")
Back to Journals » International Journal of Nanomedicine » Volume 16
Nano-Sized Hydroxyapatite Induces Apoptosis and Osteogenic Differentiation of Vascular Smooth Muscle Cells via JNK/c-JUN Pathway
Authors Liu Q, Xiang P , Chen M, Luo Y, Zhao Y, Zhu J, Jing W, Yu H
Received 28 January 2021
Accepted for publication 13 April 2021
Published 27 May 2021 Volume 2021:16 Pages 3633—3648
DOI https://doi.org/10.2147/IJN.S303714
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Yan Shen
Qi Liu,1,* Pingping Xiang,1,* Mingyao Chen,1 Yi Luo,1 Yun Zhao,1,2 Jinyun Zhu,1 Wangwei Jing,1 Hong Yu1
1Department of Cardiology, Cardiovascular Key Laboratory of Zhejiang Province, The Second Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, Zhejiang Province, 310009, People’s Republic of China; 2The Affiliated Cardiovascular Hospital of Qingdao University, Qingdao, Shandong Province, 266071, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Hong Yu
Department of Cardiology, Cardiovascular Key Laboratory of Zhejiang Province, The Second Affiliated Hospital, Zhejiang University School of Medicine, 88 JieFang Road, Hangzhou, Zhejiang Province, 310009, People’s Republic of China
Tel +86-571-87783992
Fax +86-571-88002709
Email [email protected]
Purpose: The deposition of hydroxyapatite (HAp) crystals plays an important role in the development of vascular calcification (VC). This study aimed to demonstrate the effects of nanosized HAp (nHAp) on vascular smooth muscle cells (VSMCs) and VC progression.
Methods: Transmission electron microscopy (TEM) was used to examine cellular uptake of nHAp. Cell viability was determined using CCK-8 assay kit. Mitochondrial impairment and reactive oxygen species were detected by TEM and fluorescence dye staining, respectively. Cell apoptosis was detected by Western blot analysis and Annexin V staining. Mouse model of VC was built via applying nHAp on the surface of abdominal aorta. Calcification was visualized by Alizarin red and von Kossa staining.
Results: We found that nHAp could promote osteogenic transformation of VSMCs by elevating expression of runt-related factor 2 (Runx2), osteopontin (OPN) and alkaline phosphatase (ALP), impairing function and morphology of mitochondria and inducing apoptosis of VSMCs. More phosphorylation of c-Jun N-terminal protein kinase/c-JUN (JNK/c-JUN) in VSMCs was detected after mixing nHAp with VSMCs. HAp-induced osteogenic transformation of VSMCs was blocked by JNK inhibitor SP600125, resulted in decreased ALP activity, less Runx2 and OPN expressions. SP600125 also inhibited apoptosis of VSMCs. Application of nHAp to outside of aorta induced osteogenic transformation and apoptosis of VSMCs, and significant deposition of calcium on the vessel walls of mice, which can be effectively attenuated by SP600125.
Conclusion: JNK/c-JUN signaling pathway is critical for nHAp-induced calcification, which could be a potential therapeutic target for controlling the progression of VC.
Keywords: vascular calcification, hydroxyapatite, apoptosis, mitochondria, osteogenic transformation, JNK
Introduction
Vascular calcification (VC) is associated with an increased risk of stroke, heart attack, and rupture of atherosclerotic plaque.1 The development of VC involves in the deposit of hydroxyapatite (HAp) in the vessel wall. According to previous studies, VC occurs mainly in patients with metabolic syndrome, type 1 and 2 diabetes, chronic kidney disease, atherosclerosis, and aging.2 Vascular smooth muscle cells (VSMCs) are a key cell type involved in VC. Osteogenic transdifferentiation, apoptosis, cellular senescence, calcium overload in SMCs and their release of extracellular vesicles will all accelerate the progression of VC.3,4
The main component of VC is hydroxyapatite with calcium and phosphate. High-Pi environment can stimulate VSMCs to release matrix vesicles (MVs), forming a microenvironment which can induce nucleation of nanosized HAp (nano-HAp or nHAp).5 Evidence showed that nanocrystals can affect a number of cell functions, such as stimulation of endocytotic activity,6 proliferation, activation of matrix metalloproteinases, and induction of proinflammatory cytokines.7,8 However, whether nHAp can stimulate these activities and change of VSMC phenotype remains to be proved. Therefore, the main purpose of the study is to figure out the effects of such formed nHAp on VSMCs and VC progression.
Demer et al demonstrated that hyper-phosphatemia-induced nano-crystals enhanced expressions of bone morphogenetic protein-2 (BMP-2) and osteopontin (OPN) in SMCs. Huang et al also showed that treating rat SMCs with nHAp induced not only osteoblast transformation with elevated expressions of osteogenesis-related genes runt-related factor (Runx2), osteocalcin (OCN) and BMP-2, but also apoptosis of SMCs, promoting more calcium deposits.9 Thus, HAp deposition on the cell surface may play a crucial role in VC progression. For the calcium oxalate renal stone formation, it has been proposed that after initial crystal deposition on the cell surface, CaOx crystals grow and aggregate to form large crystals and ultimately leading to the formation of renal stone.10 HAp could play a similar role as calcium oxalate crystal and induce osteogenic gene expression during VC progression. However, the exact mechanism of HAp-induced VC formation is still unclear.
Apoptosis of SMCs plays an essential role in VC.2 Either release of apoptotic signals from mitochondria or stimulation of death receptors on cell surface may initiate apoptosis.11 The release of mitochondrial cytochrome c initiated the mitochondrial pathway, forming a large multi-protein complex composed of cytochrome c, Apaf-1, and pro-caspase-9. Caspases-9 then activates downstream caspases by proteolysis, particularly caspases 3 and 7, which are the major causes of cell apoptosis.12 However, the signaling pathway involved in nHAp-induced apoptosis has not been fully investigated.
In this study, we aim to illuminate the cellular mechanisms by which nHAp crystals affect gene expression in VSMCs. Evidence from previous studies showed that nanoparticles can activate mitogen-activated protein kinase (MAPK) signaling cascade7 in cells which include macrophages,8 bone marrow stromal cells (BMSCs),13 and endothelial cells (ECs).14 We hypothesized that extracellular nHAp stimulated MAPK pathways which caused gene expression changes in SMCs. We found that JNK/c-JUN signaling pathway is critical for nHAp-induced VC. A better understanding of the response of VSMCs to nHAp will provide new strategy for the treatment of VC.
Methods
Cell Culture
Vascular SMCs were isolated from aortas of 8-week-old wild-type C57BL/6 mice (Slac Laboratory Animal, Shanghai, China) according to previous studies.15 Cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) (Hyclone) containing 20% fetal bovine serum (FBS) (Hyclone), and 100 U/mL penicillin, 100 mg/mL streptomycin in a 37°C incubator supplemented with 5% CO2. VSMCs between passages 3 and 5 were used for the following experiments.
Characterization of HAp Crystals
HAp was purchased from Aladdin Biochemical Technology Company (Shanghai, China). The CAS number is 1306–06-5. The size of the particle is less than 100 nm. X-ray diffraction (XRD, Bruker D8 Advance) with Cu-Kα radiation and Fourier transform infrared (FT-IR) spectroscopy (FITR-8400S, SHIMADZU, Japan) were used to identify the crystalline phase and phase composition of nHAp. The size and morphology of the nHAp crystals were characterized via the scanning electron microscope (SU8010, HITACHI, Japan).
Preparation of Fluorescence‐Labeled HAp
HAp crystals were labeled via fluorexon staining. Briefly, 100mg nHAp crystals and 10 mL fluorexon solution (10mg/mL) were mixed and stirred for 30 min at room temperature. Subsequently, the HAp crystals were collected via centrifugation, washed three times with anhydrous ethanol and distilled water to make sure that there was no free fluorexon residue, and dried.
Cell Viability Assay
The Cell Counting Kit-8 (CCK-8, Beyotime Biotechnology, China) assay was used to evaluate cell viability of VSMCs after nHAp treatment. The cell density seeded in each well was 2000 per well. Cells were cultured with different concentrations (0, 25, 50, 100, 200 and 400 μg/mL) of HAp for 72 h at 5% CO2, 37°C on a 96-well plate. After incubation, the mixture of CCK-8 solution (consisting of 90% growth medium and 10% CCK-8) was added into each well. Then, the absorbance (OD) was measured at 450 nm after incubation for 3 h at 37°C.
Intracellular Reactive Oxygen Species (ROS) Assay
Cell suspension of VSMCs at concentration of 1×105 cells/mL was seeded 2 mL per well in six-well plates. VSMCs were then divided into two groups: (A) control group: only DMEM was added; (B) nHAp group: 100 µg/mL nHAp (mixed with DMEM) was added; After 24 h of incubation, A 500 μL dichloro-dihydrofluorescein diacetate (DCFH-DA) (Beyotime Biotechnology, Shanghai, China) diluted with serum-free medium was added and stained for 20–30 min. After washing with serum-free medium for three times, the cells were detected by flow cytometry, and the fluorescence intensity was quantitatively detected.
Transmission Electron Microscopy (TEM)
TEM was used to detect the internalization of nHAp and mitochondrial ultrastructure of VSMCs. Briefly, after HAp treatment, the cells were digested with 0.25% trypsin for 1–2 minutes. The culture medium containing FBS was used to terminate the digestion. Then the cells were centrifuged for 5 minutes. After the supernatant was abandoned, the cells in FBS were centrifuged again. Then the cells were fixed in 2.5% glutaraldehyde overnight. After washing three times with phosphate-buffered saline (PBS), the specimens were fixed with 1% OsO4 for 1–2 h. The specimens were then dehydrated with a gradient of ethanol and soaked overnight in acetone. In addition, the specimens were implanted in Spurr resin and sectioned in Leica EM UC7 (Leica, Wetzlar, Germany). The sections were stained with uranyl acetate and alkaline lead citrate, and the pictures were acquired by Hitachi Model H-7650 TEM.
Measurement of Mitochondrial Membrane Potential (Δψm)
Δψm was measured after VSMCs were stained with tetramethyl rhodamine methyl ester (TMRM, 200 nmol/L) for 30 min in a 37°C incubator supplemented with 5% CO2. The cells were also incubated with oligomycin (10 μM) and FCCP (50 μM) for 30 min as a positive and negative controls, respectively. Cells were washed with PBS three times and covered with 200μL PBS. Images were obtained via fluorescence microscope (200×). The mean relative fluorescence intensity (MFI) of control or nano-HAp divided by the MFI difference between Oligomycin and FCCP was acquired using ImageJ software.
Activity Measurements of Alkaline Phosphatase (ALP)
The ALP activity of VSMCs was detected via an ALP detection kit according to the manufacturer’s instructions (Beyotime Biotechnology, Shanghai, China). After SMCs were cultured with HAp for 7 days, SMCs were washed with cold PBS, scraped into 100 μL of lysis buffer (0.2% NP-40 with l mM MgCl2) and incubated for 30 min on ice. The lysis was centrifugated at 5000x rpm for 5 min and the supernatant was collected. All samples were stored under −80°C until use for ALP assay which was performed following the manufacturer’s instructions (Beyotime Biotechnology, Shanghai, China). By catalyzing p-nitrophenyl phosphate (pNPP) to pNP, ALP activity is referred as the change of absorbance at 405 nm, and then normalized to the protein level. Unit Definition: One ALP DEA unit equals hydrolyzation of 1 mole of p-nitrophenyl phosphate per minute at pH 9.8 at 37°C.
Mitotracker Staining
VSMCs were grown on coverslips inside a 12-well plate filled with DMEM. When the cells were treated with or without 100 μg/mL nHAp for 24 h, the culture medium was removed from the dish, and prewarmed (37°C) staining solution containing 100 nM MitoTracker® probe (Invitrogen) was added. After incubation for 30 minutes, the staining solution was replaced with fresh prewarmed media. Cells were observed under a fluorescence microscope.
Annexin V/PI Staining to Detect Apoptosis
After washing with PBS, the harvested cells were incubated with Annexin V-APC for 30 min according to the manufacturer’s instruction (Dojindo, Minato-ku, Tokyo, Japan). After treatment, the cells were detected by flow cytometry (BD Biosciences, San Jose, CA, USA).
Western Blot
Cells were lysed by the RIPA lysis buffer (Beyotime, China) on ice. Protein concentrations were quantified by BCA protein assay (Beyotime, China). The immunoblots were probed with appropriate primary overnight at 4°C followed by incubation with the corresponding secondary antibodies at room temperature for 1 h.
The blots were infiltrated with ECL (Bio-Rad, USA) and detected by ChemiDoc™MP Imaging System (Bio-Rad, USA). The densitometry of target bands was normalized to internal control β-actin. The antibodies are listed as follow up: Runx2 (1:1000, ET1612-47, Huabio, Hangzhou, China); OPN (1:1000, 0806–6, Huabio, Hangzhou, China); cleaved-caspase 3 (1:1000, 9664S, Cell Signaling Technology, USA); cleaved-caspase 9 (1:1000, 9509S, Cell Signaling Technology, USA); TOM20 (1:1000, 42406S, Cell Signaling Technology, USA); cytochrome c (1:1000, 12,963, Cell Signaling Technology, USA); T-JNK (1:1000, ab179461, Abcam, UK); p-JNK (1:1000, ab124956, Abcam, UK); T-c-JUN (1:1000; ET1608-3, Huabio, Hangzhou, China); p-c-JUN (1:1000; ET-1608-4, Huabio, Hangzhou, China); β-actin (1:3000, R1102-1, Huabio, Hangzhou, China); tubulin (1:3000, M1305-2, Huabio, Hangzhou, China); anti-rabbit, anti-mouse secondary antibody (1:3000, #HA1011, #HA1006, Huabio, Hangzhou, China).
TUNEL Staining
Tissue was fixed with 4% paraformaldehyde and permeabilized with 0.25% Triton for 10 min each, and then, incubated with TUNEL reaction compound for 60 min at 37°C in the dark according to the manufacturer’s instructions (Beyotime Biotechnology, Shanghai, China). Nuclei were stained with Hoechst33342 (Beyotime Biotechnology). The apoptotic rate was calculated by the proportion of TUNEL-positive cells in total cells/nuclei.
Alizarin Red Staining
After cultured at specified conditions for 14 days, SMCs on dishes were fixed with 10% formalin for 30 min, then washed with double distilled water (ddH2O) twice and incubated with Alizarin Red (Solarbio Life Sciences, China) for five minutes and washed with ddH2O twice to remove the excessive dye. After examination and photography under a microscopy, the dye on the cells was extracted with 100µl Hexadecyl Pyridinium chloride (Sigma-Aldrich) and the OD at 560 nm was measured using a microplate reader (Spectra MAX 190, Molecular Devices, USA).
Von Kossa Staining
A segment of the abdominal aorta was excised and fixed in 10% formalin for 24 h, then dehydrated, embedded in paraffin, and cut into 5-µm -thick sections. The slides were deparaffinized, dehydrated and washed in distilled water (ddH2O),the samples were incubated with reagent A of Von Kossa staining Kit (Solarbio Life Sciences, China) and placed under ultraviolet light for 10 min, and then placed into reagent B for 2 min followed by staining with hematoxylin for the demonstration of cell nucleus.
Calcium Quantification
To determine the calcium deposition in vitro, cells that were cultured with 100 μg/mL nano-HAp for 2 weeks were washed in cold PBS twice and dissolved in 0.6 N HCl at 4°C for 24 hours. Calcium was measured by Calcium Assay Kit (Cayman Chemical, USA). The amount of calcium was normalized to the total amount of protein.
Animals and Treatment
All animal experiments were performed with approval of the Animal Use and Care Committee of Zhejiang University, which complies with the Guide for the Care and Use of Laboratory Animals, 8th edition published by the USA National Institutes of Health. The approval for the study was granted by the Ethics Review Committee from the Second Affiliated Hospital of Zhejiang University. Mice (C57BL/6, 3-month old) were anesthetized with isoflurane. All mice had free access to a normal diet and water. The mice were shaved and the skin was disinfected with 75% ethanol prior to operation. A longitudinal skin incision about 2 cm in length on the abdominal surface of a mouse was made, and the abdominal aorta was isolated (between renal artery and the bifurcation of common iliac artery), the vascular adventitia was torn off. Sterile cotton gauze presoaked with 0.9% NaCl or nano-HAp (25 mg/mL 0.9% NaCl) was placed on the surface of aorta for 15 min with the aid of Stereo Microscope (SZ61, Olympus, Japan). Sufficient care was taken not to contaminate surrounding tissues with nano-HAp. The abdominal cavity was then washed with sterile saline. The incisions were closed with surgical sewing. The JNK inhibitor SP600125 (30 mg/kg, i.p.) was administered 30 min before the surgery. And then SP600125 was given (30 mg/kg, i.p.) per day.
Statistical Analysis
All experimental data are presented as mean ± standard deviation (SD) and analyzed by Program Graph-Pad Prism version 6.0. For comparisons between two groups, significance was determined via Student’s t-test. For comparisons among multiple groups, data were performed by one-way ANOVA. A value of P <0.05 was considered statistically significant.
Results
Characterization and Fluorescence Labeling of nHAp
Scan electronic microscopy (SEM) revealed that nHAp crystals were homogeneous, rod-like nanoparticles (Figure 1A). Spectrum of nHAp crystals under X-ray diffraction (XRD) showed the diffraction peaks (25.8°, 31.8°, 32.9°, 34.0°, 39.8°, 46.7°,49.4°) (Figure 1B), which conform to (002), (211), (112), (202), (310), (222), and (213) crystal plane diffraction peaks of the standard card of HAp (JCPDS no. 09–0432), respectively. Fourier transform infrared (FT-IR) spectrum showed the vibration peaks of O−H stretching in HAp at 3,566 and 3,432 cm−1, and the vibration peaks of asymmetric stretching P−O in PO43− groups at 564 and 603 cm−1 (Figure 1C). Both XRD and FT-IR spectra verified that the crystals are pure-phase nHAp crystals. The nHAp crystals were dyed with green fluorescent molecule fluorexon (GF) (Figure 1D–E).
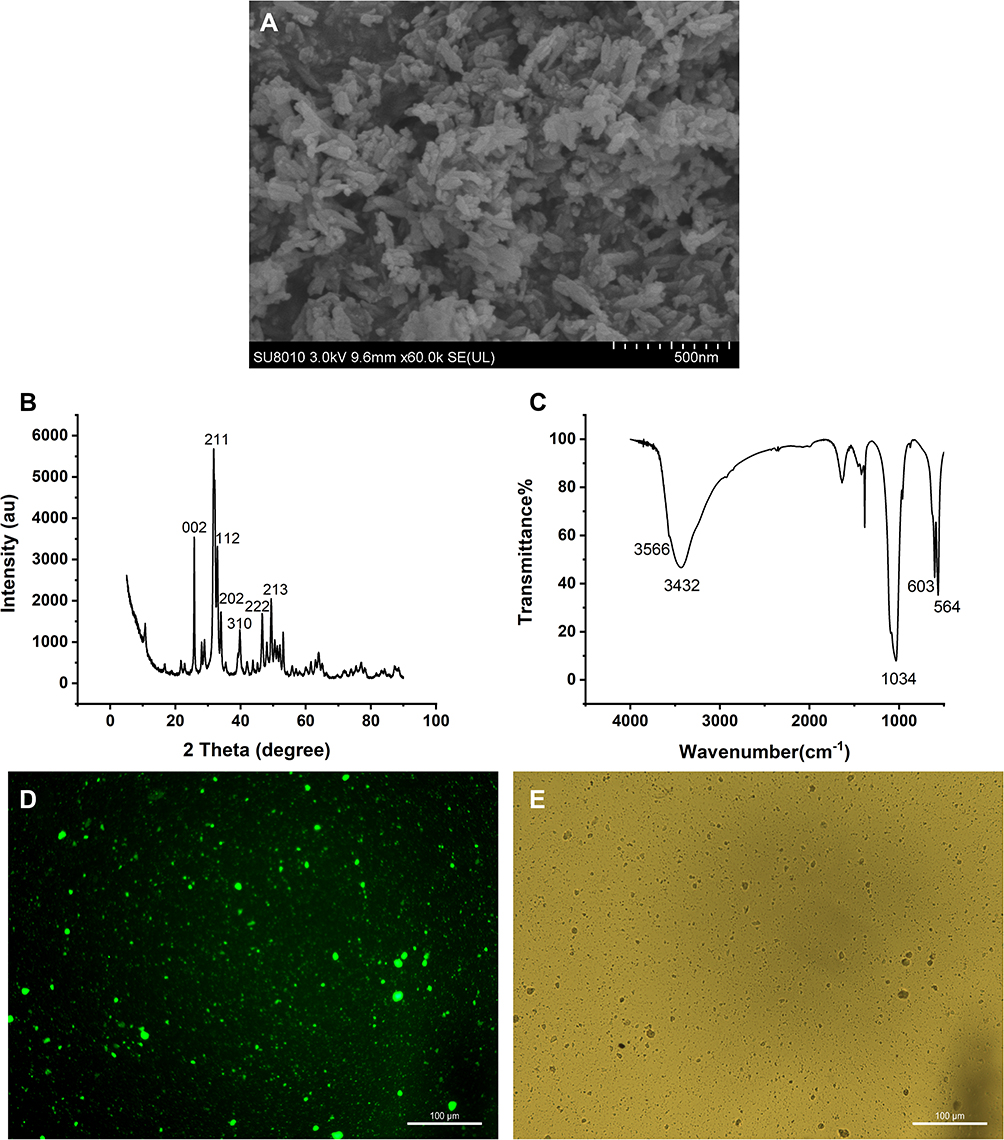 |
Figure 1 Characterization and staining of nano-HAp crystals. (A) Image of nHAp crystals under Scan Electron Microscopy (SEM); (B) XRD spectrum of nHAp; (C) FT-IR spectrum of nHAp; (D) fluorescent image of nHAp after fluorexon dyeing; (E) bright field microscopic image of nHAp. Scale bars: 100 μm. |
Nano-HAp Was Internalized into VSMCs and Caused Cytotoxicity
After GF-labeled nHAp was mixed with VSMCs, internalization of nHAp into cells were observed (Figure 2A), and electron-dense particles were accumulated in the cytoplasm and enclosed in vesicles as well (Figure 2B). Cell viability was significantly decreased after VSMCs were treated with the nHAp at different doses (25, 50, 100, 200, and 400 μg/mL) for 72 h (Figure 2C), indicating the cytotoxicity of nHAp to VSMCs.
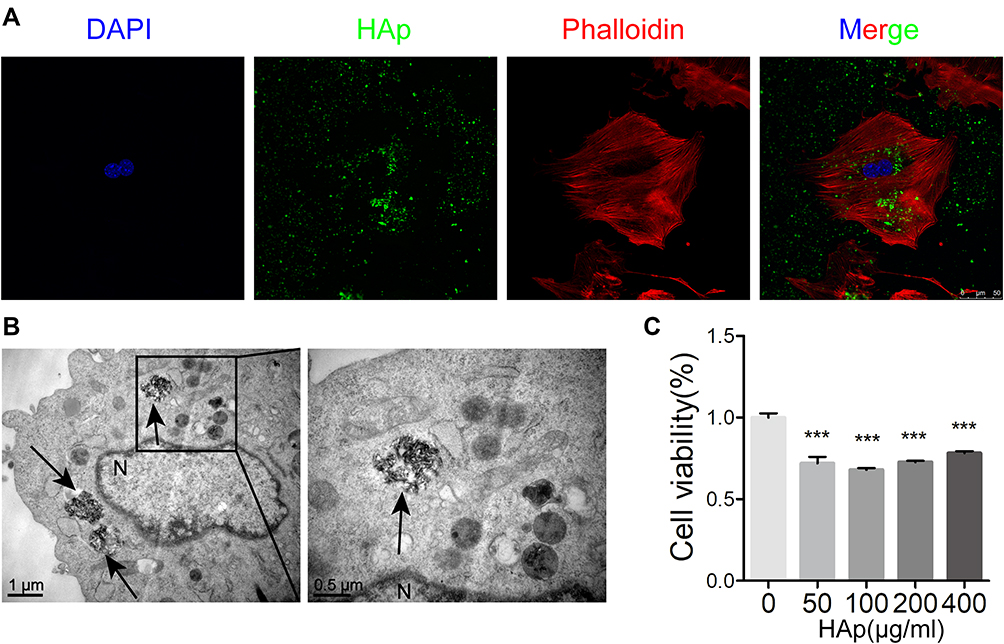 |
Figure 2 Internalization of nano-HAp by VSMCs and cell viability. (A) Laser scanning confocal microscopy (LSCM) image of VSMCs after incubation for 24 hours with green fluorescent-labeled nHAp. Cellular actins were stained with Phalloidin-Tritc (red), and nucleus were stained with Dapi (blue). (B) TEM image of VSMCs after incubation for 24 hours with 100 μg/mL nHAp. Black arrows point nHAp, N for nucleus. (C) Cell viability of VSMCs was measured with CCK8 assay after VSMCs were exposed to nHAp at different concentrations for 3 days. N=5, ***P < 0.001, compared with the control group. |
Nano-HAp Impaired Mitochondrial Function
Production of reactive oxygen species (ROS) was significantly increased after VSMCs were mixed with nHAp (VSMCsHAp) as compared with VSMCsCtrl (Figure 3A). Mitochondria became condensed and showed a spotty aggregation in VSMCsHAp, as compared with the evenly distributed signals in VSMCsCtrl (Figure 3B), indicating that nHAp disrupted the distribution of mitochondria. Morphology of mitochondria in the control cells was elongated and tubular-like shape with clear cristae, whereas those in VSMCsHAp were roundly shaped with reduced matrix density and disappearance of cristae (Figure 3C). The mitochondrial membrane potential was significantly lower in VSMCsHAp than that in VSMCsCtrl (Figure 3D). These data indicated that nHAp induced mitochondrial dysfunction.
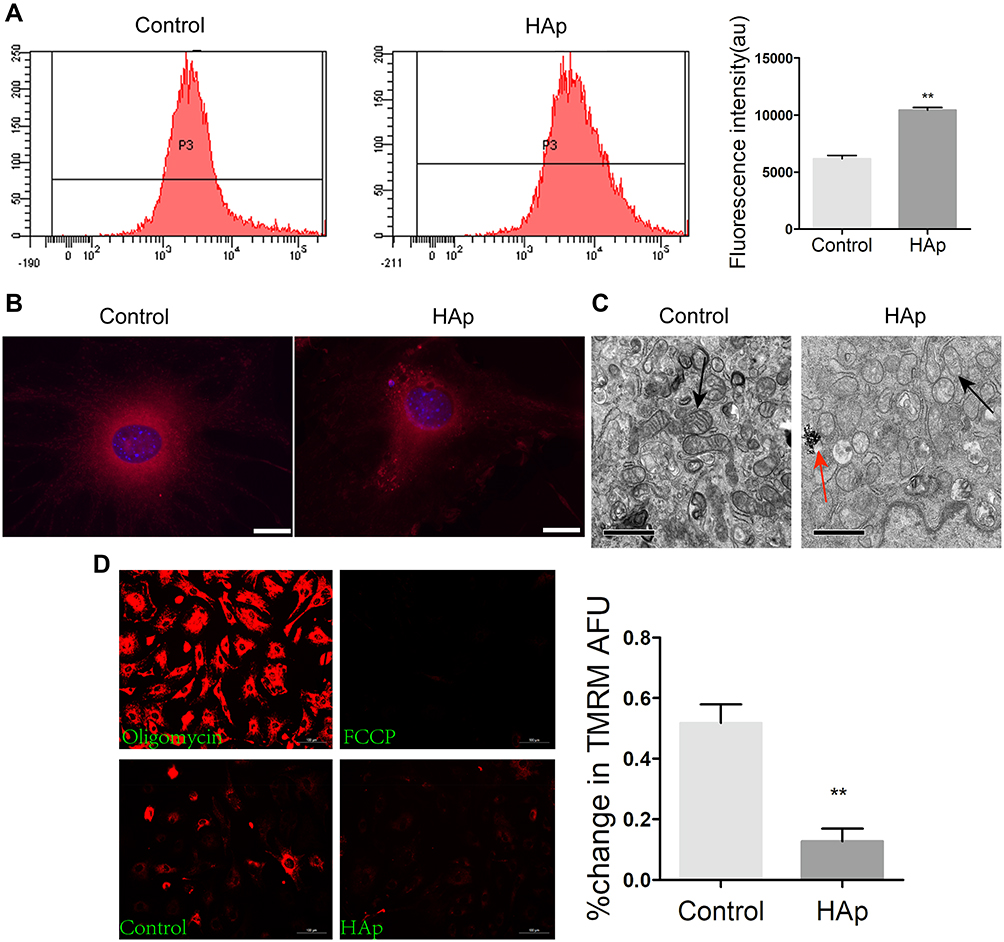 |
Figure 3 Nano-HAp impaired mitochondria of VSMCs. VSMCs were treated with 100 μg/mL nHAp for 24 h, and the following experiments were carried out. (A) VSMCs were labeled with DCFH-DA for ROS and analyzed by flow cytometry. Quantitative data were shown on the right panel. N=3. (B) Fluorescence images of SMCs after stained with mitotracker for mitochondria (red) and DAPI for nucleus (blue). (C) TEM images show the morphology of mitochondria. Black arrows point mitochondria, red arrow points nHAp. (D) Fluorescence images of VSMCs stained with TMRM to measure mitochondrial membrane potential (Δψm). VSMCs were treated with oligomycin or FCCP for positive or negative control, respectively. The quantitative Δψm data were shown on the right panel. Data are expressed as mean ± SD. N=3 (** p<0.01). |
Nano-HAp Induced Apoptosis of VSMCs
To further identify the effect of nHAp on VSMCs, cell apoptosis was detected by Annexin V-APC/PI double staining. VSMCsHAp had significant higher apoptosis rate (41.7 ± 3.8%) than VSMCsCtrl (14.8 ± 0.8%) (Figure 4A and B). More cleaved caspase 9 and cleaved caspases 3 were detected in VSMCsHAp than that in VSMCsCtrl (Figure 4C and D), confirming the increased apoptosis in VSMCsHAp. By separation of mitochondria and cytoplasm, more cytochrome c was found in cytoplasm in VSMCsHAp than that in VSMCsCtrl (Figure 4E and F), supporting that nHAp promoted the release of cytochrome c from mitochondria to cytoplasm to initiate the apoptosis.
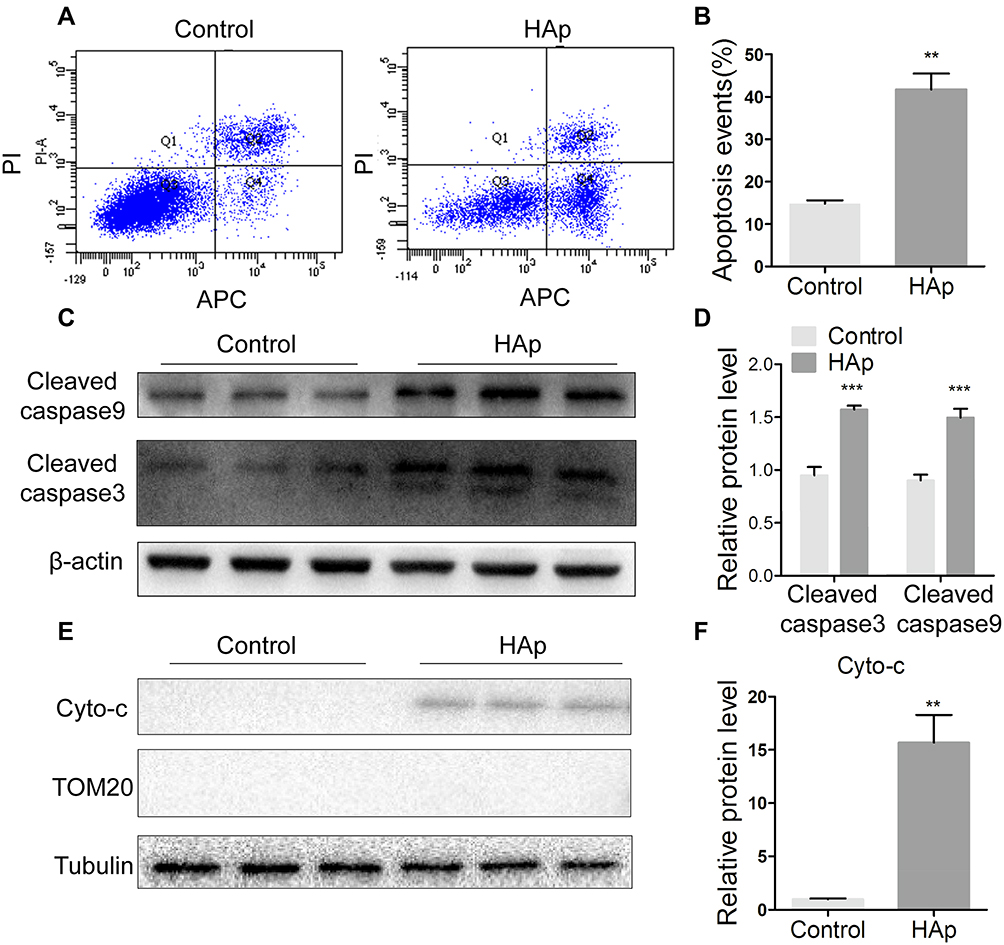 |
Figure 4 Nano-HAp induced apoptosis of VSMCs. After VSMCs were treated with 100 μg/mL nHAp for 5 days, the following assays were conducted. (A) Cells were stained with Annexin V (APC) and PI, and then analyzed by flow cytometry. (B) Apoptotic cells were quantified. Data are expressed as mean ± SD. N=3. (C) Western blot analysis was applied to detect the cleaved caspase 3 and cleaved caspase 9. (D) Protein levels were quantified. N=3. (E) Cytochrome c (cyto-c) in mitochondria-free cytoplasm was detected by Western blot. Mitochondrial protein TOM20 was used as a control to show free of mitochondria. (F) Quantification of cyto-c in E. (N=3, ** p<0.01, *** P<0.001). |
Nano-HAp Activated JNK/c-JUN Signaling Pathway
Mitogen-activated protein kinases (MAPKs), represented by p38, JNK, and ERK1/2, are a common signaling pathway cascades in responding to extracellular stimulation. Since previous studies have shown that JNK phosphorylation is related with calcification in SMC,16,17 we examined if nHAp activated JNK signaling pathway in SMCs. Significantly more phosphorylated JNK was detected after VSMCs were treated with nHAp for 15 min (Figure 5A). This was confirmed with immunofluorescent staining showing that more p-JNK was found in VSMCsHAp as compared to the control (Figure 5B). Immunofluorescence staining of JNK also revealed that majority of JNK was in cytoplasm in VSMCsCtrl, but was translocated to nucleus after the cells were treated with nHAp (Figure 5C). Both phosphorylation of JNK and translocation of p-JNK into nuclear in response to HAp treatment were blocked by SP600125, an inhibitor of JNK phosphorylation (Figure 5D and E). To further investigate the possible targets of nHAp, the expression of c-JUN, a downstream target of JNK, was analyzed. Significantly more c-JUN was phosphorylated after nHAp treatment. Similarly, the effects of nHAp on c-JUN activation were blocked by JNK inhibitor SP600125 (Figure 5F). These results indicated that nHAp activated JNK/c-JUN signaling pathway in VSMCs.
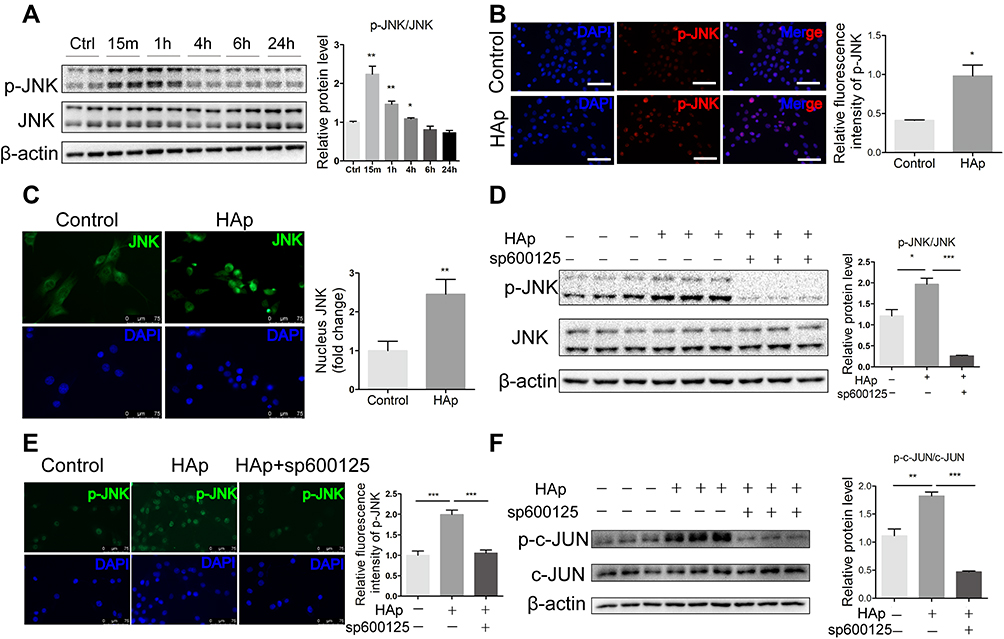 |
Figure 5 Nano-HAp activated JNK/c-JUN signaling pathway. (A) Western blot analysis of phosphorylated JNK (p-JNK) and total JNK in VSMCs that were treated with nHAp for the specified time. The ratio of p-JNK/JNK was quantified. N=4. (B) Immunofluorescence images show the distribution patterns of p-JNK in VSMCs that were stained with anti-p-JNK antibody (red) and DAPI for nuclei (blue). Scale bar: 100 μm. Relative amount of p-JNK in SMCs was quantified by measuring fluorescence intensity. N=3. (C) Immunofluorescence staining for subcellular localization of JNK (green) in VSMCs cultured with or without nHAp. Scale bars: 75 μm. The right panel shows quantification of nuclear-localized JNK. N=8. (D) Western blot analysis of p-JNK and total JNK in VSMCs that were pretreated with inhibitor for JNK (SP600125,10 μM) followed by treatment with nHAp for 15 min. N=3. (E) Immunofluorescence staining of p-JNK (green) in VSMC that were pretreated with inhibitor for JNK (SP600125,10 μM) followed by treatment with nHAp for 15 min. The right panel shows statistical analysis of p-JNK immunofluorescent staining. N=8. (F) Western blot analysis of phosphorylated c-JUN (p-c-JUN) and total c-JUN in VSMCs that were pretreated with inhibitor for JNK (SP600125,10 μM) followed by treatment with nHAp for 15 min. Data are expressed as the mean ± SD. N=3. (* p<0.05, ** p<0.01,*** p<0.001). |
JNK Inhibitor Inhibited Apoptosis and Osteogenic Trans-Differentiation of SMCs Induced by nHAp
To confirm the relationship between JNK phosphorylation and calcification of SMCs, JNK inhibitor SP600125 was used to block JNK activation in VSMCs. HAp-elevated rate of apoptosis was significantly reduced by SP600125 (Figure 6A). Less cleaved caspase 9 and cleaved caspase 3 were detected in VSMCsHAp with SP600125 as compared with VSMCsHAp only (Figure 6B), confirming the reduced apoptosis by SP600125. Compared with VSMCsCtrl, the expressions of osteogenesis markers Runx2 and OPN (Figure 6C) and the ALP activity in VSMCsHAp were significantly increased (Figure 6D), indicating that nHAp promoted osteogenic differentiation of VSMCs. In the presence of SP600125, nHAp-induced increase of Runx2 and OPN and ALP activity was blocked, confirming that nHAp-induced osteogenic transformation was associated with JNK activation (Figure 6C and D). In addition, calcified nodule formation and calcium deposition in VSMCsHAp were markedly decreased by SP600125 (Figure 6E). These results confirmed that JNK signaling pathway played an essential role in nHAp-induced transdifferentiation of VSMCs and vascular calcification.
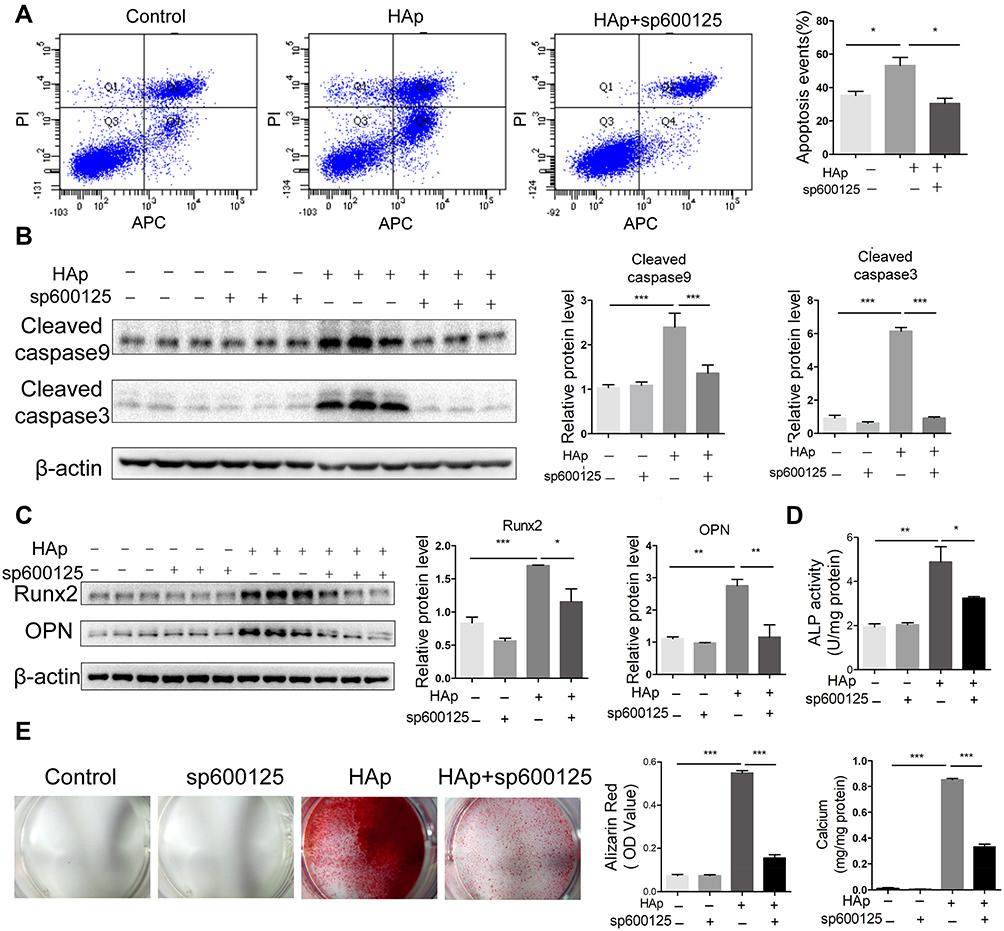 |
Figure 6 JNK pathway was involved in apoptosis and osteogenic transdifferentiation of VSMCs induced by nano-HAp. After VSMCs were treated with 100 μg/mL nHAp or 10 μm/L SP600125 or both for 5 days, the following assays were conducted. (A) Flow cytometry analysis of apoptosis rate of VSMCs stained with Annexin V. Annexin V positive cells (Q2+Q4) were considered as apoptotic cells. N=3. (B) Apoptosis-related proteins of cleaved caspase 3 and cleaved caspase 9 were detected by Western blot analysis. Right panel shows the quantification data. N=3. (C) Western blot analysis and quantification of Runx2, and OPN in VSMCs. N=3. (D) Quantification of ALP activity in VSMCs. N=3. (E) Calcification was visualized by Alizarin Red staining after cells were treated as indicated for 14 days. Alizarin Red was extracted from cells and quantified by densitometry. Total calcium on the dishes was quantified. Data are expressed as the mean ± SD. N=3, * p<0.05, ** p<0.01, *** p<0.001. |
HAp-Induced Osteogenic Differentiation and Apoptosis in vivo Was Inhibited by JNK Inhibitor
VC in mice was induced via applying nHAp on the surface of the abdominal aorta of mice. Obvious calcium deposition in the aorta treated with nHAp was detected by von Kossa staining of aortic sections, while no calcification was found in the control mice (Figure 7A). On the other hand, when mice received SP600125 (30mg/kg/day) after nHAp treatment, calcium deposition in the aorta was significantly decreased as compared with the mice treated with nHAp only (Figure 7A and B), which means JNK inhibition might block the newly formed calcium phosphate induced by nHAp. In addition, Hematoxylin eosin (HE) staining (Supplemental methods) was used to evaluate whether nHAp had effects on other organs. Results showed that there was no difference between the NaCl group and nHAp group in the aspect of histological culture of the heart, liver, spleen, lung and kidney (Supplemental figure). As expected, more TUNEL+ cells were found in the vessel wall treated with nHAp in comparison with NaCl-treated control, while SP600125 significantly reduced apoptosis of vascular cells (Figure 7C and D). In addition, immunofluorescent staining demonstrated that significantly more Runx2 was co-localized with SMCs marker α-SMA on the aorta treated with nHAp as compared to that treated with NaCl. JNK inhibition with SP600125 attenuated such enhanced expression of Runx2 (Figure 7E and F). These results support the conclusion that JNK signaling pathway was involved in the nHAp-induced VC in mice.
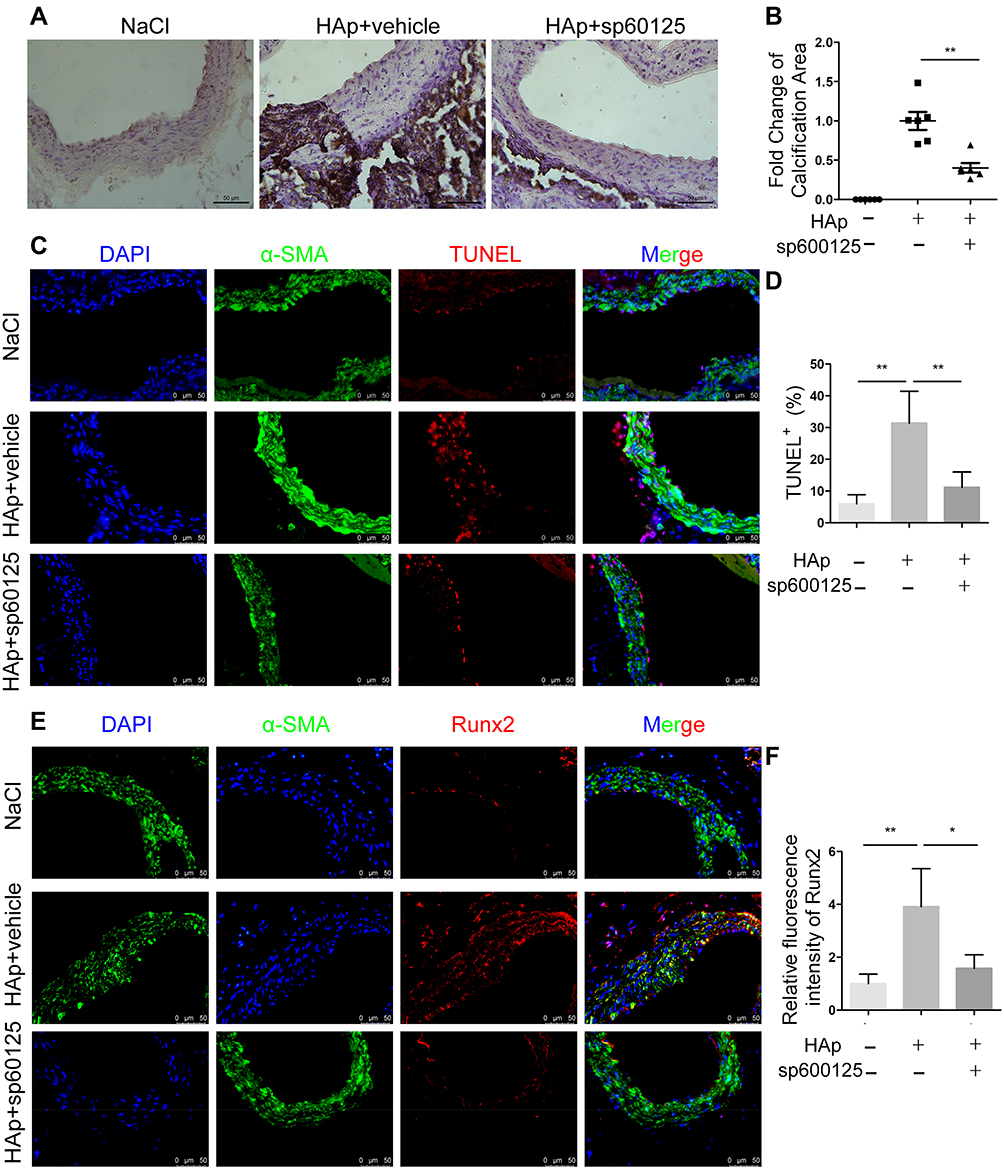 |
Figure 7 Nano-HAp-induced osteogenic differentiation and apoptosis in vivo was inhibited by JNK inhibitor. Mice aorta were exposed to either NaCl (negative control) or nHAp to induce VC. The mice of nHAp group were treated with vehicle (10% DMSO) or SP600125. Aorta were recovered 14 days later. (A) von Kossa staining of aortic sections to show calcium deposition (brown) in the aorta (magnification: 400×); (B) Statistics of the calcification area that were positive for von Kossa staining. N=6. (C) TUNEL staining to detect apoptotic cells (red) in vessel. Vascular smooth muscle cells were stained with Ab against α-SMA (green), nuclei were stained with DAPI (blue). Scale bar, 50 μm. (D) Quantification of apoptotic cells by TUNEL-positive nuclei. N=6. (E) Representative images of the aorta sections after immunofluorescent staining for Runx2 and α-SMA. Scale bar: 50 μm. (F) Quantification of Runx2 expression on the vessel. N=6; Data are expressed as mean ± SD, * p < 0.05, ** p < 0.01. |
Discussion
Vascular calcification is considered to be an active process involving VSMC apoptosis, transdifferentiation from contractile to osteochondrogenic phenotype, vesicle release, and imbalance of inhibitors and promoters, which leads to HAp deposition at extracellular matrix.18 VC has been shown to be associated with the presence of HAp on a scale of hundreds of nanometers.4 The size of the calcium- and phosphorus-containing particles had been identified in mineral tissue ranges from 100 nm to 5 μm using nano-analytical electron microscopy.19 These studies strongly suggested that the presence of HAp particles contribute to osteoblast transformation of cardiovascular cells and may play a fundamental role in calcific lesion formation.
In this study, we proved that nHAp crystals induced generation of ROS, mitochondria impairment, apoptosis and osteogenic differentiation of VSMCs; the process was JNK signaling dependent. Our results demonstrated that JNK pathway plays an important role in nHAp-induced vascular calcification. JNK inhibitor sp600125 blocked the effect of nHAp on osteogenic gene expression and calcified nodule, confirming that nHAp triggered phosphorylation of JNK, promoted translocation of JNK, enhanced phosphorylation of c-JUN, and finally promoted the expression of Runx2. In addition, nHAp-induced VC on mouse aorta was effectively inhibited by JNK inhibitor. It is the first time to demonstrate that nHAp induces apoptosis of VSMCs via JNK signaling dependent cascade, and enhanced calcium deposition on vessels.
Treatment of nHAp resulted in cell membrane damage and enhanced membrane permeability.20 Therefore, nHAp reduced cell viability (Figure 2C). It has been reported that uptake of small calcium phosphate crystals induced intracellular Ca burst, promoted ROS production through activation of NADPH oxidase and resulted in cell apoptosis after human VSMCs were exposed to Ca-P nanocrystals.21,22 Our study proved that nHAp were internalized into VSMCs when nHAp were mixed with VSMCs (Figure 2A and B), and promoted the production of ROS (Figure 3A) and increased VSMC apoptosis (Figure 4). The results are also consistent with previous studies showing that nHAp increased ROS production and lactic dehydrogenase release, and induced apoptosis of different SMCs.9,23
As reported, oxidative stress per se stimulates VC by activating specific signaling cascade to initiate and accelerate VC progression.24 Different sources of ROS may contribute to vascular dysfunction25 through damaging mitochondria and decreasing ATP production.26 It has been demonstrated that ROS activates JNK, one of MAPK members, and such activation promotes the localization of Bax on mitochondria, leading to increased permeability of mitochondrial outer membrane. The release of cytochrome c from the mitochondria with the reduced membrane potential (Δψm) attracts the caspase cascade, finally causing apoptosis.27 Previous study proved that nHAp caused mitochondrial membrane permeability and mitochondrial dysfunction.23 In addition, nHAp could decrease the Δψm and induced mitochondria-dependent apoptosis in tumor cells such as human lung cancer cells (A549)28 and mouse mammary tumor cells (4T1).29 Our study showed that nHAp impaired mitochondria with decreased Δψm (Figure 3D), promoted ROS production (Figure 3A), activated JNK pathway (Figure 5) and induced the release of cytochrome c (Figure 4E). Inhibition of JNK attenuated the apoptosis (Figure 6A and B). These results identified that nHAp induced apoptosis of VSMCs via JNK signaling dependent cascade, confirmed that uptake of the pre-existing HAp crystals by VSMCs leads to cell apoptosis. The apoptotic bodies produced by VSMCs act as nucleating sites for the calcium deposits.30 This can be supported by evidence from an in vitro model using Ca/P-treated human vessels in which VSMCs underwent a rapid wave of apoptosis while forming the first calcified crystalline nodules under electron microscopy.31
As we know, BMP/Runx2/Osterix networks and the interacting Wnt signaling cascades are major regulatory pathways controlling osteoblast differentiation, chondrogenesis and ALP expression.32 VSMCs can differentiate into osteoblast-like cells and mediate the deposition of bone matrix in blood vessels. In calcified arteries, osteogenic transcription factors, including Msx2, Sox9, Runx2, and Osterix, have been detected in VSMCs.4 Runx2 is the most important upstream transcription factor for osteoblast differentiation. It regulates the expression of Osterix, a crucial transcription factor for osteoblast differentiation.33,34 When VSMCs are exposed to procalcifying conditions, expression of the smooth muscle contractile proteins SM22α and SMα-actin is lost and expression of the calcification markers Runx2, OPN, OCN, BMP-2 and ALP is enhanced.35 Our study confirmed that HAp crystals induced expression of Runx2 and OPN and enhanced activity of ALP in VSMCs in vitro (Figure 6). HAp-induced osteogenic differentiation and mineral deposition with the increased expression of Runx2, OCN and ALP have been reported in different kinds of cells, like mesenchymal stem cells (MSCs) and osteoblasts.13,36–39 VSMC osteogenic differentiation can also be induced with high Pi-created nanocrystals, resulting in upregulated expression of BMP-2 and OPN.40
Hunter et al reported that calcifying nanoparticles (CNPs) extracted from human serum could induce accumulation of apoptotic bodies and may accelerate vascular calcification.41 Liu et al separated CaP crystals from human uremic serum, and found they could promote expression of Runx2, BMP2 and OPN in SMCs, and finally induced mineral deposition.42 Consistently, our results also showed that nHAp could induce direct calcium deposits of VSMCs in vitro (Figure 6). Whether nHAp could promote VC in vivo has not been shown in the previous studies. We demonstrated for the first time that nHAp directly caused VC in mice (Figure 7A). As reported, nanocrystals with 30–500 nm in diameter were the active substance to induce VSMC phenotypic change.43 Previous researches have also demonstrated that different sizes of crystals had different effects on target cells.21,44 This indicates that calcium deposits are destructive in its earliest stages, before crystals grow or aggregate, making the initial stage the most important prevention target.
It has been proved that intracellular MAPK signaling pathways are involved in the pathological processes of cardiovascular disease.45 Previous studies demonstrated that JNK/MAPK can act as a molecular switch to balance the differentiation of osteoblasts and adipocytes lineages.46 Activation of JNK can enhance osteogenic differentiation.47–49 JNK/AP-1 is widely believed to be an important signaling pathway in the regulation of osteogenesis. The downstream regulator of JNK, c-JUN, is the major regulator of AP-1 transcription factor family. JNK/c-JUN pathway has also been demonstrated to mediate Runx2 expression in MSCs.50 Oxidative stress and ROS production represent a unifying mechanism and act as signaling molecules to promote VC51 by activating JNK and enhancing expression of Runx2.24,52 Therefore, we assumed that nHAp may modulate osteogenic differentiation via ROS/JNK/c-JUN/Runx2 pathway. We proved that nHAp triggered phosphorylation of JNK, promoted translocation of JNK to nuclear, enhanced phosphorylation of c-JUN, and finally promoted the expression of Runx2 (Figures 5 and 6). Thus, JNK/c-JUN pathway is involved in the osteogenic transformation of VSMCs induced by nHAp. This is the first time we elucidate that JNK/c-JUN modulates Runx2 expression in VSMCs. Other reports also show that nHAp can activate other MAPK members, like p38 and ERK1/2 pathways.16,17,53 Whether they are also involved in the process of calcification caused by nHAp remains to be proved.
Extrapolating from those studies, we proposed that when the initial nano-HAp deposits on the vessel wall, such nano-HAp further accelerates VC by inducing apoptosis and osteogenic differentiation of SMCs via JNK/c-JUN pathway. This progression of VC can be attenuated by JNK inhibitor sp600125.
Conclusions
Nano-HAp crystals induced ROS production and then activated JNK signaling pathway, which induced both the osteogenic differentiation and apoptosis of VSMCs. JNK inhibitor effectively blocked these effects of nHAp on VSMCs, and inhibited nHAp-induced calcium deposits (Figure 8). These results elucidate the mechanism of VC development caused by the initially formed nHAp and put a new sight into the control of VC progression.
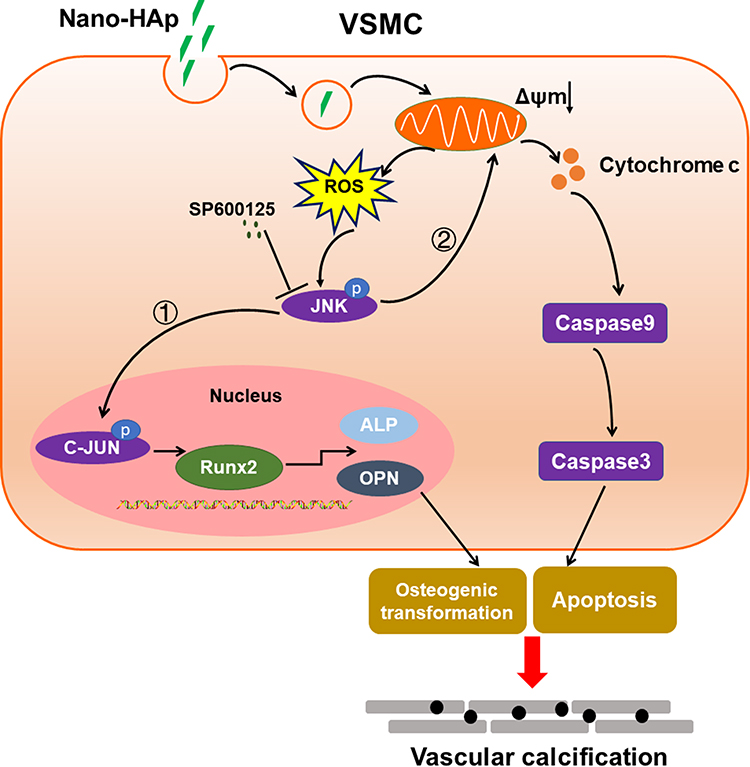 |
Figure 8 Schematic illustration of proposed cellular and molecular mechanism of vascular calcification induced by nano-HAp crystals. After HAp crystals are internalized into VSMC, more ROS will be generated, which will activate JNK phosphorylation. ① Activation of JNK/c-JUN signaling cascade will induce RunX2, OPN, and ALP gene expression and osteogenic differentiation of VSMC. ② JNK pathway promotes the release of cytochrome c, which will activate the caspase cascade, finally inducing apoptosis. Both osteogenic differentiation and apoptosis of VSMC will result in VC. JNK inhibitor effectively reduced these effects of HAp on VSMCs, and inhibited HAp-induced VC in vivo. Abbreviations: VSMC, vascular smooth muscle cell; nano-HAp, nanosized hydroxyapatite; ROS, reactive oxygen species; Runx2, runt-related factor 2; OPN, osteopontin; ALP, alkaline phosphatase. |
Acknowledgments
This research work was granted by the National Natural Science Foundation of China (No. 81570251 to HY).
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Nicoll R, Henein MY. The predictive value of arterial and valvular calcification for mortality and cardiovascular events. Int J Cardiol. 2014;3:1–5.
2. Durham AL, Speer MY, Scatena M, Giachelli CM, Shanahan CM. Role of smooth muscle cells in vascular calcification: implications in atherosclerosis and arterial stiffness. Cardiovasc Res. 2018;114:590–600.
3. Jaminon A, Reesink K, Kroon A, Schurgers L. The role of vascular smooth muscle cells in arterial remodeling: focus on calcification-related processes. Int J Mol Sci. 2019;20.
4. Leopold JA. Vascular calcification: mechanisms of vascular smooth muscle cell calcification. Trends Cardiovasc Med. 2015;25:267–274.
5. Ha SW, Jang HL, Nam KT, Beck GR
6. Sun Y, Zeng XR, Wenger L, Cheung HS. Basic calcium phosphate crystals stimulate the endocytotic activity of cells–inhibition by anti-calcification agents. Biochem Biophys Res Commun. 2003;312:1053–1059.
7. Morgan MP, McCarthy GM. Signaling mechanisms involved in crystal-induced tissue damage. Curr Opin Rheumatol. 2002;14:292–297.
8. Nadra I, Mason JC, Philippidis P, et al. Proinflammatory activation of macrophages by basic calcium phosphate crystals via protein kinase C and MAP kinase pathways: a vicious cycle of inflammation and arterial calcification? Circ Res. 2005;96:1248–1256.
9. Huang LH, Liu H, Chen JY, et al. Seaweed Porphyra yezoensis polysaccharides with different molecular weights inhibit hydroxyapatite damage and osteoblast differentiation of A7R5 cells. Food Funct. 2020;11:3393–3409.
10. Tsujihata M. Mechanism of calcium oxalate renal stone formation and renal tubular cell injury. Int J Urol. 2008;15:115–120.
11. Zimmermann KC. Bonzon C and Green DR. The machinery of programmed cell death. Pharmacol Ther. 2001;92:57–70.
12. Hengartner MO. The biochemistry of apoptosis. Nature. 2000;407:770–776.
13. Ha SW, Park J, Habib MM, Beck GR
14. Liu X, Sun J. Potential proinflammatory effects of hydroxyapatite nanoparticles on endothelial cells in a monocyte-endothelial cell coculture model. Int J Nanomedicine. 2014;9:1261–1273.
15. Shen J, Zhang N, Lin YN, et al. Regulation of vascular calcification by growth hormone-releasing hormone and its agonists. Circ Res. 2018;122:1395–1408.
16. Chang JF, Hsieh CY, Liou JC, et al. Scavenging intracellular ROS attenuates p-cresyl sulfate-triggered osteogenesis through MAPK signaling pathway and NF-κB activation in human arterial smooth muscle cells. Toxins (Basel). 2020:12.
17. Hou M, Song Y, Li Z, et al. Curcumin attenuates osteogenic differentiation and calcification of rat vascular smooth muscle cells. Mol Cell Biochem. 2016;420:151–160.
18. Wang C, Xu W, An J, et al. Poly(ADP-ribose) polymerase 1 accelerates vascular calcification by upregulating Runx2. Nat Commun. 2019;10:1203.
19. Bertazzo S, Gentleman E, Cloyd KL, Chester AH, Yacoub MH, Stevens MM. Nano-analytical electron microscopy reveals fundamental insights into human cardiovascular tissue calcification. Nat Mater. 2013;12:576–583.
20. Draeger A, Monastyrskaya K, Babiychuk EB. Plasma membrane repair and cellular damage control: the annexin survival kit. Biochem Pharmacol. 2011;81:703–712.
21. Ewence AE, Bootman M, Roderick HL, et al. Calcium phosphate crystals induce cell death in human vascular smooth muscle cells: a potential mechanism in atherosclerotic plaque destabilization. Circ Res. 2008;103:e28–34.
22. Lobascio AM, De Felici M, Anibaldi M, Greco P, Minasi MG, Greco E. Involvement of seminal leukocytes, reactive oxygen species, and sperm mitochondrial membrane potential in the DNA damage of the human spermatozoa. Andrology. 2015;3:265–270.
23. Zhang CY, Sun XY, Ouyang JM, Gui BS. Diethyl citrate and sodium citrate reduce the cytotoxic effects of nanosized hydroxyapatite crystals on mouse vascular smooth muscle cells. Int J Nanomedicine. 2017;12:8511–8525.
24. Byon CH, Javed A, Dai Q, et al. Oxidative stress induces vascular calcification through modulation of the osteogenic transcription factor Runx2 by AKT signaling. J Biol Chem. 2008;283:15319–15327.
25. Donato AJ, Machin DR, Lesniewski LA. Mechanisms of dysfunction in the aging vasculature and role in age-related disease. Circ Res. 2018;123:825–848.
26. Otera H, Ishihara N, Mihara K. New insights into the function and regulation of mitochondrial fission. Biochim Biophys Acta. 2013;1833:1256–1268.
27. Wang K, Gong Q, Zhan Y, et al. Blockage of autophagic flux and induction of mitochondria fragmentation by paroxetine hydrochloride in lung cancer cells promotes apoptosis via the ROS-MAPK pathway. Front Cell Dev Biol. 2019;7:397.
28. Sun Y, Chen Y, Ma X, et al. Mitochondria-targeted hydroxyapatite nanoparticles for selective growth inhibition of lung cancer in vitro and in vivo. ACS Appl Mater Interfaces. 2016;8:25680–25690.
29. Zhao H, Wu C, Gao D, et al. Antitumor effect by hydroxyapatite nanospheres: activation of mitochondria-dependent apoptosis and negative regulation of Phosphatidylinositol-3-Kinase/Protein Kinase B pathway. ACS Nano. 2018;12:7838–7854.
30. Proudfoot D, Skepper JN, Hegyi L, Bennett MR, Shanahan CM, Weissberg PL. Apoptosis regulates human vascular calcification in vitro: evidence for initiation of vascular calcification by apoptotic bodies. Circ Res. 2000;87:1055–1062.
31. Shroff RC, McNair R, Skepper JN, et al. Chronic mineral dysregulation promotes vascular smooth muscle cell adaptation and extracellular matrix calcification. J Am Soc Nephrol. 2010;21:103–112.
32. Salazar VS, Gamer LW, Rosen V. BMP signalling in skeletal development, disease and repair. Nat Rev Endocrinol. 2016;12:203–221.
33. Cao L, Liu W, Zhong Y, et al. Linc02349 promotes osteogenesis of human umbilical cord-derived stem cells by acting as a competing endogenous RNA for miR-25-3p and miR-33b-5p. Cell Prolif. 2020;53:e12814.
34. He S, Yang S, Zhang Y, et al. LncRNA ODIR1 inhibits osteogenic differentiation of hUC-MSCs through the FBXO25/H2BK120ub/H3K4me3/OSX axis. Cell Death Dis. 2019;10:947.
35. Steitz SA, Speer MY, Curinga G, et al. Smooth muscle cell phenotypic transition associated with calcification: upregulation of Cbfa1 and downregulation of smooth muscle lineage markers. Circ Res. 2001;89:1147–1154.
36. Campi G, Cristofaro F, Pani G, et al. Heterogeneous and self-organizing mineralization of bone matrix promoted by hydroxyapatite nanoparticles. Nanoscale. 2017;9:17274–17283.
37. Liu C, Zhai H, Zhang Z, Li Y, Xu X, Tang R. Cells recognize and prefer bone-like hydroxyapatite: biochemical understanding of ultrathin mineral platelets in bone. ACS Appl Mater Interfaces. 2016;8:29997–30004.
38. Bezerra F, Ferreira MR, Fontes GN, da Costa Fernandes CJ, Andia DC, Cruz NC, da Silva RA, Zambuzzi WF. Nano hydroxyapatite-blasted titanium surface affects pre-osteoblast morphology by modulating critical intracellular pathways. Biotechnol Bioeng. 2017;114:1888–1898.
39. Cao S, Li H, Li K, Lu J, Zhang L. In vitro mineralization of MC3T3-E1 osteoblast-like cells on collagen/nano-hydroxyapatite scaffolds coated carbon/carbon composites. J Biomed Mater Res A. 2016;104:533–543.
40. Sage AP, Lu J, Tintut Y, Demer LL. Hyperphosphatemia-induced nanocrystals upregulate the expression of bone morphogenetic protein-2 and osteopontin genes in mouse smooth muscle cells in vitro. Kidney Int. 2011;79:414–422.
41. Hunter LW, Charlesworth JE, Yu S, Lieske JC, Miller VM. Calcifying nanoparticles promote mineralization in vascular smooth muscle cells: implications for atherosclerosis. Int J Nanomedicine. 2014;9:2689–2698.
42. Liu Y, Zhang L, Ni Z, Qian J, Fang W. Calcium phosphate crystals from uremic serum promote osteogenic differentiation in human aortic smooth muscle cells. Calcif Tissue Int. 2016;99:543–555.
43. Proudfoot D, Shanahan CM. Nanocrystals seed calcification in more ways than one. Kidney Int. 2011;79:379–382.
44. Nadra I, Boccaccini AR, Philippidis P, Whelan LC, McCarthy GM, Haskard DO, Landis RC. Effect of particle size on hydroxyapatite crystal-induced tumor necrosis factor alpha secretion by macrophages. Atherosclerosis. 2008;196:98–105.
45. Muslin AJ. MAPK signalling in cardiovascular health and disease: molecular mechanisms and therapeutic targets. Clin Sci (Lond). 2008;115:203–218.
46. Yang S, Guo L, Su Y, et al. Nitric oxide balances osteoblast and adipocyte lineage differentiation via the JNK/MAPK signaling pathway in periodontal ligament stem cells. Stem Cell Res Ther. 2018;9:118.
47. Kusuyama J, Amir MS, Albertson BG, et al. JNK inactivation suppresses osteogenic differentiation, but robustly induces osteopontin expression in osteoblasts through the induction of inhibitor of DNA binding 4 (Id4). FASEB J. 2019;33:7331–7347.
48. Tang Y, Liu L, Wang P, Chen D, Wu Z, Tang C. Periostin promotes migration and osteogenic differentiation of human periodontal ligament mesenchymal stem cells via the Jun amino-terminal kinases (JNK) pathway under inflammatory conditions. Cell Prolif. 2017:50.
49. Xu R, Zhang C, Shin DY, et al. c-Jun N-Terminal Kinases (JNKs) are critical mediators of osteoblast activity in vivo. J Bone Miner Res. 2017;32:1811–1815.
50. Fu L, Peng S, Wu W, Ouyang Y, Tan D, LncRNA FX. HOTAIRM1 promotes osteogenesis by controlling JNK/AP-1 signalling-mediated RUNX2 expression. J Cell Mol Med. 2019;23:7517–7524.
51. Pescatore LA, Gamarra LF, Liberman M. Multifaceted mechanisms of vascular calcification in aging. Arterioscler Thromb Vasc Biol. 2019;39:1307–1316.
52. Liberman M, Johnson RC, Handy DE, Loscalzo J, Leopold JA. Bone morphogenetic protein-2 activates NADPH oxidase to increase endoplasmic reticulum stress and human coronary artery smooth muscle cell calcification. Biochem Biophys Res Commun. 2011;413:436–441.
53. Wang B, Li F, Zhang C. High-mobility group box-1 protein induces osteogenic phenotype changes in aortic valve interstitial cells. J Thorac Cardiovasc Surg. 2016;151:255–262.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.