein (ABCB1)-mediated multidrug resistance in cancer cells. Biochem Pharmacol. 2010;80(10):1497–1506. | ||
Zhao D, Ma S, Yi X, et al. Reversible core-crosslinked nanocarriers with pH-modulated targeting and redox-controlled drug release for overcoming drug resistance. J Mater Chem B. 2017;5(42):8399–8407. | ||
Shiraki N, Okamura K, Tokunaga J, et al. Bromocriptine reverses P-glycoprotein-mediated multidrug resistance in tumor cells. Jpn J Cancer Res. 2002;93(2):209–215. | ||
Wang J, Wang F, Li F, et al. A multifunctional poly(curcumin) nanomedicine for dual-modal targeted delivery, intracellular responsive release, dual-drug treatment and imaging of multidrug resistant cancer cells. J Mater Chem B. 2016;4(17):2954–2962. | ||
Xiao-ai L, Bei W, Xiao-hong X, et al. Curcumin re-sensitizes multidrug resistant (MDR) breast cancer to cisplatin through inducing autophagy by decreasing CCAT1 expression. RSC Advances. 2017;7(53):33572–33579. | ||
Ma X, Huang X, Moore Z, et al. Esterase-activatable β-lapachone prodrug micelles for NQO1-targeted lung cancer therapy. J Control Release. 2015;200:201–211. | ||
Choi BT, Cheong J, Choi YH. Beta-Lapachone-induced apoptosis is associated with activation of caspase-3 and inactivation of NF-kappaB in human colon cancer HCT-116 cells. Anticancer Drugs. 2003;14(10):845–850. | ||
Planchon SM, Wuerzberger S, Frydman B, et al. Beta-lapachone-mediated apoptosis in human promyelocytic leukemia (HL-60) and human prostate cancer cells: a p53-independent response. Cancer Res. 1995;55(17):3706–3711. | ||
Zhang L, Chen Z, Yang K, et al. β-Lapachone and paclitaxel combination micelles with improved drug encapsulation and therapeutic synergy as novel nanotherapeutics for NQO1-targeted cancer therapy. Mol Pharm. 2015;12(11):3999–4010. | ||
Ye M, Han Y, Tang J, et al. A tumors-specific cascade amplification drug release nanoparticle for overcoming multidrug resistance in cancers. Adv Mater. 2017;29(38). | ||
Wang C, Chen S, Yu Q, Hu F, Yuan H. Taking advantage of the disadvantage: employing the high aqueous instability of amorphous calcium carbonate to realize burst drug release within cancer cells. J Mater Chem B. 2017;5(11):2068–2073. | ||
Zhao Z, Lou S, Hu Y, Zhu J, Zhang C. A nano-in-nano polymer-dendrimer nanoparticle-based nanosystem for controlled multidrug delivery. Mol Pharm. 2017;14(8):2697–2710. | ||
Vogus DR, Pusuluri A, Chen R, Mitragotri S. Schedule dependent synergy of gemcitabine and doxorubicin: improvement of in vitro efficacy and lack of in vitro-in vivo correlation. Bioeng Transl Med. 2018;3(1):49–57. | ||
Hu FQ, Jiang SP, Du YZ, et al. Preparation and characteristics of monostearin nanostructured lipid carriers. Int J Pharm. 2006;314(1):83–89. | ||
Yuan H, Wang LL, Du YZ, et al. Preparation and characteristics of nanostructured lipid carriers for control-releasing progesterone by melt-emulsification. Colloids Surf B Biointerfaces. 2007;60(2):174–179. | ||
Iqbal MA, Md S, Sahni JK, et al. Nanostructured lipid carriers system: recent advances in drug delivery. J Drug Target. 2012;20(10):813–830. | ||
Weber S, Zimmer A, Pardeike J. Solid lipid nanoparticles (SLN) and nanostructured lipid carriers (NLC) for pulmonary application: a review of the state of the art. Eur J Pharm Biopharm. 2014;86(1):7–22. | ||
Wang Y, Zhang H, Hao J, et al. Lung cancer combination therapy: co-delivery of paclitaxel and doxorubicin by nanostructured lipid carriers for synergistic effect. Drug Deliv. 2016;23(4):1398–1403. | ||
Bai J, Xu J, Zhao J, Zhang R. Hyaluronan and calcium carbonate hybrid nanoparticles for colorectal cancer chemotherapy. Mater Res Express. 2017;4(9):095401. | ||
Xia Q, Li L, Zhao L. Silica nanoparticle-based dual-responsive nanoprodrug system for liver cancer therapy. Exp Ther Med. 2017;14(3):2071–2077. | ||
Tang D, Zhao X, Yang T, Wang C. Paclitaxel prodrug based mixed micelles for tumor-targeted chemotherapy. RSC Adv. 2018;8(1):380–389. | ||
Zheng T, Wang A, Hu D, Wang Y. Tumor-targeting templated silica nanoparticles as a dual-drug delivery system for anti-angiogenic ovarian cancer therapy. Exp Ther Med. 2017;14(3):2162–2170. | ||
Wan J, Wu W, Zhang R, Liu S, Huang Y. Anti-EGFR antibody conjugated silica nanoparticles as probes for lung cancer detection. Exp Ther Med. 2017;14(4):3407–3412. | ||
Wang C, Bao X, Ding X, et al. A multifunctional self-dissociative polyethyleneimine derivative coating polymer for enhancing the gene transfection efficiency of DNA/polyethyleneimine polyplexes in vitro and in vivo. Polym Chem. 2015;6(5):780–796. | ||
Abbad S, Wang C, Waddad AY, Lv H, Zhou J. Preparation, in vitro and in vivo evaluation of polymeric nanoparticles based on hyaluronic acid-poly (butyl cyanoacrylate) and D-alpha-tocopheryl polyethylene glycol 1000 succinate for tumor-targeted delivery of morin hydrate. Int J Nanomedicine. 2015;10:305–320. | ||
Wang C, Li M, Yang T, et al. A self-assembled system for tumor-targeted co-delivery of drug and gene. Mater Sci Eng C Mater Biol Appl. 2015;56:280–285. | ||
Ding X, Xu X, Zhao Y, et al. Tumor targeted nanostructured lipid carrier co-delivering paclitaxel and indocyanine green for laser triggered synergetic therapy of cancer. RSC Adv. 2017;7(56):35086–35095. | ||
Dong X, Mattingly CA, Tseng MT, et al. Doxorubicin and paclitaxel-loaded lipid-based nanoparticles overcome multidrug resistance by inhibiting P-glycoprotein and depleting ATP. Cancer Res. 2009;69(9):3918–3926. | ||
Wu M, Meng Q, Chen Y, et al. Large pore-sized hollow mesoporous organosilica for redox-responsive gene delivery and synergistic cancer chemotherapy. Adv Mater. 2016;28(10):1963–1969. | ||
Shiah SG, Chuang SE, Chau YP, Shen SC, Kuo ML. Activation of c-Jun NH2-terminal kinase and subsequent CPP32/Yama during topoisomerase inhibitor beta-lapachone-induced apoptosis through an oxidation-dependent pathway. Cancer Res. 1999;59(2):391–398. | ||
Calviello G, Di Nicuolo F, Serini S, et al. Docosahexaenoic acid enhances the susceptibility of human colorectal cancer cells to 5-fluorouracil. Cancer Chemother Pharmacol. 2005;55(1):12–20. | ||
Cheng C, Zhang X, Meng Y, Chen L, Zhang Q. Development of a dual drug-loaded hydrogel delivery system for enhanced cancer therapy: in situ formation, degradation and synergistic antitumor efficiency. J Mater Chem B. 2017;5(43):8487–8497. | ||
Xu L, Zhao M, Yang Y, et al. A reactive oxygen species (ROS)-responsive low molecular weight gel co-loaded with doxorubicin and Zn(ii) phthalocyanine tetrasulfonic acid for combined chemo-photodynamic therapy. J Mater Chem B. 2017;5(46):9157–9164. | ||
Wang C, Chen S, Wang Y, et al. Lipase-Triggered Water-Responsive “Pandora’s Box” for Cancer Therapy: Toward Induced Neighboring Effect and Enhanced Drug Penetration. Advanced Materials. 2018;30(14):1706407. | ||
Bai J, Xu J, Zhao J, Zhang R. Hyaluronan and calcium carbonate hybrid nanoparticles for colorectal cancer chemotherapy. Materials Research Express. 2017;4(9):095401. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.