")
Back to Journals » International Journal of Nanomedicine » Volume 9 » Issue 1
Nanoformulated cell-penetrating survivin mutant and its dual actions
Authors Sriramoju B, Kanwar R, Kanwar J
Received 6 January 2014
Accepted for publication 15 February 2014
Published 10 July 2014 Volume 2014:9(1) Pages 3279—3298
DOI https://doi.org/10.2147/IJN.S60169
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Bhasker Sriramoju, Rupinder K Kanwar, Jagat R Kanwar
Nanomedicine Laboratory of Immunology and Molecular Biomedical Research (NLIMBR), School of Medicine, Faculty of Health, Deakin University, Geelong, Australia
Abstract: In this study, we investigated the differential actions of a dominant-negative survivin mutant (SurR9-C84A) against cancerous SK-N-SH neuroblastoma cell lines and differentiated SK-N-SH neurons. In both the cases, the mutant protein displayed dual actions, where its effects were cytotoxic toward cancerous cells and proliferative toward the differentiated neurons. This can be explained by the fact that tumorous (undifferentiated SK-N-SH) cells have a high endogenous survivin pool and upon treatment with mutant SuR9-C84A causes forceful survivin expression. These events significantly lowered the microtubule dynamics and stability, eventually leading to apoptosis. In the case of differentiated SK-N-SH neurons that express negligible levels of wild-type survivin, the mutant indistinguishably behaved in a wild-type fashion.
It also favored cell-cycle progression, forming the chromosome-passenger complex, and stabilized the microtubule-organizing center. Therefore, mutant SurR9-C84A represents a novel therapeutic with its dual actions (cytotoxic toward tumor cells and protective and proliferative toward neuronal cells), and hence finds potential applications against a variety of neurological disorders. In this study, we also developed a novel poly(lactic-co-glycolic acid) nanoparticulate formulation to surmount the hurdles associated with the delivery of SurR9-C84A, thus enhancing its effective therapeutic outcome.
Keywords: survivin mutant, neurological disorders, protein therapeutics, inhibitor of apoptosis protein family, poly(lactic-co-glycolic acid)
Introduction
Neurotherapeutics has always been a rich area of research, considering the restricted potential of neuronal cells for proliferation and their inability to renew. Therefore, developing the strategies of neuronal proliferation and neuron protection are the prime ways of achieving effective therapeutics for neurological disorders.1–3 Several studies have aimed to treat neurological disorders, including cancers targeting the family of IAP proteins. Among these, the role of survivin in premature brain development has been very well documented.4–7 Supporting cell division, it plays an intriguing role in forming the chromosome-passenger complex (CPC), consisting of survivin, borealin, INCENP, and aurora B kinase. Survivin and borealin dimerize translocating the CPC to the central spindle. This event improves the stability of the microtubules facilitating the proper chromosome alignment onto the spindle fibers.8,9 It also acts against apoptosis, complexing with the HBXIP protein and its cofactor XIAP. This complex interacts with procaspase-9, avoiding the recruitment of APAF-1, and thereby prevents apoptosis.10 On the other hand, survivin significantly lowers microtubule dynamics and stability, eventually leading to apoptosis upon forceful expression.11 Considering the potential of survivin, we synthesized a mutant variety of it that carries an R9 peptide (polymeric arginine carrier) termed dominant-negative survivin (SurR9-C84A); the R9 peptide facilitates rapid uptake into the cells.12 SurR9-C84A belongs to the family of IAPs, and tends to act by dual mechanisms against cancerous and normal neuronal cells.
Cancer cells overexpress wild-type survivin, and when treated, SurR9-C84A displaces wild-type survivin, leading to increased levels of free Caspase 3 (Cas-3) and thereby inducing cell death.13 In contrast, SurR9-C84A tends to promote proliferation, enhancing the CPC organization in neurons that have lower endogenous survivin expression.
These results were substantiated with our own data, where SurR9-C84A showed apoptotic effects toward prostate cancer cells and proliferating effects toward retinoic acid-differentiated SK-N-SH neurons.12,14 The former effect of the mutant protein would be highly beneficial in the death-inducing events of degeneration and inflammation predominantly seen in Alzheimer’s disease and multiple sclerosis.15 On the other hand, protein and peptide therapy always suffer from the serious limitations of their half-life, physical and chemical instability, inefficient oral delivery, enzymatic degradation in the gastrointestinal tract, and poor permeation through biological barriers.16 Therefore, we tried to develop a novel nanoparticulate formulation for the delivery of SurR9-C84A and surmount the hurdles associated with protein delivery. Polymeric NPs made of poly(lactic-co-glycolic acid) (PLGA) have gained extreme popularity in formulating several drugs, as they are biodegradable and also approved by the US Food and Drug Administration for human use.17 PLGA is available in different varieties, of which PLGA with a copolymer ratio of 50:50 lactide:glycolide was used for this study. This study is the first report on the encapsulation of SurR9-C84A in PLGA NPs using the modified double-emulsion technique. Polyvinyl alcohol (PVA; 1%) proved to be an effective emulsifier, yielding NPs with desirable size range and smooth spherical surface morphology. All the process parameters were optimized to achieve enhanced encapsulation and drug loading.
The integrity and intactness of the protein inside the polymer matrix were studied using Fourier-transform infrared spectroscopy (FTIR), sodium dodecyl sulfate (SDS) polyacrylamide gel electrophoresis (PAGE), and differential scanning calorimetry (DSC) techniques. The efficacy of the protein-loaded NPs was studied in the undifferentiated and differentiated (retinoic induced) human neuroblastoma (SK-N-SH) cell lines.
Materials and methods
The polymer poly(D,l-lactide-co-glycolide) Resomer® RG 503 H (PLGA 50:50, inherent viscosity 0.32–0.44 dL/g, molecular weight 24,000–38,000), and PVA (molecular weight 13,000–23,000, 87%–89% hydrolyzed) were procured from Sigma-Aldrich (St Louis, MO, USA). All other solvents and chemicals used in the study were of analytical grade. SK-N-SH neuroblastoma cell lines were obtained from the American Type Culture Collection (Manassas, VA, USA), and were grown as a monolayer in Eagle’s minimum essential medium supplemented with 10% fetal bovine serum and 1% penicillin and streptomycin at 37°C in a saturated humid atmosphere with 5% CO2.
Construction of mutant variety of survivin and its purification
Mutant dominant-negative survivin (SurR9-C84A) was constructed as described previously,18 where cysteine was replaced with alanine at 84th position in the zinc-coordination site. Also, the protein was endowed with enhanced cellular permeability, as the amino terminus of the protein was modified with nine arginine (R9) residues, hence the name SurR9-C84A. The Escherichia coli BL21 strain was transfected with the SurR9-C84A-bearing plasmid, and protein expression was induced by incubating the E. coli bacteria in Luria–Bertani broth media containing 0.01% weight/volume (w/v) ampicillin at 37°C. The incubation was terminated once the optical density of the broth medium reached 0.7 at 620 nm.
Then, protein expression was induced with 0.7 mM isopropylthiogalactoside by incubation for 3 hours. After this period, the bacterial cells were collected by centrifugation at 4,500 rpm for 45 minutes at 4°C. The protein was collected by lysing the cell walls of the bacteria after treatment with a freshly prepared lysis buffer composed of (Milli-Q® [EMD Millipore, Billerica, MA, USA], 150 mM NaCl, 20% SDS, 50 mM Tris, lysozyme 0.1 mg/mL, 1% Triton™ X-100 [Sigma-Aldrich], and a protease inhibitor), followed by sonication at a 40-second pulse and 70 amplitude for 7 minutes. The crude protein was collected after centrifugation, and then purified using the glutathione agarose column. Purification of the protein was based on the principle of affinity chromatography, where the glutathione S-transferase-tagged protein bound to the column was eluted by running an elution buffer composed of 10 mM glutathione in 50 mM Tris-HCI, pH 7.5. The pure protein collected was frozen at −80°C for further experiments (Figure 1A).
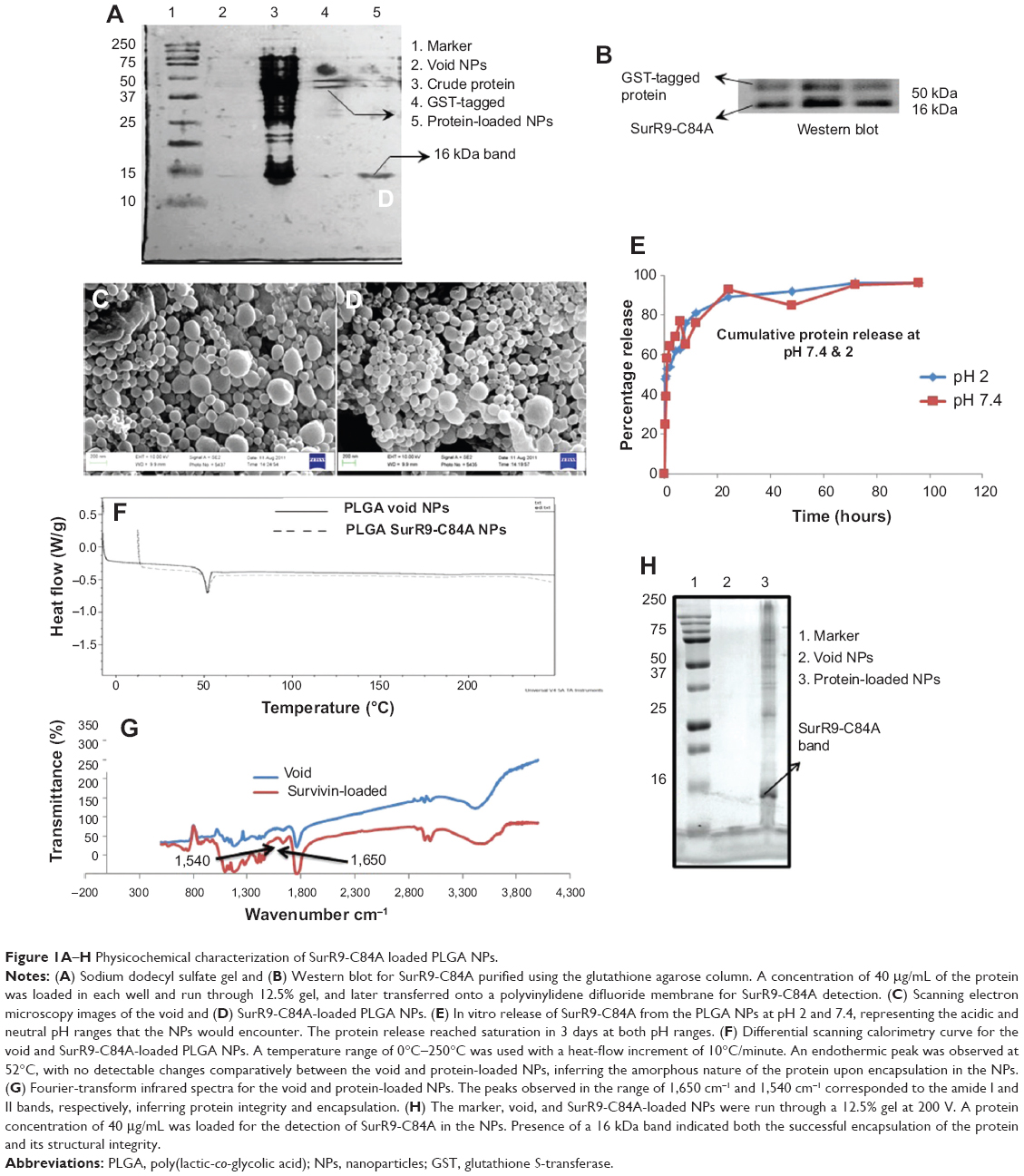 | Figure 1A–H Physicochemical characterization of SurR9-C84A loaded PLGA NPs. |
Preparation of SurR9-C84A-loaded PLGA nanoparticles
The NPs were prepared following the principle of the modified double-emulsion technique followed by evaporation. The polymer (1% w/v) was dissolved in ethyl acetate, and two equal halves of it were taken in separate tubes. Protein (10 mg/mL) was prepared in 1% PVA and equally mixed with the polymer solution prepared, followed by sonication for 30 seconds, yielding the primary w/o emulsion. Then, the final volume was made to 15 mL with 1% PVA in each of the tubes, followed by sonication to yield the final water/oil/water (w/o/w) emulsion. The final emulsion was then stirred overnight at 800 rpm at 4°C to remove the organic solvent. The protein-loaded NPs were collected after washing with distilled water followed by centrifugation at 4,500 rpm for 1 hour. Washing was done three times to ensure the collected NPs were pure, and they were immediately freeze-dried for 48 hours and stored at 4°C for further experiments.
Blank NPs were prepared following the same protocol, except the protein was replaced with Milli-Q water. The fluorescence-labeled NPs were prepared using 4% rhodamine (R6G; Sigma-Aldrich). The R6G was added to the 1% w/v polymer solution prepared initially, and the rest of the procedure was the same as described earlier.19
Characterization of nanoparticles
Particle size and surface morphology
Size analysis was performed by dynamic light scattering using a Malvern Zetasizer (Malvern Instruments, Malvern, UK). In brief, 1 mg of the freeze-dried NPs was dispersed in 1 mL of Milli-Q water and measurements were read at a scattering angle of 90° maintained at 25°C. Nanosphere morphology was ascertained by scanning electron microscopy (SEM) (Supra 55VP; Carl Zeiss Meditec, Jena, Germany), wherein the NPs suspended in water were subjected to sonication and mounted on the carbon tape followed by air-drying. Then, the stubs were gold-coated and left in the vacuum chamber overnight prior to SEM imaging at an accelerated voltage of 10 kV and a working distance of 12 and 9.9 mm.
Encapsulation efficiency and percentage loading
The prepared NPs were centrifuged at 4,500 rpm for 45 minutes at 4°C, and the supernatant was collected. Protein content in the supernatant was estimated using a Bradford protein-assay kit (Thermo Fisher Scientific, Scoresby, Victoria, Australia), and absorbance was measured at 620 nm. The amount of protein present was obtained from the concentration versus absorbance calibration curves of the known standards. Encapsulation efficiency and percentage loading were calculated using the following equations:
|
|
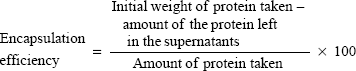
|
|

Polymer–protein interaction
To evaluate the possible polymer–protein interactions, DSC study was conducted for void and protein-loaded NPs. The corresponding DSC curves were obtained on a scanning calorimeter embedded with the thermal analysis-data system. In brief, void and protein-loaded NPs were measured (≥4 mg) and sealed in aluminum pans. A blank aluminum pan was used as a reference, and the samples were scanned at a temperature ranging from −20°C to 250°C with a heat-flow increment of 10°C/minute and nitrogen purging at 20 mL/minute.
Release kinetics
The in vitro kinetics of the SurR9-C84A-loaded PLGA NPs was evaluated in 10 mM phosphate-buffered saline (PBS) at 7.4 pH and an acidic pH 2, mimicking the in vivo conditions that NPs might encounter. A predetermined amount of the freeze-dried NPs was suspended in the PBS buffer, incubated at 37°C, and subjected to a constant rotation at 70 rpm. At regular intervals (0, 0.25, 0.5, 1, 2, 4, 6, 8, 12, 24, 48, and 96 hours), aliquots were collected and the protein concentration was estimated using a spectrophotometer at 620 nm.
Fourier-transform infrared spectroscopy
A predetermined quantity of micronized infrared-grade KBr (400 mg) was mixed separately with the void and SurR9-C84A-loaded NPs (1 mg dry weight) to prepare the pellets using a manually operated tablet press. A pressure of 5.5 metric tons for 5 minutes was applied, and the pellets were scanned 16 times over the mid-IR region (4,000–400 cm−1 wave-number range). Background spectra were subtracted from the samples, and the spectral data values were graphically represented using Excel (Microsoft, Redmond, WA, USA).
SDS-PAGE for protein integrity
The integrity of the protein encapsulated within the NPs was assessed by SDS-PAGE. The freeze-dried NPs were stored at 4°C for 1 week and run through the 12.5% SDS gel under reducing conditions (β-mercaptoethanol) including the void NPs as control. The gel was run in Tris/glycine/SDS buffer at a constant 100 V for 50 minutes using the Mini-Protean® electrophoresis system (Bio-Rad Laboratories Inc., Hercules, CA, USA). Coomassie blue (0.1%) was used as the gel stain for 30 minutes, followed by destaining with a high-destain solution (70% Milli-Q water, 20% methanol, and 10% acetic acid) and later the gel was visualized using the Gel Doc™ imaging system (Bio-Rad Laboratories Inc.).
In vitro nanoparticle-uptake studies
For the in vitro NP-uptake study, both differentiated and undifferentiated SK-N-SH cells were grown in Falcon™ eight-well culture slides (BD Biosciences, San Jose, CA, USA) at a density of 2×104 cells/well and incubated with rhodamine-labeled NPs at time points of 30 minutes and 1, 2, and 4 hours. After the specified time, the media were removed and cells washed thrice with 1× PBS to remove the free NPs that were neither taken up nor adhered to the cells.
Cells were immediately fixed with 4% paraformaldehyde for 15 minutes, followed by washing and mounted with a coverslip using a Fluoroshield™ with 4′,6-diamidino-2-phenylindole (DAPI; Sigma-Aldrich). Cellular uptake was determined using an SP5 confocal microscope (Leica Microsystems, Wetzlar, Germany), and all the images were taken at 40× magnification. The percentage internalization of NPs are expressed as means ± standard deviation obtained by counting cells in five or more different fields.
NP-uptake mechanism
To determine the precise NP-uptake mechanism, differentiated SK-N-SH cells were seeded at 2×104 cells per well in a 96-well plate. Then, they were pretreated with the endocytic inhibitors chlorpromazine (10 μg/mL, clathrin-mediated endocytosis), indomethacin (10 μg/mL, caveolae-mediated endocytosis), sodium azide (2 μg/mL), and colchicine (5 μg/mL, pinocytosis) for 1 hour, followed by rhodamine-labeled NP incubations for 4 hours at 1 mg/mL concentration. After treatment, cells were washed with PBS to remove free NPs, and then the cells were lysed with 1% Triton X-100 in 0.2 M NaOH. The resulting fluorescence was measured at 530 excitation and 630 emission, and the corresponding weight of the NPs was calculated from the standard graph plotted for rhodamine-labeled NPs. Flow-cytometry analysis was also performed, where SK-N-SH cells were plated in six-well plates and were treated with endocytic inhibitors as described above. After treatment, cells were washed with 1× PBS, detached gently by pipetting, and underwent flow-cytometry analysis.
Retinoic acid-induced differentiation in SK-N-SH cells
Undifferentiated SK-N-SH cells are generated from bone marrow metastasis, and are subdivided into neuroblast (N) cells, substrate adherent nonneuronal (S) cells, and intermediate (I) cells.
N-type cells exhibit short neuronal processes, while the S-type cells are rather flat and strongly bound to the substrate. The T-type cells display intermediate properties of both N and S cells with or without neuronal processes and substrate adherence. In order to induce differentiation in neuroblastoma cells, 20 μM retinoic acid (Sigma-Aldrich) was added to the culture media, with periodic replacement of the conditioned media every 2 days.20
Wild-type survivin expression in differentiated and undifferentiated SK-N-SH cells
In order to determine the differential expression of wild-type survivin, both differentiated and undifferentiated SK-N-SH cells were plated in eight-well culture slides at 2×104 per well and underwent immunofluorescence detection. Cells were permeabilized with 0.1% Triton X-100 in PBS, followed by blocking with 2% rabbit serum. Then, undifferentiated and differentiated cells were incubated with a mouse monoclonal antisurvivin (Santa Cruz Biotechnology, Dallas, TX, USA) 1:100 dilution for 1 hour, followed by fluorescein isothiocyanate (FITC)-labeled rabbit antimouse (1:100; Sigma-Aldrich). The cell nucleus was stained with propidium iodide (PI; 1:500; Sigma-Aldrich) for 30 minutes and mounted with mounting media. Further imaging was carried out using the Leica SP5 confocal microscope at 40× magnification.
Cytotoxicity in undifferentiated SK-N-SH cells
Live and dead cell assay
Undifferentiated SK-N-SH cells were plated in a 96-well plate at a density of 2×104 cells/well, followed by treatment with void and SurR9-C84A-loaded NPs (25, 50, 100, and 200 μg of protein/mL concentration) for 24 hours. Untreated controls, including positive controls (treated with 20% fetal bovine serum [FBS]) and negative controls (treated with 1% Triton X-100), were included in the study for the calculation of percentage live and dead cells. The entire procedure was followed as per the manufacturer’s protocol (Live/Dead® viability/cytotoxicity kit; Thermo Fisher Scientific, Waltham, MA, USA). The live/dead cell readings posttreatment were recorded using a fluorescence microplate reader at 530 nm excitation and 645 nm emission.
Cell-proliferation study
In order to evaluate the proliferative potential of SurR9-C84A on undifferentiated SK-N-SH cells, a CyQuant® (Thermo Fisher Scientific) assay was performed. SK-N-SH cells were plated in the 96-well culture plate at a density of 2×104 cells/well and incubated with NP treatments corresponding to protein concentrations of 25, 50, 100, and 200 μg/mL for 24 hours. An untreated control, void NP-treated (control for protein-loaded NPs), and 20% FBS-treated (negative control) cells were also included in the study. After treatment, the cells were washed and frozen overnight at −80°C, followed by the addition of 200 μL/well of CyQuant dye reaction mixture prepared as per the manufacturer’s protocol. Readings were taken at 480 nm excitation and 520 nm emission in a fluorescence microplate reader, and results were analyzed in Excel.
Expression of PCNA and Caspase-3
Further to the evaluation of the inhibitory effects of SurR9-C84A on undifferentiated SK-N-SH cells, immunofluorescence for the proliferative marker PCNA and the apoptotic marker Caspase-3 was studied. Cells were seeded at 105 cells/well in the BD Falcon eight-well culture slide following the protocol, as described previously.14 Cells were synchronized by the addition of nocodazole (70 ng/mL) for 12 hours, washed, and then incubated for 65 minutes to enrich the anaphase and telophase population. Then, the cells were subjected to NP treatment corresponding to protein concentrations of 25, 50, 100, and 200 μg/mL for 24 hours. In order to compare the effect, cells treated with non-nano-SurR9-C84A (75 μg/mL) were included in the study along with the void and untreated controls. After treatment, cells were washed thrice in PBS, followed by fixation in 4% paraformaldehyde for 15 minutes. Cells were permeabilized with 0.1% Triton X-100 in PBS, followed by blocking with 2% rabbit serum.
Then, the undifferentiated cells were incubated with the mouse monoclonal anti-PCNA (1:100; Sigma-Aldrich) and rabbit anti-Caspase-3 (1:100; Cell Signaling Technology, Danvers, MA, USA) for 1 hour. FITC-labeled rabbit antimouse (1:100; Sigma Aldrich) and goat anti rabbit (1:100) were used as secondary antibodies. The cell nucleus was stained with PI (1:500) for 30 minutes and mounted with mounting media. Further imaging was carried out using the Leica SP5 confocal microscope at 40× magnification.
In vitro proliferation in differentiated SK-N-SH cells
Expression of PCNA and Ki67
To evaluate the proliferative potential of SurR9-C84A on differentiated SK-N-SH cells, immunofluorescence for the proliferative markers PCNA and Ki67 were studied. Cells were seeded at 105 cells/well in BD Falcon eight-well culture slides following the protocol described for the undifferentiated cells. After treatment with SurR9-C84A, the differentiated cells were incubated with mouse monoclonal anti-PCNA and mouse anti-Ki67 (1:100; Sigma-Aldrich) for 1 hour at 37°C. FITC-labeled rabbit antimouse and goat antirabbit (1:100) were used as secondary antibodies. The cell nucleus was stained with PI (1:500) for 30 minutes, and further imaging was carried out using the Leica SP5 confocal microscope at 40× magnification.
Bromodeoxyuridine (BrdU)-labeling assay
The proliferating potential of NP-loaded SurR9-C84A was evaluated on retinoic acid-differentiated SK-N-SH cells. Cells were seeded at a density of 2×104 cells/well in a 96-well plate, and differentiation was induced for 12–14 days and was confirmed with the detection of neurite processes. Then, the cells were treated with SurR9-C84A-loaded NPs corresponding to protein concentrations of 50, 100, and 200 μg/mL for 24 hours. Void treatment and untreated controls were also included. The entire protocol was followed as per the manufacturer’s indications for the BrdU Labeling and Detection Kit III (Roche, Basel, Switzerland). In brief, after treatment, cells were incubated with the BrdU-labeling solution for 6 hours, followed by fixation with a precooled fixative (ethanol in HCl). Then, the cells were incubated with peroxidase-conjugated anti-BrdU for 30 minutes, after which the peroxidase substrate was introduced. The absorbance was measured using a microplate reader at 405 nm, keeping the wavelength 490 nm as a reference.
Gene-expression study
To define the dual mechanistic actions of SurR9-C84A (apoptotic effect on undifferentiated cells and proliferative effect on differentiated cells), quantitative real-time polymerase chain reaction (qRT-PCR) analysis was conducted for wild-type survivin, Caspase-9, Caspase-3, Caspase-8, Caspase-7, Cytochrome-C, TRAIL, β-tubulin III, p53, and cyclin D1. Undifferentiated and differentiated SK-N-SH cells were grown confluent in six-well plates and then treated with void and SurR9-C84A-loaded NPs (corresponding to protein concentrations of 50, 100, and 200 μg/mL). An untreated control and pure protein-treated wells were also included in the study. After treatment, ribonucleic acid (RNA) was isolated from the cells following the protocol supplied with the reagent (TRIzol®; Thermo Fisher Scientific). Complementary deoxyribonucleic acid (cDNA) was prepared from the total RNA extracted using SuperScript III reverse transcriptase, oligo-deoxy-thymine, and RNase-free water (Thermo Fisher Scientific). The prepared cDNA was used for gene-expression studies as described previously.14 In brief, 15 μL of the reaction mixture was prepared using the SYBR green PCR master mix (Bio-Rad Laboratories Inc.,) and underwent qRT-PCR using an iCycler™ (Bio-Rad Laboratories Inc.,). In this study, duplicate samples were run for the said genes, and a mean threshold-cycle value was used for the calculation.
The 2-ΔΔCt method was adopted for quantification, while gene amplification was verified by melt-curve analysis and visualized by ethidium bromide-stained 1% agarose gel.14 A complete list of the primer sequences is given in Table 1. Gene expression was studied with actin as an internal reference.
 | Table 1 List of primer sequences used in the study |
Flow cytometry and protein expression
Propidium iodide staining
The percentage of dead cells after treatment with SurR9-C84A-loaded NPs was evaluated by flow-cytometry analysis of PI-stained cells. In brief, both the undifferentiated and differentiated SK-N-SH cells were plated in six-well culture plates and were grown confluent, followed by treatment with protein-loaded NPs at concentrations of 25, 50, 100, and 200 μg/mL for 24 hrs. An untreated control and void NP-treated cells were also included in the study. After treatment, the cells were washed with ice-cold PBS (pH 7.4), trypsinized, and centrifuged at 800 rpm for 5 minutes. Cell pellets were collected in six separate centrifuge tubes, corresponding to the treatments, and were added to 500 μL of 1× PBS containing 2% FBS. Cells were stained with 25 μL of a 50 μg/mL concentration of PI and incubated at 4°C for 30 minutes, and fluorescence was recorded using a FACS™ cell sorter (BD Biosciences) and data acquired using FACSDiva™ software (BD Biosciences).
Annexin V FITC staining
SK-N-SH cells (both undifferentiated and differentiated) grown confluent in the six-well plates were treated with SurR9-C84A-loaded NPs at concentrations corresponding to 50, 100, and 200 μg/mL of the pure protein for 24 hours. An untreated control, void-, and pure survivin (75 μg/mL)-treated wells were also included in the study.
After treatment, cells were washed in ice-cold PBS, trypsinized, and incubated separately for 10 minutes with the reaction mixture (consisting of incubation buffer, Annexin-V-Fluos labeling reagent, and propidium iodide), prepared as per the manufacturer’s protocol (Annexin-V-Fluos staining kit; Roche). Fluorescence signals were detected at excitations of 488 nm and emissions at 510±30 and 600±30 for annexin and PI, respectively, using the flow cytometer (BD Biosciences) equipped with FACSDiva software.
Protein expression
After treatment with undifferentiated and differentiated SurR9-C84A, SK-N-SH cells were evaluated for the expression of apoptotic and proliferative markers through flow-cytometry analysis. Initially, undifferentiated SK-N-SH cells were studied for the expression of BAX, Cyt-C, and Cas-3 to determine the intrinsic apoptotic cascade. Following this, endogenous levels of survivin, β-tubulin, PCNA, and Ki67 were also estimated. Considering the proliferative potential of SurR9-C84A, differentiated SK-N-SH cells were mainly evaluated for the cell-division markers PCNA, β-tubulin, endogenous survivin, and Ki67. Also, the apoptotic proteins described earlier were also included for the differentiated cells.
Statistics
Statistical differences were calculated using Student’s t-test, and results were considered to be statistically significant with P<0.05.
Results
The mutant variety of SurR9-C84A competitively antagonized the actions of cell division and apoptosis exhibited by wild-type survivin overexpressed in cancers. It also has significant therapeutic potential in promoting the proliferation of differentiated neuroblastoma cells (neuronal origin) that have no/negligible expression of survivin.14 Therefore, SurR9-C84A can be suitably applied to cut down on proliferation in brain cancers (neuroblastoma in this case) and provoke proliferation in differentiated neurons. Considering the limitations of protein delivery, the prime goal of this study was to formulate a novel polymeric nanodelivery system that would enhance protein stability and thereby its half-life. The use of PLGA offered the safest delivery on account of its biodegradability, and physicochemical characterization revealed salient features of this novel nanoformulation. The technique of modified double emulsion provided enough control on the process parameters, with a size range not exceeding 150 nm and a protein encapsulation of 81%.21
Physicochemical characterization: protein purification, particle size, surface morphology, and drug encapsulation
Protein purification was performed using column chromatography, and purity was checked against SDS-PAGE electrophoresis and later confirmed with Western blot (Figure 1, A and B). Average particle size determined by dynamic light scattering was around 100–150 nm, a range ideal for the delivery of drugs through the blood–brain barrier. Surface morphology was smooth and spherical with uniform size distribution, as determined by SEM for both the void (Figure 1C) and protein-loaded NPs (Figure 1D). As seen in Table 2, the average encapsulation efficiency was found to be 80.9%, with a loading of 3.2%.
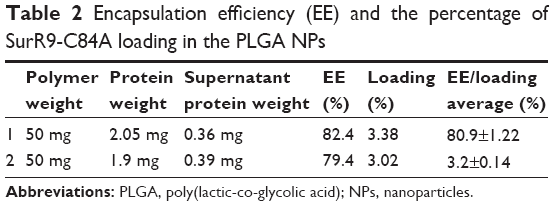 | Table 2 Encapsulation efficiency (EE) and the percentage of SurR9-C84A loading in the PLGA NPs |
The protein encapsulation is in agreement with approximately 1 mg of the drug-loaded NP powder corresponding to 25 μg of SurR9-C84A that is encapsulated. In general, peptides and proteins frequently display hydrophilic behavior, and entrapping them in the hydrophobic polymer matrix is often challenging. In our study, a reasonably high encapsulation of 81% was achieved employing the principle of modified double emulsion, where the protein was initially solubilized in the internal aqueous phase, avoiding protein dispersion should the single-emulsion technique be employed.22 Also, the ionic interactions generated between the positively charged amino groups (NH2+) and the negatively charged carboxyl groups (COO−) of the acid-terminated surface of the PLGA polymer accounted for the enhanced drug entrapment.23,24
Release kinetics
Protein release from the nanoformulation is represented in Figure 1E. A biphasic protein-release pattern was observed at different pH values. A burst release of 63% was observed within 6 hours at pH 7.4, while the release rate remained sustained after 12 hours and reached saturation in 4 days. At pH 2, the burst release was observed to be 77% within 6 hours, and the release pattern did not maintain similar consistency to that found at pH 7.4. The initial high burst effect was due to drug diffusion from the polymer matrix (reduced diffusion-path length), and also because NPs in the small size range of 150 nm offer a higher surface-to-volume ratio, facilitating the enhanced release of protein that is near or on the NP surface.25 This can also be explained as the drug release from the PLGA polymer being influenced by the ratio of the lactide and glycolide content. The lower ratio in PLGA (50:50) imparts hydrophilicity, water absorbance, and faster degradation rates compared to the slower drug-release profiles with lactide-rich PLGA (85:15).
In addition, acidic conditions hasten PLGA degradation, which in turn enhances autocatalysis due to the accumulation of lactic and glycolic acids. Therefore, the burst release at acidic pH was higher compared to the neutral pH.17,26 The sustained effect observed after 12 hours requires the combined effect of both diffusion and degradation of the polymer matrix for protein release.25 On the whole, it was shown that the molecular weight, copolymer ratio, pH, and surface area of the NPs strongly influenced protein release.
Polymer–protein interaction
Figure 1F shows the DSC-curve patterns for the freeze-dried void and protein-loaded PLGA NPs. A peak was observed at 52°C for both void and protein-loaded NPs, indicating the endothermic peak for the PLGA polymer. The following inference was made from the DSC curve obtained. The absence of a discrete melting point suggested the amorphous nature of the polymer, and also its thermal stability was observed up to 250°C. No significant difference was observed for the DSC curves of the void and protein-loaded NPs, indicating that the protein was encapsulated within the polymer without affecting its integrity. The absence of a crystalline peak also suggested the amorphous phase of the protein encapsulated inside the NPs.27,28
Fourier-transform infrared spectroscopy
FTIR spectroscopy is a valuable tool in characterizing protein binding and integrity once encapsulated within the NPs. Figure 1G shows the FTIR spectra for the void and SurR9-C84A-loaded PLGA nanoparticles. As observed, the bands detected at 1,650 cm−1 and 1,540 cm−1 confirm protein encapsulation within the NPs and represent the amide I and II bands, respectively. These bands arise due to the corresponding C=O and N–H stretch vibrations in the amide linkages of the protein. FTIR analysis also confirmed the intactness of the protein secondary structure during the process of NP formation and that the protein integrity was preserved.29
SDS-PAGE for protein integrity
Figure 1H shows the presence of a 16 KDa band characteristic of SurR9-C84A. The harsh parameters involved in the NP-preparation process, such as the organic solvents, sonication, and emulsification, might have resulted in the denaturation of the protein.
As observed in the figure, the structure of the protein was intact, and no additional bands were detected, though there are studies reporting the degradation and protein hydrolysis when encapsulated in PLGA polymer.30–32 The modified double-emulsion technique employed in this study proved to be effective in successful encapsulation of the protein without denaturation.
Determination of NP-internalization and -uptake mechanism
In vitro NP-uptake studies
Figure 2A–D shows the uptake of rhodamine-labeled NPs in differentiated and undifferentiated SK-N-SH cells. The histogram (Figure 2E) shows a time-dependent increase of NP internalization in both types of cells. The idea behind the NP-uptake study in SK-N-SH cells was that it serves as a useful model for evaluating the in vitro efficacy of SurR9-C84A for neurological diseases.33 As observed in Figure 2A–D, the number of NPs interacting with the cell membrane increased with time, as did their internalization. The significant feature of these NPs was the rapid uptake by the cells, where 40%–45% of them were internalized within 30 minutes of incubation. This could be explained by the small size range (100–150 nm) of the NPs. As observed from the confocal images, the presence of rhodamine-labeled NPs (red) around the nucleus (blue, stained by DAPI) confirmed the presence of NPs in the cytoplasm and their internalization.
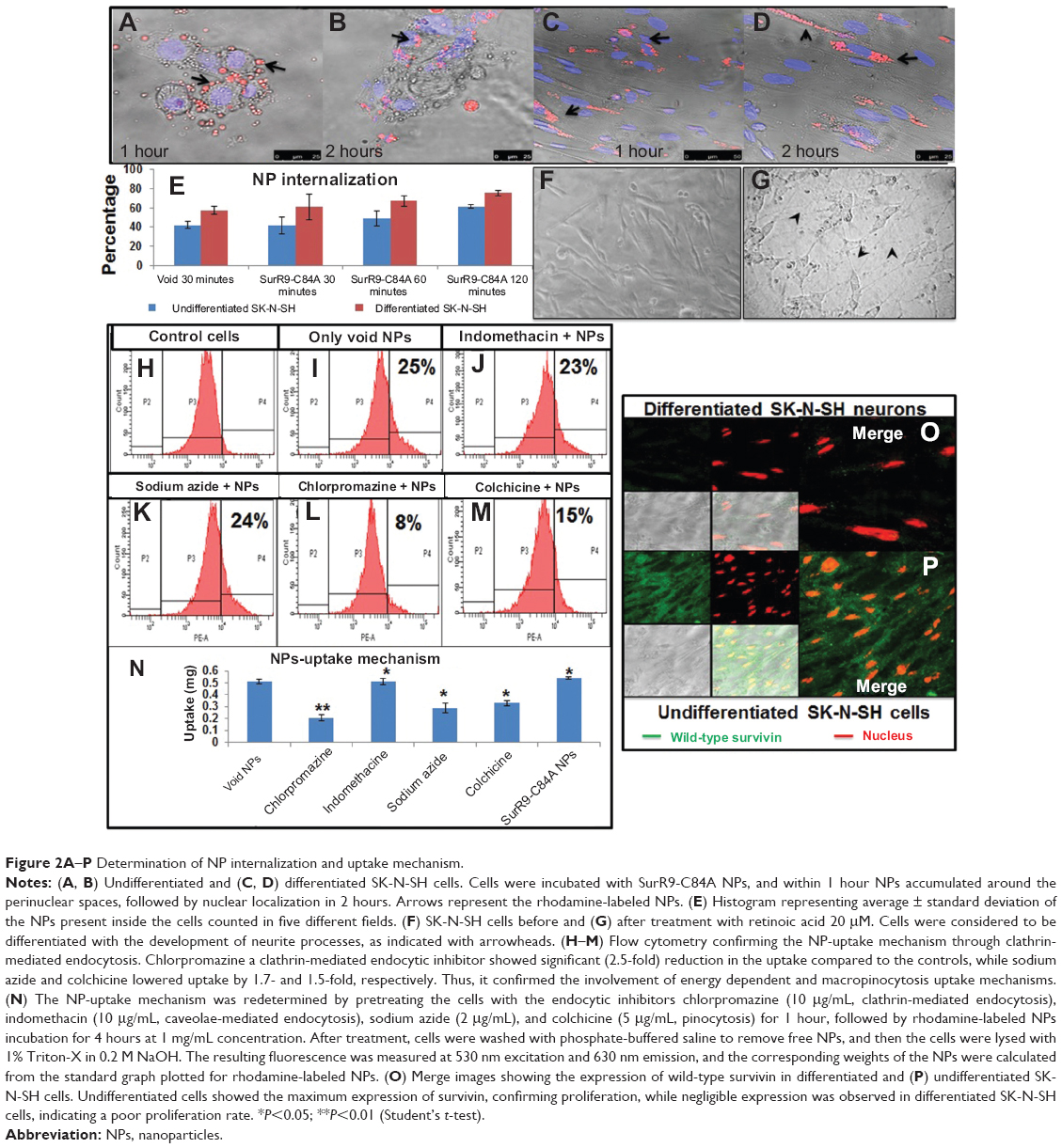 | Figure 2A–P Determination of NP internalization and uptake mechanism. |
Retinoic acid-induced differentiation in SK-N-SH cells
Retinoic acid (20 μM) induced morphological changes from day 2 of its addition, followed by distinct induction of neurite processes in N-subtype cells. Cells were considered to be fully differentiated if they developed neurite processes longer than their cell bodies. Figure 2F shows the undifferentiated cells, and differentiation was achieved after 12 days of treatment with conditioned media (Figure 2G).
NP-uptake mechanism
Cellular uptake of NPs involves a variety of endocytic mechanisms, including clathrin-mediated, caveolae-dependent, and independent endocytosis and macropinocytosis. In order to determine the precise mechanism of uptake, incubation with the corresponding inhibitors revealed interesting results. Chlorpromazine, a clathrin endocytic inhibitor, showed a 40% reduction in uptake compared to the void and SurR9-C84A-loaded NPs, while indomethacin showed no difference. Slight reductions in uptake were observed after sodium azide (44.2%) and colchicine (35.5%) treatment, indicating that the uptake of PLGA NPs in differentiated SK-N-SH was through clathrin-mediated endocytosis along with macropinocytosis and energy involvement (Figure 2N). Macropinocytosis is a physiological function utilized by cells for extracellular fluid turnover, and involvement of this process in uptake indicates that a few percent of NPs were internalized within the cells devoid of cell-surface interaction. This also signifies escape from macrophage phagocytosis.34
Flow-cytometric analysis (Figure 2H–M) also revealed similar results, where cells pretreated with chlorpromazine showed only 8.1% uptake compared to 24.8% of only PLGA-treated controls. Pretreatments with indomethacin (Figure 2J) showed no change, while colchicine reduced uptake to 14.7%, confirming the involvement of clathrin and macropinocytosis-uptake mechanisms.
Wild-type survivin expression in differentiated and undifferentiated SK-N-SH cells
As observed in Figure 2, undifferentiated SK-N-SH cells showed almost 95% wild-type survivin expression, indicating the substantial role of survivin in proliferation, while the differentiated cells (Figure 2P) showed no or negligible expression, confirming lowered proliferative potential.
In vitro cytotoxicity in undifferentiated SK-N-SH cells
Expression of PCNA and Cas-3
PCNA is a proliferation marker expressed at the G2/M cell-cycle phase marking the significant influence of wild-type survivin in provoking cell division.14 In this study, we aimed to evaluate the mechanism by which the SurR9-C84A acts and inhibits proliferation, leading to cell-cycle arrest and eventually ending up with apoptosis. In Figure 3A and B, a concentration-dependent increase in the expression of Cas-3 and vice versa with PCNA (Figure 3, C and D) was observed. This indicates the antagonistic effects of SurR9-C84A after dimerization with wild-type survivin, followed by the blockade of its functioning. Therefore, cells arrested in mitosis (G2/M) overexpress Cas-3, eventually ending up in cell death (Figure 3E).35
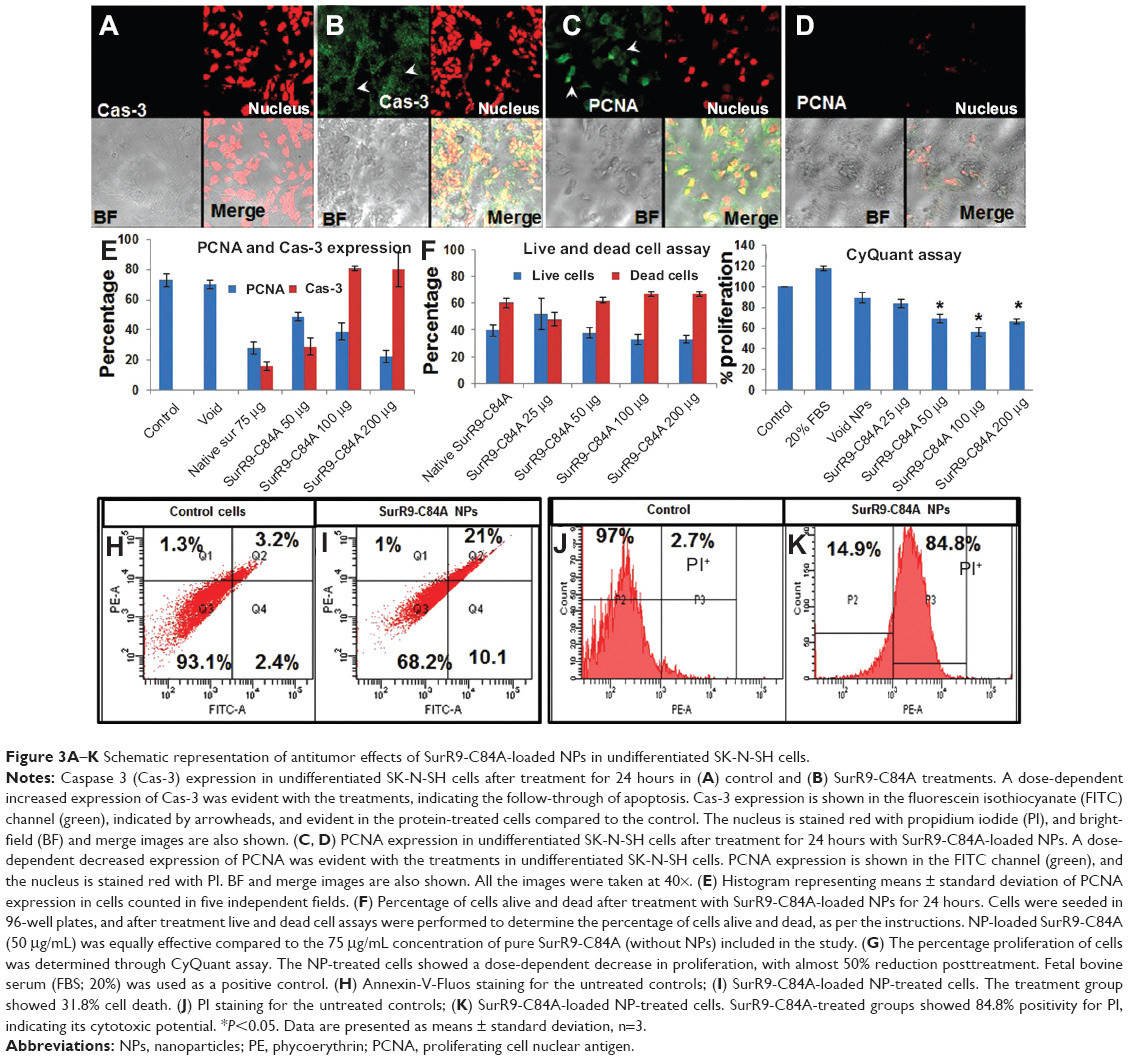 | Figure 3A–K Schematic representation of antitumor effects of SurR9-C84A-loaded NPs in undifferentiated SK-N-SH cells. |
Live and dead cell assay
This assay was conducted to determine the cytotoxic potential of the SurR9-C84A-loaded NPs toward SK-N-SH cells. As seen in Figure 3F, the protein-loaded NPs were prominent in exerting the cytotoxic effect, as 50 μg/mL of the SurR9-C84A-loaded NPs was equally effective compared to pure protein (75 μg/mL). This could have been because of the enhanced stability and half-life of the protein encapsulated in the nanoformulation compared to its free form. A dose-dependent effect of protein loaded NPs was also evident with the increased percentage of dead cells.
Cell-proliferation study
The DNA content exhibited by individual cells is constant, and hence in this assay, the fluorescence-intensity measure of DNA corresponds to the cell number, and thus gives a measure of cell proliferation.36 As seen in Figure 3G, SurR9-C84A exhibited proliferation inhibition in undifferentiated SK-N-SH (neuroblastoma) cells in a concentration-dependent manner, with almost half of the cells (56%) wiped out at 100 μg/mL concentration.
In vitro proliferation in differentiated SK-N-SH cells
Expression of proliferation markers PCNA and Ki67 and BrdU assay
The proliferative potential of SurR9-C84A on differentiated SK-N-SH cells was studied by confirming the expression of PCNA (Figure 4A and B) and Ki67 (Figure 4, D and E). PCNA was expressed at the G2/M phase of the cell cycle, while Ki67 was expressed at all active phases of cell division except at the resting phase (G0).37 Therefore, these are both excellent markers for determining proliferation in differentiated SK-N-SH cells. Dividing cells were also determined utilizing the BrdU assay, which is an excellent method of monitoring cell proliferation. BrdU is an analog of thymidine, and gets incorporated into the DNA of the dividing cells. Consequently, many studies have reported the use of BrdU labeling for determining cell proliferation, particularly neurons.38
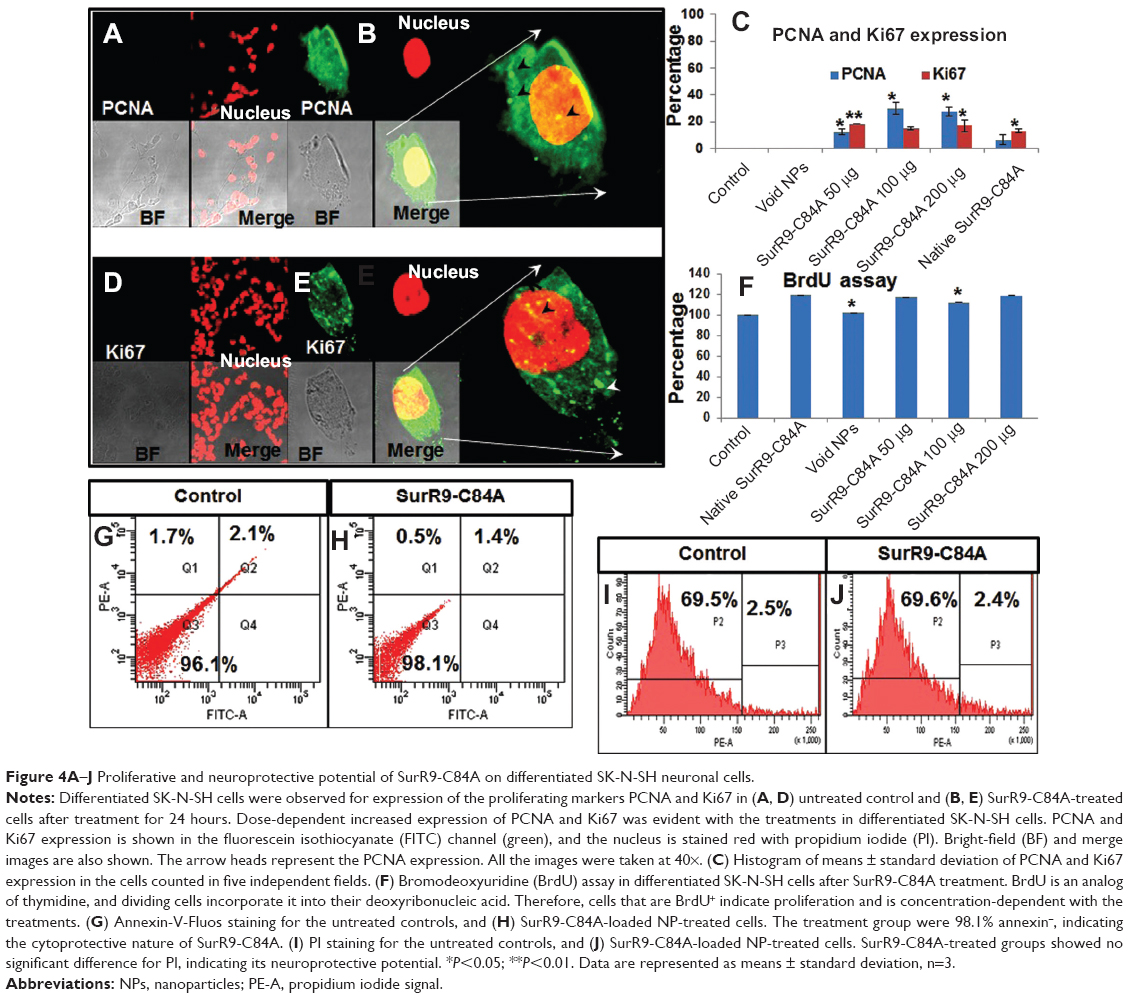 | Figure 4A–J Proliferative and neuroprotective potential of SurR9-C84A on differentiated SK-N-SH neuronal cells. |
After treatment with SurR9-C84A, 23.2% of the differentiated SK-N-SH cells expressed PCNA, while 16% of them were found positive for Ki67. Also, when compared with the controls, BrdU labeling was found to be increased by 1.15-fold after SurR9-C84A treatment (Figure 4F). Together, these results confirm that the differentiated SK-N-SH cells, which were deficient in wild-type survivin, regained their proliferation when treated with SurR9-C84A.
Flow-cytometry PI and annexin V FITC staining
Apoptotic cells are typically characterized by DNA fragmentation, followed by a significant loss of DNA content. PI staining is specific to DNA, and thereby provides a rapid estimation of the percentage of dead cells.39 As observed from Figure 3H–K, a significant amount of dead cells was observed with the SurR9-C84A-treated undifferentiated cells compared to the untreated controls, and the trend was concentration-dependent. Figure 3I–K, shows 30.8% of annexin+ and 84.8% of PI+ cells, respectively, indicating the percentage of apoptotic cells. In contrast, the percentage of dead cells was significantly reduced (98% annexin− and 69.5% PI− cells) after SurR9-C84A treatment in differentiated SK-N-SH cells (Figure 4G–J).
Gene-expression study
To confirm the characteristic mechanism of SurR9-C84A on undifferentiated and differentiated SK-N-SH cells, qRT-PCR was performed for the proliferative (wild-type survivin) and the apoptotic genes for Cas-3, -7, -8, and -9, Cyt-C, and p53. In the case of undifferentiated SK-N-SH cells, the apoptotic genes Cas-3, -7, -8, and -9, Cyt-C, and p53 showed 1.32-, 1.54-, 2.39-, 1.55-, 2.84-, and 1.2-fold increases, respectively, while the proliferative marker endogenous survivin showed a twofold reduction, confirming the antitumor potential of SurR9-C84A.
When studied in differentiated SK-N-SH cells, the same apoptotic genes for Cas-8, Cas-9, and p53 showed 1.53-, 1.58-, and 3.33-fold reduced expression. Further, endogenous survivin levels showed a 1.1-fold increase in expression, provoking proliferative potential (Figure 5A–D).
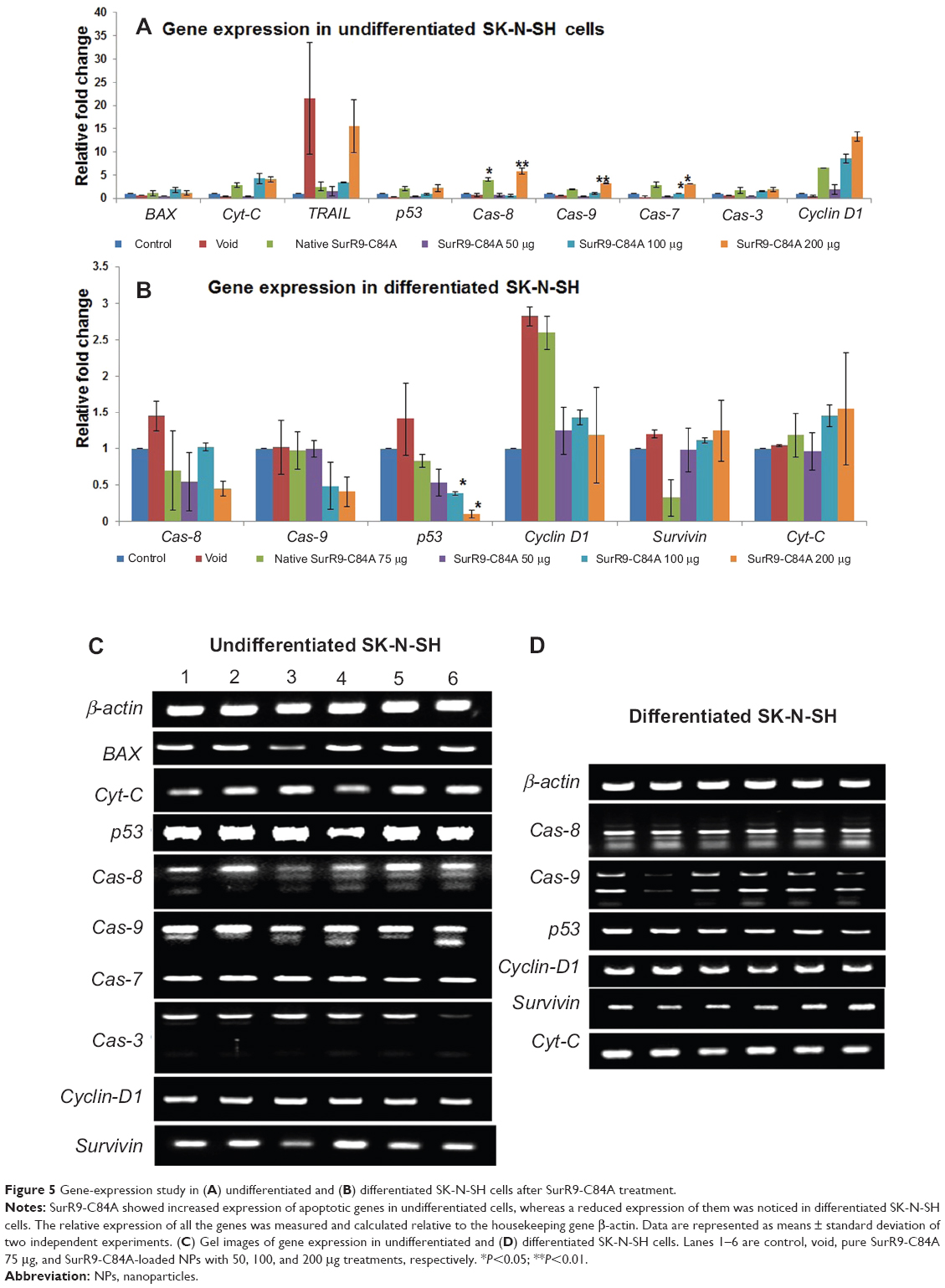 | Figure 5 Gene-expression study in (A) undifferentiated and (B) differentiated SK-N-SH cells after SurR9-C84A treatment. |
Protein expression
SurR9-C84A showed dual but distinct actions on undifferentiated and differentiated SK-N-SH cells that represented tumorous and neuronal characteristics. The apoptotic markers p53, BAX, Cyt-C, and Cas-3 were upregulated by 77.4%, 90.9%, 4.5%, and 14%, respectively, indicating the antitumor effects of SurR9-C84A. Also, the proliferative markers β-tubulin, survivin, PCNA, and Ki67 were downregulated by 34.5%, 79%, 25.88%, and 15%, respectively (Figure 6A). These results were consistent with our earlier results of the antitumor activities of SurR9-C84A.12 Owing to the proliferative potential of SurR9-C84A in neurons with a low endogenous pool of survivin, differentiated SK-N-SH cells exhibited upregulation of cell-division markers. Endogenous survivin levels increased up to 46.3%, while PCNA and Ki67 showed a 5.1% and 24.9% increment, respectively. Substantiating this, the apoptotic markers Cyt-C, Cas-3 and p53 showed a respective reduction by 65.6%, 54.5%, and 74.5%, respectively. Also, the specific neuronal differentiating marker β-tubulin III showed a 3.7% downregulation, indicating the change of differentiation phase to proliferation (Figure 6B). Given these dual actions, SurR9-C84A holds promising potential for a variety of neurological ailments. A comparative analysis of various proteins studied for undifferentiated and differentiated SK-N-SH cells is given in Table 3.
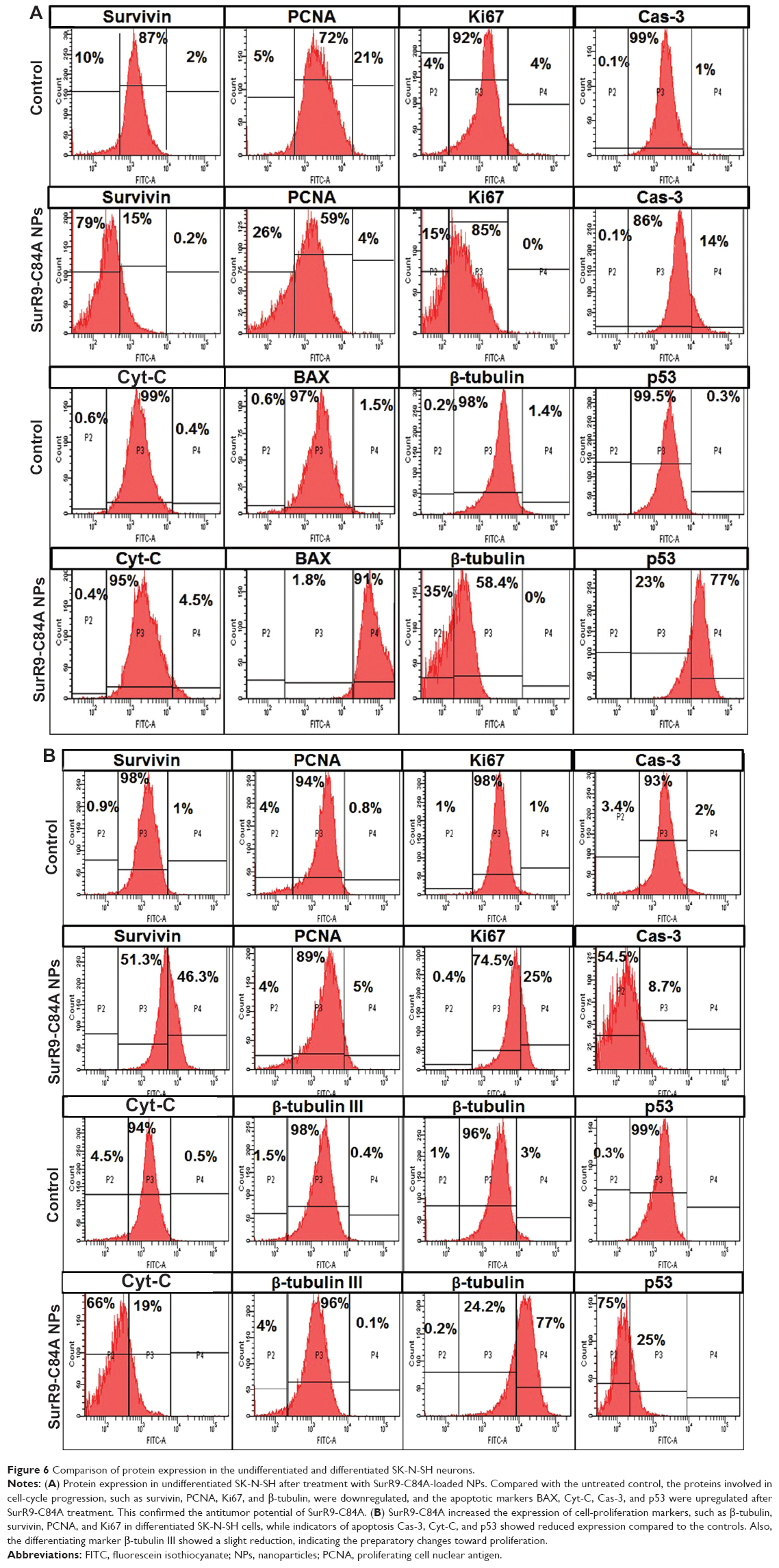 | Figure 6 Comparison of protein expression in the undifferentiated and differentiated SK-N-SH neurons. |
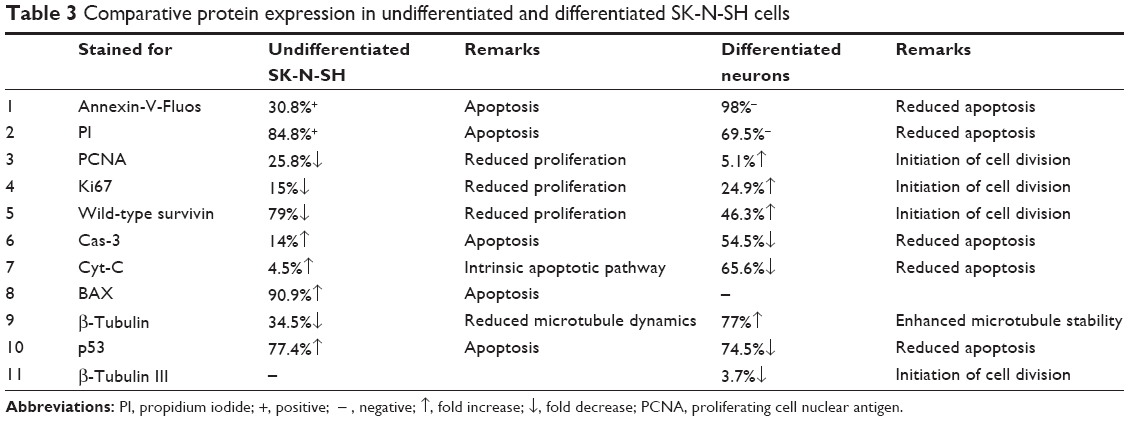 | Table 3 Comparative protein expression in undifferentiated and differentiated SK-N-SH cells |
Discussion
The understanding of various neurological disorders ranging from brain tumors to neurodegeneration has undergone rapid improvement in recent times. Despite the differences in these pathologies, the ultimate result is the irreversible death or degeneration of the neurons.40 Therefore, an ideal therapeutic differentially inducing apoptosis in brain tumor cells and provoking proliferation in neurons would potentially serve the demand. In this study, we have reported the comparative effects of SurR9-C84A on tumorous undifferentiated SK-N-SH and the neuronal differentiated SK-N-SH cells for the first time. Tumor cells due to overexpression of survivin proliferate indefinitely, while the postmitotic cells like the neurons (differentiated SK-N-SH in this case) were deficit in the endogenous pool and showed reduced or no proliferation.14,41 We also observed similar results where undifferentiated and differentiated SK-N-SH cells showed higher and lower expression of survivin, respectively. In terms of cancer therapeutics, survivin is an attractive target on account of its tumor-specific expression compared to normal cells, except the testis and endometrium. Tumor-specific expression is also highly unlikely for any of the IAPs studied so far.5,42 Wild-type survivin is overexpressed in tumor cells, with the potential to regulate cell division and apoptotic mechanisms.18,43 Several reports have confirmed that agents acting against survivin, such as small-molecule inhibitors, antagonists, and antisense agents, interfere with survivin expression or function and suppress tumor progression.12,18,44,45 Studies have also reported that survivin overexpression is responsible for the resistance developed in advanced tumors.5,46 Therefore, in a clinical setting, interfering with the functions of wild-type survivin results in strong antitumor activity, and for this purpose baculovirus inhibitory apoptotic protein repeat (BIR)-motif mutant SurR9-C84A-loaded NPs were employed against the neuroblastoma cell lines. Owing to the difficulty in delivering protein drugs in terms of their physicochemical stability, bioavailability, and biological half-life, etc,16 we tried to encapsulate SurR9-C84A in biodegradable polymeric PLGA NPs. Moreover, PLGA NPs are biodegradable and biocompatible, yielding lactic and glycolic acids that in turn are converted to nontoxic carbon dioxide and water. The PLGA polymer employed in this study had an equal copolymer polylactic acid (PLA) and polyglycolic acid (PGA) ratio, and thus it showed initial burst followed by sustained release.47–49 The significant feature of this nanoformulation was that it showed internalization within 30 minutes of incubation and was found to accumulate in the perinuclear spaces and also in the nucleus (Figure 2A–D). Interestingly, when treated with SurR9-C84A, undifferentiated SK-N-SH cells showed reduced proliferation in a concentration-dependent manner, confirmed by the reduced expression of endogenous survivin and proliferation markers PCNA and Ki67 (Figure 3). Several studies also reported similar results, where cytotoxicity of tumor cells was observed upon interfering with survivin using dominant-negative survivin mutants, antisense oligonucleotides, and small interfering RNA.18,44,45,50 In accordance with the previous results, we also observed increased expression of the apoptotic genes for Cas-3, Cas-8, and Cas-9, with a simultaneous downregulation of wild-type survivin in a dose-dependent manner in undifferentiated cells (Figure 5, A and C). Based on the data published on the selective role of wild-type survivin in antagonizing the mitochondria-mediated intrinsic apoptotic pathway,51 we chose to confirm the mechanistic SurR9-C84A apoptosis by studying the expression of Cyt-C, Cas-3, and Cas-9. In addition, molecular antagonists targeting wild-type survivin preferentially mediate the mitochondria-dependent apoptosis followed by Cas-9 activation.52–54 Therefore, we studied the expression of Cas-3 and Cas-9, and ascertained the following apoptotic mechanism for SurR9-C84A.
As indicated, SurR9-C84A displaced the overexpressed wild-type survivin, leading to mislocalization and disturbed microtubule dynamics and catastrophic effects, eventually culminating in cell death. It also interfered with the formation of the survivin–SMAC/DIABLO complex, thereby relieving its inhibitory effect on Cas-9 activity.54 Also, due to the unavailability of wild-type survivin, hepatitis B X-interacting protein cannot alone inhibit the activation of procaspase-9, and thus propagates mitochondria-mediated apoptosis (Figure 7). Overexpression of cyclin D1 was also observed, owing to the cancerous nature of the neuroblastoma, which was consistent with the earlier results (Figure 5A and C). Though cyclin D1 belongs to the category of oncogenes, overexpression of it indicates the initiation of programmed cell death. A number of studies have reported that cyclin D1 is overexpressed in postmitotic cells, including the neuroblastoma cell lines preceding apoptosis.14,55 This has been explained as a specific selection strategy being exhibited by the cells toward the expression of cyclin D1, where moderate expression leads to overgrowth, while overexpression leads to apoptotic cell death.56 Similarly, the results were in agreement with increased expression of cyclin D1 being observed in undifferentiated SK-N-SH cells and vice versa with differentiated cells. The mechanism can be explained as SurR9-C84A upon forceful expression displacing wild-type survivin from the microtubular assembly, leading to disturbed microtubular dynamics with increased expression of Cyt-C and Cas-3, as determined by the gene and protein study.
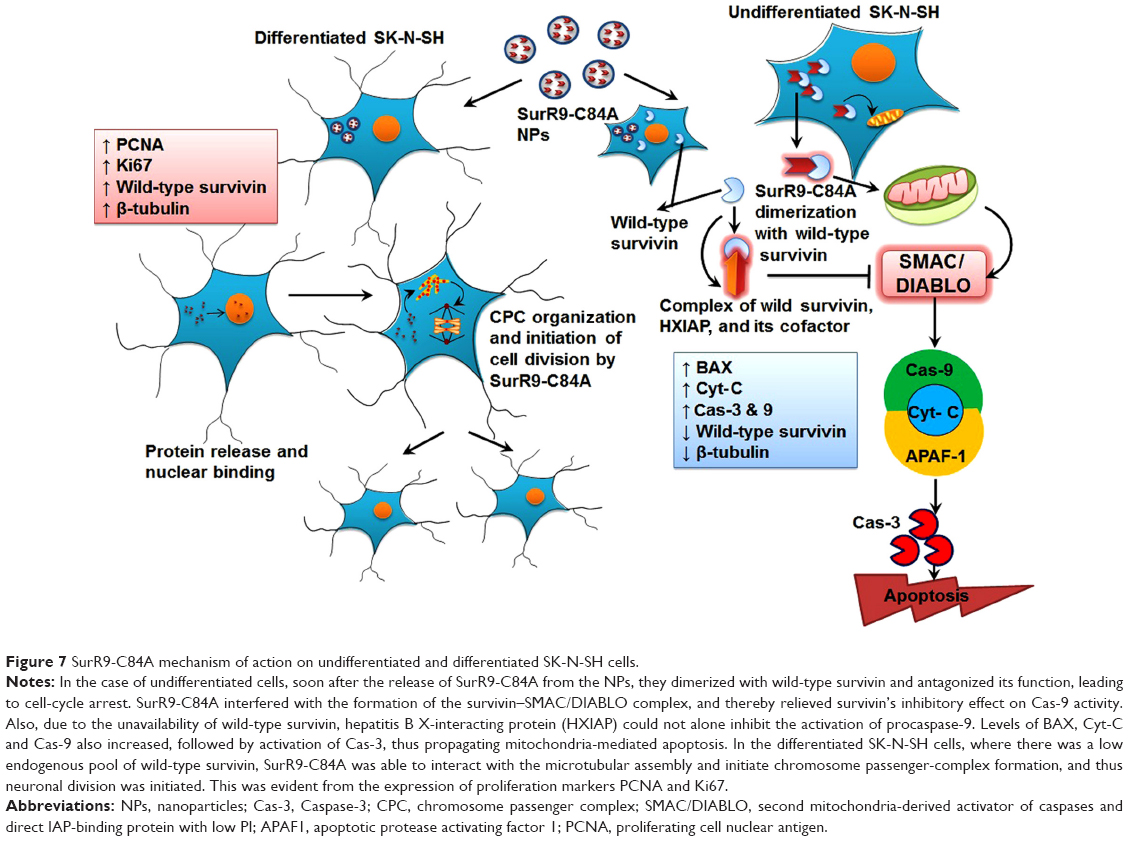 | Figure 7 SurR9-C84A mechanism of action on undifferentiated and differentiated SK-N-SH cells. |
Proliferative effects of SurR9-C84A
Apoptosis is the central mechanism of neuronal loss in various neurological diseases, and developing antiapoptotic strategies to counteract it is a prime way of rescuing the neurons and preventing damage. Among the various antiapoptotic molecules studied, survivin, belonging to the IAP family, may find utility in the treatment of these diseases.
To begin with, survivin is a 16.5 kDa protein exhibiting dual actions on cell cycle and survival. Several studies have reported the essential role of survivin in premature brain development, and that defects in its expression exhibited abnormal brain hypoplasty and immature fetal deaths.4,57 It was also reported to have neuroprotective effects where it colocalized with active Cas-3 and protected astrocytes after brain injury.58 Given the role of endogenous survivin in cell-cycle progression, it plays a prominent role in the spindle-fiber formation interacting with the microtubules and organizes the CPC, complexing with aurora B kinases and INCENP. Also, several reports exist on the role of survivin in premature brain development.4 Therefore, to address the issue of degenerative neurological diseases, a strategy of either neurogeneration or neuroprotection has to be employed. Agents acting against apoptosis like the inducers of Bcl-2 and IAPs have already gained attention for their protective activity in preclinical studies.59,60 Therefore, survivin can be considered an attractive target for treating neurological diseases, as it acts against apoptosis and provokes proliferation. Supporting this concept, one study observed that introduction of survivin restored neural progenitor cell proliferation in the brain, which was achieved by the modulating survivin expression or Wnt signaling.61 The neuroprotective activity of survivin was also confirmed in an in vitro model of multiple sclerosis, where mouse brain astrocytes were insulted with the BeAn strain of Theiler murine encephalomyelitis virus. Interesting results were reported with the upregulation of survivin expression, where it formed a complex with the caspase-3 enzyme, nullifying the apoptotic events to be followed.62
Supportive data also exist on the cytoprotective actions of dominant-negative survivin M34 against the DNA-damaging induced agents.50,53 Given its protective potential, several mutant versions of wild-type survivin have been explored, of which the BIR-motif mutant known as dominant-negative survivin C84A was employed in this study. The reason we chose this mutant is that its mechanism of action is indistinguishable from wild-type survivin, where it binds and organizes microtubular assembly during the interphase. Also, it has neither interference with the separation of chromosomes nor with cell division mimicking the actions of wild-type survivin during mitosis.41 Therefore, we report here the proliferative potential of SurR9-C84A on differentiated neurons, and added to this our previous results also confirmed the neuroprotective activity of SurR9-C84A.14 Though various mutant forms of survivin exist, such as survivin T34A, survivin C84A, survivin T34A-C84A,63 and survivin-D53A, none of them is reported to have the bifunctional activity exhibited by SurR9-C84A.14 Therefore, targeting survivin by the mutant SurR9-C84A seems to have great potential as a therapy for neuronal proliferation and neuroprotection. In order to determine the proliferative effects, SurR9-C84A-loaded NPs were treated with differentiated SK-N-SH cells that behaved like neurons. Interestingly, they showed increased incorporation of BrdU (Figure 4F) specific to cell proliferation and also increased expression of survivin and proliferation markers PCNA and Ki67 (Figure 4A–E). Differentiated SK-N-SH cells showed reduced or no expression of survivin, due to their inability to divide, while treatment with SurR9-C84A enhanced their proliferative potential. This mechanism is explained by SurR9-C84A being able to interact with the microtubular assembly and initiate the spindle formation, which is similar to the actions of endogenous survivin (Figure 7).13,41
We report here for the first time a comparative effect of nanoformulated SurR9-C84A on undifferentiated (tumorous neuroblastoma) and differentiated SK-N-SH (neuronal) cells, where it showed a dual effect on these forms. These dual effects observed with SurR9-C84A were due to the differential expression of endogenous survivin levels. A cytotoxic effect was evident in high wild-type survivin-expressing undifferentiated cells, due to the displacement of endogenous survivin by SurR9-C84A and disturbed microtubular dynamics. In contrast, a proliferative effect was reported for the differentiated cells, owing to their low endogenous survivin levels, where SurR9-C84A effects were indistinguishable from wild-type survivin. These effects were observed through the upregulation of Cas-3, Cyt-C, cyclin D1, annexin and PI+ cells in undifferentiated cells, while the reverse effects were noticed in differentiated cells.
Conclusion
We attempted to demonstrate the dual actions of SurR9-C84A, where it showed unique actions on undifferentiated (cytotoxic) and differentiated (proliferative) SK-N-SH cells. This drives the potential future application of SurR9-C84A for treating a variety of neurological disorders, and the beauty of this therapy would be the selective killing of brain tumor cells owing to the nature of high endogenous survivin expression sparing the neurons. Also, initiation of neuronal proliferation would be a tremendous application in culminating neurodegenerative diseases where neuronal loss is a classical hallmark. Further, future perspectives include the development of aptamer-conjugated brain-targeted delivery of SurR9-C84A, which would enhance bioavailability and therapeutic output.
Acknowledgments
The authors would like to thank the Australia–India Strategic Research Fund (AISRF) and National Health and Medical Research Council (NHMRC) for financial support. No writing assistance was utilized in the production of this manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
Yang Y, Geldmacher DS, Herrup K. DNA replication precedes neuronal cell death in Alzheimer’s disease. J Neurosci. 2001;21(8):2661–2668. | ||
Lindvall O, Kokaia Z, Martinez-Serrano A. Stem cell therapy for human neurodegenerative disorders – how to make it work. Nat Med. 2004;10 Suppl:S42–S50. | ||
Kanwar JR, Kanwar RK, Krissansen GW. Simultaneous neuroprotection and blockade of inflammation reverses autoimmune encephalomyelitis. Brain. 2004;127(Pt 6):1313–1331. | ||
Jiang Y, de Bruin A, Caldas H, et al. Essential role for survivin in early brain development. J Neurosci. 2005;25(30):6962–6970. | ||
Kanwar RK, Cheung CH, Chang JY, Kanwar JR. Recent advances in anti-survivin treatments for cancer. Curr Med Chem. 2010;17(15):1509–1515. | ||
Xu D, Bureau Y, McIntyre DC, et al. Attenuation of ischemia-induced cellular and behavioral deficits by X chromosome-linked inhibitor of apoptosis protein overexpression in the rat hippocampus. J Neurosci. 1999;19(12):5026–5033. | ||
Baratchi S, Kanwar RK, Kanwar JR. Survivin: a target from brain cancer to neurodegenerative disease. Crit Rev Biochem Mol Biol. 2010;45(6):535–554. | ||
Makrantoni V, Stark MJ. Efficient chromosome biorientation and the tension checkpoint in Saccharomyces cerevisiae both require Bir1. Mol Cell Biol. 2009;29(16):4552–4562. | ||
Ruchaud S, Carmena M, Earnshaw WC. The chromosomal passenger complex: one for all and all for one. Cell. 2007;131(2):230–231. | ||
Marusawa H, Matsuzawa S, Welsh K, et al. HBXIP functions as a cofactor of survivin in apoptosis suppression. EMBO J. 2003;22(11):2729–2740. | ||
Rosa J, Canovas P, Islam A, Altieri DC, Doxsey SJ. Survivin modulates microtubule dynamics and nucleation throughout the cell cycle. Mol Biol Cell. 2006;17(3):1483–1493. | ||
Cheung CH, Sun X, Kanwar JR, Bai JZ, Cheng L, Krissansen GW. A cell-permeable dominant-negative survivin protein induces apoptosis and sensitizes prostate cancer cells to TNF-α therapy. Cancer Cell Int. 2010;10:36. | ||
Li F, Ambrosini G, Chu EY, et al. Control of apoptosis and mitotic spindle checkpoint by survivin. Nature. 1998;396(6711):580–584. | ||
Baratchi S, Kanwar RK, Cheung CH, Kanwar JR. Proliferative and protective effects of SurR9-C84A on differentiated neural cells. J Neuroimmunol. 2010;227(1–2):120–132. | ||
Kanwar JR, Kanwar RK, Wang D, Krissansen GW. Prevention of a chronic progressive form of experimental autoimmune encephalomyelitis by an antibody against mucosal addressin cell adhesion molecule-1, given early in the course of disease progression. Immunol Cell Biol. 2000;78(6):641–645. | ||
Rekha MR, Sharma CP. Oral delivery of therapeutic protein/peptide for diabetes – future perspectives. Int J Pharm. 2013;440(1):48–62. | ||
Mukherjee B, Santra K, Pattnaik G, Ghosh S. Preparation, characterization and in-vitro evaluation of sustained release protein-loaded nanoparticles based on biodegradable polymers. Int J Nanomedicine. 2008;3(4):487–496. | ||
Kanwar JR, Shen WP, Kanwar RK, Berg RW, Krissansen GW. Effects of survivin antagonists on growth of established tumors and B7-1 immunogene therapy. J Natl Cancer Inst. 2001;93(20):1541–1552. | ||
Rajapaksa TE, Stover-Hamer M, Fernandez X, Eckelhoefer HA, Lo DD. Claudin 4-targeted protein incorporated into PLGA nanoparticles can mediate M cell targeted delivery. J Control Release. 2010;142(2):196–205. | ||
Messi E, Florian MC, Caccia C, Zanisi M, Maggi R. Retinoic acid reduces human neuroblastoma cell migration and invasiveness: effects on DCX, LIS1, neurofilaments-68 and vimentin expression. BMC Cancer. 2008;8:30. | ||
Jain RA. The manufacturing techniques of various drug loaded biodegradable poly(lactide-co-glycolide) (PLGA) devices. Biomaterials. 2000;21(23):2475–2490. | ||
Bilati U, Allémann E, Doelker E. Poly(D,L-lactide-co-glycolide) protein-loaded nanoparticles prepared by the double emulsion method – processing and formulation issues for enhanced entrapment efficiency. J Microencapsul. 2005;22(2):205–214. | ||
Gasper MM, Blanco D, Cruz ME, Alonso MJ. Formulation of L-asparaginase-loaded poly(lactide-co-glycolide) nanoparticles: influence of polymer properties on enzyme loading, activity and in vitro release. J Control Release. 1998;52(1–2):53–62. | ||
Nicoli S, Santi P, Couvreur P, Couarraze G, Colombo P, Fattal E. Design of triptorelin loaded nanospheres for transdermal iontophoretic administration. Int J Pharm. 2001;214(1–2):31–35. | ||
Kumar G, Sharma S, Shafiq N, Khuller GK, Malhotra S. Optimization, in vitro-in vivo evaluation, and short-term tolerability of novel levofloxacin-loaded PLGA nanoparticle formulation. J Pharm Sci. 2012;101(6):2165–2176. | ||
Betancourt T, Brown B, Brannon-Peppas L. Doxorubicin-loaded PLGA nanoparticles by nanoprecipitation: preparation, characterization and in vitro evaluation. Nanomedicine (Lond). 2007;2(2):219–232. | ||
Joshi SA, Chavhan SS, Sawant KK. Rivastigmine-loaded PLGA and PBCA nanoparticles: preparation, optimization, characterization, in vitro and pharmacodynamic studies. Eur J Pharm Biopharm. 2010;76(2):189–199. | ||
Mukerjee A, Vishwanatha JK. Formulation, characterization and evaluation of curcumin-loaded PLGA nanospheres for cancer therapy. Anticancer Res. 2009;29(10):3867–3875. | ||
Ahmad A, Mukherjee P, Senapati S, et al. Extracellular biosynthesis of silver nanoparticles using the fungus Fusarium oxysporum. Colloids Surf B Biointerfaces. 2003;28(4):313–318. | ||
Park H, Ko MY, Paik MK, Soh CT, Seo JH, Im KI. Cytotoxicity of a cysteine proteinase of adult Clonorchis sinensis. Korean J Parasitol. 1995;33(3):211–218. | ||
Pitt CG. The controlled parenteral delivery of polypeptides and proteins. Int J Pharm. 1990;59(3):173–196. | ||
Uchida T, Yagi A, Oda Y, Nakada Y, Goto S. Instability of bovine insulin in poly(lactide-co-glycolide) (PLGA) microspheres. Chem Pharm Bull (Tokyo). 1996;44(1):235–236. | ||
Chen HB, Zheng Y, Tian G, et al. Oral delivery of DMAB-modified docetaxel-loaded PLGA-TPGS nanoparticles for cancer chemotherapy. Nanoscale Res Lett. 2010;6(1):1–10. | ||
Fernando LP, Kandel PK, Yu J, McNeill J, Ackroyd PC, Christensen KA. Mechanism of cellular uptake of highly fluorescent conjugated polymer nanoparticles. Biomacromolecules. 2010;11(10):2675–2682. | ||
Lai SK, Wong CH, Lee YP, Li HY. Caspase-3-mediated degradation of condensin Cap-H regulates mitotic cell death. Cell Death Differ. 2011;18(6):996–1004. | ||
Li N, Nguyen HH, Byrom M, Ellington AD. Inhibition of cell proliferation by an anti-EGFR aptamer. PLoS One. 2011;6(6):e20299. | ||
Scholzen T, Gerdes J. The Ki-67 protein: from the known and the unknown. J Cell Physiol. 2000;182(3):311–322. | ||
Taupin P. BrdU immunohistochemistry for studying adult neurogenesis: paradigms, pitfalls, limitations, and validation. Brain Res Rev. 2007;53(1):198–214. | ||
Riccardi C, Nicoletti I. Analysis of apoptosis by propidium iodide staining and flow cytometry. Nat Protoc. 2006;1(3):1458–1461. | ||
Kanwar JR, Sriramoju B, Kanwar RK. Neurological disorders and therapeutics targeted to surmount the blood–brain barrier. Int J Nanomedicine. 2012;7:3259–3278. | ||
Skoufias DA, Mollinari C, Lacroix FB, Margolis RL. Human survivin is a kinetochore-associated passenger protein. J Cell Biol. 2000;151(7):1575–1582. | ||
Kanwar JR, Kamalapuram SK, Kanwar RK. Survivin signaling in clinical oncology: a multifaceted dragon. Med Res Rev. 2013;33(4):765–789. | ||
Cheung CH, Sun X, Kanwar JR, Bai JZ, Cheng L, Krissansen GW. Correction: A cell-permeable dominant-negative survivin protein induces apoptosis and sensitizes prostate cancer cells to TNF-α therapy. Cancer Cell Int. 2010;10:43. | ||
Williams NS, Gaynor RB, Scoggin S, et al. Identification and validation of genes involved in the pathogenesis of colorectal cancer using cDNA microarrays and RNA interference. Clin Cancer Res. 2003;9(3):931–946. | ||
Yamamoto T, Manome Y, Nakamura M, Tanigawa N. Downregulation of survivin expression by induction of the effector cell protease receptor-1 reduces tumor growth potential and results in an increased sensitivity to anticancer agents in human colon cancer. Eur J Cancer. 2002;38(17):2316–2324. | ||
Krepela E, Dankova P, Moravcikova E, et al. Increased expression of inhibitor of apoptosis proteins, survivin and XIAP, in non-small cell lung carcinoma. Int J Oncol. 2009;35(6):1449–1462. | ||
Dorta M, Munguía O, Llabres M. Effects of polymerization variables on PLGA properties: molecular weight, composition and chain structure. Int J Pharm. 1993;100(1–3):9–14. | ||
Fonseca C, Simoes S, Gaspar R. Paclitaxel-loaded PLGA nanoparticles: preparation, physicochemical characterization and in vitro anti-tumoral activity. J Control Release. 2002;83(2):273–286. | ||
Okada H, Toguchi H. Biodegradable microspheres in drug delivery. Crit Rev Ther Drug Carrier Syst. 1995;12(1):1–99. | ||
Grossman D, Kim PJ, Schechner JS, Altieri DC. Inhibition of melanoma tumor growth in vivo by survivin targeting. Proc Natl Acad Sci U S A. 2001;98(2):635–640. | ||
Grossman D, Kim PJ, Blanc-Brude OP, et al. Transgenic expression of survivin in keratinocytes counteracts UVB-induced apoptosis and cooperates with loss of p53. J Clin Invest. 2001;108(7):991–999. | ||
Du C, Fang M, Li Y, Li L, Wang X. Smac, a mitochondrial protein that promotes cytochrome C-dependent caspase activation by eliminating IAP inhibition. Cell. 2000;102(1):33–42. | ||
O’Connor DS, Grossman D, Plescia J, et al. Regulation of apoptosis at cell division by p34cdc2 phosphorylation of survivin. Proc Natl Acad Sci U S A. 2000;97(24):13103–13107. | ||
Shi Y. Mechanisms of caspase activation and inhibition during apoptosis. Mol Cell. 2002;9(3):459–470. | ||
Freeman RS, Estus S, Johnson EM Jr. Analysis of cell cycle-related gene expression in postmitotic neurons: selective induction of cyclin D1 during programmed cell death. Neuron. 1994;12(2):343–355. | ||
Kranenburg O, van der Eb AJ, Zantema A. Cyclin D1 is an essential mediator of apoptotic neuronal cell death. EMBO J. 1996;15(1):46–54. | ||
Delvaeye M, De Vriese A, Zwerts F, et al. Role of the 2 zebrafish survivin genes in vasculo-angiogenesis, neurogenesis, cardiogenesis and hematopoiesis. BMC Dev Biol. 2009;9:25. | ||
Villapol S, Acarin L, Faiz M, Castellano B, Gonzalez B. Survivin and heat shock protein 25/27 colocalize with cleaved caspase-3 in surviving reactive astrocytes following excitotoxicity to the immature brain. Neuroscience. 2008;153(1):108–119. | ||
Rami A, Bechmann I, Stehle JH. Exploiting endogenous anti-apoptotic proteins for novel therapeutic strategies in cerebral ischemia. Prog Neurobiol. 2008;85(3):273–296. | ||
Trapp T, Korhonen L, Besselmann M, Martinez R, Mercer EA, Lindholm D. Transgenic mice overexpressing XIAP in neurons show better outcome after transient cerebral ischemia. Mol Cell Neurosci. 2003;23(2):302–313. | ||
Miranda CJ, Braun L, Jiang Y, et al. Aging brain microenvironment decreases hippocampal neurogenesis through Wnt-mediated survivin signaling. Aging Cell. 2012;11(3):542–552. | ||
Rubio N, Garcia-Segura LM, Arevalo MA. Survivin prevents apoptosis by binding to caspase-3 in astrocytes infected with the BeAn strain of Theiler’s murine encephalomyelitis virus. J Neurovirol. 2012;18(5):354–363. | ||
Yan H, Thomas J, Liu T, et al. Induction of melanoma cell apoptosis and inhibition of tumor growth using a cell-permeable survivin antagonist. Oncogene. 2006;25(52):6968–6974. | ||
Wang JB, Qi LL, Zheng SD, Wu TX. Curcumin induces apoptosis through the mitochondria-mediated apoptotic pathway in HT-29 cells. J Zhejiang Univ Sci B. 2009;10(2):93–102. | ||
Kurata K, Yanagisawa R, Ohira M, Kitagawa M, Nakagawara A, Kamijo T. Stress via p53 pathway causes apoptosis by mitochondrial Noxa upregulation in doxorubicin-treated neuroblastoma cells. Oncogene. 2008;27(6):741–754. | ||
Monga J, Pandit S, Chauhan CS, Sharma M. Cytotoxicity and apoptosis induction in human breast adenocarcinoma MCF-7 cells by (+)-cyanidan-3-ol. Exp Toxicol Pathol. 2013;65(7–8):1091–1100. | ||
Ling Q, Xu X, Wei X, et al. Oxymatrine induces human pancreatic cancer PANC-1 cells apoptosis via regulating expression of Bcl-2 and IAP families, and releasing of cytochrome C. J Exp Clin Cancer Res. 2011;30:66. | ||
Li Y, Shibata Y, Zhang L, Kuboyama N, Abiko Y. Periodontal pathogen Aggregatibacter actinomycetemcomitans LPS induces mitochondria-dependent-apoptosis in human placental trophoblasts. Placenta. 2011;32(1):11–19. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.