")
Back to Journals » International Journal of Nanomedicine » Volume 14
Nanosome-Mediated Delivery Of Protein Kinase D Inhibitor Protects Chondrocytes From Interleukin-1β-Induced Stress And Apoptotic Death
Authors Cho H, Bhatti FUR, Hasty KA, Yi AK
Received 10 June 2019
Accepted for publication 30 August 2019
Published 11 November 2019 Volume 2019:14 Pages 8835—8846
DOI https://doi.org/10.2147/IJN.S218901
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Hongsik Cho,1–3,* Fazal-Ur-Rehman Bhatti,1,3,* Karen A Hasty,1–3 Ae-Kyung Yi4
1Department of Orthopaedic Surgery and Biomedical Engineering, The University of Tennessee Health Science Center, Memphis, TN, USA; 2Department of Orthopaedic Surgery, Campbell Clinic, Memphis, TN, USA; 3151 Research Service, Veterans Affairs Medical Center, Memphis, TN, USA; 4Department of Microbiology, Immunology and Biochemistry, The University of Tennessee Health Science Center, Memphis, TN, USA
*These authors contributed equally to this work
Correspondence: Hongsik Cho
Department of Orthopaedic Surgery and Biomedical Engineering, The University of Tennessee Health Science Center, Research 151, VAMC, 1030 Jefferson Ave, Memphis TN 38104 USA
Tel +1 901 523-8990 (Ext: 6456)
Fax +1 901 577-7273
Email [email protected]
Ae-Kyung Yi
Department of Microbiology, Immunology and Biochemistry, The University of Tennessee Health Science Center, 858 Madison Ave., Suite 501C, Memphis, TN 38163, USA
Tel +1 901 448-1775
Fax +1 901 448-7360
Email [email protected]
Background: Inflammatory stress caused by protein kinase D (PKD) plays a critical role in damaging chondrocytes and extracellular matrix (ECM) during osteoarthritis (OA). The PKD inhibitor (PKDi) (CRT0066101) has been used to overcome inflammation in different cell types. However, the efficacy of a therapeutic drug can be limited due to off-target distribution, slow cellular internalization, and limited lysosomal escape. In order to overcome this issue, we developed nanosomes carrying CRT0066101 (PKDi-Nano) and tested their efficacy in vitro in chondrocytes.
Methods: Chondrocytes were subjected to IL-1β-induced inflammatory stress treated with either PKDi or PKDi-Nano. Effects of treatment were measured in terms of cytotoxicity, cellular morphology, viability, apoptosis, phosphorylation of protein kinase B (Akt), and anabolic/catabolic gene expression analyses related to cartilage tissue.
Results and Discussion: The effects of PKDi-Nano treatment were more pronounced as compared to PKDi treatment. Cytotoxicity and apoptosis were significantly reduced following PKDi-Nano treatment (P < 0.001). Cellular morphology was also restored to normal size and shape. The viability of chondrocytes was significantly enhanced in PKDi-Nano-treated cells (P < 0.001). The data indicated that PKDi-Nano acted independently of the Akt pathway. Gene expression analyses revealed significant increases in the expression levels of anabolic genes with concomitant decreases in the level of catabolic genes. Our results indicate that PKDi-Nano attenuated the effects of IL-1β via the nuclear factor kappa-light-chain enhancer of activated B cells (NF-κB) pathway.
Conclusion: Taken together, these results suggest that PKDi-Nano can be used as a successful strategy to reduce IL1β-induced inflammatory stress in chondrocytes.
Keywords: CRT0066101, liposome, cartilage, osteoarthritis, cytokine
Introduction
Osteoarthritis (OA) is a disease of the articular joints characterized by the phenotypic changes in chondrocytes, loss of cartilage tissue, synovial inflammation, and the formation of osteophytes.1 There are multiple risk factors that can lead to the hypertrophy of chondrocytes that include, age, weight, gender, and other joint abnormalities.2 These risk factors trigger inflammatory mediators, such as cytokines and chemokines that are seen to be elevated within the synovial fluid of patients with OA.3
The homeostasis of normal cartilage is maintained by resident chondrocytes that provide a balance between the production of extracellular matrix (ECM) components and catabolic factors. OA begins with early focal lesions in the articular cartilage catalyzed by proinflammatory mediators that are released from hypertrophic ike chondrocytes. This balance is altered in osteoarthritis (OA) due to apoptosis of chondrocytes and inflammation. The overall result is the degradation of cartilage and joint inflammation.4 Thus, OA is characterized by loss of structural integrity and physiological failure of the synovial joints.5 Moreover, OA not only degrades the cartilage tissue but also results in the destruction of other synovial tissues such as subchondral bone, ligaments, synovium, menisci, capsule, and periarticular muscles.6
The apoptosis of chondrocytes in OA is evident by empty lacunae and hypocellularity accompanied by the production of reactive oxygen species (ROS), degradation of ECM, mechanical injury, and decreased the production of growth factors by chondrocytes.7–9 Oxidative stress due to aging increases the production of ROS, resulting in the loss of ECM with a concomitant increase in the production of chemokines, cytokines, and matrix metalloproteinases (MMPs) causing cell death leading to OA. Thus, aging plays a key etiological role in the early development of OA.10 Moreover, the interaction between ECM and chondrocytes is vital for cell survival. Thus, damage to ECM components due to trauma or mechanical load can cause apoptosis of chondrocytes that in turn cause further cause breakdown of ECM.11,12 The key mediators during apoptosis of chondrocytes in OA are caspases.13 Hence, the inhibition of caspases, such as caspase-3 activity, can be utilized to prevent cell death in OA.14–16
The inflammatory mediators in OA released by infiltrating immune cells and chondrocytes promote degradation of the ECM.17 The two most prominent cytokines interleukin 1β (IL-1β) and tumor necrosis factor-α (TNFα) play key inflammatory roles in the destruction of joint architecture.18,19 The osteoarthritic chondrocytes under the effect of IL-1β produce matrix metalloproteinase-13 (MMP-13) causing the degradation of the most prevalent collagen type II α1 (COL2A1) in the cartilage tissue, leading to irreversible loss of the joint tissue.20 The irreversible degradation of COL2A1 is a major early event in the progression of OA.21 Moreover, the induction of MMP13 production by IL-1β requires the transcription factor nuclear factor κ-light-chain enhancer of activated B cells (NF-κB).5 Altogether, the inflammatory mediators form a cascade that results in loss of cartilage and irreversible damage to the joint tissue.
PKD is a family of three serine/threonine kinase isoforms. The events that lead to the activation of PKD during oxidative stress include direct binding of diacylglycerol (DAG), phosphorylation, and caspase-mediated proteolytic cleavage.22,23 The activated PKD has been reported to activate multiple signaling pathways such as the NF-κB pathway.24 It has been reported that members of protein kinase C (PKC) modulate the expression of MMPs via a PKD-dependent manner.25 Consequently, inhibition of PKD has been reported to reduce the expression of MMPs and promote cartilage repair.26 Expression and activation of MMPs can be modulated by ligands of Toll-like receptors (TLRs) and IL-1 receptors.27,28 Previously, we have found that PKD1 is activated by TLRs and IL-1Rs via a MyD88-dependent, but PKC-independent, manner. This plays a pivotal role in TLR/IL-1R-mediated NF-κB activation and subsequent expression of inflammatory mediators in leukocytes.22,23,29,30 These findings suggest the possibility that inhibition of PKD activity will have a protective effect to reduce the MMP production and catabolic activity in hypertrophic chondrocytes.
The current challenge to treat OA through pharmacological intervention is limited by the lack of blood circulation because of the avascular nature and poor self-repair capacity of articular cartilage.31 This nanosome formulation aids in overcoming these barriers by providing stability to the drug, increasing cellular and tissue uptake, improving biodistribution, enhancing specificity to the target site, and minimizing side effects.32 In our previous studies, we have developed targeted nanosomes containing therapeutic agents that aided in targeted drug delivery in the mouse knee joint. We showed that our nanosomes can efficiently deliver the therapeutic drug.33,34 Therefore, in this study, we investigated whether a PKD inhibitor (CRT0066101) treatment can protect the cultured porcine chondrocytes against the catabolic activity of IL-1β and whether nanosome-mediated delivery of the therapeutic drug to chondrocytes potentiate drug efficacy.
Methods
Preparation Of PKD Inhibitor Nanosomes
The nanosome synthesis was performed as described previously with slight modifications.33,34 All lipids were dissolved in chloroform (Avanti, Alabaster, AL, USA). A lipid film was prepared using a molar ratio of 52:45:2.9:0.1 for 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC), cholesterol, 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethyleneglycol)-2000] (DSPE-PEG 2000), and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[maleimide (polyethylene glycol) 2000] (DSPE-PEG 2000-maleimide), respectively. The protein kinase D (PKD) inhibitor, 2-[4-[[(2R)-2-aminobutyl]amino]-2-pyrimidinyl]-4-(1-methyl-1H-pyrazol-4-yl)phenol dihydrochloride (CRT 0066101) (TOCRIS, Minneapolis, MN, USA), was added to the lipid mixture at a concentration of 1 mM and dried under nitrogen gas and vacuum. The film was rehydrated with 1× phosphate buffered saline (PBS) (Gibco, Minneapolis, MN, USA). A 200 nm porous membrane was used to extrude the rehydrated film to generate nanosomes with a mean diameter of 200 nm and 1–3 lamellar membranes (Supplement Figure 3). The size exclusion chromatography was done using the Sepharose CL-4B (Sigma-Aldrich, St. Louis, MO, USA) exclusion column to separate the extruded nanosomes from free molecules. The PKD inhibitor nanosomes were stored at 4°C until further use.
Chondrocyte Culture And Treatment
The primary chondrocytes were harvested from the articular cartilage of the femoral condyles of 3–4-month-old domestic pigs euthanized according to the approved protocol and the ethical guidelines of Institutional Animal Care and Use Committee (IACUC) at the University of Tennessee Health Science Center (UTHSC) (animal protocol number: 17-092.0, approval date: 11/14/2017). The pieces of the articular cartilage were dissected from the femoral condyles, minced in a sterile hood, and digested at 37°C and 5% CO2 in 0.05% Pronase solution (Boehringer Mannheim, Mannheim, Germany) for 2 hrs followed by overnight digestion at 37°C and 5% CO2 in 0.2% collagenase solution (Worthington Biologicals, NJ, USA). The digestion of cartilage was followed by centrifugation at 1000 rpm for 10 mins to yield a chondrocyte pellet that was suspended in modified F-12K medium (Invitrogen Carlsbad, CA, USA) supplemented with 10% fetal calf serum (FCS) (Atlanta Biologicals, Flowery Branch, GA, USA), 50 μg/mL streptomycin (Thermo Fisher Scientific, Waltham, MA, USA), 50 IU/mL penicillin G (Thermo Fisher Scientific), 2 mM L-glutamine (Thermo Fisher Scientific), and 50 μg/mL ascorbic acid (Thermo Fisher Scientific). The cells were seeded in 100 mm petri dish at a density of 20 × 106 cells/mL and incubated at 37°C in a humidified atmosphere of 5% CO2. The medium was changed every other day until the cells reached confluence. Cells at the first passage (P1) were used for subsequent experiments. Chondrocytes at P1 were randomly divided into the four groups and treated as illustrated in Table 1.
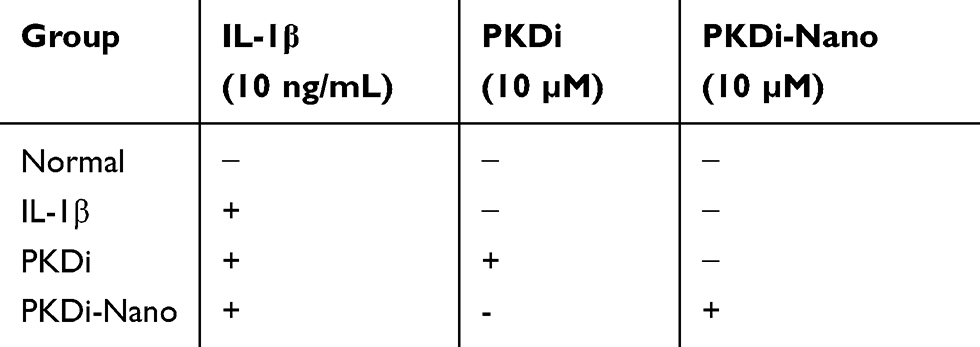 |
Table 1 Scheme Of Study Showing Control And Experimental Groups |
Evaluation Of Cellular Morphology, Live/Dead Stain, And Trypan Blue Exclusion Assay
Microscopic images (n=15 per group) were captured using the Invitrogen™ EVOS™ FL Auto Imaging System (Invitrogen) at 10× magnification. The bright-field images were used to evaluate the cell shape and number following treatment of chondrocytes. The viability of treated chondrocytes was examined by live/dead stain using the Live and Dead Cell Assay Kit (Abcam, Cambridge, MA, USA) according to the manufacturer’s instructions.38 The results of live/dead stain were visualized by fluorescence microscopy and quantified by flow cytometry. A minimum of 50,000 events/samples were collected on a BD Biosciences LSR II flow cytometer (BD Biosciences, San Diego, CA, USA) and analyzed with FlowJo flow cytometry data analysis software (FlowJo LLC, Ashland, OR, USA). Trypan blue exclusion assay was performed to quantify the percentage of live cells following treatment.
Biochemical Analyses Following Treatment Of Chondrocytes
The biochemical analyses were performed using the media collected following treatment of chondrocytes. The biochemical analyses included the following assays:
Lactate Dehydrogenase (LDH) Assay
The In Vitro Toxicology Assay Kit (Sigma-Aldrich) was used to determine cellular toxicity according to manufacturer’s instructions. Absorbance was read at 490 nm and at 690 nm (background) using SpectraMax M5 microplate reader (Molecular Devices, San Jose, CA USA). The LDH release in media per group was expressed as %control that represents untreated cells lyzed by 1% Triton X-100 (Bio-Rad, CA, USA).
Total Nitric Oxide (NO) Assay
The total NO levels in media were measured using the Nitrate/Nitrite Fluorometric Assay Kit according to the manufacturer’s instructions (Cayman Chemical Company, Ann Arbor, MI, USA) to determine oxidative stress. The fluorescence was read at 375 nm excitation and 417 nm emission using the SpectraMax M5 microplate reader. The nitrate standard curve was used to determine the concentration of total NO in each sample.
Amplex Red Assay
The levels of hydrogen peroxide (H2O2) in media were evaluated using the Amplex™ Red Hydrogen Peroxide/Peroxidase Assay Kit (Thermo Fisher Scientific) according to the manufacturer’s instructions. The fluorescence was read at 560 nm excitation and 590 nm emission using the SpectraMax M5 microplate reader. The level of H2O2 is presented in terms of fluorescence units.
Caspase-3 Activity Assay
Apoptosis was assessed in cell lysates using the EnzChek® Caspase-3 Assay Kit (Molecular Probes™, OR, USA). The fluorescence was read using SpectraMax M5 microplate reader at excitation and emission wavelengths of 496 and 520 nm, respectively. The Caspase-3 activity was presented in terms of fluorescence units.
Prostaglandin E2 (PGE2) Assay
The level of PGE2 in the medium was examined using the Parameter PGE2 Immunoassay kit (R&D Systems, Minneapolis, MN, USA) according to the manufacturer’s instructions to determine inflammation. The absorbance was measured using the SpectraMax M5 microplate reader at 450 nm. The wavelength correction was made at 570 nm. The PGE2 standard curve was drawn to determine the final concentration of PGE2 in each sample.
Total Akt/phospho-Akt Enzyme-Linked Immunosorbent Assay (ELISA)
The levels of total Akt to phospho-Akt in cell lysates were determined using Akt (pS473) + total Akt ELISA Kit according to the manufacturer’s instructions (Abcam). Absorbance was read at 450 nm and wavelength correction at 690 nm using the SpectraMax M5 microplate reader.
Real-Time RT-qPCR For Gene Expression Analysis
The RNA was extracted following treatment of chondrocytes using the GeneJET RNA Purification Kit (Thermo Fisher Scientific). The cDNA was prepared using 0.5 µg RNA by TaqMan® Reverse Transcription Reagents (Thermo Fisher Scientific). Semiquantitative gene expression (qPCR) was done using TaqMan™ Gene Expression Assays (Thermo Fisher Scientific) and Roche LC480 instrument (Roche, Basel, Switzerland) for the following genes Aggrecan (ACAN), Collagen type II alpha 1 (COL2A1), SRY (sex-determining region Y)-Box 9 (SOX9), nuclear factor kappa B (NFκB), matrix metalloproteinase 13 (MMP13), tumor protein P53 (p53) and Caspase 3 (Casp3). To calculate the data as 2−(ΔΔCT), the expression levels of target genes were normalized to the housekeeping gene.35
Statistical Analysis
The GraphPad Prism v.5.00 for Windows (GraphPad Software, San Diego, CA, USA, http://www.graphpad.com) was used to perform statistical analysis. The quantitative data were expressed as mean ± SD (n=5). Statistical analysis was performed by one-way ANOVA followed by Bonferroni’s post hoc comparison test for the comparison of group mean differences against the IL-1β-treated group. Student’s t-test was done for unpaired comparison. A P value of ≤0.05 was considered statistically significant. All the experiments were performed in triplicate and repeated thrice.
Results
PKDi-Nano Reduces IL-1β-Induced Cytotoxicity, Cellular Morphology Change, And Death Of Chondrocytes
To evaluate the effects of a PKD inhibitor on IL-1β-induced chondrocyte death and to compare the efficacy of nanosome-mediated delivery of a small molecule inhibitor to chondrocytes, porcine primary chondrocytes were treated with IL-1β, a pathogenic inflammatory mediator in OA, in the presence or absence of a PKD inhibitor (CRT0066101; PKDi) or nanosomes containing CRT0066101 (PKDi-Nano). Lactate dehydrogenase (LDH) release in the medium, an indication of cellular toxicity, was analyzed. The normal chondrocytes released low levels of LDH (5.19 ± 0.07%) (Figure 1A). The levels of LDH release were significantly increased following IL-1β treatment compared to the normal untreated chondrocytes (Supplement Figure 1). LDH release after IL-1β stimulation was significantly reduced by treatment with either PKDi or PKDi-Nano compared to the PBS-treated group. In addition, PKDi-Nano treatment significantly decreased LDH levels compared to PKDi treatment.
The normal chondrocytes showed the spindle-shaped morphology and high levels of viability (98 ± 0.97%) (Figure 1B–E). A substantial number of chondrocytes lost their normal spindle-shaped morphology after IL-1β treatment (Figure 1C). The death of chondrocytes was also significantly increased following IL-1β treatment compared to the normal untreated chondrocytes (Figure 1B, D and E). PKDi at a concentration of 10 µM did not significantly prevent chondrocytes from undergoing IL-1β-induced morphology change and death. In contrast, chondrocytes treated with IL-1β in the presence of PKDi-Nano maintained the size and shape that are identical with normal chondrocytes. In addition, the viability of IL-1β-treated chondrocytes was significantly increased in the presence of PKDi-Nano. Compared to the PKDi plus IL-1β-treated group, chondrocyte viability of PKDi-nano plus IL-1β-treated group was significantly higher. Viability of PKDi-Nano plus IL-1β-treated chondrocytes was not significantly different from the viability of untreated normal chondrocytes.
Reduced Apoptosis And Akt Activation Following PKDi Or PKDi-Nano Treatment Of Chondrocytes
To evaluate the effect of IL-1β and PKDi on apoptotic activity, caspase-3 assays were conducted. Caspase-3 activity was significantly increased by IL-1β treatment as compared to the normal group (Figure 2A). Treatment with PKDi and PKDi-Nano significantly reduced the IL-1β-induced caspase-3 activity. However, a significant decrease in caspase-3 activity was observed for PKDi-Nano-treated cells as compared to the PKDi-treated cells. Caspase-3 activity in PKDi-Nano plus IL-1β-treated chondrocytes were comparable to that in the untreated chondrocytes.
IL-1β activates Akt through a PI3K-dependent manner.36,37 Akt is a serine/threonine kinase that is known to promote cell survival.38 Therefore, we investigated whether IL-1β could affect the Akt pathway and whether PKDi could impair IL-1β-mediated signaling, leading to Akt activation in chondrocytes. The level of phosphorylated Akt, as an indication of Akt activation, increased significantly after IL-1β treatment as compared to the normal group (Figure 2B). This IL-1β-induced phosphorylation of Akt was significantly inhibited in the presence of either PKDi or PKDi-Nano. PKDi-Nano treatment more effectively inhibited IL-1β-mediated Akt activation compared to PKDi treatment. The levels of phosphorylated Akt in PKDi-Nano plus IL-1β-treated chondrocytes were comparable to those in the normal chondrocytes.
Effect Of PKDi And PKDi-Nano On Oxidative Stress In Chondrocytes
ROS production has been found to increase in osteoarthritic joints and has long been considered as the primary inducer of chondrocyte apoptosis.39 We investigated whether IL-1β induces ROS generation and whether PKDi can suppress ROS generation in chondrocytes. As compared to the normal chondrocytes, the levels of total NO and H2O2 production were significantly increased after treatment with IL-1β, indicating that IL-1β induces oxidative stress in chondrocytes (Figure 3A and B). To investigate the effects of the PKDi on IL-1β-induced ROS generation, we measured total NO level in media by a spectrophotometric assay based on the Griess reaction. The IL-1β-mediated NO production was substantially inhibited in the presence of PKDi, but the level of reduction was not statistically significant (Figure 3A). However, PKDi-Nano treatment significantly reduced the level of total NO production in IL-1β-treated chondrocytes. Treatment with either PKDi or PKDi-Nano significantly reduced the H2O2 generation in the IL-1β-treated chondrocytes (Figure 3B). No significant difference between the H2O2 levels was found among PKDi plus IL-1β-treated group, PKDi-Nano plus IL-1β-treated group, and the untreated normal group.
Effect Of PKDi And PKDi-Nano Treatment On Inflammation In Chondrocytes
To evaluate the ability of PKDi to prevent inflammatory activity in chondrocytes that catabolized by IL-1β, we measured PGE2 levels in media. The levels of PGE2 were significantly increased in the IL-1β-treated group as compared to the normal group (Figure 3C). Treatment with either PKDi or PKDi-Nano significantly reduced the levels of PGE2 production in IL-1β-treated chondrocytes. In addition, the levels of IL-1β-induced PGE2 in the presence of PKDi-Nano were significantly reduced even further compared to those in the presence of PKDi. The levels of PGE2 production in IL-1β-treated chondrocytes in the presence of PKDi-Nano were not different with those in normal untreated chondrocytes. These results suggest that PKD activation is involved in inflammation mediated by IL-1β. Consequentially, it also shows that PKDi can be used to reduce IL-1β-mediated inflammation in chondrocytes.
Effect Of PKDi And PKDi-Nano Treatment On Expression Of Genes In IL-1β-Treated Chondrocytes
We performed real-time RT-qPCR to evaluate the anabolic and catabolic activity at the gene expression level in each treatment group. The expression level of genes that are associated with anabolism of cartilage tissue (ACAN, COL2A1, and SOX9) was significantly decreased in the IL-1β-treated chondrocytes as compared to those in the normal untreated chondrocytes (Figure 4A–C). These IL-1β-mediated suppressions of ACAN, COL2A1, and SOX9 gene expressions were substantially reverted by PKDi or PKDi-Nano. An increase in expression levels of those anabolic genes was observed in IL-1β-treated chondrocytes in the presence of PKDi or PKDi-Nano. The increase was more significant in the expression level of ACAN and COL2A1 genes in the PKDi-Nano group as compared to the PKDi group. Although no statistically significant difference was found between PKDi plus IL-1β-treated group and PKDi-Nano plus IL-1β-treated group on the SOX9 gene expression, there was a tendency of increased SOX9 gene expression in PKDi-Nano group compared to the PKDi group.
Expression levels of genes associated with inflammation (NFκB) and catabolism of cartilage (MMP13) showed significant increases following IL-1β treatment in chondrocytes as compared to the normal chondrocytes (Figure 3D and E). A decrease in gene expression levels of NFκB and MMP13 was observed following the addition of PKDi or PKDi-Nano as compared to IL-1β-only group (Figure 4D and E). The decrease was more significant in PKDi-Nano group as compared to the PKDi group. In addition to this, the gene expression level of molecules associated with apoptosis, p53, and Casp3 showed a significant increase in IL-1β-treated group as compared to the normal group. A decrease in gene expression levels of p53 and Casp3 genes was observed following treatment with PKDi or PKDi-Nano as compared to the IL-1β only group. The decrease was more significant in PKDi-Nano group as compared to PKDi group (Figure 4F and G).
Discussion
In this study, we investigated the inflammatory stress caused by PKD which plays a critical role in damaging the chondrocytes and their ECM during in vitro proinflammatory cytokine (IL-1β)-induced arthritic condition. Therefore, we hypothesized that PKDi (CRT 0066101) could be used to overcome this inflammation in an arthritic model of chondrocytes. The dose of 10 µM PKDi was selected based on our previous study and the PKDi-Nano was diluted to yield a PKDi concentration at 10 µM (Supplement Figure 2). We also evaluated the feasibility of targeted delivery of PKDi by nanosomes that may have more effective delivery and better efficacy for future in vivo applications. These nanosomes have no biosafety concerns under in vitro and in vivo conditions according to toxicity tests. Our results showed that PKDi-Nano efficiently attenuated inflammation and apoptosis against the stress induced by IL-1β in chondrocytes.
Inflammation and oxidative stress deteriorate the integrity of the cartilage tissue during OA via the release of various chemokines, cytokines, prostaglandins, growth factors, ROS, and reactive nitrogen species (RNS).40 The regulation of intracellular signaling by these factors causes chondrocyte senescence and apoptosis, degradation of ECM, synovial inflammation, and dysfunction of subchondral bone.41 NFκB signaling has been associated with the generation of ROS during oxidative stress.42 NFκB signaling pathway plays a critical role in the pathogenesis of OA. For instance, activation of NFκB under the influence of IL-1β causes destruction of cartilage ECM by the release of MMPs.43 Furthermore, the activation of PKD3 has been reported to play a significant role in the proinflammatory mechanism leading to the destruction of cartilage by the release of MMPs, while inhibition of PKD has been reported to attenuate the cartilage destruction in chondrocytes.25 Because of its role in inflammation, PKD has been a key interest in treating inflammatory diseases.23,28 PKD has been previously shown to be activated by toll-like receptors (TLR) and myeloid differentiation factor 88 (MyD88), components that play a major role in proinflammatory responses by macrophages.23,28 It has been shown in previous studies that inhibiting PKD can promote cartilage repair and could possibly be used as a drug for future study in OA therapy.25 However, the effect of utilizing a small molecule to inhibit PKD and its role in relation to NFκB signaling has not been explored in chondrocytes. Therefore, in this study we utilized the PKD inhibitor, CRT 0066101, to study its effect on NFκB signaling that in turn mediates inflammation and apoptosis in chondrocytes under IL-1β-induced stress. Moreover, the PKD inhibitor was encapsulated in nanosomes for effective delivery via interaction between nanosomes and chondrocytes.
The biochemical analyses revealed that the stress induced by IL-1β caused chondrocytes to undergo apoptosis and inflammatory stress. Cell viability following IL-1β-induced stress increased significantly from compared to control PBS upon treatment with PKD inhibitor. We assume that the restoration of cell viability following treatment with PKD inhibitor was in turn related to a decrease in cytotoxicity. Furthermore, PKD inhibitor treatment significantly decreased the IL-1β-induced increase in ROS and RNS as has been observed in this study and others.44 The apoptosis of chondrocytes following IL-1β stimulation was also seen to diminish following PKD inhibitor treatment.45,46 A previous study has reported that IL-1β-induced production of NO reduced the proliferation of chondrocytes via PGE2.47 We also observed the same phenomenon and in addition observed significant attenuation of this effect following treatment with PKD inhibitor. The results obtained from the gene expression analyses clearly showed that IL-1β induces degradation of the ECM components by an increase in the production of MMP13 that is stimulated by NFκB. Furthermore, the rate of apoptosis was significantly higher in IL-1β-stressed chondrocytes. The treatment with PKD inhibitor significantly attenuated the inflammatory and apoptotic effects of IL-1β.
We also studied the possibility of involvement of the Akt signaling pathway in the survival of chondrocytes under IL-1β stimulation.48,49 Levels of phospho Akt increased after IL-1β stimulation; however, concomitant treatment with PKD inhibitor decreased the level of phospho Akt, indicating that the mechanism by which PKD inhibitor confers resistance against IL-1β is not dependent on Akt signaling mechanism, but rather PKD might modulate IL-1β receptor signal transduction leading to Akt activation. Future studies are needed to explore the role of PKD in the pathways, leading to the activation of NFκB in chondrocytes. In cells of the human and mouse immune systems, we previously found that PKD1 is activated by all TLRs that utilize MyD88 as a signaling adaptor, IL-1R via a MyD88, IRAK-dependent mechanisms, and PKD1 which plays an indispensable role on TLR/IL-1R-mediated NF-κB activation and subsequent proinflammatory gene expression and production.23,29 In addition to IL-1R, TLR1 and TLR2 are also expressed in human normal healthy chondrocytes as well as degenerated chondrocytes in OA cartilage, and the cognate ligand binding to these TLRs can lead to proinflammatory cytokines including TNFα in human chondrocytes.50 In turn, TNFα can induce the expression of TLRs in human chondrocytes.50 These findings by Nordstrom et al imply that cartilage matrix/chondrocyte-derived danger signals or degradation products may activate TLRs leading to the production of inflammatory mediators. These inflammatory mediators in turn further upregulate expression of TLRs producing more proinflammatory mediators establishing a vicious feedforward cycle of joint inflammation and cartilage damage in OA. Taken together, previous findings by others, in addition to our findings from this study, support the notion that PKD might be a very effective target for OA therapy.
Although OA represents the most common form of arthritis in the aging population, there is no effective disease-modifying drug (DMD) for OA. In addition to the lack of an effective DMD, another major challenge in OA therapy is the difficulty of effective drug delivery to the chondrocytes in the joints. We have developed a monoclonal type II collagen antibody-targeted drug delivery system using nanometer-sized liposomes, called nanosomes, to enclose the therapeutic agent. Since the collagen type II is exposed in the damaged cartilage, these nanosomes bind specifically at the sites of cartilage damage lesion and effectively release their contents without unwanted side effects.34
In summary, to the best of our knowledge, this is the first study to show that treatment of chondrocytes with PKD inhibitor impedes the catabolic activity associated with cartilage destruction. Our results imply that the concomitant sustained anabolism observed in gene expression levels was the result of a reduction in the catabolic activity (Figure 5). Moreover, PKD inhibitor encapsulated in nanosomes produced more pronounced effects as compared to PKD inhibitor in solution. Thus, the delivery of PKD inhibitor via nanosomes is a more potent way of attenuating the stress induced by IL-1β in chondrocytes. Further studies exploring the signaling pathways and validating the findings of this study in animal models of OA will be beneficial and warranted. Collectively, the findings of this study demonstrate the effectiveness of a nanosome-mediated delivery of PKD inhibitor to increase cartilage anabolism and decrease its catabolism under inflammatory stress conditions that mimic OA conditions in vitro.
Conclusion
The present study indicated that the anti-inflammatory and antiapoptotic effects of PKDi-Nano are mediated in part via Akt and NF-κB signaling pathways. Further in vitro studies are needed to understand the key players in these pathways that are affected by PKDi-Nano treatment in chondrocytes. The findings of this study suggest that nanosomes can serve as an ideal drug delivery system to inhibit PKD. Thus, this will be helpful in designing pharmacological therapeutic in diseases that require cartilage repair such as OA.
Chondrocytes were treated at P1 for 24 hrs at 37°C and 5% CO2 in a humidified environment. The treatment was carried out in serum-free medium. The dose of 10 µM PKDi (CRT 0066101) was selected based on our previously published study.30 PKDi-Nano were diluted to yield a PKDi concentration at 10 µM. Recombinant porcine IL-1β (R&D Systems) was used to induce inflammatory stress in chondrocytes. The concentrations used in each group were determined from previous studies (Supplementary Figure 1 and 2).
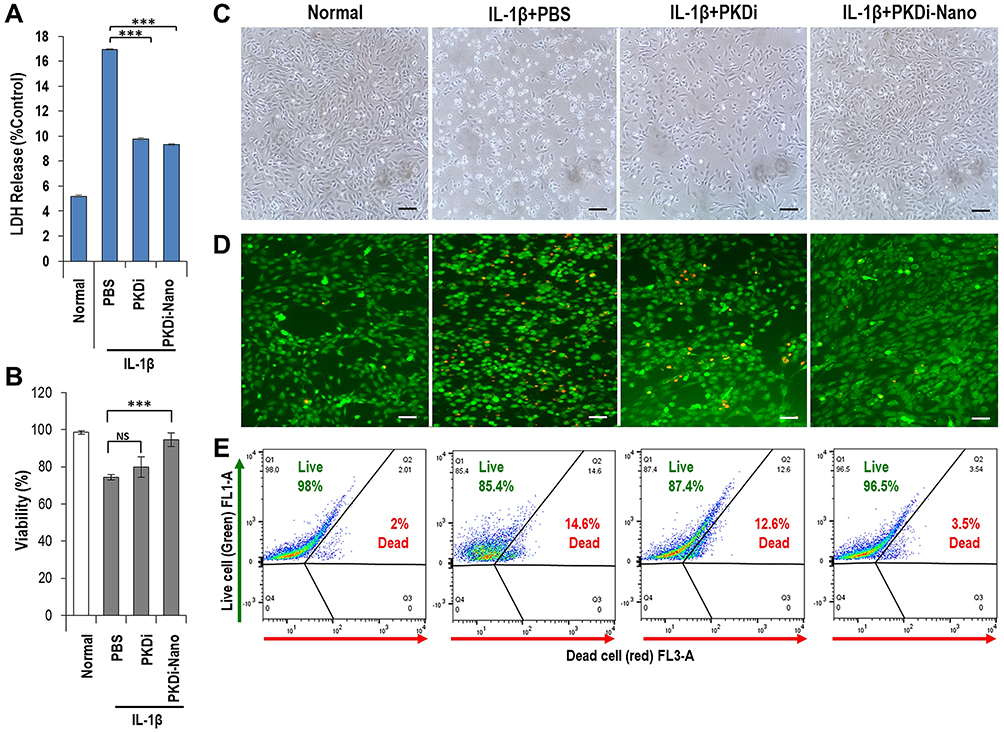 |
Figure 1 Cytotoxicity, cellular morphology, and viability. (A) Graphical representation of LDH release in the medium. Data are represented as mean ± SD (n=5). ***P < 0.001 vs PBS, (B) Trypan blue exclusion assay showing percentage of live cells. Data are represented as mean ± SD (n=5). ***P < 0.001 vs PBS, nsP > 0.05 vs PBS. (C) Morphology of chondrocytes in monolayer culture. Scale bar = 200 µm. (D) Live/dead stain fluorescence images. Live cells (green) dead cells (red). Scale bar = 200 µm. (E) Flow cytometry analysis after live/dead stain. Percentage of live cells is represented in Q1 and Q2, respectively. Data are represented as mean ± SD (n=5). |
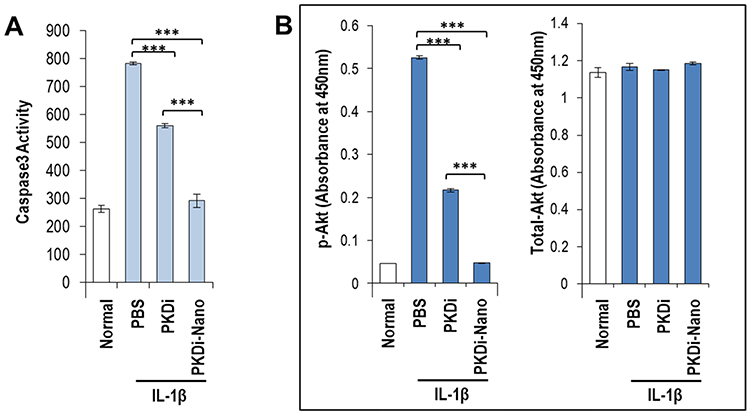 |
Figure 2 Quantification of apoptosis and phosphorylation of Akt. (A) Caspase-3 activity indicating apoptosis. (B) Comparison between total Akt to phospho Akt. Data are represented as mean ± SD (n=5). ***P < 0.001 vs PBS. |
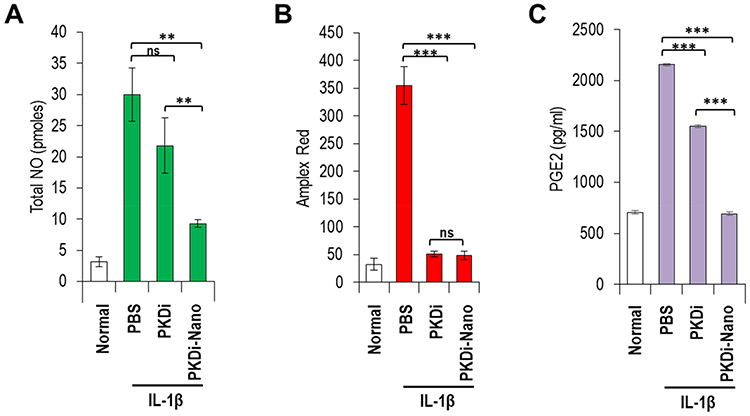 |
Figure 3 Biochemical analyses of treated chondrocytes. (A) Total NO assay demonstrating reactive nitrogen species. Data are represented as mean concentration (pmole) ± SD (n=5). **P < 0.01 vs PBS, nsP > 0.05 vs PBS. (B) Amplex Red assay indicating reactive oxygen species (H2O2). Data are represented as mean concentration (uM) or mean RFU? ± SD (n=5). ***P < 0.001 vs PBS, nsP > 0.05 vs PKDi. (C) PGE2 assay showing degree of inflammation. Data are represented as mean concentration (pg/mL) ± SD (n=5). ***P < 0.001 vs PBS. |
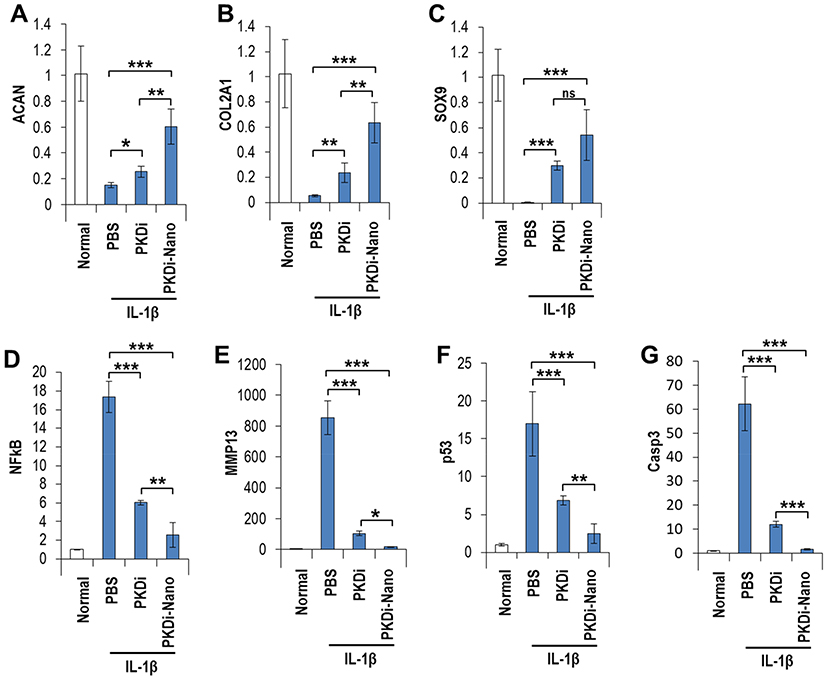 |
Figure 4 Gene expression analysis of treated chondrocytes. (A) Aggrecan (ACAN), (B) Collagen type II alpha 1 (COL2A1), (C) SRY (sex-determining region Y)-Box 9 (SOX9), (D) nuclear factor kappa B (NFκB), (E) matrix metalloproteinase 13 (MMP13), (F) tumor protein P53 (p53), and (G) caspase 3 (Casp3). Data are represented as mean of fluorescence level ± SD (n=5). ***P < 0.001 vs PBS, **P < 0.01 vs PBS, *P < 0.05 vs PBS, nsP > 0.05 vs PKDi. |
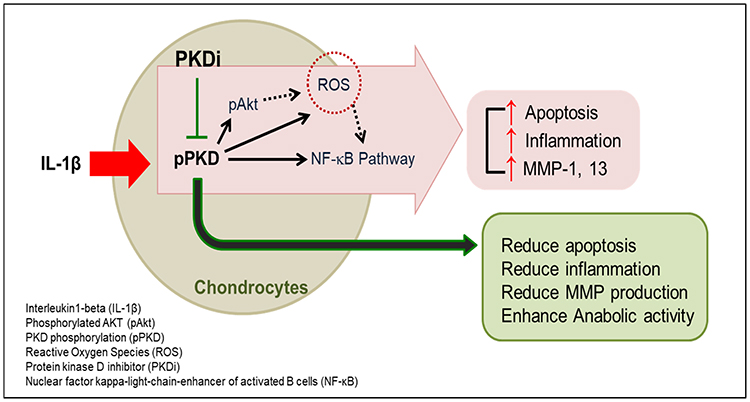 |
Figure 5 Hypothetical model of PKDi confers resistance against IL-1β-induced stress of chondrocytes. IL-1β induces activation of PKD. Activated PKD, in turn, contributes to activation of Akt, generation of ROS, and activation of NFκB. These lead chondrocytes to undergo apoptosis and inflammation that results in the degradation of ECM and hence OA. PKDi attenuates the deteriorating effects of IL-1β by inhibiting PKD activity in articular chondrocytes. |
Acknowledgment
The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of the Arthritis Foundation, Oxnard Foundation, NIH, Campbell Clinic, Department of Veterans Affairs, or University of Tennessee Health Science Center.
Author Contributions
All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
H.C. was supported by grants from the Arthritis Foundation (Discovery Award) and Oxnard Foundation (Medical Research). K.A.H. was supported by grants from the Department of Veterans Affairs (Merit Review award, Research Career Scientist Award). A.K.Y. was supported by grants from the Arthritis Foundation (Innovative Research Grant) and NIH (AR064723, AR069010). The authors declare no other conflicts of interest in this work.
References
1. Chen D, Shen J, Zhao W, et al. Osteoarthritis: toward a comprehensive understanding of pathological mechanism. Bone Res. 2017;5:16044. doi:10.1038/boneres.2016.44
2. Mandl LA. Osteoarthritis year in review 2018: clinical. Osteoarthritis Cartilage. 2019;27(3):359–364. doi:10.1016/j.joca.2018.11.001
3. Mathiessen A, Conaghan PG. Synovitis in osteoarthritis: current understanding with therapeutic implications. Arthritis Res Ther. 2017;19(1):18. doi:10.1186/s13075-017-1229-9
4. Wang X, Guo Y, Wang C, Yu H, Yu X, Yu H. MicroRNA-142-3p inhibits chondrocyte apoptosis and inflammation in osteoarthritis by targeting HMGB1. Inflammation. 2016;39(5):1718–1728. doi:10.1007/s10753-016-0406-3
5. Mengshol JA, Vincenti MP, Coon CI, Barchowsky A, Brinckerhoff CE. Interleukin-1 induction of collagenase 3 (matrix metalloproteinase 13) gene expression in chondrocytes requires p38, c-Jun N-terminal kinase, and nuclear factor KappaB: differential regulation of collagenase 1 and collagenase 3. Arthritis Rheum. 2000;43(4):801–811. doi:10.1002/1529-0131(200004)43:4<801::AID-ANR10>3.0.CO;2-4
6. Hunter DJ, Felson DT. Osteoarthritis. BMJ. 2006;332(7542):639–642. doi:10.1136/bmj.332.7542.639
7. Kim HA, Blanco FJ. Cell death and apoptosis in osteoarthritic cartilage. Curr Drug Targets. 2007;8(2):333–345. doi:10.2174/138945007779940025
8. Aigner T, Kim HA. Apoptosis and cellular vitality: issues in osteoarthritic cartilage degeneration. Arthritis Rheum. 2002;46(8):1986–1996. doi:10.1002/art.10554
9. Del Carlo M
10. Loeser RF. The role of aging in the development of osteoarthritis. Trans Am Clin Climatol Assoc. 2017;128:44–54.
11. Zemmyo M, Meharra EJ, Kuhn K, Creighton-Achermann L, Lotz M. Accelerated, aging-dependent development of osteoarthritis in alpha1 integrin-deficient mice. Arthritis Rheum. 2003;48(10):2873–2880. doi:10.1002/art.11246
12. Kunwar A, Kumar M, Singh S. Pathological perspective of chondrocyte apoptosis in osteoarthritis. J Orthopedics Traumatol Rehabil. 2017;9(1):1–5. doi:10.4103/0975-7341.207173
13. Matsuo M, Nishida K, Yoshida A, Murakami T, Inoue H. Expression of caspase-3 and −9 relevant to cartilage destruction and chondrocyte apoptosis in human osteoarthritic cartilage. Acta Med Okayama. 2001;55(6):333–340. doi:10.18926/AMO/32000
14. D’Lima DD, Hashimoto S, Chen PC, Lotz MK, Colwell CW
15. Nuttall ME, Nadeau DP, Fisher PW, et al. Inhibition of caspase-3-like activity prevents apoptosis while retaining functionality of human chondrocytes in vitro. J Orthop Res. 2000;18(3):356–363. doi:10.1002/jor.1100180306
16. D’Lima D, Hermida J, Hashimoto S, Colwell C, Lotz M. Caspase inhibitors reduce severity of cartilage lesions in experimental osteoarthritis. Arthritis Rheum. 2006;54(6):1814–1821. doi:10.1002/art.21874
17. Sakkas LI, Platsoucas CD. The role of T cells in the pathogenesis of osteoarthritis. Arthritis Rheum. 2007;56(2):409–424. doi:10.1002/art.22369
18. Koenders MI, Joosten LA, van Den Berg WB. Potential new targets in arthritis therapy: interleukin (IL)-17 and its relation to tumour necrosis factor and IL-1 in experimental arthritis. Ann Rheum Dis. 2006;65(Suppl 3):iii29–iii33. doi:10.1136/ard.2006.058529
19. Goldring MB, Goldring SR. Osteoarthritis. J Cell Physiol. 2007;213(3):626–634. doi:10.1002/jcp.21258
20. Mengshol JA, Vincenti MP, Brinckerhoff CE. IL-1 induces collagenase-3 (MMP-13) promoter activity in stably transfected chondrocytic cells: requirement for Runx-2 and activation by p38 MAPK and JNK pathways. Nucleic Acids Res. 2001;29(21):4361–4372. doi:10.1093/nar/29.21.4361
21. Billinghurst RC, Dahlberg L, Ionescu M, et al. Enhanced cleavage of type II collagen by collagenases in osteoarthritic articular cartilage. J Clin Invest. 1997;99(7):1534–1545. doi:10.1172/JCI119316
22. Kim YI, Park JE, Brand DD, Fitzpatrick EA, Yi AK. Protein kinase D1 is essential for the proinflammatory response induced by hypersensitivity pneumonitis-causing thermophilic actinomycetes Saccharopolyspora rectivirgula. J Immunol. 2010;184(6):3145–3156. doi:10.4049/jimmunol.0903718
23. Park JE, Kim YI, Yi AK. Protein kinase D1: a new component in TLR9 signaling. J Immunol. 2008;181(3):2044–2055. doi:10.4049/jimmunol.181.3.2044
24. Cobbaut M, Van Lint J. Function and regulation of protein kinase D in oxidative stress: a tale of isoforms. Oxid Med Cell Longev. 2018;2018:2138502. doi:10.1155/2018/2138502
25. Baker J, Falconer AMD, Wilkinson DJ, Europe-Finner GN, Litherland GJ, Rowan AD. Protein kinase D3 modulates MMP1 and MMP13 expression in human chondrocytes. PLoS One. 2018;13(4):e0195864. doi:10.1371/journal.pone.0195864
26. Chung R, Foster BK, Xian CJ. Inhibition of protein kinase-D promotes cartilage repair at injured growth plate in rats. Injury. 2013;44(7):914–922. doi:10.1016/j.injury.2013.01.038
27. Lee Y, Kim H, Kim S, Kim KH, Chung JH. Activation of toll-like receptors 2, 3 or 5 induces matrix metalloproteinase-1 and −9 expression with the involvement of MAPKs and NF-kappaB in human epidermal keratinocytes. Exp Dermatol. 2010;19(8):e44–e49. doi:10.1111/j.1600-0625.2009.00963.x
28. Liang KC, Lee CW, Lin WN, et al. Interleukin-1beta induces MMP-9 expression via p42/p44 MAPK, p38 MAPK, JNK, and nuclear factor-kappaB signaling pathways in human tracheal smooth muscle cells. J Cell Physiol. 2007;211(3):759–770. doi:10.1002/jcp.20992
29. Park JE, Kim YI, Yi AK. Protein kinase D1 is essential for MyD88-dependent TLR signaling pathway. J Immunol. 2009;182(10):6316–6327. doi:10.4049/jimmunol.0804239
30. Upadhyay K, Park JE, Yoon TW, et al. Group B streptococci induce proinflammatory responses via a protein kinase D1-dependent pathway. J Immunol. 2017;198(11):4448–4457. doi:10.4049/jimmunol.1601089
31. Sophia Fox AJ, Bedi A, Rodeo SA. The basic science of articular cartilage: structure, composition, and function. Sports Health. 2009;1(6):461–468. doi:10.1177/1941738109350438
32. Sercombe L, Veerati T, Moheimani F, Wu SY, Sood AK, Hua S. Advances and challenges of liposome assisted drug delivery. Front Pharmacol. 2015;6:286. doi:10.3389/fphar.2015.00286
33. Bhatti FUR, Hasty KA, Cho H. Anti-inflammatory role of TPCA-1 encapsulated nanosomes in porcine chondrocytes against TNF-alpha stimulation. Inflammopharmacology. 2019. doi:10.1007/s10787-018-0542-5
34. Cho H, Pinkhassik E, David V, Stuart JM, Hasty KA. Detection of early cartilage damage using targeted nanosomes in a post-traumatic osteoarthritis mouse model. Nanomedicine. 2015;11(4):939–946. doi:10.1016/j.nano.2015.01.011
35. Cho H, Seth A, Warmbold J, Robertson JT, Hasty KA. Aging affects response to cyclic tensile stretch: paradigm for intervertebral disc degeneration. Eur Cell Mater. 2011;22:
36. Choi EK, Jang HC, Kim JH, et al. Enhancement of cytokine-mediated NF-kappaB activation by phosphatidylinositol 3-kinase inhibitors in monocytic cells. Int Immunopharmacol. 2006;6(6):908–915. doi:10.1016/j.intimp.2006.01.007
37. Sizemore N, Leung S, Stark GR. Activation of phosphatidylinositol 3-kinase in response to interleukin-1 leads to phosphorylation and activation of the NF-kappaB p65/RelA subunit. Mol Cell Biol. 1999;19(7):4798–4805. doi:10.1128/mcb.19.7.4798
38. Gold MR, Scheid MP, Santos L, et al. The B cell antigen receptor activates the Akt (protein kinase B)/glycogen synthase kinase-3 signaling pathway via phosphatidylinositol 3-kinase. J Immunol. 1999;163(4):1894–1905.
39. Henrotin YE, Bruckner P, Pujol JP. The role of reactive oxygen species in homeostasis and degradation of cartilage. Osteoarthritis Cartilage. 2003;11(10):747–755.
40. Marchev AS, Dimitrova PA, Burns AJ, Kostov RV, Dinkova-Kostova AT, Georgiev MI. Oxidative stress and chronic inflammation in osteoarthritis: can NRF2 counteract these partners in crime? Ann N Y Acad Sci. 2017;1401(1):114–135. doi:10.1111/nyas.13407
41. Lepetsos P, Papavassiliou AG. ROS/oxidative stress signaling in osteoarthritis. Biochim Biophys Acta. 2016;1862(4):576–591. doi:10.1016/j.bbadis.2016.01.003
42. Morgan MJ, Liu ZG. Crosstalk of reactive oxygen species and NF-kappaB signaling. Cell Res. 2011;21(1):103–115. doi:10.1038/cr.2010.178
43. Rigoglou S, Papavassiliou AG. The NF-kappaB signalling pathway in osteoarthritis. Int J Biochem Cell Biol. 2013;45(11):2580–2584. doi:10.1016/j.biocel.2013.08.018
44. Yasuhara R, Miyamoto Y, Akaike T, et al. Interleukin-1beta induces death in chondrocyte-like ATDC5 cells through mitochondrial dysfunction and energy depletion in a reactive nitrogen and oxygen species-dependent manner. Biochem J. 2005;389(Pt 2):315–323. doi:10.1042/BJ20041996
45. Wang L, Gai P, Xu R, et al. Shikonin protects chondrocytes from interleukin-1beta-induced apoptosis by regulating PI3K/Akt signaling pathway. Int J Clin Exp Pathol. 2015;8(1):298–308.
46. Zhou PH, Liu SQ, Peng H. The effect of hyaluronic acid on IL-1beta-induced chondrocyte apoptosis in a rat model of osteoarthritis. J Orthop Res. 2008;26(12):1643–1648. doi:10.1002/jor.20683
47. Blanco FJ, Lotz M. IL-1-induced nitric oxide inhibits chondrocyte proliferation via PGE2. Exp Cell Res. 1995;218(1):319–325. doi:10.1006/excr.1995.1161
48. Bhatti FUR, Kim SJ, Yi AK, Hasty KA, Cho H. Cytoprotective role of vitamin E in porcine adipose-tissue-derived mesenchymal stem cells against hydrogen-peroxide-induced oxidative stress. Cell Tissue Res. 2018;374(1):111–120. doi:10.1007/s00441-018-2857-3
49. Rao Z, Wang S, Wang J. Peroxiredoxin 4 inhibits IL-1beta-induced chondrocyte apoptosis via PI3K/AKT signaling. Biomed Pharmacother. 2017;90:414–420. doi:10.1016/j.biopha.2017.03.075
50. Sillat T, Barreto G, Clarijs P, et al. Toll-like receptors in human chondrocytes and osteoarthritic cartilage. Acta Orthop. 2013;84(6):585–592. doi:10.3109/17453674.2013.854666
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.