")
Back to Journals » International Journal of Nanomedicine » Volume 11
Nanomedicine engulfed by macrophages for targeted tumor therapy
Authors Li S, Feng S, Ding L, Liu Y, Zhu Q, Qian Z, Gu Y
Received 8 April 2016
Accepted for publication 19 June 2016
Published 23 August 2016 Volume 2016:11 Pages 4107—4124
DOI https://doi.org/10.2147/IJN.S110146
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Lei Yang
Siwen Li,1,* Song Feng,1,* Li Ding,1 Yuxi Liu,1 Qiuyun Zhu,1 Zhiyu Qian,2 Yueqing Gu1
1Department of Biomedical Engineering, China Pharmaceutical University, 2Department of Biomedical Engineering, School of Automation, Nanjing University of Aeronautics and Astronautics, Nanjing, Jiangsu, People’s Republic of China
*These authors contributed equally to this work
Abstract: Macrophages, exhibiting high intrinsic accumulation and infiltration into tumor tissues, are a novel drug vehicle for directional drug delivery. However, the low drug-loading (DL) capacity and the drug cytotoxicity to the cell vehicle have limited the application of macrophages in tumor therapy. In this study, different drugs involving small molecular and nanoparticle drugs were loaded into intrinsic macrophages to find a better way to overcome these limitations. Their DL capacity and cytotoxicity to the macrophages were first compared. Furthermore, their phagocytic ratio, dynamic distributions, and tumoricidal effects were also investigated. Results indicated that more lipid-soluble molecules and DL particles can be phagocytized by macrophages than hydrophilic ones. In addition, the N-succinyl-N'-octyl chitosan (SOC) DL particles showed low cytotoxicity to the macrophage itself, while the dynamic biodistribution of macrophages engulfed with different particles/small molecules showed similar profiles, mainly excreted from liver to intestine pathway. Furthermore, macrophages loaded with SOC–paclitaxel (PTX) particles exhibited greater therapeutic efficacies than those of macrophages directly carrying small molecular drugs such as doxorubicin and PTX. Interestingly, macrophages displayed stronger targeting ability to the tumor site hypersecreting chemokine in immunocompetent mice in comparison to the tumor site secreting low levels of chemokine in immunodeficiency mice. Finally, results demonstrated that macrophages carrying SOC–PTX are a promising pharmaceutical preparation for tumor-targeted therapy.
Keywords: macrophage, drug-loading capacity, SOC–PTX, tumor-targeted therapy
Introduction
Targeted delivery of chemotherapeutic drugs to tumor sites is a major challenge in cancer chemotherapy. The use of drug delivery systems such as nanoparticles can improve the pharmacologic properties of traditional chemotherapeutics by altering drug pharmacokinetics and biodistribution.1,2 Nonetheless, these drug-loaded nanoparticles have two key shortcomings. First, these nanometer materials, as exogenous components, are usually toxic and may cause adverse effects. Second, the rapid recognition and clearance of nanoparticles from the blood stream by the reticuloendothelial system limits their usefulness as drug carriers.3 Therefore, a wide variety of alternative drug delivery systems are currently being studied by researchers around the world, using intrinsic functional cells in living subjects as drug vehicles.4–7
As reported, various kinds of cells, including mesenchymal stem cells,4,5 tumor cells,6 and natural killer cells,7 can effectively deliver chemotherapeutic drug-loaded nanoparticles to the tumor because of the homing ability of these cells, leading to improved therapeutic efficacy. However, the drug-loading (DL) efficiency of these cells was restricted largely due to the high cytotoxicity of drugs. Macrophage as an intrinsic cell exhibits a strong tolerance of anticancer drugs and unique phagocytic capacity due to its specific immune properties.8 Furthermore, macrophage possesses a quick directional migration ability to the pathological sites secreting specific chemokine factors, which allows it to serve as a vehicle for the targeted delivery of drugs.9 Solid tumor is a high chemokine-secreting and percolating tissue, which allows the macrophage to directionally accumulate and immerse into the tumor environment.10 These merits of macrophage encourage us to use it as an ideal intrinsic drug carrier.
Macrophage plays an important physiological function in removing foreign materials, cellular debris, and pathogens from circulation.11 A number of chemokines, such as CCL2 and CCL5, are believed to be hypersecreted in tumor sites, ensuring the recruitment of macrophages to the tumor sites.12 Up to date, most studies have used macrophages as cellular vehicles for gene therapy.13–15 A limited number of studies have demonstrated the feasibility of delivering drug-loaded particles or anticancer drugs themselves to tumors using macrophages, including carrying gold nanorod to target breast cancer cells,16 carrying adenovirus to prostate tumors,17 carrying gold nanoshells to gliomas,18 and carrying doxorubicin (DOX) hydrochloride directly to breast tumor.19 However, until now, no drug-loaded macrophages have displayed excellent antitumor efficacy due to the low DL capacity. Furthermore, the high DL capacity may destroy the cell vehicle, resulting in the decrease of homing ability. To our knowledge, no research has been carried out to compare the DL capacity of macrophages with different drugs and drug-loaded nanoparticles. Most importantly, no study has been reported regarding the cytotoxicity of engulfed contents to the macrophages. These unknown key issues motivated us to systematically investigate the DL capacity and cytotoxicity of macrophages.
Therefore, in this study, we used macrophages to deliver chemotherapeutic drugs directly or as drug-loaded particles to tumor cells. The five model drugs with different solubility, Cypate (poorly soluble), fluorescein (slightly soluble), paclitaxel (slightly soluble), DOX hydrochloride (soluble), and fluorescein sodium (highly soluble), were encapsulated into macrophages to compare the achievable DL and encapsulation efficiencies. N-succinyl-N′-octyl Chitosan (SOC) and nanostructured lipid carriers (NLCs),20–23 consisting of polysaccharide and partial crystallized lipid particles are useful delivery systems with high DL efficiency, encapsulation efficiency (EE), and stability. By possessing high cell permeability, these nanoparticles can effectively improve the bioavailability of drugs with poor water solubility. Their uptake and release in the macrophage drug-carrying system were compared with small molecules. The dynamic behavior and tumor-targeting capability of drug-loaded macrophages were investigated. Finally, the therapeutic efficacies of the different macrophage–drug delivery systems were investigated and compared. The schematic illustration is shown in Figure 1.
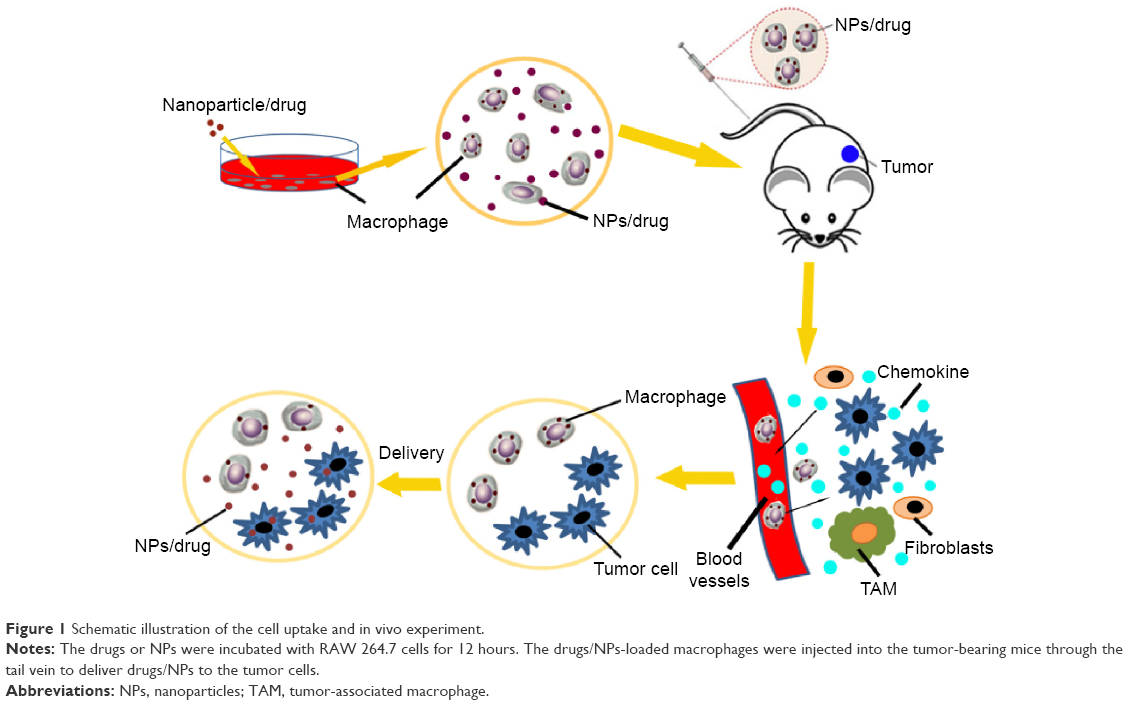 | Figure 1 Schematic illustration of the cell uptake and in vivo experiment. |
Experimental section
Materials
Hydrophobic near-infrared (NIR) dye ICG-Der-01 (Cypate) was prepared in our laboratory.24,25 L-15, Dulbecco’s Modified Eagle’s Medium (DMEM), fetal bovine serum, penicillin, streptomycin, trypsin–ethylenediaminetetraacetic acid, and Hoechst 33342 were purchased from Thermo Fisher Scientific (Waltham, MA, USA). All other reagents used in the study were certified analytical reagent grade (Shanghai Chemical Reagent Company, Shanghai, People’s Republic of China). All chemicals were analytical grade and used without further purification.
In vitro studies
Cell cultures
The cell lines Raw 264.7 (Mus musculus macrophage), MDA-MB-231 (human breast cancer), and MCF-7 (human breast adenocarcinoma) were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). All the cell lines were cultured in DMEM supplemented with 10% (v/v) calf serum, penicillin (80 U/mL), and (0.08 mg/mL) streptomycin. Cell lines were maintained at 37°C in a humidified atmosphere containing 5% CO2.
Loading and encapsulation of drugs directly swallowed by macrophage
Raw 264.7 cells were treated with each drug (100–800 μmol/mL) for 24 hours. Then, the medium was collected and measured by ultraviolet spectrophotometry. Five model drugs with different solubility, lipid soluble (PTX, Cypate, and fluorescein) dissolved in dimethyl sulfoxide, water soluble (DOX and fluorescein sodium) dissolved in water, and two nanoparticle drug carriers, SOC and NLCs (the synthesis and characteristics of SOC and NCL particles were reported in our previous work.1,26), were used to determine the DL concentration (μmol/104 cells) and EE by Lambda 25 UV/Vis Spectrophotometer (PerkinElmer Inc., Waltham, MA, USA), at λ=227 nm (PTX), 490 nm (fluorescein and fluorescein sodium), 520 nm (DOX), and 793 nm (Cypate). The concentration of each drug was calculated using calibration curve. DL and EE were calculated according to Equations 1 and 2, respectively.
|
|
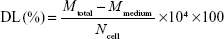
|
|
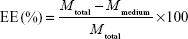
where Mtotal, Mmedium, and Ncell are the total amount of drugs, the amount of drugs in culture mediums, and the total cell counts, respectively. The total cell counts were measured by using cell counting chamber.
In vitro drug release
Kinetic analysis of each drug release was performed as follows: 5 mL of fresh cell culture medium was added into the culture dish of drug-loaded macrophages (104 cells). At regular time intervals (0.5 hours, 1 hour, 2 hours, 4 hours, 6 hours, 8 hours, 12 hours, and 24 hours), 0.1 mL of samples were collected and replaced by an equal volume of the medium. The amount of drug released into the medium was calculated according to an absorption standard curve at 227 nm, 470 nm, 520 nm, and 793 nm. Each releasing experiment was performed in triplicate (n=3).
Drug uptake by macrophages
Macrophage cells, Raw 264.7, were in the confocal dishes at a density of 3×105 cells/dish. After 12 hours of cell attachment, 100 μL of nanoparticle solution (containing 1 mg/mL nanoparticles and same fluorescence intensity) was added in the culture medium and incubated for different times (0.5 hours, 2 hours, 6 hours, and 12 hours). Cell nuclei were stained with Hoechst 33342 (10 μg/mL) for 30 minutes. After washing three times with phosphate-buffered saline (PBS), the cells were imaged by a laser confocal microscope (Olympus FV1100, Japan).
Flow cytometer (BD™ LSR II, BD, USA) was used to detect the mean fluorescent intensity of the cells, thus allowing a quantitative determination of the particle uptake ability of macrophage. The slope values of the uptake/time profiles were calculated to indicate the uptake speed.
Drug cytotoxicity to the nanoparticle carrier assay
PTX (100 μmol/L) and SOC–PTX (100 μmol/L) were added to the cultured macrophages in 96-well plates (n=6). The cells were observed under a microscope after 6 hours of co-incubation, and then the medium was replaced with new one. Afterward, tumor cell viability assays were carried out by 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT) assay after co-incubating for 24 hours. Meanwhile, nontreated macrophages were cultured as control (n=6). MTT solution was added into each well after incubation, and the absorbance of the solution in each well was measured at 490 nm with a multi-well plate reader. The cell viability was calculated as follows:
|
|
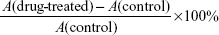
where A represents the absorption of MTT.
In vitro antitumor activity assessment of nanoparticles/drug-loaded macrophages
DOX-loaded macrophages (at the maximal loading of 1.25 μg/mL DOX, respectively, loaded in 3×104/mL macrophages), PTX- and SOC–PTX-loaded macrophages (at the maximal loading of 1.6 μg/mL and 2.2 μg/mL PTX, respectively, loaded in 3×104/mL macrophages), and non-loaded macrophages (3×104/mL) were added to the cultured tumor cells (MCF-7 and MDA-MB-231) in 96-well plates (n=6). The cells were observed under a microscope after 12-hour co-incubation. Afterwards, tumor cell viability assays were carried out by MTT assay after co-incubating for 48 hours. Meanwhile, nontreated tumor cells were cultured as control, and macrophages alone or loading different packages were individually cultured as blank (n=6). MTT solution was added into each well after incubation, and the absorbance of the solution in each well was measured at 490 nm with a multi-well plate reader. The cell viability was calculated as:
|
|
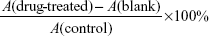
Animal experiments
Animal models
Female Kunming mice and nude mice (25 g, 6–8 weeks old) used in this study were purchased from Laboratory Animal Resources of China Pharmaceutical University. All experiments were carried out in compliance with the Animal Management Rules of the Ministry of Health of the People’s Republic of China and the guidelines for the Care and Use of Laboratory Animals of China Pharmaceutical University. All procedures involving animals and their care in this study were in strict accordance with protocols approved by the Ethics Committee of China Pharmaceutical University.
The EAC (mice of Ehrlich ascites carcinoma) tumor model was established by subcutaneously inoculating EAC cells (5×106) into the upper left axillary fossa in the mice (n=6). The mice were investigated when the tumor grew to a diameter of 5–7 mm.
In vivo dynamic distribution of macrophage
To investigate the dynamic distribution of macrophage delivery system, different molecules/particles-loaded macrophage systems were administered into the subject mouse (nude mice or denuded Kunming mice) via the tail vein. The lipid-soluble dye, Cypate, was used as the monitor tracer. The NIR dyes were swallowed by macrophages for in vivo imaging. In addition, the dynamic distributions of macrophages in normal nude mice and denuded Kunming mice were compared. The fluorescence imaging of the mice post injection was acquired at designated time points. After 24-hour injection, the mice were sacrificed. Different organs were separated and washed by saline, and then they were put together for fluorescence imaging.
To confirm the dynamic biodistribution of each molecule/nanoparticle-loaded macrophage in different organs, the injected Kunming mice were sacrificed at regular time intervals (0.5 hours, 1 hour, 2 hours, 4 hours, 6 hours, 8 hours, 12 hours, and 24 hours) post injection. Different organs were separated and washed by saline, and then they were put together for fluorescence imaging. The contrast ratios were analyzed and compared by using the analysis of regions of interest function. Data are expressed as mean ± SD (n=3).
Tumor-targeting ability of macrophage delivery system in tumor-bearing mice
To investigate the tumor-targeting ability of macrophage system, the Cypate-loaded macrophages were used as representative to be intravenously injected into the EAC tumor-bearing mice (nude mice or denuded Kunming mice). Fluorescence images of the mice were acquired by NIR imaging system at different time points post injection. As control, free Cypate was administered to EAC tumor-bearing mice by a similar procedure. The tumor/normal (T/N) tissue ratios were analyzed and compared by using the analysis of regions of interest function.
Immunoresponse and organ toxicity
To evaluate the immunoreaction and organ toxicity of macrophages in mice, a total of 20 healthy Kunming mice (male and female, half and half) were randomly assigned in four groups and intravenously injected with 200 μL of blank macrophages (0 macrophages/kg, 1.5×108 macrophages/kg, 7.5×108 macrophages/kg, 1.5×109 macrophages/kg), respectively. The mice were weighed every day, and their survival rates were recorded. After 7 days of administration, blood samples were collected from the orbital sinus by quickly removing the eyeball from the mice, in which the immune-associated cells (lymphocyte, monocyte, neutrophil, and basophilic granulocyte) were quantitated. Afterward, the mice were sacrificed, and their livers and lungs were processed for histologic examination.
In vivo antitumor efficacy
EAC tumor-bearing Kunming mice were randomly divided into seven groups (n=6 per group). The mice in each group were treated every other day for 15 days via tail vein injection with different solutions (0.2 mL): 1) saline (control group, nothing injected); 2) only macrophages PBS solution (1.2×108 macrophages/kg); 3) free DOX (10 μmol/kg); 4) DOX-packaging macrophages PBS solution (DOX:10 μmol/kg, macrophages: 1.2×108 macrophages/kg); 5) free PTX (12 μmol/kg); 6) PTX-packaging macrophages PBS solution (PTX:12 μmol/kg, macrophages: 1.2×108 macrophages/kg) and 7) SOC-PTX-packaging macrophages PBS solution (PTX:12 μmol/kg, macrophages: 1.2×108 cells/kg). The therapeutic efficacies and systemic toxicities of each solution on these tumor-bearing mice were assessed by everyday tumor volume and body weight measurement. After completing the treatment (15 days), the mice were sacrificed and the resected tumors were imaged.
Statistical analysis
All the data were expressed as mean ± standard deviation. Statistical analysis was conducted by Student’s t-test, with statistical significance assigned to a P-value of <0.05.
Results
Loading, encapsulation, and release profiles of drugs swallowed by macrophages
DL capacity is an important issue for drug carriers. In this study, lipid-soluble (PTX, Cypate, fluorescein) and water-soluble (DOX, fluorescein sodium) model drugs and DL particles (SOC, NLC) were used to investigate the phagocytic capacity of macrophages. As shown in Figure 2A, the DL contents of macrophages increased with the elevation of drug concentration and reached the maximum up to ~600 nmol/mL for hydrophilic drugs, whereas the loading rate of the hydrophobic drugs tardily increased after 600 nmol/mL (Figure 2A). The highest DL contents for hydrophobic Cypate, PTX, and fluorescein were 1.11 μmol/104 cells, 1.08 μmol/104 cells, and 1.02 μmol/104 cells, respectively, which were higher than that of water-soluble DOX (0.98 μmol/104 cells) and fluorescein sodium (0.70 μmol/104 cells), indicating that the drug solubility has a great impact on the phagocytosis. Importantly, the nanoparticle-entrapped drugs can be engulfed more in the macrophages. To further investigate the loading efficiency of the small molecular drugs and nanoparticle-loaded drugs, we compared the loading rate of PTX and SOC–PTX by macrophages at the same concentration, 600 nmol/mL. Obviously, the loading rate of SOC–PTX is higher than PTX (Figure S1A), confirming that SOC–PTX would be more preferred by the macrophage. As expected, the EE of all the drugs decreased with the increasing of drug concentrations, with extremely low encapsulation for the high-soluble drug model, fluorescein sodium (Figure 2B). The release profiles of each drug (Figure 2C) indicated that the release rate has no obvious difference among all the small molecular drugs; however, the release rate of the SOC–PTX is much slower than the free PTX. The results demonstrated that lipid-soluble drugs have higher loading efficiency than water-soluble ones, and the SOC nano-packaging could help the PTX improve the loading rate and slow down the speed of drug release.
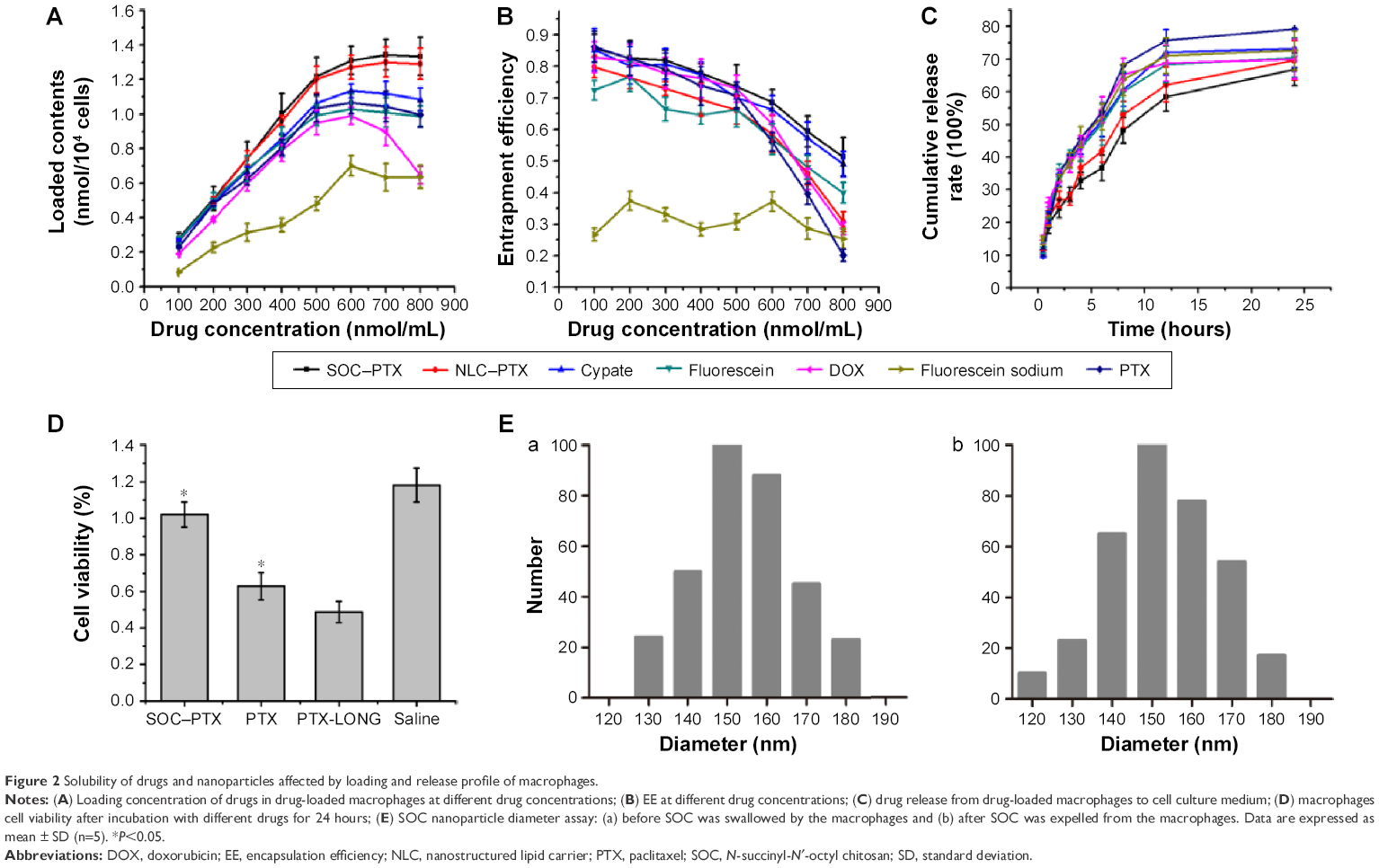 | Figure 2 Solubility of drugs and nanoparticles affected by loading and release profile of macrophages. |
Further, we compared the toxicity of the engulfed PTX and SOC–PTX with the macrophage itself. We controlled the same amount of PTX to be engulfed into the macrophages. Figure 2D shows that the SOC–PTX group exhibits almost no toxicity, but the cell viability of free PTX group is only 39% even though they were treated with the same concentration of PTX. In contrast, after incubation of the macrophage with PTX for 48 hours without changing the medium (PTX-LONG), the MTT assay showed that the viability of macrophage was only 20%. This indicated that the free PTX displayed severe toxicity to the macrophage, while the SOC–PTX could be safe. To investigate whether the SOC nanoparticle could remain intact when it was transferred to tumor cells from the macrophage, the particle size was measured before getting in and after coming out from the macrophage. As shown in Figure 2E, no obvious change was observed between the two groups. Both two groups could be kept at 150 nm size. It was suggested that the SOC nanoparticle could remain intact when released from the macrophage. In addition, we investigated the PTX-releasing rate from the SOC carrier in vitro. Only <20% PTX was released from the SOC carrier after 12 hours with most PTX in SOC set out after 24 hours (Figure S1B).
Phagocytosis of macrophage to different drug-loaded nanoparticles
To assess the nanoparticle uptake ability of macrophage, different nanoparticles or drugs were incubated with RAW 264.7 cells, which were monitored at different incubation time points by using confocal microscopy, with blue for nucleus stain, red for DOX, and green for fluorescein (Figure 3). The fluorescence from fluorescein showed the obvious stronger signal than from DOX with the same incubation time, and then we used the NLC and SOC to load the same amount of fluorescein. As shown in Figure 3A, the cellular uptake processes of NLC and SOC nanoparticles (in same size range) loaded with the same amount of lipophilic fluorescein were almost the same, with a great amount of fluorescence observed in the nucleus, but both of them have stronger fluorescence signal than the free fluorescein. The results implied that the nanoparticle-loading drug could be taken up more easily than the small molecular drug.
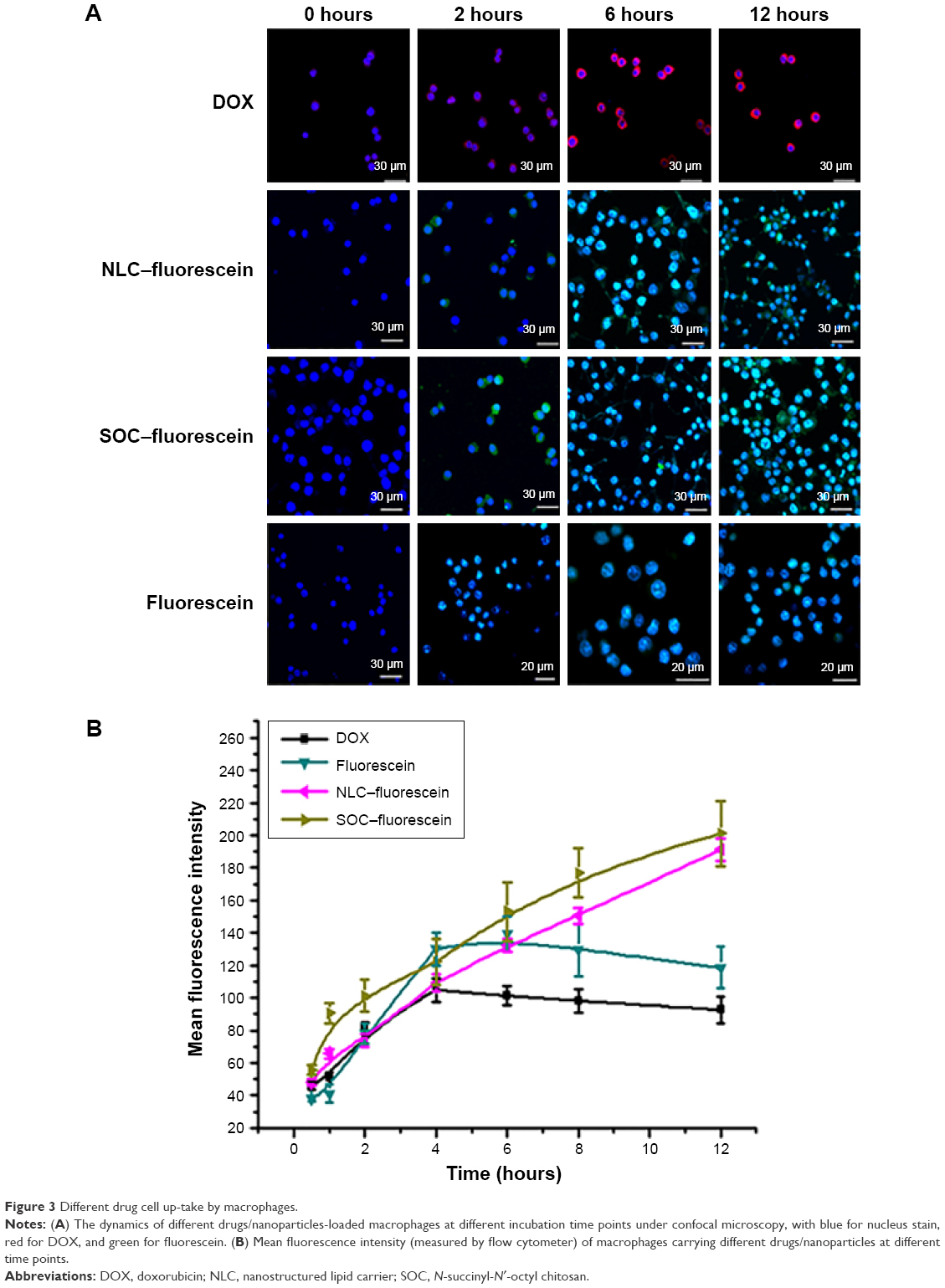 | Figure 3 Different drug cell up-take by macrophages. |
To study the quantitative uptake of macrophage to different nanoparticles or drugs, the fluorescence signals of cells were detected by flow cytometer (Figure 3B). The maximal fluorescence intensity of fluorescein in macrophage was higher than that of DOX. The NLC and SOC package also enhanced the fluorescein uptake, and the enhancing rate was almost the same.
Drug transfer process from macrophages to tumor cells
To explore the drug transfer process from nanoparticles/drug-loaded macrophages to the co-incubating tumor cells, the drugs- or nanoparticles-loaded macrophages were prepared and added to the cultured MCF-7 and MDA-MB-231 cells for different periods. The obvious drug transfer was observed under a laser confocal microscope (Figure 4A). The fluorescence was initially observed only in macrophages, later it was found in both macrophages and MCF-7 cells 4 hours later of co-incubation, but for the signal in the tumor cells, the free fluorescein group is stronger than the SOC–fluorescein group. In addition, the fluorescent intensity of tumor cells was increasing during the co-incubation, and reached the maximum at 12 hours (Figure 4B), whereas that of macrophages was decreasing within the co-incubating period (Figure 4C). It indicated that the drug engulfed by macrophages can gradually be transferred to the co-incubating tumor cells, causing the toxicity to the tumor cells. The results were same in the MDA-MB-231 group as shown in Figure S2. Furthermore, Figure 4B also shows that the speed of free fluorescein transfer from the macrophage to tumor cells is higher than that of SCO fluorescein.
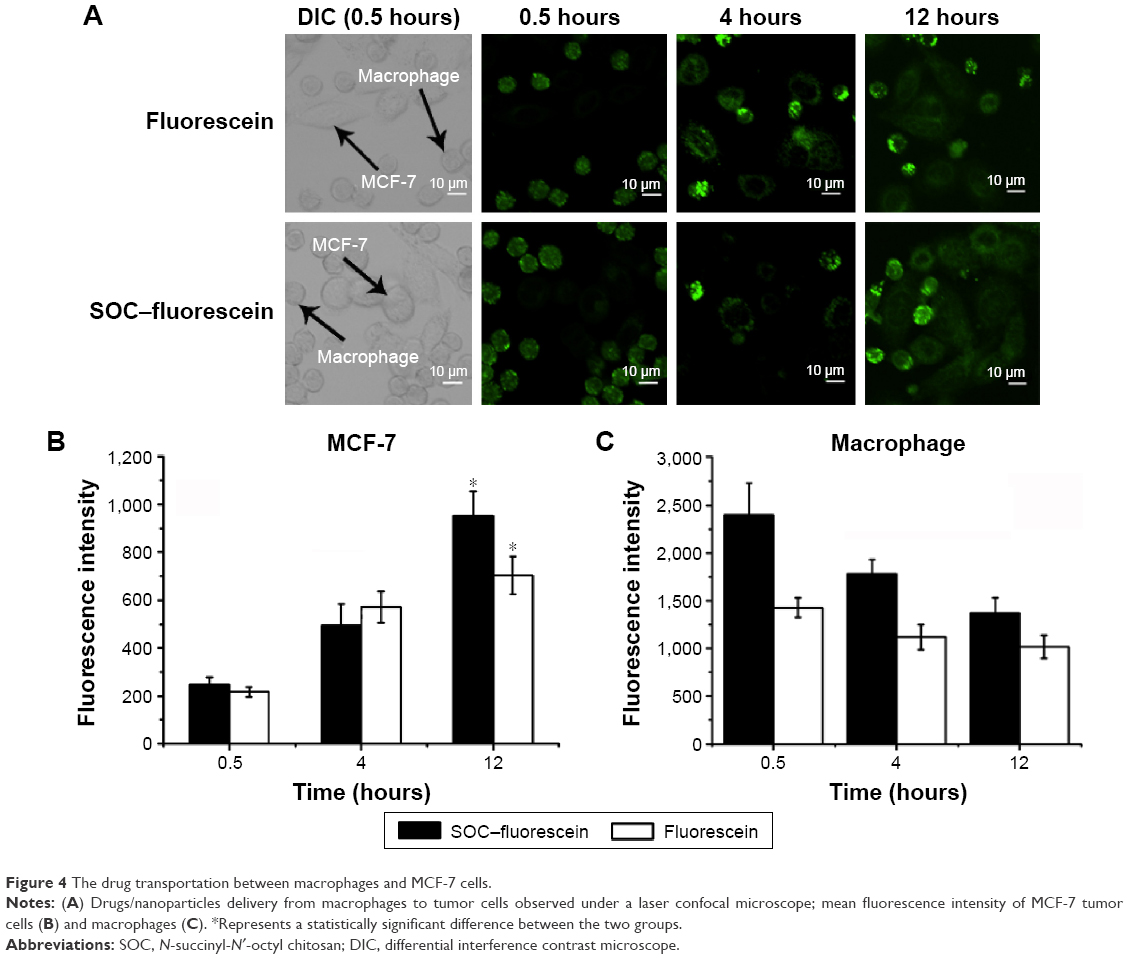 | Figure 4 The drug transportation between macrophages and MCF-7 cells. |
Cytotoxicity of nanoparticles/drug-loaded macrophages
As shown in Figure 5A, the amount of cell indicated that the live cell left after 48 hours of drug incubation and PTX- and SOC–PTX-loaded macrophages significantly inhibited the growth of both MCF-7 and MDA-MB-231 and displayed greater tumoricidal effects than DOX-carrying macrophages. In the histogram, the efficiency of SOC–PTX was also obvious (Figure 5B and C). These findings demonstrated that macrophages are indeed capable of encapsulating drugs and nanoparticles, and more lipid-soluble drugs could be loaded by macrophages and delivered into tumor cells than water-soluble ones, thus inducing a higher antitumor efficacy.
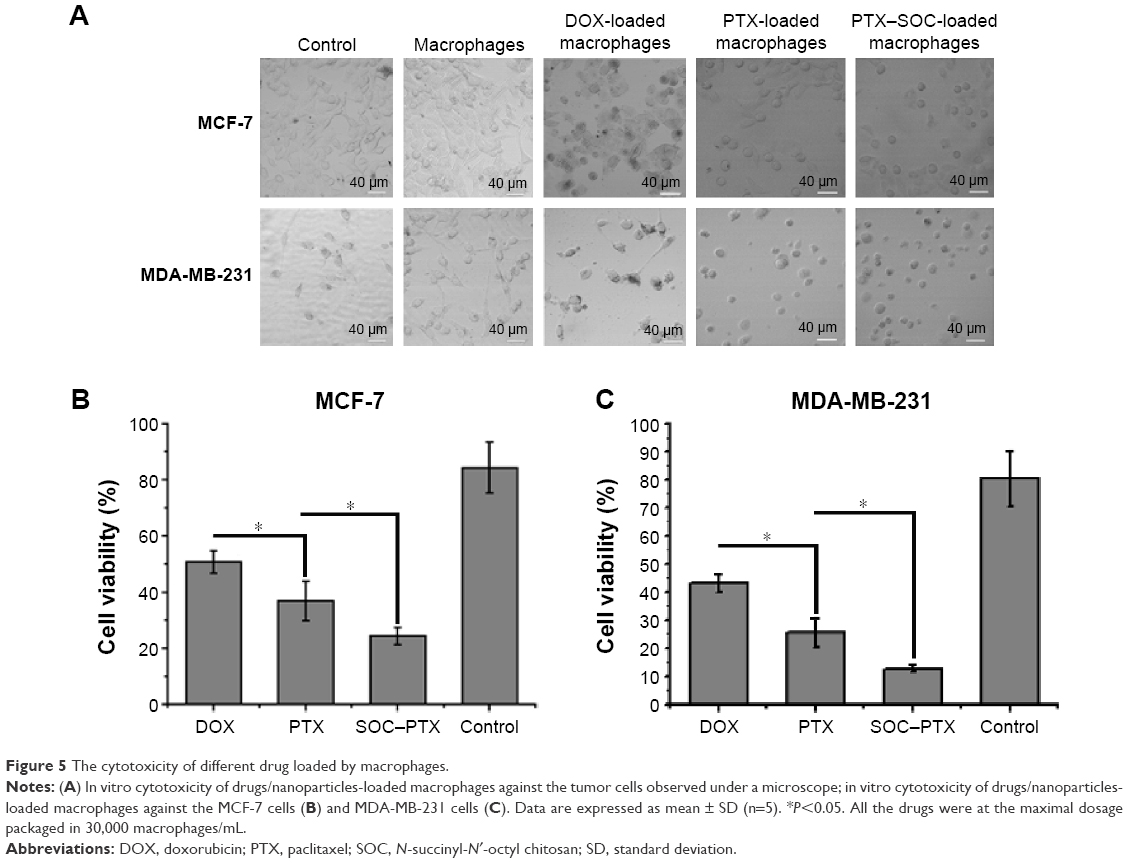 | Figure 5 The cytotoxicity of different drug loaded by macrophages. |
In vivo dynamic distribution of macrophage
To investigate the in vivo physiological behavior of macrophage, the dynamics of the NIR dye, Cypate-loaded macrophages as a representative, were investigated in nude mice and denuded Kunming mice. Nude mice injected with Cypate-loaded macrophages were monitored for 24 hours, and the typical fluorescence distributions are shown in Figure 6A. Cypate-loaded macrophages quickly reached the liver within 1 hour and then passed into the intestine within 2 hours post injection. After 24 hours post injection, the fluorescence of macrophages had mostly cleared from the body. Similar to macrophage-injected nude mice, Cypate-loaded macrophages showed a rapid clearing rate through the liver–intestine pathway within 24 hours in the Kunming mice27 (Figure 6B and C).
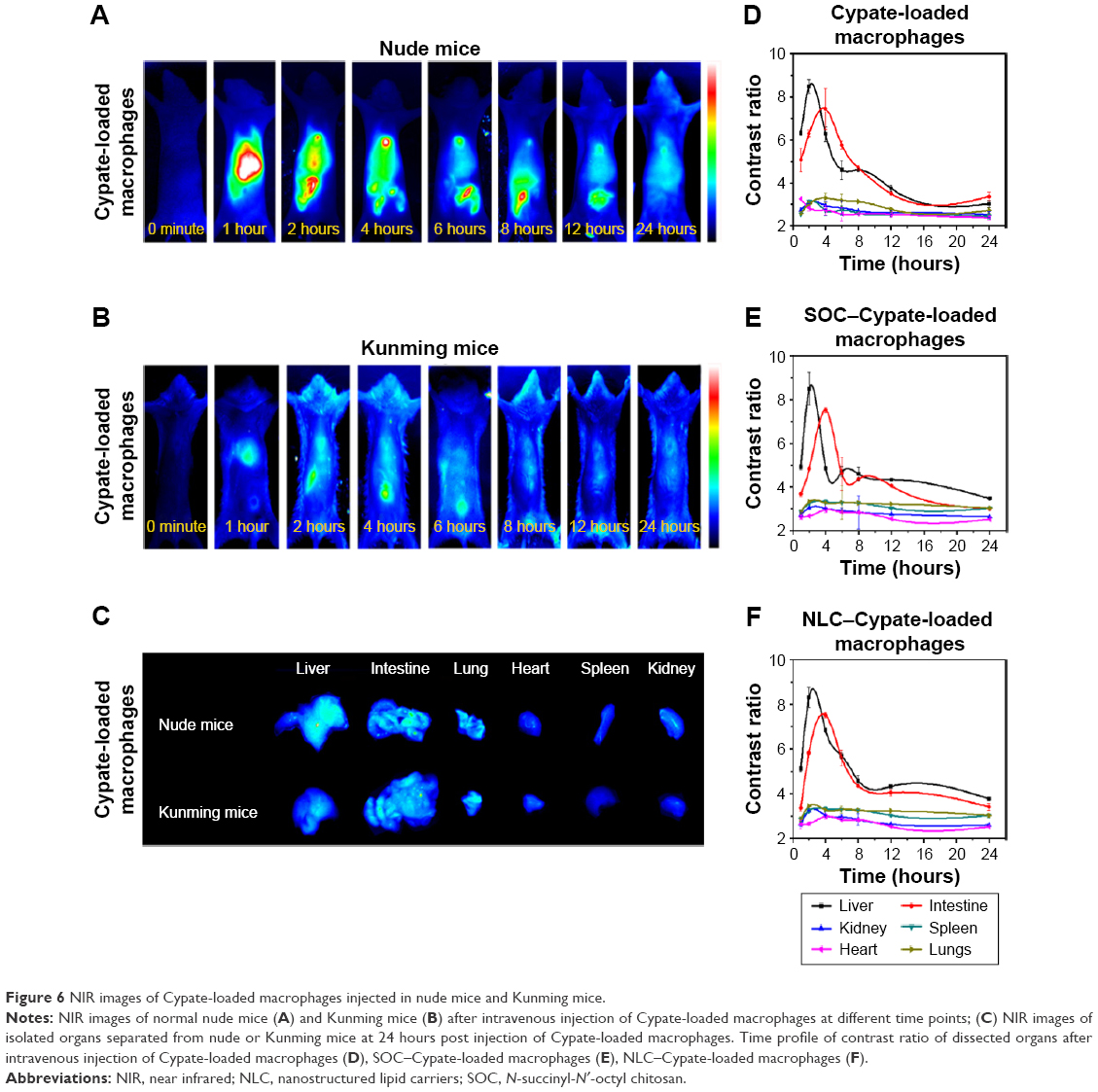 | Figure 6 NIR images of Cypate-loaded macrophages injected in nude mice and Kunming mice. |
To further investigate the effect of drugs/nanoparticles on the dynamic distribution of macrophage, Kunming mice were injected with different drugs/nanoparticles-loaded macrophages and sacrificed at regular time intervals (0.5 hours, 1 hour, 2 hours, 4 hours, 6 hours, 8 hours, 12 hours, and 24 hours) post injection. The organs were removed and subjected to NIR imaging. The control ratio of each organ was shown in Figure 6D–F. No obvious differences were observed in the dynamics and biodistribution of the drugs/nanoparticles-loaded macrophages, further indicating that different nanoparticles-loaded macrophages had the same clearance pathway. Therefore, the properties of nanoparticles were not obviously related to the dynamic distribution of macrophages.
Tumor-targeting ability of macrophage
To evaluate the tumor-targeting ability of macrophage, nude mice bearing human breast tumor (MCF-7) models and Kunming mice bearing EAC tumor models were used. In vivo behavior of Cypate-loaded macrophages was assessed following their intravenous injection through the tail vein of the mice, with free Cypate as control. At different time points post administration, NIR images of mice were captured (Figure 7A–C). Within 2 hours after injection of Cypate-loaded macrophages, EAC xenografts in immunocompetent Kunming mice were identifiable, and the intense signal at tumor sites was maintained up to 48 hours after injection (Figure 7A). In contrast, weaker fluorescence was observed at the tumor site of immunodeficiency mice (Figure 7B), with maximum T/N tissue ratios of 3.2, which was much lower than that of 6.8 in immunocompetent mice (Figure 7D). This distinction of targeting ability was due to the higher chemotactic cytokine (such as CCL2) expression in the tumor tissues of immunocompetent mice than that of immunodeficiency mice (Figure 7E), since the overexpressing CCL2 chemokine is the dominant factor responsible for the homing ability of macrophages, inducing the macrophages for accumulation.28,29 As expected, no obvious fluorescence signal was observed in the Kunming mice injected with the negative control of free Cypate.
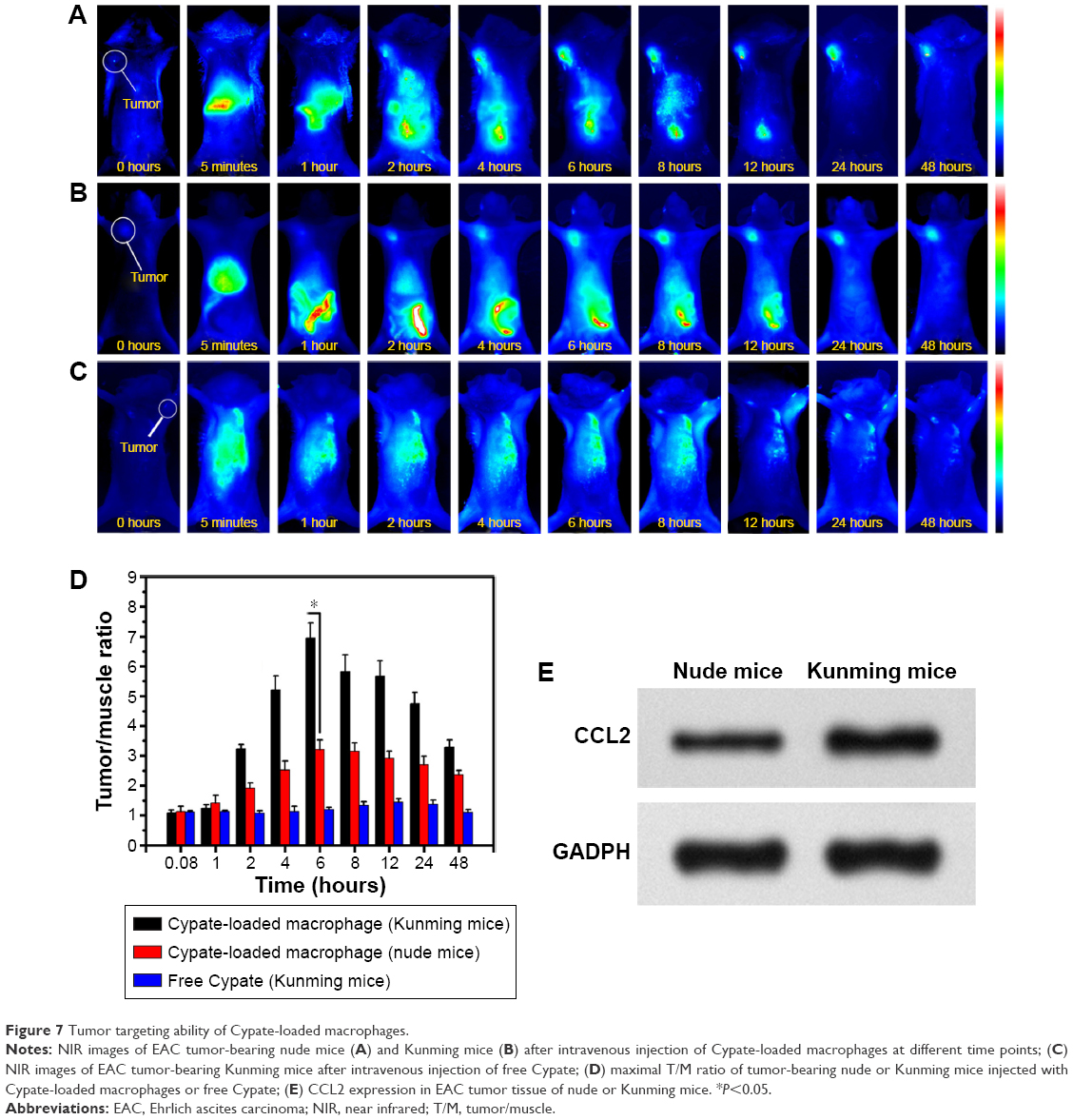 | Figure 7 Tumor targeting ability of Cypate-loaded macrophages. |
Systemic toxicity of macrophages
To investigate the systemic toxicity of macrophages, after injection with different amounts of macrophages, the body weight of mice was measured, and the immune-associated cells in blood were quantitated, while the pathological assay of the main organs (liver and lung) affected by macrophages was performed.30 As shown in Figure 8A, the body weight of mice in both the control and macrophages-treated groups displayed similar trends, indicating no physical toxicity caused by macrophages. After 7-day administration, the number of immune-associated cells, including lymphocytes, monocytes, neutrophils, and basophilic granulocytes, did not show any obvious changes even under different dosages of macrophages compared with that of the control group (Figure 8B), indicating no obvious immunoreaction. The tissue slices of liver and lung are shown in Figure 8C, suggesting that animals treated with different dosages of macrophages did not exhibit any noticeable liver and lung toxicity within 7 days of treatment, although the lung slices of mice that received 1.5×109 macrophages exhibited slight tissue damage. These results demonstrated that no obvious immunoreaction and organ toxicity were caused by macrophages in the dosage (1.5×108 macrophages/kg), which would be similar with the in vivo antitumor investigation.
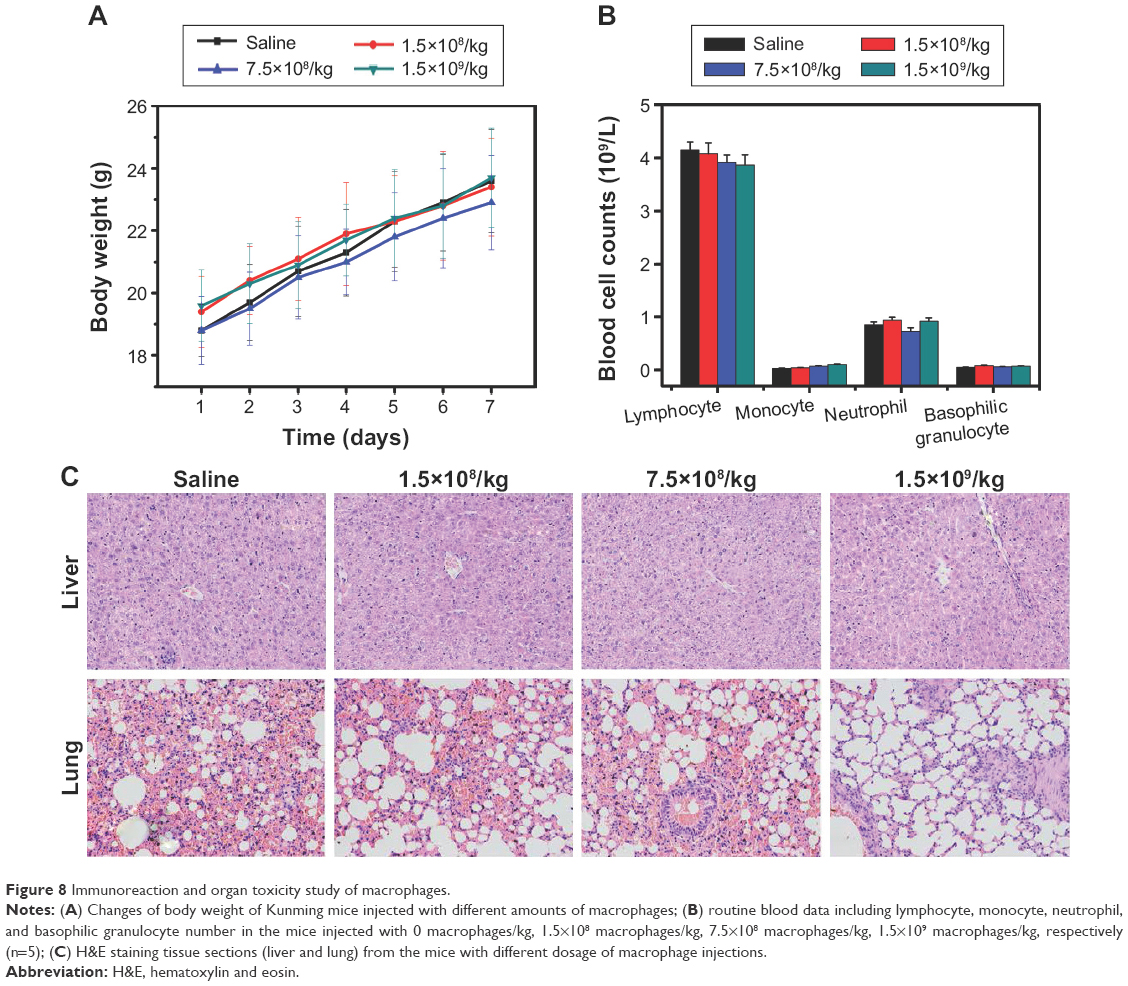 | Figure 8 Immunoreaction and organ toxicity study of macrophages. |
In vivo antitumor efficacy
In vivo antitumor efficacy of macrophage–drug delivery system was evaluated in EAC-bearing mice by measuring the tumor growth rate and the body weight of the mice. As shown in Figure 9A and B, tumors in saline-treated mice grew faster than those of any other drug-treated mice. The administration of DOX-loaded macrophages reduced the tumor volume by ~66% after 15 days of treatment. This decrease was higher than that of free DOX-treated mice (~52%) as well as non-loaded macrophages (~32%). Interestingly, the treatment of PTX- and SOC–PTX-loaded macrophages reduced the tumor volume by ~69% and ~93%, respectively, the greatest extent of tumor reduction in any of the groups. Comparing with free PTX (~60%), their antitumor efficacy was increased due to the tumor-targeting ability of macrophages.
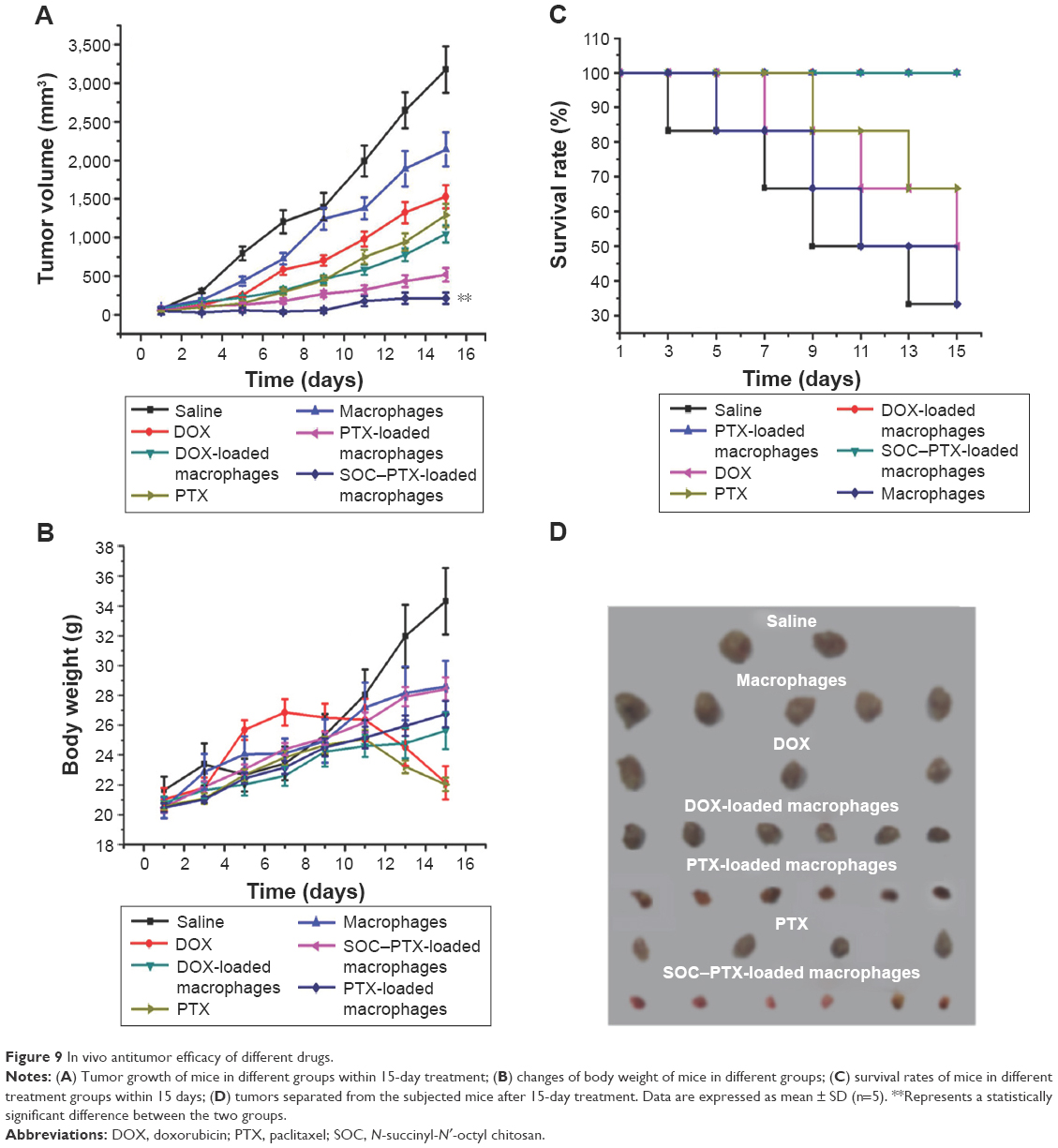 | Figure 9 In vivo antitumor efficacy of different drugs. |
Furthermore, the body weights of mice in different groups gradually increased within the treatment period, indicating no apparent physical toxicity. But a decrease in body weight was observed in the free DOX- and PTX-treated groups after 7 days and 11 days of treatment, respectively (Figure 9B), revealing the side effect of DOX and PTX without macrophages carrying. The 15-day survival rates of mice in the DOX-, PTX-, and SOC–PTX-loaded macrophages were 100%, and those of free DOX and free PTX groups were 50% and 66.7%, respectively, whereas only two members of the saline group and blank macrophages survived (Figure 9C). In Figure 9D, the tumor tissues of different groups were removed and the tumor size represented the treatment efficacy.
Discussion
Malignant tumors remain a challenge to scientists and clinicians alike. Chemotherapy is still considered as a first line of defense against most cancers. However, the biggest limitation of anticancer chemotherapy lies in its adverse effects. To resolve the problem, innovative approaches to deliver chemotherapeutic drugs to pathological sites possess great promise to reduce the systemic toxicity. With many unique advantages, macrophage has great potential to be a drug carrier for targeted cancer therapy.13–17 First, macrophage is a typical immune cell, which will cause less immunogenicity after intravenous administration. Second, as a dominated phagocyte, macrophage has the high capability to identify and phagocytize extrinsic materials such as large particles, making it possess high DL ability and tolerance to toxic drugs. Most importantly, macrophages possess high homing ability to the pathological sites with hypersecretion of chemokine, leading to the directional drug delivery. Therefore, macrophages can be developed as an ideal drug carrier and a novel and patient friendly approach for targeted chemotherapeutics.
Macrophage has demonstrated its ability to directly engulf small molecular drugs. The influence of drug solubility on the DL and release properties was investigated (Figure 2). The hydrophobic drug such as PTX displayed higher loading efficiency into macrophages than hydrophilic ones, mostly due to the high bilayer membrane permeability of lipid soluble drugs. Based on the principle of “like dissolves like”, hydrophobic drugs can be easily engulfed by macrophages in a simple penetrative diffusion mode, accompanying little energy consumption.31 This makes macrophage to swallow more lipid-soluble drugs such as PTX than water-soluble DOX, thus delivering more PTX to tumor cells for increased tumor cell toxicity compared with DOX macrophage system (Figure 5).
Chitosan, an abundant natural biopolymer, has shown many advantages such as high biocompatibility, biodegradation, and low toxicity toward mammalian cells. Active hydroxyl and amine groups in the backbone of chitosan allow simple chemical modification to form a large variety of multifunctional structures including micelles. In our previous studies, SOC micelles were synthesized successfully.26 In the experiment of DL rate assay, we can clearly see that the hydrophobic drug, PTX, could be more loaded in the macrophage than the hydrophilic DOX. Furthermore, the SOC carrier could help the free PTX to improve the loading rate. It is suggested that the SOC nanocarrier could help more drug uptake by the macrophage cells. So, besides the micelle structure providing a good DL ability, the large size of SOC also contributes to its high loading efficiency into macrophages. Larger particles can be easily identified by matrix metalloproteinases, which activate the phagocytosis of macrophage,32,33 indicating that SOC is the optimal nanocarrier and an important component of the macrophage delivery system. However, the cell toxicity of the chemical drug to macrophage cells must be considered, because the chemical drug could not recognize which cells should be killed and which cells should be protected. As the drug carrier, the macrophage should not be destroyed by the loading drug. In Figure 2D, it is shown that the cell viability in the SOC–PTX group is obviously higher than other groups. It indicates that the chemical drug, PTX, also has the cell toxicity to the macrophage. But the SOC nanocarrier could protect the macrophage from the PTX toxicity. However, in the results of treatment efficiency in vitro and in vivo, the SOC–PTX-loaded macrophage also showed a higher ability of killing of tumor cells than the free PTX in the macrophage. For explaining this phenomenon, we designed several experiments. We studied the release rate of both SOC–PTX and free PTX from macrophage and found that the release of SOC–PTX is slower than the free PTX. Only <40% of SOC-PTX were released from the macrophages after 12 hours (Figure 2C). At the same time, the ability of drug release from the SOC nanocarrier was also studied. However, in the 5.3 pH medium the release of PTX could only be <40% (Figure S1B). This implies that the time of PTX released from the SOC nanocarrier is longer than the time of SOC–PTX released from the macrophage. So when the SOC–PTX DL nanoparticle was set out from the macrophage, most of the nanoparticles still remained in the original and intact style (Figure 2E). We all know that when the PTX released from the nanocarrier, the cell toxicity would show. Therefore, this could be the reason for SOC–PTX being safe to the macrophage carrier.
We also compared the time of drug release (Figure 2C) with the homing time of the macrophage in vivo (Figure 7D). In the results, we found that there was >50% PTX released from the macrophage before it arrived at the tumor site in vivo in the free PTX-loading group. However, only 26% of SOC–PTX was set out from the macrophage in the SOC–PTX loading group. It is obviously indicated that most of the SOC–PTX could be released after the macrophage arrived at the tumor site, but almost half of the PTX would be set out from the macrophage with the fluid circulation before getting into the tumor tissue. The ideal chemical drug treatment situation is that the drug could be set out directly at the tumor site but not in the circulation in vivo, so the SOC nanocarrier could be a better choice. So, the high DL capacity, transmembrane efficiency, and good biocompatibility highlight that SOC can be used as an excellent nanoparticle for ideal drug-delivery and therapeutic efficacy.
Tumor-targeting ability is another important evaluation criterion for the availability of drug delivery system. Since the different drug/nanoparticle-loaded macrophages displayed similar biodistribution in mouse subjects (Figure 6), the NIR dye, Cypate, was used as a tracer agent swallowed by macrophages. NIR images (Figure 7) indicated that macrophages displayed a stronger targeting ability to EAC xenografts in Kunming mice than in nude mice, in which different CCL2 expression levels strongly supported our hypothesis that CCL2 is the dominating factor for the homing ability of macrophages. Tumor cells may stimulate the chemokine secretion (such as CCL2) of the surrounding cells in immunocompetent mice, but the immunodeficiency mice lack the capacity to produce more CCL2 chemokine. Owing to the high chemokine level of most tumor tissues, macrophage has promising advantages in tumor targeting, which along with the few immunoreactions and physical toxicities (Figure 8) indicated that macrophage-carrying drugs/nanoparticles are a potential pharmaceutical preparation applicable to the investigation of in vivo anticancer activity.
Based on the homing ability of macrophages, DOX macrophages displayed a superior therapeutic efficacy to free DOX, so as the PTX-macrophages to free PTX. Partly due to the lipid solubility, PTX-loaded macrophages exhibited higher inhibitory effect of tumor growth than hydrophilic DOX. On the other hand, with the introduction of SOC carrier, the antitumor efficacy of PTX macrophages also showed a significant increase, further confirming the importance of the lipotropy of drugs and particles to the efficacy of macrophage-based tumor-targeting delivery. In contrast to the saline-treated control, PTX macrophages and SOC–PTX macrophages efficiently inhibited tumor growth, thus prolonging survival time. Both in vitro and in vivo studies confirmed the therapeutic efficiency of these drugs.
Overall, we have used both in vitro and in vivo approaches to investigate the tumor-targeting drug delivery and anticancer capability of macrophage-based system loading lipid-soluble drugs and nanoparticles. However, further studies are needed for the pharmaceutic preparation to clinical applicability. Biocompatibility is an important index to estimate the clinical feasibility of drugs. Although it was initially investigated in mice, the biocompatibility should be comprehensively studied in other animal models (such as monkeys) possessing high immune homology with humans. For the human clinical application there is still a long way to go, especially to obtain the safe dose size and to determine what the possible side effects of macrophages in vivo are. In addition, it is also a problem that the macrophages would cause the cytokinesis storm the patients’ body. Also, our future work would include the meticulous investigation of the mechanism of macrophages swallowing different drugs and particles, optimization of the formulation of macrophages-based system for optimal administration, and the study of the pharmacokinetics in different in vivo model systems.
Conclusion
Macrophages-based system exhibited significant tumor-targeting capability in chemokine-positive tumors and represents a potential antitumor agent in chemokine overexpressing tumor tissues. In vitro studies indicated that macrophages can load and deliver more lipophilic drugs/particles than hydrophilic ones. At the same time, SOC nanocarrier exhibits the better drug package rate and low toxicity to the macrophage. So the high efficiency of drug-loaded macrophages, SOC–PTX macrophages, was successfully developed and showed greater in vitro and in vivo antitumor efficacy. Overall, our results demonstrate that the nanoparticles-macrophage lipophilic drugs carried by macrophages are a promising medicinal preparation for targeted drug delivery and cancer therapy.
Acknowledgments
The authors are grateful to Natural Science Foundation Committee of China (NSFC 81220108012, 61335007, 81371684, 81000666, 81171395, and 81328012) for their financial support. Siwen Li and Song Feng were regarded as co-first authors.
Disclosure
The authors report no conflicts of interest in this work.
References
Chen J, Chen H, Cui S, et al. Glucosamine derivative modified nanostructured lipid carriers for targeted tumor delivery. J Mater Chem. 2012;22:5770–5783. | ||
Du C, Deng D, Shan L, et al. A pH-sensitive doxorubicin prodrug based on folate-conjugated BSA for tumor-targeted drug delivery. Biomaterials. 2013;34(12):3087–3097. | ||
Petros RA, DeSimone JM. Strategies in the design of nanoparticles for therapeutic applications. Nat Rev Drug Discov. 2010;9(8):615–627. | ||
Gao Z, Zhang L, Hu J, Sun Y. Mesenchymal stem cells: a potential targeted-delivery vehicle for anti-cancer drug, loaded nanoparticles. Nanomedicine. 2013;9(2):174–184. | ||
Wei X, Yang X, Han Z, Qu F, Shao L, Shi YF. Mesenchymal stem cells: a new trend for cell therapy. Acta Pharmacol Sin. 2013;34(6):747–754. | ||
García-Castro J, Martínez-Palacio J, Lillo R, et al. Tumor cells as cellular vehicles to deliver gene therapies to metastatic tumors. Cancer Gene Ther. 2005;12(4):341–349. | ||
Jang ES, Shin JH, Ren G, et al. The manipulation of natural killer cells to target tumor sites using magnetic nanoparticles. Biomaterials. 2012;33(22):5584–5592. | ||
Martinez FO, Sica A, Mantovani A, Locati M. Macrophage activation and polarization. Front Biosci. 2008;13:453–461. | ||
Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004;25(12):677–686. | ||
Raman D, Baugher PJ, Thu YM, Richmond A. Role of chemokines in tumor growth. Cancer Lett. 2007;256(2):137–165. | ||
MacMicking J, Xie Q, Nathan C. Nitric oxide and macrophage function. Annu Rev Immunol. 1997;15:323–350. | ||
Qian BZ, Li J, Zhang H, et al. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature. 2011;475(7355):222–225. | ||
He W, Qiang M, Ma W, et al. Development of a synthetic promoter for macrophage gene therapy. Hum Gene Ther. 2006;17(9):949–959. | ||
Giffiths L, Binley K, Iqball S, et al. The macrophage – a novel system to deliver gene therapy to pathological hypoxia. Gene Ther. 2000;7(3):255–262. | ||
Burke B, Sumner S, Maitland N, Lewis CE. Macrophages in gene therapy: cellular deliver vehicles and in vivo targets. J Leukoc Biol. 2002;72(3):417–428. | ||
Dreaden EC, Mwakwari SC, Austin LA, Kieffer MJ, Oyelere AK, El-Sayed MA. Small molecule–gold nanorod conjugates selectively target and induce macrophage cytotoxicity towards breast cancer cells. Small. 2012;8(18):2819–2822. | ||
Muthana M, Giannoudis A, Scott SD, et al. Use of macrophages to target therapeutic adenovirus to human prostate tumors. Cancer Res. 2011;71(5):1805–1815. | ||
Baek SK, Makkouk AR, Krasieva T, Sun CH, Madsen SJ, Hirschberg H. Photothermal treatment of glioma; an in vitro study of macrophage-mediated delivery of gold nanoshells. J Neurooncol. 2011;104(2):439–448. | ||
Fu J, Wang D, Mei D, et al. Macrophages mediated biomimetic delivery system for the treatment of lung metastasis of breast cancer. J Control Release. 2015;204:11–19. | ||
Zhang X, Qiao H, Zhang T, Shi Y, Ni J. Enhancement of gastrointestinal absorption of isoliquiritigenin by nanostructured lipid carrier. Adv Powder Technol. 2014;25(3):1060–1068. | ||
Beloqui A, Solinís MÁ, Gascón AR, del Pozo-Rodríguez A, des Rieux A, Préat V. Mechanism of transport of saquinavir-loaded nanostructured lipid carriers across the intestinal barrier. J Control Release. 2013;166(2):115–123. | ||
Tamjidi F, Shahedi M, Varshosaz J, Nasirpour A. Nanostructured lipid carriers (NLC): a potential delivery system for bioactive food molecules. Innovat Food Sci Emerg Technol. 2013;19:29–43. | ||
Müller RH, Radtke M, Wissing SA. Nanostructured lipid matrices for improved microencapsulation of drugs. Int J Pharm. 2002;242(1–2):121–128. | ||
Cao J, Wan S, Tian J, et al. Fast clearing RGD-based near-infrared fluorescent probes for in vivo tumor diagnosis. Contrast Media Mol Imaging. 2012;7(4):390–402. | ||
Achilefu S, Dorshow RB, Bugaj JE, Rajagopalan R. Novel receptor-targeted fluorescent contrast agents for in vivo tumor imaging. Invest Radiol. 2000;35(8):479–485. | ||
Zhu H, Cao J, Cui S, Qian Z, Gu Y. Enhanced tumor targeting and antitumor efficacy via hydroxycamptothecin-encapsulated folate-modified N-succinyl-N′-octyl chitosan micelles. J Pharm Sci. 2013;102(4):1318–1332. | ||
Feng S, Cui S, Jin J, Gu Y. Macrophage as cellular vehicles for delivery of nanoparticles. J Innovat Opt Heal Sci. 2014;7:1450023–1450030. | ||
Eardley KS, Zehnder D, Quinkler M, et al. The relationship between albuminuria, MCP-1/CCL2, and interstitial macrophages in chronic kidney disease. Kidney Int. 2006;69(7):1189–1197. | ||
Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3(1):23–35. | ||
Serody JS, Burkett SE, Panoskaltsis-Mortari A, et al. T-lymphocyte production of macrophage inflammatory protein-1α is critical to the recruitment of CD8(+) T cells to the liver, lung, and spleen during graft-versus-host disease. Blood. 2000;96(9):2973–2980. | ||
Higgins CF. Flip-flop: the transmembrane translocation of lipids. Cell. 1994;79(3):393–395. | ||
Shah PK, Falk E, Badimon JJ, et al. Human monocyte-derived macrophages induce collagen breakdown in fibrous caps of atherosclerotic plaques. Potential role of matrix-degrading metalloproteinases and implications for plaque rupture. Circulation. 1995;92(6):1565–1569. | ||
Marx N, Sukhova G, Murphy C, Libby P, Plutzky J. Macrophages in human atheroma contain PPARγ: differentiation-dependent peroxisomal proliferator-activated receptor γ (PPARγ) expression and reduction of MMP-9 activity through PPARγ activation in mononuclear phagocytes in vitro. Am J Pathol. 1998;153(1):17–23. |
Supplementary materials
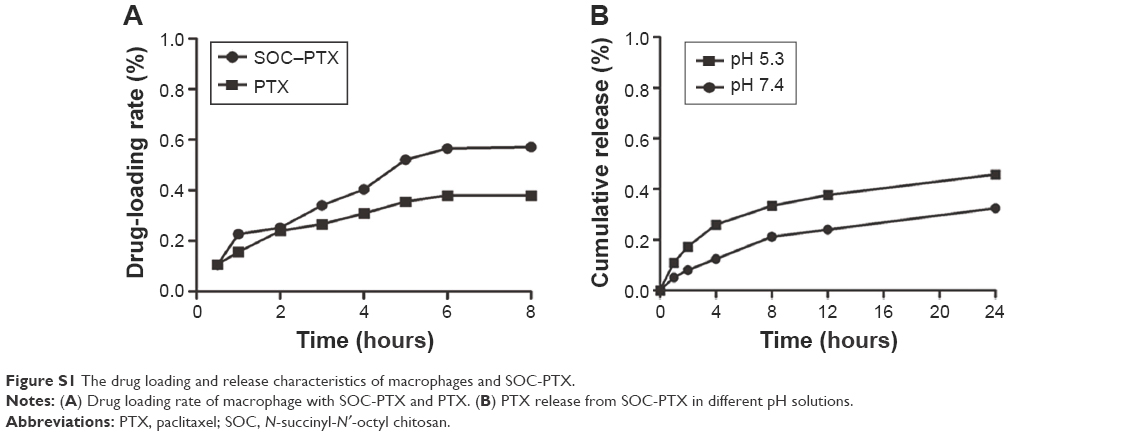 | Figure S1 The drug loading and release characteristics of macrophages and SOC-PTX. |
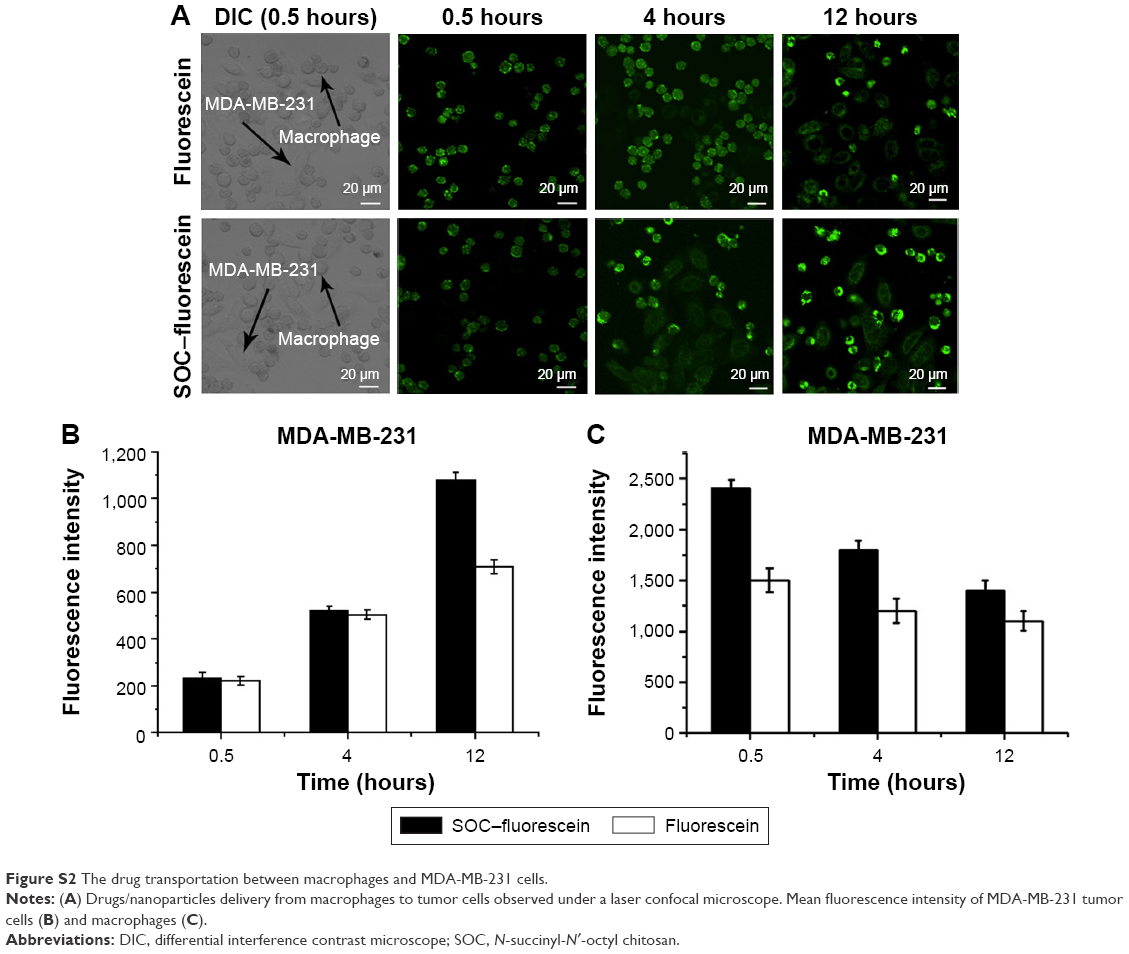 | Figure S2 The drug transportation between macrophages and MDA-MB-231 cells. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.