")
Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Nanomedicine strategies for treatment of secondary spinal cord injury
Authors White-Schenk D, Shi R, Leary JF
Received 11 October 2014
Accepted for publication 28 November 2014
Published 29 January 2015 Volume 2015:10(1) Pages 923—938
DOI https://doi.org/10.2147/IJN.S75686
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Carlos Rinaldi
Désirée White-Schenk,1,4 Riyi Shi,1–3 James F Leary1–4
1Interdisciplinary Biomedical Sciences Program, 2Weldon School of Biomedical Engineering, 3Department of Basic Medical Sciences, Lynn School of Veterinary Medicine, 4Birck Nanotechnology Center, Discovery Park, Purdue University, West Lafayette, IN, USA
Abstract: Neurological injury, such as spinal cord injury, has a secondary injury associated with it. The secondary injury results from the biological cascade after the primary injury and affects previous uninjured, healthy tissue. Therefore, the mitigation of such a cascade would benefit patients suffering a primary injury and allow the body to recover more quickly. Unfortunately, the delivery of effective therapeutics is quite limited. Due to the inefficient delivery of therapeutic drugs, nanoparticles have become a major field of exploration for medical applications. Based on their material properties, they can help treat disease by delivering drugs to specific tissues, enhancing detection methods, or a mixture of both. Incorporating nanomedicine into the treatment of neuronal injury and disease would likely push nanomedicine into a new light. This review highlights the various pathological issues involved in secondary spinal cord injury, current treatment options, and the improvements that could be made using a nanomedical approach.
Keywords: spinal cord injury, acrolein, drug delivery, methylprednisolone, secondary injury
Introduction
Spinal cord injury
Neurological injury often results from biological damage, such as multiple sclerosis, or mechanical damage, such as compression in spinal cord injury (SCI). Regardless of the affliction, a patient’s quality of life is likely reduced due to the loss of motor, sensory, or cognitive function. Motor vehicle accidents most heavily contribute to the occurrence of central nervous system (CNS) injuries. Since young adults and teens are most likely to be involved in these types of accidents, their age group is significantly affected.1 Aside from possible mortality, injury usually results in impaired motor capabilities (paralysis), impaired sensory capabilities (hypersensitivity and hyposensitivity), and/or neurologically-based pain. This can drastically reduce a patient’s quality of life and place a large burden on society, both in health care costs and lost productivity. Regardless of how the CNS is damaged, it has adverse effects on a patient. Neuronal injury is a major field of exploration, but this review will focus on aspects of treatment after SCI. Various therapies have been explored, but as discussed later, the treatments may have little effect or adverse side effects, which calls for a new approach to treating such trauma.
It is well established that physical impact is not solely responsible for the severe tissue damage resulting from SCI. Rather, mechanical trauma induces a cascade of chemical reactions leading to a delayed secondary neurological injury that amplifies the effects of the initial injury and expands the damage throughout the cord.2,3 Due to the delayed nature of this pathology, which offers a window of intervention, the inhibition of secondary injury processes has emerged as an important therapeutic strategy to deter further degeneration and promote functional recovery.3 Therefore, ascertaining the secondary injury and identifying key therapeutic targets is warranted.
Nanomedicine: a new medical approach
Nanotechnology holds promise in aiding the treatment of chronic disease and medical conditions. In biology and medicine, the field has developed rapidly in the last decade to create a new application of nanotechnology in medicine known as nanomedicine (for reviews, see Haglund et al4 Seale-Goldsmith and Leary,5 and Leary6). Major goals in the field involve the improvement of targeted drug delivery, improved diagnostic or imaging techniques, and simultaneous therapeutics and diagnostics (“theranosis”), which is a combination of both therapeutics and diagnostics in the same nanomedical drug/device. Nanomedicine gives rise to potential therapies that can be tailored to diagnose and treat for specific disease states while greatly diminishing the side effects of traditional medicine through both the targeting process and greatly lowered total systemic doses due to that targeting process, and also longer circulation times created by stealth layers on the nanomedical device that prevent opsonification and decrease uptake by the kidneys and liver. Additionally, as the field broadens, it expands to include nanopharmacology and nanotoxicity for exploration of how the body reacts to the new nanosized structures. While sometimes criticized as offering as yet unfulfilled promise, much of the delay is due to uncertainties in the evaluation process on the part of regulatory agencies such as the US Food and Drug Administration (FDA), which is still coming to terms with nanotechnology.7 Pharmaceutical firms worry about the twin hurdles of FDA approval as a “combo” device, since nanomedicine involves both a nanodevice and a drug. However, early and simpler forms of nanomedical systems have been approved by the FDA, and more are in the approval process pipeline.
Composition of nanoparticles
The composition of nanoparticles is extremely diverse. They have been synthesized using a wide range of materials, including metal (eg, gold, silver, cadmium), metal oxides (iron oxide, titanium oxide, zinc oxide), silica, polymers, and biological molecules (peptides and DNA), among others.8–11 As the field has progressed, more nanoparticles are being made from mixtures of these materials, which provide new benefits and functions for the various applications that a single material will not possess. Many times, the amalgamation allows both the diagnostic and therapeutic effects to occur either simultaneously or in a time-dependent manner. This is achieved by making nanoparticles one layer at a time, called layer-by-layer synthesis. Each layer changes the characteristics of the nanoparticle and adds functionality, essentially making the nanoparticle programmable. Such multifunctional nanoparticles become a unique nanomedical system used to address a biological problem.
Neurological disease pathology: overview
Neurological degeneration in SCI
Although it can recover somewhat, generally the CNS recovers very little after injury. On the other hand, the peripheral nervous system recovers more efficiently. The enzymatic and molecular composition of the neurons involved contributes heavily to the differences in their regeneration. The CNS myelin, composed of oligodendrocytes, contains direct inhibitors of axonal growth, such as the aptly named Nogo receptor, that interact with myelin proteins.12–16 In the peripheral nervous system, protective Schwann cells form a scaffold that allows the renewed growth of injured axons.17–19 Injury to the CNS also affects a patient more critically. The brain and spinal cord are the control centers for the body’s many functions. When messages cannot be transported back and forth, the brain cannot relay appropriate responses to incoming sensory information, or it does not receive the information at all. In the peripheral nervous system, although the loss of information may be detrimental, it is usually both locally concentrated and less critical than the copious amounts of information the brain and spinal cord both send and receive. In the CNS, the axons of the neurons can be quite long, and once damaged or destroyed, the information from a large part of the body is lost.
In places in and around the injury site, cells often suffer from membrane and myelin damage.20 Neuronal membrane integrity is critical to cellular function. The membrane not only retains chemicals and proteins but also prevents them from entering. The function of a neuron depends on specific ion concentration gradients between the extracellular and intracellular spaces. Extracellularly, sodium and calcium ions are abundant in comparison with those in the cytosol. Intracellularly, the potassium ion concentration is much higher inside the cell. In a normal cell, the differences in concentration create a voltage potential across the membrane and polarize the cell. When the membrane is compromised, allowing free movement of ions, the gradients diminish or disappear. The cell depolarizes, and the leaky membrane prevents it from repolarizing, thereby destroying its ability to generate an action potential.21 Beyond that, other chemicals, such as reactive species and proteins, can invade and damage the cell, generating secondary injury and necrosis in a cycle. The cycle of stress and death impedes the ability of cells to recover from the inundation of toxins and return to homeostasis.
In the CNS, oligodendrocytes are responsible for myelinating neuronal axons. The oligodendrocytes form segments of sheaths around the axons. Spaces between the segments are known as the nodes of Ranvier. The sheaths allow action potentials to “jump” quickly from node to node. Further, the junctions contain distinct enzymes and signaling molecules to keep different ion channels segregated. The nodes of Ranvier contain potassium channels, and the sodium channels congregate in the adjacent juxtanodal regions. The channels open and close at different points during an action potential, and the separation of the ion channels not only allows movement of the action potential, but also facilitates faster depolarization along the axon.22,23 When the ion channels do not remain separated, neuronal function is impaired.22 Therefore, after demyelination, when the ion channels are desegregated, the ability of the neuron to transmit action potentials is hindered or destroyed. Beyond SCI, demyelination is a large issue in the progression of other neurodegenerative diseases.24,25
At a deeper level, initial SCI results in a concentrated site of necrotic cell death, membrane damage, and, often, myelin damage. In contrast with apoptosis, where cell contents are packaged and recycled before cell death, the contents of necrotic cells are released haphazardly into their environment. Normally, the body uses many of the molecules that are released during necrosis for various mechanisms, including signaling, degradation, and synthesis.26–30 Hydrogen peroxide and nitric oxide, both potent oxidative species, are secondary messengers for different cellular pathways.27,29 For instance, nitric oxide is a secondary messenger that results in vasodilation of smooth muscle, among other functions.27 To help destroy invading pathogens, hydrogen peroxide is secreted by cells responsible for immunity.28 The superoxide anion is produced in the mitochondria during the electron transport chain. The combination of superoxide and nitric oxide results in peroxynitrite; its formation and sequestration by uric acid after SCI was studied and found to ameliorate some effects of peroxynitrite formation, but the major mechanism seemed to stem from attenuating the inflammatory response from immune cells.31 Vitamin B12 works specifically through a radical reaction with cobalt, creating upstream products for DNA synthesis.26 Upon uncontrolled release, though, many of these molecules and enzymes, specifically reactive oxygen species and reactive nitrogen species (Figure 1), travel to neighboring cells, including healthy ones, and cause secondary damage through effects such as lipid peroxidation and inflammation.31–33
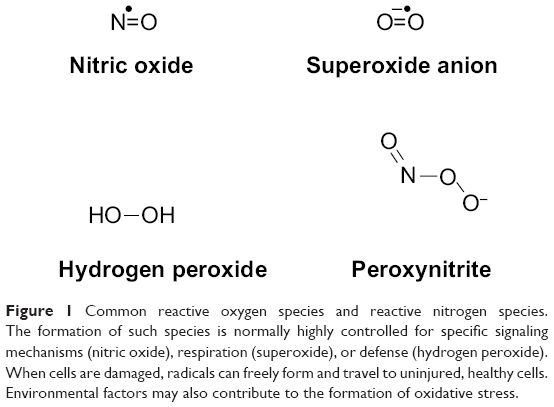 | Figure 1 Common reactive oxygen species and reactive nitrogen species. The formation of such species is normally highly controlled for specific signaling mechanisms (nitric oxide), respiration (superoxide), or defense (hydrogen peroxide). When cells are damaged, radicals can freely form and travel to uninjured, healthy cells. Environmental factors may also contribute to the formation of oxidative stress. |
Biochemical targets of SCI
In SCI, inflammation plays a role in the health and progression of CNS neurons. In vitro, activated microglial cells induce oxidative stress when cultured with neuronal cells.34,35 Although the response is natural, the oxidative stress can be detrimental to cells that are already undergoing stress. Figure 2 highlights the role inflammation can have in neuronal injury.
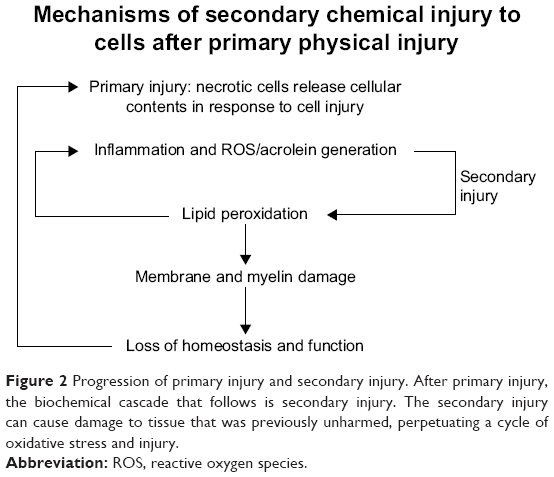 | Figure 2 Progression of primary injury and secondary injury. After primary injury, the biochemical cascade that follows is secondary injury. The secondary injury can cause damage to tissue that was previously unharmed, perpetuating a cycle of oxidative stress and injury. |
Acrolein (Figure 3), a reactive oxygen species, has been implicated in secondary injury in neuronal tissues.36,37 Both studies found increased lipid peroxidation specifically caused by acrolein. Luo et al monitored the protein adducts of acrolein after SCI in vivo. They found that acrolein levels peaked 24 hours after injury, but stayed elevated for up to a week.37 Later studies explored the various biochemical changes caused by acrolein in neuronal tissues, including lipid peroxidation, myelin damage, and mitochondrial damage.38–40 The primary carbon of acrolein can undergo nucleophilic (“nucleus-loving”) attack, but contributing to its destructive nature, the electrophilic (“electron-loving” or electron-deficient) carbons of the π-bond (second bond of C=C) can undergo attack by nucleophiles in the cell. Specifically, acrolein reacts with amine-containing lysine residues, histidines, and electrophilic unsaturated fatty acids. Changes to protein residues upon reacting with acrolein can cause functional and conformational detriments in proteins. When acrolein attacks fatty acids, more acrolein is generated in a cycle of uncontrolled lipid peroxidation.41 Environmentally, acrolein is a byproduct of the burning of plastics, overheated cooking oil, and cigarette smoke, which makes it an important environmental source of oxidative stress that can adversely affect the body.42–44
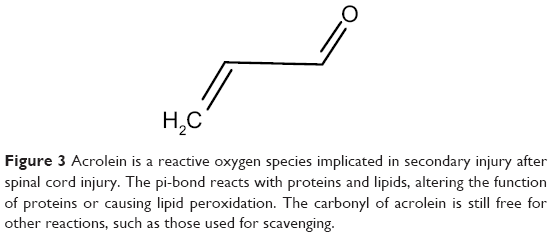 | Figure 3 Acrolein is a reactive oxygen species implicated in secondary injury after spinal cord injury. The pi-bond reacts with proteins and lipids, altering the function of proteins or causing lipid peroxidation. The carbonyl of acrolein is still free for other reactions, such as those used for scavenging. |
When acrolein reacts with lipids, such as those in the cell membrane, the membrane is compromised, and more acrolein is released to attack other tissue in a feed-forward process. It creates a cascade of oxidative stress to a particular area of injury. In this way, acrolein can increase stress and damage healthy tissues surrounding the injury site.32,38,45 The large amount of oxidative stress overwhelms the body’s natural antioxidant, glutathione.46 Normally, glutathione is oxidized, which stops reactive oxygen species and reactive nitrogen species from oxidizing proteins and structures in the cell. It then would be reduced to its original form by glutathione reductase. In a large area of oxidation, glutathione cannot be reduced rapidly enough to account for the overwhelming, cyclic, and unchecked production of acrolein.
Acrolein is a small molecule target in SCI, but inherent biological systems also play a role in SCI. Calpain, a calcium-dependent cysteine protease, cleaves various proteins associated with the cell membrane. The protease is strongly conserved across different species. In humans, it has two main isoforms, calpain-1 and calpain-2, also referred to as μ-calpain and m-calpain, respectively, where the latter notation refers to the concentration of calcium (micromolar and millimolar) necessary for optimal activation. Cysteine proteases use a nucleophilic thiol group of the cysteine residue to cleave peptide bonds. Peptide bonds contain an electrophilic carbonyl where cleavage takes place, breaking the bond between the carboxylic acid portion of one amino acid and the amine of the neighboring residue. The reaction requires water, and the cysteine of the active site only catalyzes the break, thereby being recycled.47 Because calpain cleaves numerous proteins with seemingly little order, various studies have attempted to deduce and deconstruct substrate cleavage sites.48,49 For example, Betts et al constructed a series of synthetic peptides and tested calpain activity against known substrates for the purpose of analyzing, residue by residue, the construction of the inherent inhibitor, calpastatin.48
Calpain expression has been implicated in different disease states and become a therapeutic target because of its role in disease states, apoptosis, and, mainly, necrosis.50–55 In damaged neuronal tissue, large influxes of calcium can cause widespread activation of calpains. In these states, its natural inhibitor, calpastatin, is underexpressed, leading to an imbalance of calpain activity (Figure 4).56 Additionally, calpastatin specifically inhibits calpain.57 Various efforts have been made to create new calpain inhibitors, but they often target other proteases as well.58–62 Calpeptin, a short peptide, for example, targets both forms of calpain very well, but it also inhibits papain, although at higher concentrations.59
 | Figure 4 Example of the calpain-calpastatin molecular mechanisms in a damaged cell. Calcium influx causes mass activation of calpain, which cleaves protein substrates and regulators of its inhibitor, calpastatin. An efficient delivery of calpastatin or other calpain inhibitors may hinder the damage caused by the extensive activation of calpain after injury. Calpain may also serve as a protein target for nanomedical systems. Cell is not drawn to scale. For a review of explored calpain inhibitors, see Donkor.62 |
In 1989, Maki et al determined the domain of calpastatin involved in the inhibition of calpain. It consists of a 27-mer portion of the protein known as B-peptide, with the sequence DPMSSTYIEELGKREVTIPPKYRELLA.63 Later, it became known as the calpastatin B-27 peptide, based on its sequence location in exon 1B. Since then, the crystal structure of the two proteins has been published and discussed.64 Calpastatin’s α-helix in the B-peptide turns away from the reactive cysteine, while the other portions of the protein facilitate binding of the B-peptide. Modeling and analysis has been performed to determine the important morphology of the calpain-calpastatin system and to provide insight for designing new targets.49,65
Although other biological factors may play a role in SCI, such as various molecular pathways, inflammation, calpain, and acrolein are targets that the current clinical therapies (namely methyprednisolone) do not sufficiently address. In particular, the persistence of acrolein in vivo leads to more oxidative stress that can travel to healthy tissues, causing the injury site to grow beyond the initial trauma point. Further, unlike other oxidative species, its levels remain high, which hinders the ability of tissues to recover effectively. Specifically, both acrolein and inflammation can lead to necrosis, which negatively impacts not only the dying tissue, but also the surrounding tissue. In a sensitive area like the spinal cord, necrosis and damage can affect the entire body, not just a concentrated area.
Therapeutic explorations in treating SCI
Immunosuppression
Although methylprednisolone, a steroid, is the most common drug used for SCI, it has been the subject of criticism.66 Hugenholtz et al concluded that the drug is too expensive to prescribe with the current clinical data that are scattered and inconclusive.66 Bracken et al recommend that the drug be given within the first 8 hours post-injury to be effective.67 Regardless, the drug aims to suppress the immune system, reducing inflammation around the injury site. Although it may attenuate oxidative stress and cytokines from immune cells, it does not address the process of stress from dead or dying cells. Methylprednisolone is the only treatment used clinically, but its mechanism of action and questionable efficacy warrants re-evaluation and decidedly better alternatives. On the other hand, a new and better vehicle may help to mitigate its negatives, but its ability to stop secondary injury is focused on preventing stress from one of the many sources of stress. Therefore, more work needs to be done to find new and more effective therapies for SCI.
Glutathione
A seemingly easy approach for improving the body’s stores of antioxidants includes glutathione supplementation (Figure 5). The molecule is made from cysteine, glycine, and glutamate, and its antioxidant properties stem from the nucleophilic thiol group of the cysteine. Oxidative species are electrophilic and the extra valence electrons of the thiol group reduce the oxidative species and create water in the process. In other words, the antioxidant is oxidized rather than allowing essential parts of the cell to be oxidized. Unfortunately, orally administered glutathione itself is poorly absorbed.68 Studies of glutathione levels have found that it forms an equilibrium with its amino acid components through synthesis of glutathione and uptake of cysteine.69,70 To increase glutathione levels, rather than absorbing glutathione, increasing the uptake of its amino acid components shifts the equilibrium toward synthesis of glutathione.
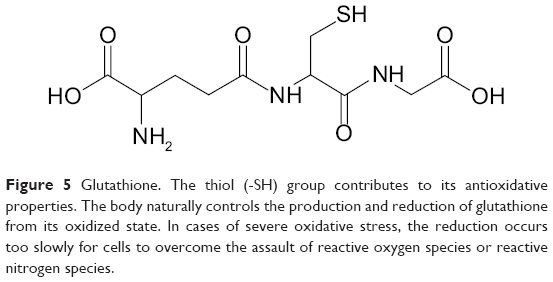 | Figure 5 Glutathione. The thiol (-SH) group contributes to its antioxidative properties. The body naturally controls the production and reduction of glutathione from its oxidized state. In cases of severe oxidative stress, the reduction occurs too slowly for cells to overcome the assault of reactive oxygen species or reactive nitrogen species. |
Due to its limited bioavailability, effectively delivering glutathione directly to the site of injury could help increase the antioxidant capabilities of cells suffering from secondary injury. Since glutathione is the cell’s primary defense, its efficacy is likely to be higher with fewer side effects. Cells already contain a system to process, use, and dispose of glutathione effectively. Using such a treatment would act as a supplement to the process already in place.
Calpain inhibition
Based on its role in neurological injury, some groups have used calpain inhibitors to target the apoptosis and necrosis of neurons in various disease models.50,52,60,71–74 In a notable in vivo study that used both intravenous and intraperitoneal injections, Yu and Geddes found that only when the two types of injections were combined (intravenously after injury and then intraperitoneally daily for a week after the injury) did the pathology of the injury change significantly.74 Otherwise, they found that the lesion increased and tissues died similar to the levels of the control injury. The authors admit that their results could be improved with better delivery and availability of the inhibitor, which is less of an issue in vitro.74 In in vitro systems, the cells of interest can be studied directly without numerous other factors that may affect the delivery of therapeutic molecules in vivo. Although the group had positive therapeutic results in vivo, the increase in the lesion site was likely due to other secondary injury factors not addressed by the calpain inhibitor.
One group attempted to address the issue by combining methylprednisolone and calpeptin.73 Although this approach is creative, it does not target the cascade of secondary injury caused by inherent oxidative stress. The animals were sacrificed after 48 hours, their motor capacities were not reported, and pathology was not examined. If the study had continued and the pathology was determined, it would have given better insight into the mechanisms of SCI that the authors were attempting to study.
Calpain inhibition is a critical aspect of secondary injury in need of address. Calpain activation is likely to affect living, but compromised, cells. Preventing their necrotic death could temper the spread of the injury site to previously uninjured tissues. Like most therapeutics, specificity is difficult to obtain in small molecules, and bioinspired therapeutics generally require protection from degradation. Like the study described above, a more effective treatment will likely require a combination therapy technique.
Hydralazine as an acrolein scavenger
The drug hydralazine (Figure 6) has been found to “scavenge” for acrolein and other oxidative species.3,32,38,75–77 In particular, the drug will scavenge and react with acrolein that has reacted with proteins.38 As mentioned earlier, the carbonyl group in acrolein is free for nucleophilic attack. Once acrolein reacts with a protein, the carbonyl is free, and hydralazine can attack it. The reaction results in a Schiff-base and imine (C=N bond) formation, which the body uses regularly for amino acid synthesis. When the drug reacts with free acrolein, it reduces the amount of acrolein that can induce lipid peroxidation, arresting the feed-forward production of acrolein. Ultimately, stopping this process would allow the cells to recover more effectively.
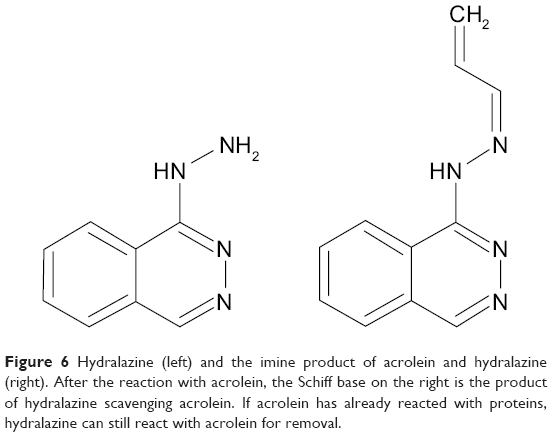 | Figure 6 Hydralazine (left) and the imine product of acrolein and hydralazine (right). After the reaction with acrolein, the Schiff base on the right is the product of hydralazine scavenging acrolein. If acrolein has already reacted with proteins, hydralazine can still react with acrolein for removal. |
Currently, hydralazine is used for hypertension. It activates a G-protein coupled receptor, guanylate cyclase, in smooth muscle, causing a downstream signal to relax the smooth muscle.78 Due to this mechanism, administering hydralazine for acrolein scavenging could critically lower an injured patient’s blood pressure. Further, hydralazine has a fairly short half-life of about 2 hours in vivo, and genetic factors influence the metabolism and side effects of the drug. Some patients who metabolize the compound rapidly have been shown to exhibit lupus-like symptoms.79 Therefore, a therapy that could concentrate hydralazine locally without lowering blood pressure or invading other tissues would have great potential for SCI patients. Hydralazine treatment would lack the need for regulatory approval, but its specificity limits its translation for SCI. Unlike glutathione, hydralazine is limited in its scavenging of a broad range of oxidative species, but being a small molecule, it is inexpensive and a prime candidate for delivery manipulation.
Polymeric therapeutic molecules
Long-chain polymers have been also studied for SCI. Poly(ethylene) glycol (PEG) and chitosan have both been studied for their effects in treating SCI and other neurological diseases.3,45,77,80 The polymers first plug the damaged membrane by associating with the holes. Interaction of the polymers then causes the two separated parts of the membrane to associate with each other, effectively closing the hole and stopping the invasion of unwanted ions and molecules.45,80
The two polymers have a few distinct differences. The molecular weight of PEG ranges from very small (molecular weight 250) to very large (molecular weight 500,000), depending on the application. It also comes with a range of functional groups for different modifications or can be modified for functionality. The applications and functionalities of PEG are discussed in the next section. Chitosan, on the other hand, is derived from the exoskeletons of shell fish, such as crabs and shrimp.81 Chitin, the polymeric material from the shells, is deacetylated in various degrees to create chitosan. Chitosan alone is not water-soluble, so must be modified to become water-soluble.10 The properties of PEG/chitosan blends have also been studied to combine the biodegradation and biocompatibility effects of both polymers.82–84 De Campos et al compared polymeric particles coated with PEG and those coated with chitosan for therapeutic delivery to the cornea. They found that PEG had better delivery, but chitosan had a better drug retention effect.85
Polymers such as PEG and chitosan could play an important role in SCI therapy. PEG is a stealth polymer used to extend the half-life of therapeutics (discussed later in this review). Membrane sealing is a primary step in helping cells to achieve homeostasis and stopping an onslaught of unwanted extracellular material, which would ideally allow cells to eventually obtain a membrane potential. Membrane sealing alone will not stop the spread of damage, and only addresses a small aspect of the secondary injury problem.
Treatment summary
A number of different molecules have been used to target the symptoms of SCI. All of them attempt to address some, although not all, characteristics of SCI. Table 1 summarizes the origin and action of many of the common therapies. It is evident that a new approach is necessary to address the complex processes that occur during and after SCI. Since the major explorations involve controlling one of the aforementioned aspects of SCI, a therapy that could combat all of them would be a large step in helping SCI patients. Granted, axonal regeneration is another factor after SCI, but oxidative stress and proteolysis are direct pathways to apoptosis and necrosis. Therefore, controlling and preventing cell death is extremely important before the tissues can repair themselves. Additionally, the major problem in inflammation is induction of oxidative stress through cytokines. Rather than affecting a patient’s entire immune system, it is more beneficial to control local inflammation and oxidative stress.
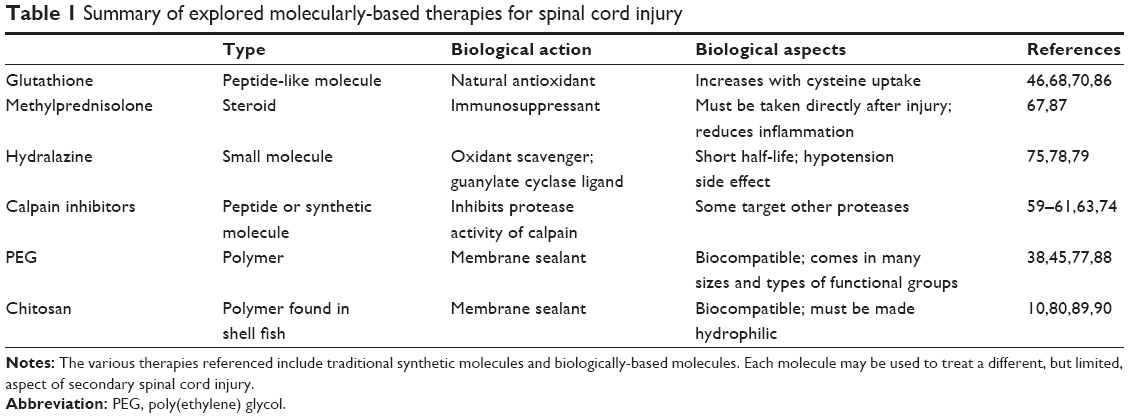 | Table 1 Summary of explored molecularly-based therapies for spinal cord injury |
Nanomedicine strategies
Due to current therapeutic limitations, a nanomedical approach may help to ameliorate poor drug availability and the biochemical effects of secondary injury. In current therapies, drugs are administered and, hopefully, arrive at their intended location. Nanoparticles can act as carriers and increase the circulation time of a therapy, which is critical for hydrophobic and short-lived therapies, thereby improving the therapy’s chance of reaching the cells of interest. Further, biomolecules (natural or mimetic) can be added to the nanoparticle assemblies to help with cell-uptake specificity. After circulation and accumulation, the biomolecule hopefully increases the likelihood of nanoparticles being internalized by the intended cells.91 Peptides, DNA, RNA, and receptor ligands have been used for their affinity for a particular biomarker or characteristic, such as an increase in folate receptors on cancer cells.92–96 Unfortunately, the disease state must have a studied and reliable biomarker to target, which may not always be the case. Without these moieties, nanoparticles may accumulate in different areas, for instance, by changing their shape or size.97,98 Different clearance organs, such as the spleen, liver, and kidneys, will filter out particles based on their size. He et al specifically found that smaller particles (<200 nm) were less likely to accumulate in the spleen and liver compared with larger particles.97
Table 2 summarizes many of the common materials used to make nanoparticles. The list includes inorganic material (silica and iron oxide), transition metals (gold and quantum dots), and organic materials (chitosan, DNA, PEG). Each material harbors certain advantages and disadvantages versus the others. Efforts have also explored hybrid particles, merging the advantages of the different materials.
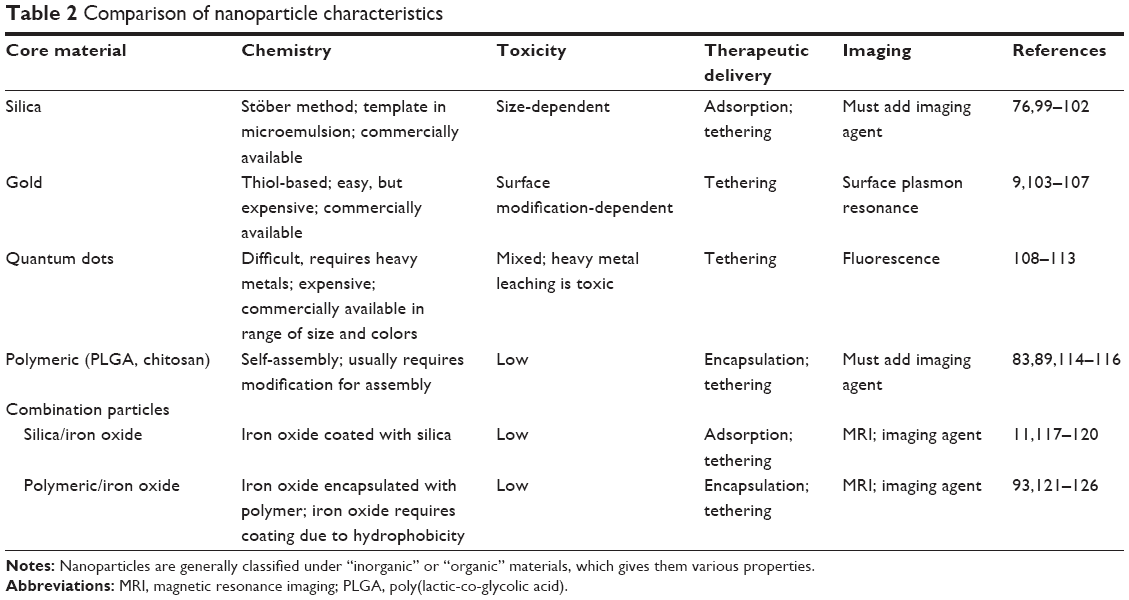 | Table 2 Comparison of nanoparticle characteristics |
Table 2 indicates that both silica and polymeric nanoparticles provide essential features to ameliorate the limitations of current SCI therapies, most notably they have an ability to adsorb and encapsulate drugs. The distinct and overwhelming advantage that silica nanoparticles provide is the ability to form a mesoporous network. This porosity makes them especially useful for various medical applications, including drug and gene delivery. In particular, the tunable mesoporous structure of silica networks gives them qualities of zero-order drug release. The drug is released at a constant rate, rather than at an exponential or logarithmic rate, which could prevent side effects from high drug doses and decrease the amount of drug necessary for effective delivery. The silica encapsulates the drug until reaching the cell or cells of interest. The method can be applied to potent drugs, such as chemotherapy agents, or environmentally-sensitive agents.
In polymeric particles, drugs are usually encapsulated via self-assembly.115 Polymeric particles are usually created via sonication or mixing, which causes them to assemble into nanostructures.8,127 When the nanoparticles disassemble in biological systems, the drugs are released in a burst, which is usually the cause of adverse side effects.116 When therapies are released rapidly, a high local concentration can be toxic to cells, especially when delivering very potent drugs (eg, chemotherapy agents). Avgoustakis et al synthesized polymeric nanoparticles, and in initial release, about 20% of the anticancer drug cisplatin was released.8 Zhang and Feng also reported an initial burst of the anticancer therapy paclitaxel of about 20%.127 In the other types of nanoparticles, the therapies are usually added by a form of conjugation method, which can be limiting when needing to deliver a small molecule.
Various examples of using silica to deliver therapeutic molecules have recently been demonstrated. Chauhan et al have used silica hydrogels to load hydrophobic drugs and monitor their release profile from contact lenses used for glaucoma. They found ways to change the release profile and induce release for weeks or months.128–131 The therapeutic agents can be loaded by incubating them with the porous silica. Molecular interactions, generally Van der Waal’s forces, cause the drug to adsorb onto the surface of the silica.76,132 Yu et al successfully coated iron oxide nanoparticles with silica for the purpose of protein adsorption.120
Silica nanoparticles can be prepared and functionalized in various ways. The starting material, or precursor, is generally a small silicate, such as tetramethylorthosilicate or tetraethylorthosilicate. Upon addition of an acid or base, the precursor hydrolyzes and eventually condenses to form a silica network at room temperature (Figure 7). Although many reaction types exist, many of these stem from the Stöber method published in 1968.102 The method requires use of a light alcohol (methanol or ethanol) and ammonium hydroxide for catalysis.102 A common variation on this method includes the water-in-oil microemulsion, which uses a nonmiscible solvent (an alkane) and a surfactant.133 Other reagents explored include an array of bases, solvents, and even templates for silica nucleation. The templates are usually an organic salt, such as cetyltrimethylammonium bromide, which creates a surfactant layer for nanoparticle nucleation in the two-solvent system. In a later step, though, the template must be removed to create the silica shell structure. Because the template is usually a salt, it is removed using strongly acidified alcohol, which adds an additional step to the overall synthesis. A similar method for nucleation has been employed to coat iron oxide nanoparticles with silica.11 Depending on the various reaction conditions, the pore size, surface area, size, and shape of the particles will change.101,134–137 Starting material concentration, catalyst concentration, and the solvent system also play an important role in the silica formation.134,138
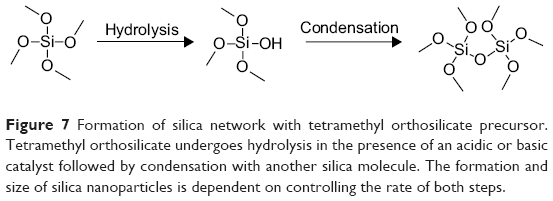 | Figure 7 Formation of silica network with tetramethyl orthosilicate precursor. Tetramethyl orthosilicate undergoes hydrolysis in the presence of an acidic or basic catalyst followed by condensation with another silica molecule. The formation and size of silica nanoparticles is dependent on controlling the rate of both steps. |
Different functional groups can be introduced on the surface of the silica by adding an additional precursor with a functional group of interest, which is known as the cocondensation method. Using this method, the functionality and application of silica nanoparticles has greatly improved. Organosiloxane is introduced into the original mixture or grafted onto the particles in a later reaction. In grafting reactions, it is possible that the functional groups will become part of the silica network or nucleate, creating new nanoparticles.137,139 Therefore, not only does grafting create a separate step in synthesis of the nanoparticles, but it may also create diverse populations of nanoparticles in one mixture. This adds both time and cost to the overall process if the particles must fit within a defined range of criteria. Otherwise, organosiloxanes come with a variety of groups, such as carboxylic acids, amines, and polymers, which are especially useful for biological tethering.
PEG is a biocompatible polymer that is commonly used for a “stealth” layer on nanoparticles.138,140–143 PEG prevents protein adsorption on the surface of nanoparticles during circulation (opsonization), which “hides” the nanoparticles from attack by immune cells. It can be combined with silica to improve aqueous stability and retention, and to reduce toxicity both in vivo and in vitro.141,143–145 Similar to other reaction conditions, even the amount of PEG seems to affect the size of silica nanoparticles.141 Like organosiloxanes, PEG is available in numerous sizes and may have a number of functional moieties for various applications. Further, PEG provides additional targeting for both localization and therapy for SCI applications. Local injection of PEG helped to alleviate systems of oxidative stress in neuronal cells by aiding in the repair of damage to the membrane.45
Cytotoxicity has been a concern with silica nanoparticles. Studies using these nanoparticles have found that both very small and very large particles tend to create toxicity problems. On the other hand, particles in the submicron range (50–300 nm) show less toxicity.99,100,146 In in vivo studies, there are mixed results. Table 3 shows a few often-cited studies on the toxicity of silica nanoparticles. For comparison purposes, this table only highlights single-dose toxicity studies in mice. Hudson et al found their particles to be the most toxic using both intraperitoneal and intravenous injections. Based on particle characterization, their results seem to be attributed to the dose and size of their particles. Although all the studies used both dynamic light scattering (DLS) and electron microscopy to confirm and corroborate their size results, Hudson et al found disparities in the size of nanoparticles achieved using each method.
 | Table 3 In vivo studies of silica nanoparticle toxicity |
DLS derives the radius a particle appears to have in solution. Components of a solvent or buffer form a layer that moves with the nanoparticle, forming the hydrodynamic radius. As a particle moves through solution (Brownian motion), it scatters light that can be collected. A large particle moves more slowly and scatters less light, while a smaller particle moves more quickly in comparison.147 Obviously, since the particles are moving in a fluid, the solvent properties, specifically viscosity, change their motion in the fluid system. On electron microscopy, the particles appear dried and may be sputtered before being imaged, which can cause aggregation.148 Therefore, DLS is likely a truer measurement of how silica nanoparticles behave in a fluid biological system. Hudson et al had very large particles (470±252 nm) based on DLS measurements, while scanning electron microscopy showed particles of only 100–150 nm. In their preparation for DLS, they reported that the samples were sonicated and vortexed for 2 hours prior to measurement, which is a good indication that their particles were probably both large and aggregated since such forces were needed to separate them. Further, they mentioned that, even after preparation, the particles would fall out of suspension.149 The size of the particles, coupled with the large dose, likely contributed to the erratic behavior and mortality of the mice used in their study. Granted, both Hudson et al and Liu et al found that high doses (>1,000 mg/kg) of the nanoparticles had some toxic effects.
Integrating neurological disease and nanomedicine
Methylprednisolone and inflammation
Because methylprednisolone is already used to treat SCI, various groups have attempted to improve its local delivery with the goal of circumventing its side effects and improving efficacy. An early study used poly(lactic-co-glycolic acid)-based nanoparticles and observed 65% encapsulation efficiency.151 The study showed improvements over methylprednisolone alone in vivothrough reduction of the lesion site, but the large nanoparticles (200–700 nm) exhibited a burst release and stopped releasing after 4 days in a saline-based experiment. Another group used smaller (~109 nm) carboxymethylchitosan/polyamidoamine dendrimer nanoparticles and were able to observe release for 14 days after an initial burst within the first 24 hours.152 The study’s in vivo results showed an improved locomotor score with the methylprednisolone-loaded nanoparticles that was not seen with methylprednisolone alone.
In models of multiple sclerosis and ocular inflammation, researchers constructed liposomal nanoparticles using a lipid-conjugated PEG, also conjugated with glutathione, and phosphatidyl choline to encapsulate a methylprednisolone prodrug.153,154 The approach uses both the delivery capability of the nanoparticles and their ability to reduce inflammation. Glutathione serves as a target and antioxidant, and the authors found improved crossing across the blood-brain barrier when gluathione was conjugated to the liposomes compared with lipoposomes without gluathione when administered intravenously.153 In both studies, the group found an improvement in the in vivo inflammation models.
Antioxidants
Many antioxidants show promising results in vitro, but their translation in vivo is limited by short half-lives, limited efficacy, and metabolism. One of the major drives of nanomedicine is improving the delivery of compounds that already demonstrate efficacy but have limited capabilities. Ideally, nanomedicine could turn the “scrapping” of a promising therapeutic into “repackaging” a promising therapeutic, and this is particularly true for natural products and their synthetic derivatives.
Oxidative stress is a common pathology in various conditions. Therefore, the amount of natural and derived antioxidants being studied for various uses is quite extensive, including compounds from grapefruit, strawberries, and green tea. 35,155–157 Dube et al packaged epigallocatechin gallate, a natural product in green tea, into chitosan nanoparticles to improve its bioavailability, which is known to be quite low.158 Another group attempted to improve bioavailability via lipid-based nanoparticles for use in Alzheimer’s disease, and found a two-fold increase in bioavailability.159 Others have encapsulated epigallocatechin gallate and other polyphenols to study how the loaded nanoparticles affect their antitumor effects.160–164
The potential of both synthetic and natural antioxidants has been studied, but they are hindered by poor stability, availability, and/or delivery. The field of nanomedicine has great potential to improve these products beyond their current stage. Further, many of them are GRAS (“generally regarded as safe”) products, which removes the hurdle of needing a compound to be approved by regulatory agencies.
Targeting acrolein
Although antioxidants may help reduce oxidative stress, some researchers have focused particularly on massively destructive aldehydes, such as acrolein. Cho et al synthesized silica nanoparticles to deliver hydralazine to healthy cells that had been exposed to acrolein.76 They used a template-based synthesis of their silica nanoparticles, grafted an additional silica layer, and attached a PEG layer. In the end, the particles were around 126 nm. They were able to deliver hydralazine to PC-12 cells (rat neurons) over a period of 5 days in vitro but not in a linear fashion. The bulk of the drug was released in the first few hours. Based on numerous cell assays that measure cell viability and therapeutic activity, they concluded that their nanoparticles were able to rescue the neurons without inducing further toxicity. In a separate study, they used similar nanoparticles ex vivoafter crushing the spinal cord.165 The particles did not contain hydralazine for scavenging of acrolein and appeared to recover their ability to conduct action potentials nearly to the precrush level. They speculate that the recovery originated from the PEG coating on the outside of the nanoparticles.
Later, Cho et al attempted to use chitosan-based nanoparticles to study acrolein scavenging. Hydralazine was encapsulated by the assembled nanoparticles, which had a diameter of around 350 nm. They found that the chitosan nanoparticles had lower encapsulation efficiency, but they were still able to decrease the death of healthy cells exposed to acrolein.89 Tysseling-Mattiace et al used self-assembling peptides to try to promote axonal regeneration after SCI. Their mice improved in motor capabilities and they found decreased apoptosis in and around the injury site.166
Conclusion
Upon devising an appropriate strategy for treating SCI with nanomedicine, the first method of delivery will likely require direct administration near a patient’s site of injury, which is the current standard for methylprednisolone. Using nanomaterials that cross the blood–brain barrier without compromising its integrity would be an ideal strategy. Such an approach may then prove useful for other neural injuries and disease without a major overhaul of the nanomedical system. Further, increasing the bioavailability of a therapeutic in general has direct benefits for many products with the same limitation. Knowing the pathology of SCI leads to integrating stress sensors or switches into nanoparticles to ensure they deliver their payload to the appropriate cells. Unlike many biological traumas, cells damaged in SCI cannot easily keep nanoparticles out of a cell, but they do not necessarily contain clear biological markers for targeting, as in other conditions.
Using nanoparticles to address some of the secondary injury problems demonstrates the potential of using a nanomedical approach, but both the delivery systems and therapeutics require improvement. In terms of secondary SCI, many of the problems have been identified, but the optimal treatment has yet to be determined, and methylprednisolone, even with its problems and side effects, is the only treatment in clinical use specifically for SCI. Therefore, attempting to mitigate its side effects using nanomedicine has a distinct advantage in terms of pushing both the treatment of SCI and nanomedicine forward. On the other hand, the need for better treatments is self-evident, but those treatments are waylaid by a lack of clinical results. The research route of administering an unapproved treatment via nanomedicine is decidedly difficult but likely promising in clinical translation.
Finally, scientists have much work in convincing government agencies, the pharmaceutical industry, and the public regarding the toxicity of nanotechnology in a general sense. Beyond that, regulatory agencies and industry must then be convinced that nanomedical systems are beneficial to expand the current research efforts and drive improvements in the technology through research.
Acknowledgments
This work was supported by an NSF Graduate Research Fellowship (DGE-1333468) to DW-S, Endowed Professorship funds to JFL, and a grant from the National Institutes of Health (NS073636) to RS.
Disclosure
The authors report no conflicts of interest in this work.
References
Burke D, Linden R, Zhang Y, Maiste A, Shields C. Incidence rates and populations at risk for spinal cord injury: a regional study. Spinal Cord. 2001;39:274–278. | ||
Hall ED, Braughler JM. Central nervous system trauma and stroke: II. Physiological and pharmacological evidence for involvement of oxygen radicals and lipid peroxidation. Free Radic Biol Med. 1989;6:303–313. | ||
Hamann K, Shi R. Acrolein scavenging: a potential novel mechanism of attenuating oxidative stress following spinal cord injury. J Neurochem. 2009;111:1348–1356. | ||
Haglund E, Seale-Goldsmith M-M, Leary JF. Design of multifunctional nanomedical systems. Ann Biomed Eng. 2009;37:2048–2063. | ||
Seale-Goldsmith M-M, Leary JF. Nanobiosystems. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2009;1:553–567. | ||
Leary JF. Nanotechnology: what is it and why is small so big? Can J Ophthalmol. 2010;45:449–456. | ||
Leary JF. Nanomedicine – reality will trump hype! J Nanomedicine Biotherapeutic Discov. 2013;4(1):e125. | ||
Avgoustakis K, Beletsi A, Panagi Z, Klepetsanis P, Karydas A, Ithakissios D. PLGA-mPEG nanoparticles of cisplatin: in vitro nanoparticle degradation, in vitro drug release and in vivo drug residence in blood properties. J Control Release. 2002;79:123–135. | ||
Niidome T, Yamagata M, Okamoto Y, et al. PEG-modified gold nanorods with a stealth character for in vivo applications. J Control Release. 2006;114:343–347. | ||
Sashiwa H, Yajima H, Aiba S. Synthesis of a chitosan-dendrimer hybrid and its biodegradation. Biomacromolecules. 2003;4:1244–1249. | ||
Zhang C, Wängler B, Morgenstern B, et al. Silica- and alkoxysilane-coated ultrasmall superparamagnetic iron oxide particles: a promising tool to label cells for magnetic resonance imaging. Langmuir. 2006;23:1427–1434. | ||
Cafferty WB, Duffy P, Huebner E, Strittmatter SM. MAG and OMgp synergize with Nogo-A to restrict axonal growth and neurological recovery after spinal cord trauma. J Neurosci. 2010;30:6825–6837. | ||
GrandPre T, Nakamura F, Vartanian T, Strittmatter SM. Identification of the Nogo inhibitor of axon regeneration as a Reticulon protein. Nature. 2000;403:439–444. | ||
Kottis V, Thibault P, Mikol D, et al. Oligodendrocyte-myelin glycoprotein (OMgp) is an inhibitor of neurite outgrowth. J Neurochem. 2002;82:1566–1569. | ||
Mukhopadhyay G, Doherty P, Walsh FS, Crocker PR, Filbin MT. A novel role for myelin-associated glycoprotein as an inhibitor of axonal regeneration. Neuron. 1994;13:757–767. | ||
Winzeler AM, Mandemakers WJ, Sun MZ, Stafford M, Phillips CT, Barres BA. The lipid sulfatide is a novel myelin-associated inhibitor of CNS axon outgrowth. J Neurosci. 2011;31:6481–6492. | ||
Ide C, Tohyama K, Yokota R, Nitatori T, Onodera S. Schwann cell basal lamina and nerve regeneration. Brain Res. 1983;288:61–75. | ||
Son Y-J, Thompson WJ. Schwann cell processes guide regeneration of peripheral axons. Neuron. 1995;14:125–132. | ||
Xu XM, Chen A, Guenard V, Kleitman N, Bunge MB. Bridging Schwann cell transplants promote axonal regeneration from both the rostral and caudal stumps of transected adult rat spinal cord. J Neurocytol. 1997;26:1–16. | ||
Simon CM, Sharif S, Tan RP, LaPlaca MC. Spinal cord contusion causes acute plasma membrane damage. J Neurotrauma. 2009;26:563–574. | ||
Hall J, Guyton A. Organization of the nervous system, basic functions of synapses, and neurotransmitters. In: Guyton and Hall Textbook of Medical Physiology. 12th ed. Philadelphia, PA, USA: Saunders/Elsevier; 2011. | ||
Bhat MA, Rios JC, Lu Y, et al. Axon-glia interactions and the domain organization of myelinated axons requires neurexin IV/Caspr/Paranodin. Neuron. 2001;30:369–383. | ||
Sasaki M. Molecular reconstruction of nodes of Ranvier after remyelination by transplanted olfactory ensheathing cells in the demyelinated spinal cord. J Neurosci. 2006;26:1803–1812. | ||
Olsson T. White matter disease: roles of anti-MOG antibodies in demyelinating diseases. Nat Rev Neurol. 2011;7:248–249. | ||
Wallström E, Khademi M, Andersson M, Weissert R, Linington C, Olsson T. Increased reactivity to myelin oligodendrocyte glycoprotein peptides and epitope mapping in HLA DR2(15)+ multiple sclerosis. Eur J Immunol. 1998;28:3329–3335. | ||
Metz J, Kelly A, Swett VC, Waxman S, Herbert V. Deranged DNA synthesis by bone marrow from vitamin B12-deficient humans. Br J Haematol. 1968;14:575–592. | ||
Radomski MW, Palmer RM, Moncada S. The anti-aggregating properties of vascular endothelium: interactions between prostacyclin and nitric oxide. Br J Pharmacol. 1987;92:639–646. | ||
Thorne KJ, Svvennsen RJ, Franks D. Role of hydrogen peroxide in the cytotoxic reaction of T lymphocytes. Clin Exp Immunol. 1980;39:486–495. | ||
White AA, Crawford KM, Patt CS, Lad PJ. Activation of soluble guanylate cyclase from rat lung by incubation or by hydrogen peroxide. J Biol Chem. 1976;251:7304–7312. | ||
Wollowitz S, Halpern J. Free radical rearrangement involving the 1,2-migration of a thioester group. Model for the coenzyme B12-dependent methylmalonyl-CoA mutase reaction. J Am Chem Soc. 1984;106:8319–8321. | ||
Scott GS, Cuzzocrea S, Genovese T, Koprowski H, Hooper DC. Uric acid protects against secondary damage after spinal cord injury. Proc Natl Acad Sci U S A. 2005;102:3483–3488. | ||
Hamann K, Durkes A, Ouyang H, Uchida K, Pond A, Shi R. Critical role of acrolein in secondary injury following ex vivo spinal cord trauma. J Neurochem. 2008;107:712–721. | ||
Lopez-Vales R, Redensek A, Skinner TAA, et al. Fenretinide promotes functional recovery and tissue protection after spinal cord contusion injury in mice. J Neurosci. 2010;30:3220–3226. | ||
Markowitz AJ, White MG, Kolson DL, Jordan-Sciutto KL. Cellular interplay between neurons and glia: toward a comprehensive mechanism for excitotoxic neuronal loss in neurodegeneration. Cellscience. 2007;4:111–146. | ||
Zheng LT, Ock J, Kwon B-M, Suk K. Suppressive effects of flavonoid fisetin on lipopolysaccharide-induced microglial activation and neurotoxicity. Int Immunopharmacol. 2008;8:484–494. | ||
Hamann K, Nehrt G, Ouyang H, Duerstock B, Shi R. Hydralazine inhibits compression and acrolein-mediated injuries in ex vivo spinal cord. J Neurochem. 2008;104:708–718. | ||
Luo J, Uchida K, Shi R. Accumulation of acrolein-protein adducts after traumatic spinal cord injury. Neurochem Res. 2005;30:291–295. | ||
Liu-Snyder P, McNally H, Shi R, Borgens RB. Acrolein-mediated mechanisms of neuronal death. J Neurosci Res. 2006;84:209–218. | ||
Luo J, Shi R. Acrolein induces axolemmal disruption, oxidative stress, and mitochondrial impairment in spinal cord tissue. Neurochem Int. 2004;44:475–486. | ||
Shi R, Luo J, Peasley M. Acrolein inflicts axonal membrane disruption and conduction loss in isolated guinea-pig spinal cord. Neuroscience. 2002;115:337–340. | ||
Uchida K, Kanematsu M, Morimitsu Y, Osawa T, Noguchi N, Niki E. Acrolein is a product of lipid peroxidation reaction. J Biol Chem. 1998;273:16058–16066. | ||
Ayer HE, Yeager DW. Irritants in cigarette smoke plumes. Am J Public Health. 1982;72:1283–1285. | ||
Mottram DS, Wedzicha BL, Dodson AT. Food chemistry: acrylamide is formed in the Maillard reaction. Nature. 2002;419:448–449. | ||
Tsai C-J, Chen M-L, Chang K-F, Chang F-K, Mao I-F. The pollution characteristics of odor, volatile organochlorinated compounds and polycyclic aromatic hydrocarbons emitted from plastic waste recycling plants. Chemosphere. 2009;74:1104–1110. | ||
Liu-Snyder P, Logan MP, Shi R, Smith DT, Borgens RB. Neuroprotection from secondary injury by polyethylene glycol requires its internalization. J Exp Biol. 2007;210:1455–1462. | ||
Zitting A, Heinonen T. Decrease of reduced glutathione in isolated rat hepatocytes caused by acrolein, acrylonitrile, and the thermal degradation products of styrene copolymers. Toxicology. 1980;17:333–341. | ||
Silverman RB. The Organic Chemistry of Enzyme-Catalyzed Reactions. San Diego, CA, USA: Academic Press; 2002. | ||
Betts R, Weinsheimer S, Blouse GE, Anagli J. Structural determinants of the calpain inhibitory activity of calpastatin peptide B27-WT. J Biol Chem. 2003;278:7800–7809. | ||
Tompa P, Buzder-Lantos P, Tantos A, et al. On the sequential determinants of calpain cleavage. J Biol Chem. 2004;279:20775–20785. | ||
Crocker SJ, Smith PD, Jackson-Lewis V, et al. Inhibition of calpains prevents neuronal and behavioral deficits in an MPTP mouse model of Parkinson’s disease. J Neurosci. 2003;23:4081–4091. | ||
Ray SK, Fidan M, Nowak MW, Wilford GG, Hogan EL, Banik NL. Oxidative stress and Ca2+ influx upregulate calpain and induce apoptosis in PC12 cells. Brain Res. 2000;852:326–334. | ||
Ray SK, Matzelle DD, Wilford GG, Hogan EL, Banik NL. Increased calpain expression is associated with apoptosis in rat spinal cord injury: calpain inhibitor provides neuroprotection. Neurochem Res. 2000;25:1191–1198. | ||
Shields DC, Tyor WR, Deibler GE, Hogan EL, Banik NL. Increased calpain expression in activated glial and inflammatory cells in experimental allergic encephalomyelitis. Proc Natl Acad Sci U S A. 1998;95:5768–5772. | ||
Squier MK, Sehnert AJ, Sellins KS, Malkinson AM, Takano E, Cohen JJ. Calpain and calpastatin regulate neutrophil apoptosis. J Cell Physiol. 1999;178:311–319. | ||
Trinchese F, Fa’ M, Liu S, et al. Inhibition of calpains improves memory and synaptic transmission in a mouse model of Alzheimer disease. J Clin Invest. 2008;118:2796–2807. | ||
Averna M, De Tullio R, Capini P, Salamino F, Pontremoli S, Melloni E. Changes in calpastatin localization and expression during calpain activation: a new mechanism for the regulation of intracellular Ca2+-dependent proteolysis. Cell Mol Life Sci. 2003;60:2669–2678. | ||
Murachi T, Tanaka K, Hatanaka M, Murakami T. Intracellular Ca2+-dependent protease (CALPAIN) and its high-molecular-weight endogenous inhibitor (CALPASTATIN). Adv Enzyme Regul. 1981;19:407–424. | ||
Sasaki T, Kishi M, Saito M, et al. Inhibitory effect of di- and tripeptidyl aldehydes on calpains and cathepsins. J Enzyme Inhib Med Chem. 1990;3:195–201. | ||
Tsujinaka T, Kajiwara Y, Kambayashi J, et al. Synthesis of a new cell penetrating calpain inhibitor (calpeptin). Biochem Biophys Res Commun. 1988;153:1201–1208. | ||
Wang KK, Nath R, Posner A, et al. An alpha-mercaptoacrylic acid derivative is a selective nonpeptide cell-permeable calpain inhibitor and is neuroprotective. Proc Natl Acad Sci U S A. 1996;93:6687–6692. | ||
Wu H-Y, Tomizawa K, Matsushita M, Lu Y-F, Li S-T, Matsui H. Poly-arginine-fused calpastatin peptide, a living cell membrane-permeable and specific inhibitor for calpain. Neurosci Res. 2003;47:131–135. | ||
Donkor IO. Calpain inhibitors: a survey of compounds reported in the patent and scientific literature. Expert Opin Ther Pat. 2011;21:601–636. | ||
Maki M, Bagci H, Hamaguchi K, Ueda M, Murachi T, Hatanaka M. Inhibition of calpain by a synthetic oligopeptide corresponding to an exon of the human calpastatin gene. J Biol Chem. 1989;264:18866–18869. | ||
Hanna RA, Campbell RL, Davies PL. Calcium-bound structure of calpain and its mechanism of inhibition by calpastatin. Nature. 2008;456:409–412. | ||
Jiao W, McDonald DQ, Coxon JM, Parker EJ. Molecular modeling studies of peptide inhibitors highlight the importance of conformational prearrangement for inhibition of calpain. Biochemistry. 2010;49:5533–5539. | ||
Hugenholtz H, Cass DE, Dvorak MF, et al. High-dose methylprednisolone for acute closed spinal cord injury – only a treatment option. Can J Neurol Sci. 2002;29:227–235. | ||
Bracken MB, Shepard MJ, Collins WF, et al. A randomized, controlled trial of methylprednisolone or naloxone in the treatment of acute spinal-cord injury. N Engl J Med. 1990;322:1405–1411. | ||
Witschi A, Reddy S, Stofer B, Lauterburg B. The systemic availability of oral glutathione. Eur J Clin Pharmacol. 1992;43:667–669. | ||
Aebi S, Assereto R, Lauterburg BH. High-dose intravenous glutathione in man. Pharmacokinetics and effects on cyst(e)ine in plasma and urine. Eur J Clin Invest. 1991;21:103–110. | ||
Tateishi N, Higashi T, Naruse A, Nakashima K, Shiozaki H. Rat liver glutathione: possible role as a reservoir of cysteine. J Nutr. 1977;107:51–60. | ||
Das A, Sribnick EA, Wingrave JM, et al. Calpain activation in apoptosis of ventral spinal cord 4.1 (VSC4.1) motoneurons exposed to glutamate: calpain inhibition provides functional neuroprotection. J Neurosci Res. 2005;81:551–562. | ||
Guyton MK, Das A, Samantaray S, et al. Calpeptin attenuated inflammation, cell death, and axonal damage in animal model of multiple sclerosis. J Neurosci Res. 2010;88:2398–2408. | ||
Ray SK, Wilford GG, Matzelle DC, Hogan EL, Banik NL. Calpeptin and methylprednisolone inhibit apoptosis in rat spinal cord injury. Ann N Y Acad Sci. 1999;890:261–269. | ||
Yu C-G, Geddes J. Sustained calpain inhibition improves locomotor function and tissue sparing following contusive spinal cord injury. Neurochem Res. 2007;32:2046–2053. | ||
Liu-Snyder P, Borgens RB, Shi R. Hydralazine rescues PC12 cells from acrolein-mediated death. J Neurosci Res. 2006;84:219–227. | ||
Cho Y, Shi R, Borgens RB, Ivanisevic A. Functionalized mesoporous silica nanoparticle-based drug delivery system to rescue acrolein-mediated cell death. Nanomedicine (Lond). 2008;3:507–519. | ||
Leung G, Sun W, Zheng L, Brookes S, Tully M, Shi R. Anti-acrolein treatment improves behavioral outcome and alleviates myelin damage in experimental autoimmune encephalomyelitis mouse. Neuroscience. 2011;173:150–155. | ||
Leitch IM, Read MA, Boura AL, Walters WA. Effect of inhibition of nitric oxide synthase and guanylate cyclase on hydralazine-induced vasodilatation of the human fetal placental circulation. Clin Exp Pharmacol Physiol. 1994;21:615–622. | ||
Shen DD, Hosler JP, Schroder RL, Azarnoff DL. Pharmacokinetics of hydralazine and its acid-labile hydrazone metabolites in relation to acetylator phenotype. J Pharmacokinet Pharmacodyn. 1980;8:53–68. | ||
Cho Y, Shi R, Borgens RB. Chitosan produces potent neuroprotection and physiological recovery following traumatic spinal cord injury. J Exp Biol. 2010;213:1513–1520. | ||
Shahidi F, Synowiecki J. Isolation and characterization of nutrients and value-added products from snow crab (Chionoecetes opilio) and shrimp (Pandalus borealis) processing discards. J Agric Food Chem. 1991;39:1527–1532. | ||
Kolhe P, Kannan RM. Improvement in ductility of chitosan through blending and copolymerization with PEG: FTIR investigation of molecular interactions. Biomacromolecules. 2002;4:173–180. | ||
Mao S, Shuai X, Unger F, Wittmar M, Xie X, Kissel T. Synthesis, characterization and cytotoxicity of poly(ethylene glycol)-graft-trimethyl chitosan block copolymers. Biomaterials. 2005;26:6343–6356. | ||
Zhang M, Li XH, Gong YD, Zhao NM, Zhang XF. Properties and biocompatibility of chitosan films modified by blending with PEG. Biomaterials. 2002;23:2641–2648. | ||
De Campos AM, Sánchez A, Gref R, Calvo P, Alonso MJ. The effect of a PEG versus a chitosan coating on the interaction of drug colloidal carriers with the ocular mucosa. Eur J Pharm Sci. 2003;20:73–81. | ||
Abbott WA, Bridges RJ, Meister A. Extracellular metabolism of glutathione accounts for its disappearance from the basolateral circulation of the kidney. J Biol Chem. 1984;259:15393–15400. | ||
Barth J, Winkler J, Schumann R, et al. Population pharmacokinetics of methylprednisolone in accident victims with spinal cord injury. Int J Clin Pharmacol Ther. 2004;42:504–511. | ||
Smyth HF, Carpenter CP, Weil CS. The chronic oral toxicology of the polyethylene glycols. J Am Pharm Assoc. 1955;44:27–30. | ||
Cho Y, Shi R, Borgens RB. Chitosan nanoparticle-based neuronal membrane sealing and neuroprotection following acrolein-induced cell injury. J Biol Eng. 2010;4:2. | ||
Onishi H, Machida Y. Biodegradation and distribution of water-soluble chitosan in mice. Biomaterials. 1999;20:175–182. | ||
Bae YH, Park K. Targeted drug delivery to tumors: myths, reality and possibility. J Control Release. 2011;153:198–205. | ||
Farokhzad OC, Cheng J, Teply BA, et al. Targeted nanoparticle-aptamer bioconjugates for cancer chemotherapy in vivo. Proc Natl Acad Sci U S A. 2006;103:6315–6320. | ||
Key J, Dhawan D, Knapp DW, et al. Multimodal in vivo MRI and NIRF imaging of bladder tumor using peptide conjugated glycol chitosan nanoparticles. Proc SPIE. 2012;8225:82251F–82251F. | ||
Kukowska-Latallo JF, Candido KA, Cao Z, et al. Nanoparticle targeting of anticancer drug improves therapeutic response in animal model of human epithelial cancer. Cancer Res. 2005;65:5317–5324. | ||
Schiffelers RM, Ansari A, Xu J, et al. Cancer siRNA therapy by tumor selective delivery with ligand-targeted sterically stabilized nanoparticle. Nucleic Acids Res. 2004;32:e149. | ||
Yu D-H, Lu Q, Xie J, Fang C, Chen H-Z. Peptide-conjugated biodegradable nanoparticles as a carrier to target paclitaxel to tumor neovasculature. Biomaterials. 2010;31:2278–2292. | ||
He Q, Zhang Z, Gao F, Li Y, Shi J. In vivo biodistribution and urinary excretion of mesoporous silica nanoparticles: effects of particle size and PEGylation. Small. 2011;7:271–280. | ||
Huang X, Li L, Liu T, et al. The shape effect of mesoporous silica nanoparticles on biodistribution, clearance, and biocompatibility in vivo. ACS Nano. 2011;5:5390–5399. | ||
Corbalan JJ, Medina C, Jacoby A, Malinski T, Radomski MW. Amorphous silica nanoparticles trigger nitric oxide/peroxynitrite imbalance in human endothelial cells: inflammatory and cytotoxic effects. Int J Nanomedicine. 2011;6:2821–2835. | ||
Napierska D, Thomassen LC, Rabolli V, et al. Size-dependent cytotoxicity of monodisperse silica nanoparticles in human endothelial cells. Small. 2009;5:846–853. | ||
Nooney RI, Thirunavukkarasu D, Chen Y, Josephs R, Ostafin AE. Synthesis of nanoscale mesoporous silica spheres with controlled particle size. Chem Mater. 2002;14:4721–4728. | ||
Stöber W, Fink A, Bohn E. Controlled growth of monodisperse silica spheres in the micron size range. J Colloid Interface Sci. 1968;26:62–69. | ||
Connor EE, Mwamuka J, Gole A, Murphy CJ, Wyatt MD. Gold nanoparticles are taken up by human cells but do not cause acute cytotoxicity. Small. 2005;1:325–327. | ||
Elghanian R, Storhoff JJ, Mucic RC, Letsinger RL, Mirkin CA. Selective colorimetric detection of polynucleotides based on the distance-dependent optical properties of gold nanoparticles. Science. 1997;277:1078–1081. | ||
El-Sayed IH, Huang X, El-Sayed MA. Surface plasmon resonance scattering and absorption of anti-EGFR antibody conjugated gold nanoparticles in cancer diagnostics: applications in oral cancer. Nano Lett. 2005;5:829–834. | ||
Goodman CM, McCusker CD, Yilmaz T, Rotello VM. Toxicity of gold nanoparticles functionalized with cationic and anionic side chains. Bioconjug Chem. 2004;15:897–900. | ||
Storhoff JJ, Elghanian R, Mucic RC, Mirkin CA, Letsinger RL. One-pot colorimetric differentiation of polynucleotides with single base imperfections using gold nanoparticle probes. J Am Chem Soc. 1998;120:1959–1964. | ||
Ballou B, Lagerholm BC, Ernst LA, Bruchez MP, Waggoner AS. Noninvasive imaging of quantum dots in mice. Bioconjug Chem. 2003;15:79–86. | ||
Gao X, Cui Y, Levenson RM, Chung LW, Nie S. In vivo cancer targeting and imaging with semiconductor quantum dots. Nat Biotechnol. 2004;22:969–976. | ||
Hauck TS, Anderson RE, Fischer HC, Newbigging S, Chan WC. In vivo quantum-dot toxicity assessment. Small. 2010;6:138–144. | ||
Hoshino A, Fujioka K, Oku T, et al. Physicochemical properties and cellular toxicity of nanocrystal quantum dots depend on their surface modification. Nano Lett. 2004;4:2163–2169. | ||
Lovrić J, Bazzi HS, Cuie Y, Fortin GR, Winnik FM, Maysinger D. Differences in subcellular distribution and toxicity of green and red emitting CdTe quantum dots. J Mol Med. 2005;83:377–385. | ||
Yu WW, Chang E, Drezek R, Colvin VL. Water-soluble quantum dots for biomedical applications. Biochem Biophys Res Commun. 2006;348:781–786. | ||
Hyung Park J, Kwon S, Lee M, et al. Self-assembled nanoparticles based on glycol chitosan bearing hydrophobic moieties as carriers for doxorubicin: in vivo biodistribution and anti-tumor activity. Biomaterials. 2006;27:119–126. | ||
Min KH, Park K, Kim Y-S, et al. Hydrophobically modified glycol chitosan nanoparticles-encapsulated camptothecin enhance the drug stability and tumor targeting in cancer therapy. J Control Release. 2008;127:208–218. | ||
Xu P, Gullotti E, Tong L, et al. Intracellular drug delivery by poly(lactic-co-glycolic acid) nanoparticles, revisited. Mol Pharm. 2008;6:190–201. | ||
Ku S, Yan F, Wang Y, Sun Y, Yang N, Ye L. The blood–brain barrier penetration and distribution of PEGylated fluorescein-doped magnetic silica nanoparticles in rat brain. Biochem Biophys Res Commun. 2010;394:871–876. | ||
Lu Y, Yin Y, Mayers BT, Xia Y. Modifying the surface properties of superparamagnetic iron oxide nanoparticles through a sol-gel approach. Nano Lett. 2002;2:183–186. | ||
Santra S, Tapec R, Theodoropoulou N, Dobson J, Hebard A, Tan W. Synthesis and characterization of silica-coated iron oxide nanoparticles in microemulsion: the effect of nonionic surfactants. Langmuir. 2001;17:2900–2906. | ||
Yu CH, Al-Saadi A, Shih S-J, Qiu L, Tam KY, Tsang SC. Immobilization of BSA on silica-coated magnetic iron oxide nanoparticle. J Phys Chem C. 2008;113:537–543. | ||
Arbab AS, Wilson LB, Ashari P, Jordan EK, Lewis BK, Frank JA. A model of lysosomal metabolism of dextran coated superparamagnetic iron oxide (SPIO) nanoparticles: implications for cellular magnetic resonance imaging. NMR Biomed. 2005;18:383–389. | ||
Mahmoudi M, Simchi A, Imani M, Milani AS, Stroeve P. An in vitro study of bare and poly(ethylene glycol)-co-fumarate-coated superparamagnetic iron oxide nanoparticles: a new toxicity identification procedure. Nanotechnology. 2009;20:225104. | ||
Pisanic TR 2nd, Blackwell JD, Shubayev VI, Fiñones RR, Jin S. Nanotoxicity of iron oxide nanoparticle internalization in growing neurons. Biomaterials. 2007;28:2572–2581. | ||
Talelli M, Rijcken CJF, Lammers T, et al. Superparamagnetic iron oxide nanoparticles encapsulated in biodegradable thermosensitive polymeric micelles: toward a targeted nanomedicine suitable for image-guided drug delivery. Langmuir. 2009;25:2060–2067. | ||
Xu Z, Wang C, Yang W, Deng Y, Fu S. Encapsulation of nanosized magnetic iron oxide by polyacrylamide via inverse miniemulsion polymerization. J Magn Magn Mater. 2004;277:136–143. | ||
Yu MK, Jeong YY, Park J, et al. Drug-loaded superparamagnetic iron oxide nanoparticles for combined cancer imaging and therapy in vivo. Angew Chem Int Ed. 2008;47:5362–5365. | ||
Zhang Z, Feng S-S. The drug encapsulation efficiency, in vitro drug release, cellular uptake and cytotoxicity of paclitaxel-loaded poly(lactide)-tocopheryl polyethylene glycol succinate nanoparticles. Biomaterials. 2006;27:4025–4033. | ||
Gulsen D, Chauhan A. Ophthalmic drug delivery through contact lenses. Invest Ophthalmol Vis Sci. 2004;45:2342–2347. | ||
Gulsen D, Chauhan A. Dispersion of microemulsion drops in HEMA hydrogel: a potential ophthalmic drug delivery vehicle. Int J Pharm. 2005;292:95–117. | ||
Kim J, Conway A, Chauhan A. Extended delivery of ophthalmic drugs by silicone hydrogel contact lenses. Biomaterials. 2008;29:2259–2269. | ||
Peng C-C, Chauhan A. Extended cyclosporine delivery by silicone-hydrogel contact lenses. J Control Release. 2011;154:267–274. | ||
Yang J, Lee J, Kang J, et al. Hollow silica nanocontainers as drug delivery vehicles. Langmuir. 2008;24:3417–3421. | ||
Finnie KS, Bartlett JR, Barbé CJ, Kong L. Formation of silica nanoparticles in microemulsions. Langmuir. 2007;23:3017–3024. | ||
Cai Q, Luo Z-S, Pang W-Q, Fan Y-W, Chen X-H, Cui F-Z. Dilute solution routes to various controllable morphologies of MCM-41 silica with a basic medium. Chem Mater. 2001;13:258–263. | ||
Huh S, Wiench JW, Yoo J-C, Pruski M, Lin VS-Y. Organic functionalization and morphology control of mesoporous silicas via a co-condensation synthesis method. Chem Mater. 2003;15:4247–4256. | ||
Green D, Lin J, Lam Y-F, Hu MZ-C, Schaefer DW, Harris M. Size, volume fraction, and nucleation of Stober silica nanoparticles. J Colloid Interface Sci. 2003;266:346–358. | ||
Sadasivan S, Khushalani D, Mann S. Synthesis and shape modification of organo-functionalised silica nanoparticles with ordered mesostructured interiors. J Mater Chem. 2003;13:1023–1029. | ||
Xu H, Yan F, Monson EE, Kopelman R. Room-temperature preparation and characterization of poly(ethylene glycol)-coated silica nanoparticles for biomedical applications. J Biomed Mater Res A. 2003;66A:870–879. | ||
Radu DR, Lai C-Y, Huang J, Shu X, Lin VS-Y. Fine-tuning the degree of organic functionalization of mesoporous silica nanosphere materials via an interfacially designed co-condensation method. Chem Commun (Camb). 2005;10:1264–1266. | ||
Bagwe RP, Hilliard LR, Tan W. Surface modification of silica nanoparticles to reduce aggregation and nonspecific binding. Langmuir. 2006;22:4357–4362. | ||
He Q, Zhang J, Shi J, et al. The effect of PEGylation of mesoporous silica nanoparticles on nonspecific binding of serum proteins and cellular responses. Biomaterials. 2010;31:1085–1092. | ||
Lesot P, Chapuis S, Bayle JP, et al. Structural-dynamical relationship in silica PEG hybrid gels. J Mater Chem. 1998;8:147–151. | ||
Peracchia M, Fattal E, Desmaële D, et al. Stealth® PEGylated polycyanoacrylate nanoparticles for intravenous administration and splenic targeting. J Control Release. 1999;60:121–128. | ||
Branda F, Silvestri B, Luciani G, Costantini A, Tescione F. Synthesis structure and stability of amino functionalized PEGylated silica nanoparticles. Colloids Surf Physicochem Eng Asp. 2010;367:12–16. | ||
Zhang Z, Berns AE, Willbold S, Buitenhuis J. Synthesis of poly(ethylene glycol) (PEG)-grafted colloidal silica particles with improved stability in aqueous solvents. J Colloid Interface Sci. 2007;310:446–455. | ||
Park J-H, Gu L, von Maltzahn G, Ruoslahti E, Bhatia SN, Sailor MJ. Biodegradable luminescent porous silicon nanoparticles for in vivo applications. Nat Mater. 2009;8:331–336. | ||
Pusey PN, Tough RJ. Dynamic light scattering, a probe of brownian particle dynamics. Adv Colloid Interface Sci. 1982;16:143–159. | ||
Berriozabal G, de Miguel YR. Synthesis and characterisation of silica nanoparticles bearing different functional groups obtained via a two-stage method. Phys Status Solidi C. 2010;7:2692–2696. | ||
Hudson S, Padera RF, Langer R, Kohane DS. The biocompatibility of mesoporous silicates. Biomaterials. 2008;29:4045–4055. | ||
Liu T, Li L, Teng X, et al. Single and repeated dose toxicity of mesoporous hollow silica nanoparticles in intravenously exposed mice. Biomaterials. 2011;32:1657–1668. | ||
Kim Y, Caldwell J-M, Bellamkonda RV. Nanoparticle-mediated local delivery of methylprednisolone after spinal cord injury. Biomaterials. 2009;30:2582–2590. | ||
Cerqueira SR, Oliveira JM, Silva NA, et al. Microglia response and in vivo therapeutic potential of methylprednisolone-loaded dendrimer nanoparticles in spinal cord injury. Small. 2013;9(5):738–749. | ||
Gaillard PJ, Appeldoorn CC, Rip J, et al. Enhanced brain delivery of liposomal methylprednisolone improved therapeutic efficacy in a model of neuroinflammation. J Control Release. 2012;164:364–369. | ||
Reijerkerk A, Appeldoorn CC, Rip J, Boer M de, Gaillard PJ. Systemic treatment with glutathione PEGylated liposomal methylprednisolone (2B3-201) improves therapeutic efficacy in a model of ocular inflammation. Invest Ophthalmol Vis Sci. 2014;55(4):2788–2794. | ||
Ansari MA, Keller JN, Scheff SW. Protective effect of Pycnogenol in human neuroblastoma SH-SY5Y cells following acrolein-induced cytotoxicity. Free Radic Biol Med. 2008;45:1510–1519. | ||
Singh M, Arseneault M, Sanderson T, Murthy V, Ramassamy C. Challenges for research on polyphenols from foods in Alzheimer’s disease: bioavailability, metabolism, and cellular and molecular mechanisms. J Agric Food Chem. 2008;56:4855–4873. | ||
Nam Y, Choi M, Hwang H, et al. Natural flavone jaceosidin is a neuroinflammation inhibitor. Phytother Res. 2013;27(3):404–411. | ||
Dube A, Nicolazzo JA, Larson I. Chitosan nanoparticles enhance the plasma exposure of (-)-epigallocatechin gallate in mice through an enhancement in intestinal stability. Eur J Pharm Sci. 2011;44:422–426. | ||
Smith A, Giunta B, Bickford PC, Fountain M, Tan J, Shytle RD. Nanolipidic particles improve the bioavailability and α-secretase inducing ability of epigallocatechin-3-gallate (EGCG) for the treatment of Alzheimer’s disease. Int J Pharm. 2010;389:207–212. | ||
Shutava TG, Balkundi SS, Vangala P, et al. Layer-by-layer-coated gelatin nanoparticles as a vehicle for delivery of natural polyphenols. ACS Nano. 2009;3:1877–1885. | ||
Sanna V, Pintus G, Roggio AM, et al. Targeted biocompatible nanoparticles for the delivery of (-)-epigallocatechin 3-gallate to prostate cancer cells. J Med Chem. 2011;54:1321–1332. | ||
Shutava TG, Lvov YM. Encapsulation of natural polyphenols with antioxidant properties in polyelectrolyte capsules and nanoparticles. In: Diederich M, Noworyta K, editors. Natural Compounds as Inducers of Cell Death. Dordrecht, The Netherlands: Springer; 2012. Available from: http://link.springer.com/chapter/10.1007/978-94-007-4575-9_9. Accessed November 21, 2013. | ||
Khan N, Bharali DJ, Adhami VM, et al. Oral administration of naturally occurring chitosan based nanoformulated green tea polyphenol EGCG effectively inhibits prostate cancer cell growth in a xenograft model. Carcinogenesis. 2013:bgt321. | ||
Lemarié F, Chang CW, Blatchford DR, et al. Antitumor activity of the tea polyphenol epigallocatechin-3-gallate encapsulated in targeted vesicles after intravenous administration. Nanomedicine (Lond). 2013;8:181–192. | ||
Cho Y, Shi R, Borgens R, Ivanisevic A. Repairing the damaged spinal cord and brain with nanomedicine. Small. 2008;4:1676–1681. | ||
Tysseling-Mattiace VM, Sahni V, Niece KL, et al. Self-Assembling nanofibers inhibit glial scar formation and promote axon elongation after spinal cord injury. J Neurosci. 2008;28:3814–3823. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.