")
Back to Journals » International Journal of Nanomedicine » Volume 12
Nano-sized cytochrome P450 3A4 inhibitors to block hepatic metabolism of docetaxel
Authors Paolini M, Poul L, Berjaud C, Germain M, Darmon A, Bergère M, Pottier A, Levy L, Vibert E
Received 5 May 2017
Accepted for publication 7 June 2017
Published 2 August 2017 Volume 2017:12 Pages 5537—5556
DOI https://doi.org/10.2147/IJN.S141145
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Marion Paolini,1,2 Laurence Poul,1 Céline Berjaud,1 Matthieu Germain,1 Audrey Darmon,1 Maxime Bergère,1 Agnès Pottier,1 Laurent Levy,1 Eric Vibert2
1Nanobiotix, Paris, 2UMR-S 1193 INSERM/Paris-Sud University, Centre Hépato-Biliaire, Hôpital Paul Brousse, Villejuif, France
Abstract: Most drugs are metabolized by hepatic cytochrome P450 3A4 (CYP3A4), resulting in their reduced bioavailability. In this study, we present the design and evaluation of biocompatible nanocarriers trapping a natural CYP3A4-inhibiting compound. Our aim in using nanocarriers was to target the natural CYP3A4-inhibiting agent to hepatic CYP3A4 and leave drug-metabolizing enzymes in other organs undisturbed. In the design of such nanocarriers, we took advantage of the nonspecific accumulation of small nanoparticles in the liver. Specific targeting functionalization was added to direct nanocarriers toward hepatocytes. Nanocarriers were evaluated in vitro for their CYP3A4 inhibition capacity and in vivo for their biodistribution, and finally injected 24 hours prior to the drug docetaxel, for their ability to improve the efficiency of the drug docetaxel. Nanoparticles of poly(lactic-co-glycolic) acid (PLGA) with a hydrodynamic diameter of 63 nm, functionalized with galactosamine, showed efficient in vitro CYP3A4 inhibition and the highest accumulation in hepatocytes. When compared to docetaxel alone, in nude mice bearing the human breast cancer, MDA-MB-231 model, they significantly improved the delay in tumor growth (treated group versus docetaxel alone, percent treated versus control ratio [%T/C] of 32%) and demonstrated a major improvement in overall survival (survival rate of 67% versus 0% at day 55).
Keywords: hepatic metabolism, CYP3A4, PLGA nanoparticles, hepatocyte targeting, galactosamine
Introduction
Taxotere® (docetaxel) is a chemotherapy agent used in clinic, injected intravenously, for several indications, such as non-small cell lung cancer, breast cancer, gastric cancer and prostate cancer.1,2 Docetaxel is extensively metabolized by cytochrome P450 3A4 (CYP3A4), resulting in low systemic bioavailability.3–5 Recently, docetaxel was administered with oral drugs that are known to be potent CYP3A4 inhibitors to increase its bioavailability and to enable its oral administration, especially by the inhibition of intestinal CYP34A.6,7 However, on top of their own pharmacological activity, these orally administered inhibitors of CYP3A4 generally have a wide biodistribution. For instance, they can reach the blood–brain barrier and inhibit the P-glycoprotein, which can be beneficial in the indication of HIV, but could present a source of unwanted toxicity for other treatments, like cytotoxic agents such as docetaxel.8
What if nanocarriers could deliver CYP3A4-inhibiting compounds to hepatocytes, responsible for hepatic metabolism?
Nanocarriers-based drug delivery systems have emerged as a solution to modify the biodistribution of drugs’ classical formulations. These drug delivery systems have specific attributes: composition, structure, size, shape and surface, which determine their interactions with biological entities. One major obstacle in the development of such systems is their sequestration by the mononuclear phagocyte system (MPS), preventing their accumulation at target site. Interestingly, the place of the liver in the circulation and its vascularization make it the main organ responsible for the clearance of materials from the blood stream. An observation made by Tsoi et al9 is that the reduced velocity of the blood flow by a factor of 1,000 within the liver sinusoids favored the interactions of hard nanoparticles with surrounding hepatic cells. Moreover, once the nanocarriers, with a size typically <100 nm, are in the liver sinusoids, the 150–175 nm fenestrae of the liver sinusoids occupying 6%–8% of the endothelial surface allow the nanocarriers to reach the hepatocytes.10 Finally, specific receptors expressed by hepatocytes – such as asialoglycoprotein receptors (ASGPR) − may be exploited to enhance the recognition and capture of nanocarriers.
Taking advantage of these interesting features, we designed biocompatible and biodegradable organic-based nanocarriers − micelles and polymeric nanoparticles − with size <100 nm, with various composition and structure to modulate their stability and payload release in vivo and with different surfaces to specifically or nonspecifically reach hepatocyte membrane receptors. We screened their ability to biodistribute and target hepatocytes. In parallel, these nanocarriers, loaded with bergamottin (BM), a natural CYP3A4-inhibiting agent,11–13 were tested for their ability to interact with hepatocytes in vitro, modulate CYP3A4 activity and increase the antitumor efficacy of docetaxel.
Materials and methods
Materials
Acetone, absolute ethanol (99.5%, extra dry, AcroSeal®), N-acetylgalactosamine and galactosamine (Ga), N-(3-Dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC) and N-hydroxysulfosuccinimide (NHS) were all purchased from Acros Organics (Geel, Belgium). 1,2-Distearoyl-sn-glycero-3-phosphoethanolamine-N-(carboxy(polyethylene glycol)-2000) (DSPE-PEG) and 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-lactosyl (lactosyl-PE) were purchased from Avanti Polar Lipids (Alabaster, AL, USA). Docetaxel was purchased from Accord (London, UK). Di(benzyloxymethoxy)fluorescein (DBOMF), 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindodicarbocyanine (DilC18) and all cell culture media were purchased from Thermo Fisher Scientific (Waltham, MA, USA). All other reagents were purchased from Sigma-Aldrich Co. (St Louis, MO, USA) unless otherwise specified.
Synthesis of BM-loaded (and/or DilC18-loaded) nanocarriers
A schematic representation of the nanocarriers and their synthesis are presented in Figure S1. For qualitative cellular uptake and biodistribution studies, the fluorescent probe DilC18 was used in place of BM.
BM-loaded polysorbate 80 (PS80)–ethanol micelles (PS80–EtOH BM micelles)
Micelles of BM in polysorbate 80–ethanol (ie, PS80–EtOH BM micelles) were formed by self-assembly. The surfactant (PS80) and absolute ethanol were mixed in the ratio of 1:1 (v/v) to form a PS80–EtOH solution. BM was then weighted and dissolved in the PS80–EtOH solution at 15 mM. Once the dissolution was complete, micelles were formed by the addition of NaCl 1% (w/w) in a ratio equal to 9:1 (v/v) to the PS80–EtOH solution containing BM. Under these conditions, the resulting final concentration of BM in micelles was 1.5 mM. Nonencapsulated BM was removed by filtration in polyethersulfone (PES) Vivaspin® ultrafiltration units, with 106 Da molecular weight cutoff (Sartorius, Goettingen, Germany). All the PS80 introduced in solution was considered to form micelles, and no titration of PS80 was performed. Titration of BM in samples was performed on filtrates by fluorescence and compared to the fluorescence (excitation: 290–300 nm/emission: 475–490 nm) of a solution at the same BM concentration introduced and 5% ethanol in NaCl 0.95% (w/v). For in vivo injections, the micelles were injected as such (in 0.9% v/v NaCl final).
BM-loaded DSPE-PEG micelles (DSPE-PEG BM micelles)
Micelles were prepared by lipid film hydration. Briefly, DSPE-PEG and lactosyl-PE were dissolved in chloroform. Untargeted and targeted micelles were composed of DSPE-PEG (100 mol%) and DSPE-PEG/lactosyl-PE (80/20 mol%), respectively, both using a total phospholipid concentration of 25 mM. For samples encapsulating BM, a solution of BM (2.5 mM) in ethanol was added in phospholipid solution. Phospholipid solution was dried under nitrogen stream and residual organic solvent removed by placing samples overnight in a vacuum pump. Then, samples were hydrated using a PES 0.22 μm filtered solution of HEPES (25 mM)/NaCl (145 mM) pH 7.4 heated to 60°C. Samples were vigorously vortexed until full hydration of phospholipid films. After micelle hydration, nonencapsulated BM was removed by centrifugation in a 106 Da PES Vivaspin® ultrafiltration units at 70 g for 30 minutes. Titration of phospholipids in samples was performed on filtrates by Bartlett method,14,15 and titration of BM was performed by fluorescence in water/ethanol (50:50, v/v), compared to the fluorescence of a solution at the same BM concentration introduced in water/ethanol (50:50, v/v). For in vivo injections, glucose was added to the micelle solution (5% v/v final).
BM-loaded poly(lactic-co-glycolic) acid nanoparticles (PLGA BM nanoparticles)
Blank nanoparticles were obtained by a modified solvent diffusion (nanoprecipitation) technique.16 Briefly, PLGA (10 mg) was solubilized in acetone (850 μL), and then 150 μL ethanol was added. This organic phase was quickly poured into 10 mL deionized water (aqueous phase) and kept stirring at 1,000 rpm for 3 hours. BM-loaded nanoparticles were prepared using the same procedure, and BM was added in ethanol to the organic phase. N-acetylgalactosamine or alternatively Ga coatings were performed via carbodiimide chemistry, using the EDC/NHS cross-linking procedure (Figure S2). Typically, to 40 mL of 0.6 g/L PLGA nanoparticle suspension at pH 5, aqueous solutions containing 10.6 mg of EDC and 15.9 mg of NHS were added. After 30 minutes of incubation at room temperature, the pH was raised to 7.4 and an aqueous solution containing 2.97 mg of Ga was added. The suspensions were then kept at 4°C, purified and sterilized by filtration through 0.22 μm PES filters. When required, the so-obtained nanoparticle suspensions were concentrated by 3–10 times, using 10 kDa PES Vivaspin® ultrafiltration units under 192× g centrifugation and washed again using dialysis membranes (regenerated cellulose, molecular weight cutoff: 12–14 kDa; minimum 4 hours, twice). Titration of BM in samples was performed by ultraviolet (UV) absorbance at 310 nm in water/ethanol (50:50, v/v), compared to the UV absorbance of a solution at the same BM concentration introduced in water/ethanol (50:50, v/v). Titration of lactic acids (D and L) was performed with colorimetric assay kits (Sigma-Aldrich Co.) according to the manufacturer’s instructions. For in vivo injections, glucose was added to the nanoparticle suspension (5% v/v final).
DilC18-loaded hyaluronic acid (HA) nanoparticles (HA DilC18 nanoparticles)
Nanoparticles were synthesized by adapting a technique described by Hu et al.17 Briefly, nanoparticles were synthesized based on cross-linking polymer chains through their carboxyl groups. Briefly, aqueous solution of polymer (HA) was prepared by mixing the polymer in water (2.5 g/L, 5.4 mL). Then, 17 mL of acetone was added and stirred with a mechanical agitation for 25 minutes (320 rpm) to make sure all the components in the solution were well dissolved. DilC18 was added in the organic phase. Then, 0.125 mL of a solution of EDC (50 mg/mL) in water was added to the flask, and followed 5 minutes later by an addition into the mix of 0.35 mL of NHS in water (27.5 mg/mL). After mixing the solution for 5 minutes, 21.5 mL of acetone was added to the solution, and stirring was continued for ~16 hours. Then, the reaction was stopped by dialysis of the solution against reverse osmosis water using dialysis membranes (regenerated cellulose, molecular weight cutoff: 12–14 kDa; minimum 4 hours, twice). Finally, the nanoparticle suspension was washed again and concentrated with an Amicon® system (Biomax®; 50 kDa; d =25 mm; PES) and stored at 4°C. All the HA introduced in solution was considered to form nanoparticles, and no titration of HA was performed. For in vivo injections, glucose was added to the nanoparticle suspension (5% v/v final).
Characterizations of nanocarriers
Size
The hydrodynamic diameter and size polydispersity of the micelles and nanoparticles were determined by dynamic light scattering (DLS). Measurements were performed on a Zetasizer Nano-ZS device (Malvern Instruments, Malvern, UK) with a laser operating at 633 nm and a photodiode detector at an angle of 173°. The mean of three measurements of 11 runs each in Z-average, along with the polydispersity index, is presented. For field emission gun-scanning electron microscope (FEG-SEM), imaging was performed on an SU-70 Hitachi FEG-SEM. Substrates used for sample deposition were silicon thin slides. A drop of particle solution was deposited on the slides either directly and vacuum dried at room temperature or previously spun for optimal particle repartition on the surface. For observation, low voltage (500 V, 1 kV, 2 kV) and low current beam (about a few picoamperes) conditions were chosen to prevent damaging of the polymer.
Charge
The surface charge of the nanocarriers was evaluated by zeta potential measurement on a Zetasizer Nano-ZS device, in water at neutral pH (7–8). The mean of three zeta potential measurements of 14 runs each, along with zeta deviation measurements, is presented.
Encapsulation
Encapsulation efficiencies (%EE) were calculated with the following formula: %EE = (concentration of BM in final solution)/(concentration of BM introduced*dilution factor)*100. BM concentration was evaluated by fluorescence (excitation: 290–300 nm/emission: 475–490 nm) or UV absorbance at 310 nm. Fluorescence was read at 700 V on a Cary Eclipse fluorimeter equiped with 96-well plate reader (Agilent Technologies, Santa Clara, Ca, USA) and UV absorbance was read on a Cary 100 Scan uv-visible spectrophotometer in UV cuvettes (Agilent Technologies, Santa Clara, CA, USA).
In vitro assays
Cell culture
The human breast adenocarcinoma MDA-MB-231 cell line was purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA) and was cultured in minimum essential media with Earle’s salts supplemented with Glutamax, 10% fetal bovine serum, 1% nonessential amino acids, 1% penicillin–streptomycin and 1% sodium pyruvate. HepaRG cell line was purchased from Thermo Fisher Scientific and cultured in Williams E medium + Glutamax supplemented with 10% fetal calf serum, 5 μg/mL bovine insulin, 50 μM hydrocortisone 21-hemisuccinate, 50 U/mL penicillin and 50 μg/mL streptomycin. HepaRG cells were cultivated to induce CYP3A4. They were allowed to grow in a T75 flask for a week in an incubator after thawing (37°C, 5% CO2). Cells were then detached with Trypsin 0.05% and counted by Trypan blue exclusion. Cell viability >80% was considered adequate. Cells were resuspended in the same culture medium and seeded on collagen-coated 96-well plates at a density of 33,000 cells/well (0.22×106 cells/mL) and allowed to grow in an incubator (37°C, 5% CO2). Cells were allowed to reach full confluence for 2 weeks and cultivated 2 more weeks in the presence of a final concentration of 2% dimethyl sulfoxide (DMSO). Culture medium was renewed every 2–3 days. Cells were used for experiments from passages 2–10.
In vitro CYP3A4 inhibition assays
Half-maximal inhibitory concentration (IC50) was determined on CYP3A4-expressing HepaRG cells, after incubation of the fluorogenic substrate DBOMF, specific for CYP3A4. DBOMF was prepared in acetonitrile at a stock concentration of 2 mM. To be incubated on cells, DBOMF was diluted at a concentration of 10 μM in a solution of PBS, 10 mM HEPES and 10 mM glucose with pH adjusted to 7.3. For the 3-hour incubation, 100 μL/well of inhibitors (BM-loaded micelles or nanoparticles) was incubated on cells 30 minutes before adding the substrate (100 μL/well of 10 μM DBOMF) for 2.5 additional hours. Then, 75 μL/well of the substrate incubated on cells was taken and mixed on a white 96-well plate with 25 μL of enzymatic solution: beta-glucuronidase/arylsulfatase diluted by 100 M and 0.5 M sodium acetate pH 4.5 (3:2, v/v).18,19 The solution was incubated for 1 hour at 37°C. Once the incubation completed, 100 μL/well of 2 N sodium hydroxide was added. Fluorescence (excitation: 490 nm/emission: 520 nm) was read at a voltage set between 450 V and 550 V on a Cary Eclipse fluorimeter. Cytotoxicity was measured using a lactate dehydrogenase (LDH) assay according to the manufacturer’s instructions. UV absorbance for the LDH assay was measured on a UV 96-well plate reader PowerWave 340.
In vivo assays
In vivo imaging system (IVIS)
IVIS manipulation was performed on adult female mice (Naval Medical Research Institute [NMRI]-Fox1nu/Foxn1nu; Le Genest-Saint-Isle, Janvier, France) at the Institut Gustave Roussy (Villejuif, France), according to its ethic committee policy, following approval. Animals received good care and humane treatments. Mice were injected intravenously in the caudal vein with DilC18-loaded nanocarriers (micelles or nanoparticles). All solutions were injected at 10 mL/kg, in 5% (v/v) glucose for PLGA and HA nanoparticles and DSPE-PEG micelles and 0.9% NaCl (w/v) for PS80–EtOH micelles. Imaging was launched right before injection. Images were acquired on an IVIS Spectrum (PerkinElmer, Courtaboeuf, France) with the following settings: 640 nm excitation, 680 nm emission, binning factor of 8 with a fixed field of view (13.2 cm). The exposure time was automatically computed by the software to raise a minimal threshold of 6,000 counts within 1 minute. Acquisitions were repeated each minute during 1 hour after nanocarriers’ injection. For each kinetic, data were represented in radiance efficiency which is a normalized unit according to the acquisition parameters. Fluorescence of the spleen or of the liver was normalized per the organ surface. Mice were euthanized, and selected organs (liver, spleen, intestines and stomach) were imaged ex vivo.
Flow cytometry analysis of hepatocytes
Experiments were performed on adult female mice (NMRI-Fox1nu/Foxn1nu) at the Ecole Nationale Veterinaire d’Alfort (National Veterinary School of Alfort) (Maisons-Alfort, France), according to its ethic committee policy, following approval. Animals received good care and humane treatments. Mice were injected intravenously in the caudal vein with DilC18-loaded nanocarriers. All solutions were injected at 10 mL/kg, in 5% (v/v) glucose for PLGA and HA nanoparticles and 0.9% NaCl (w/v) for PS80–EtOH micelles. Then, 1 hour or 20 hours after intravenous injection of DilC18-loaded nanocarriers, mice were euthanized with the purpose of recovering their hepatocytes. After opening the body cavity, a catheter was placed in the portal vein.20,21 A few milliliters (~5–10 mL) of Liver Perfusion Medium, heated to 37°C, were injected very slowly in the portal vein, and the inferior vena cava was cut a few seconds after the beginning of the perfusion. Then, collagenase (0.5 mg/mL collagenase type IV in PBS), also heated to 37°C, was injected in the portal vein. Perfusion of Liver Perfusion Medium was continued until all the collagenase has gone from the catheter to the liver. Then, the liver was isolated, kept in collagenase (0.5 mg/mL in PBS), minced with scalpels and homogenized by pipetting. The cells in solution were filtered through a cell strainer (70 μM mesh size). Cells were rinsed by centrifugation (50× g, 5 minutes). Cell counts and viability of isolated hepatocytes were performed, and cells were fixed in 4% paraformaldehyde. The fluorescence was then detected and quantified with a BD Accuri C6 flow cytometer (BD Biosciences, San Jose, CA, USA) with the filters fluorescein isothiocyanate (FITC; laser 488 nm, 533/30 nm) and allophycocyanin (APC) (laser 640 nm, filter 675/25 nm). The data were analyzed using BD CSampler Plus software (BD Biosciences).
Antitumor efficacy studies
Antitumor efficacy studies were performed on adult female mice (NMRI-Fox1nu/Foxn1nu) at the Ecole Nationale Veterinaire d’Alfort (Maisons-Alfort, France), according to its ethic committee policy, following approval. Animals received good care and humane treatments. Mice were xenografted with MDA-MB-231 cells: 5 million cells in 50 μL were injected subcutaneously in the lower right flank. Tumor volume (mm3) was measured with a digital caliper and calculated by the following formula: tumor volume = length*width2/2. Mice were randomized on the day of experiment when the mean tumor volume was equal to 170 mm3 (SD: 34%): four animals per group in the control groups and six animals per group in the treated groups. Groups were treated as shown in Table S1. One vial of Taxotere (PS80-based docetaxel formulation), was diluted in NaCl 1% (1:9 v/v) prior to injection. Mice were followed up for clinical signs, body weight and tumor size at least twice a week. The treatment efficacy was determined using the optimal percent treated versus control ratio (%T/C),22 corresponding to the ratio of the mean tumor volume of treated groups versus control group and a Kaplan–Meier survival diagram.
Statistical analysis
Data were analyzed and plotted on Excel (Microsoft 2013). In vitro and in vivo results were analyzed using GraphPad Prism 5.00 version (GraphPad Software, Inc., La Jolla, CA, USA), IC50 values were calculated with the “log(inhibitor) versus response” model and statistical analyses of tumor growth delay studies were obtained with a two-way ANOVA, followed by Bonferroni post tests.
Results
Characterizations of nanocarriers
Selection of materials for the design of the nanocarriers – surfactants PS80, DSPE-PEG and the polymers PLGA and HA – was driven by their proven ability to be used in human. From these materials, we designed several biocompatible and biodegradable nanocarriers, with size below 100 nm, and specific physicochemical properties (Table 1). The key parameters tested were their composition and structure, which impacted on their blood stability and on the kinetic of release of their payload,23 and their ability to target hepatocytes (nonspecific addressing via negative surface charge versus specific addressing of functionalized nanocarriers).
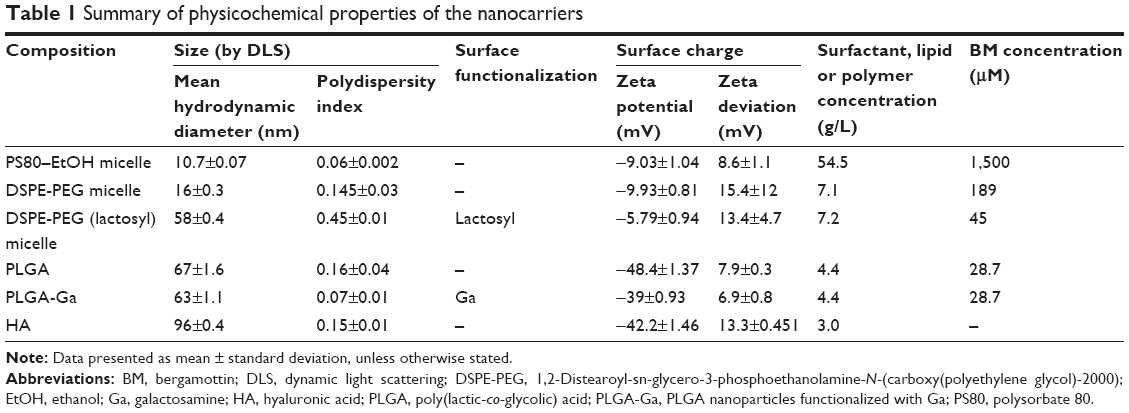 | Table 1 Summary of physicochemical properties of the nanocarriers |
PS80–EtOH micelles have a high critical micelle concentration (CMC) value of 10−5 M and a known poor stability in blood.24 DSPE-PEG micelles have a lower CMC value because block copolymers with lipids are more hydrophobic than surfactants, which stabilizes their micelle structure, and therefore such micelles present a higher stability in blood. Polymeric PLGA and HA nanoparticles have been reported to be stable in blood although the smaller the particles are, the fastest they release their payload due to an increased surface area.
The size of the nanocarrier remains one of the main drivers of clearance by the MPS: beyond 150 nm, the larger the particle’s diameter is, the more readily it is uptaken by macrophages of the liver and the spleen.25 All the nanocarriers presented in this study were <100 nm, allowing them to cross the space of Disse to be in contact with hepatocytes. Measured by DLS, PS80–EtOH, DSPE-PEG, DSPE-PEG (lactosyl) micelles, PLGA, PLGA nanoparticles functionalized with galactosamine (PLGA-Ga) and HA nanoparticles, respectively, have a hydrodynamic diameter (Z-average) of 11 nm, 16 nm, 58 nm, 67 nm, 63 nm and 96 nm, confirmed by observations in FEG-SEM for PLGA nanoparticles (Table 1; Figure 1).
 | Figure 1 FEG-SEM image of PLGA nanoparticles. |
Surface charge is also a fundamental physicochemical parameter involved in recognition by the MPS. BM-loaded micelles present quasi-neutral surface charges (<|10| mV). BM-loaded PLGA and PLGA-Ga nanoparticles and HA nanoparticles present negative surface charges with the following respective zeta potentials: −48.4 mV, −39 mV and −42.2 mV (Table 1). The absolute value of the surface charge of PLGA particles after coating with Ga decreases by 10 mV (Table 2), which tends to support the efficiency of the surface functionalization. Size measurements by DLS of PLGA and PLGA-Ga nanoparticles were also performed in 150 mM NaCl confirming a gain in stability due to functionalization.
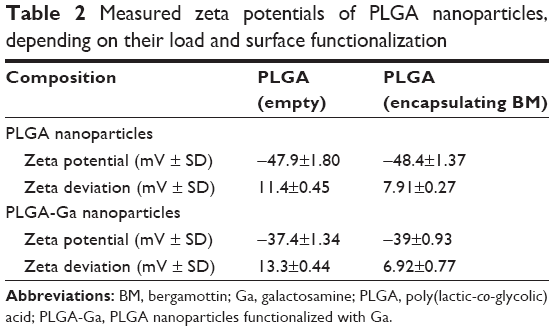 | Table 2 Measured zeta potentials of PLGA nanoparticles, depending on their load and surface functionalization |
Charged nanoparticles are known to be recognized by the MPS and to be trapped in the liver, contrary to neutral and stealth nanoparticles. Negative surface charges are generally recognized to be less toxic to the cells than positively charged nanoparticles, which tend to be trapped more rapidly by macrophages of the spleen, liver and lungs.26 Size inferior to 150 nm, combined to a negative surface charge, was chosen for a rapid clearance from the blood stream, and once in the liver sinusoids, to diffuse through the space of Disse. In addition, functionalization with lactosyl and Ga were used to enhance recognition by hepatocytes.
BM loading in nanocarriers
For PS80–EtOH and the two DSPE-PEG micelles, a good %EE (>65%) of BM was observed. Of note, for the PS80–EtOH, BM concentration was close to its solubility limit. The ratio, BM (g/L)/surfactant or lipid (g/L), is 0.9% for both micelle types. For polymeric PLGA nanoparticles, a lower %EE was observed with a maximum of 8 mg/L of BM encapsulated, corresponding to a ratio, BM (g/L)/polymer (g/L), of 0.2%, which may be explained by the polymeric nature of the particles limiting their loading capacity (Table 1).23
In vitro: CYP3A4-inhibiting nanocarriers
In vitro IC50 values of CYP3A4 by BM in different nanocarriers were measured on the HepaRG cell line,27,28 using a fluorogenic substrate: DBOMF (Table 3).18 All nanocarriers showed <20% cytotoxicity on the HepaRG cell line at the doses tested to evaluate the IC50.
 | Table 3 In vitro IC50 of BM in different nanocarriers, measured with the fluorogenic DBOMF probe, after 3 hours of co-incubation on HepaRG cells; results are the mean of at least three independent experiments performed in triplicate, presented with 95% confidence interval |
IC50 values were relatively close for nonencapsulated BM in ethanol and BM in PS80–EtOH, DSPE-PEG and DSPE-PEG (lactosyl) micelles, respectively, at 2.1 μM (1.3–3.4 μM), 1.0 μM (0.6–1.8 μM), 2.9 μM (2.1–3.9 μM) and 1.4 μM (1.0–2.1 μM). IC50 values of BM in PLGA nanoparticles were lower: 0.26 μM (0.18–0.37 μM) for PLGA and 0.56 μM (0.40–0.79 μM) for PLGA-Ga.
In vitro uptake of nanocarriers
Cellular uptake of nanocarriers containing DilC18, a near-infrared dye used in place of BM for better fluorescent resolution, was confirmed by confocal imaging of fixed HepaRG cells, after incubation with nanocarriers for 3 hours (Figures S3 and S4). Qualitatively, cell uptake analysis by confocal imaging at fixed parameters allows us to see that, after 3 hours of incubation on HepaRG cells in vitro, the uptake of lactosyl functionalized micelles is higher than the uptake of DSPE-PEG micelles. This is also the case for DilC18-loaded PLGA-Ga nanoparticles compared to DilC18-loaded PLGA nanoparticles after 3 hours and 24 hours of incubation.
In vivo: hepatocyte-targeting nanocarriers
To monitor the effective liver targeting at a macroscopic level, the biodistribution of the different DilC18-loaded nanocarriers was studied by IVIS, after intravenous injection in the tail vein of mice.
Nanocarriers were first injected at iso-fluorescence, ie, at a DilC18 concentration for which fluorescence of the solutions before injection was of the same level. Figure 2A shows typical IVIS of mice 10 minutes after injection with the different micelles loaded with DilC18. Micelles of PS80–EtOH, DSPE-PEG and DSPE-PEG (lactosyl) do not accumulate specifically in the liver. The reason may be their insufficient stability in plasma or, on the opposite, their prolonged circulation in blood.23 The “free” DilC18 (in EtOH–H2O 1:1, v/v) accumulates rapidly in the lungs (Figure S5), which tends to support the hypothesis of a prolonged circulation of the micelles in blood. Figure 2B represents typical IVIS of mice 10 minutes after injection with PLGA, PLGA-Ga and HA nanoparticles loaded with DilC18. PLGA nanoparticles accumulate in the liver. PLGA (coated and uncoated) nanoparticles reach a peak of accumulation in a few minutes, which then decreases progressively. Compared to PLGA nanoparticles, HA nanoparticles tend to circulate, while a part accumulates progressively in the liver (Figure S6). Then, to ensure that the fluorescence seen in the liver did not hide other potential sites of accumulation, the concentrations of PLGA, PLGA-Ga and HA nanoparticles were lowered from 4 g/L to 0.25 g/L in polymer (Figure S7). Then, 1 hour after this “low-dose” injection (concentration in polymer PLGA or HA of 0.25 g/L), mice were euthanized and their livers and spleens were imaged ex vivo (Figure 2C). PLGA-Ga nanoparticles and HA nanoparticles accumulate less in the spleen than uncoated PLGA nanoparticles. The mean ratio (fluorescence of the spleen)/(fluorescence of the liver) measured ex vivo, 1 hour after injection, for mice injected with uncoated PLGA is 15%, compared to 4% for mice injected with coated PLGA-Ga nanoparticles and 6% for mice injected with HA nanoparticles. For PLGA-Ga and HA nanoparticles, the chemical functions at their surface help enhancing rapid capture by the liver, avoiding their capture by the spleen.
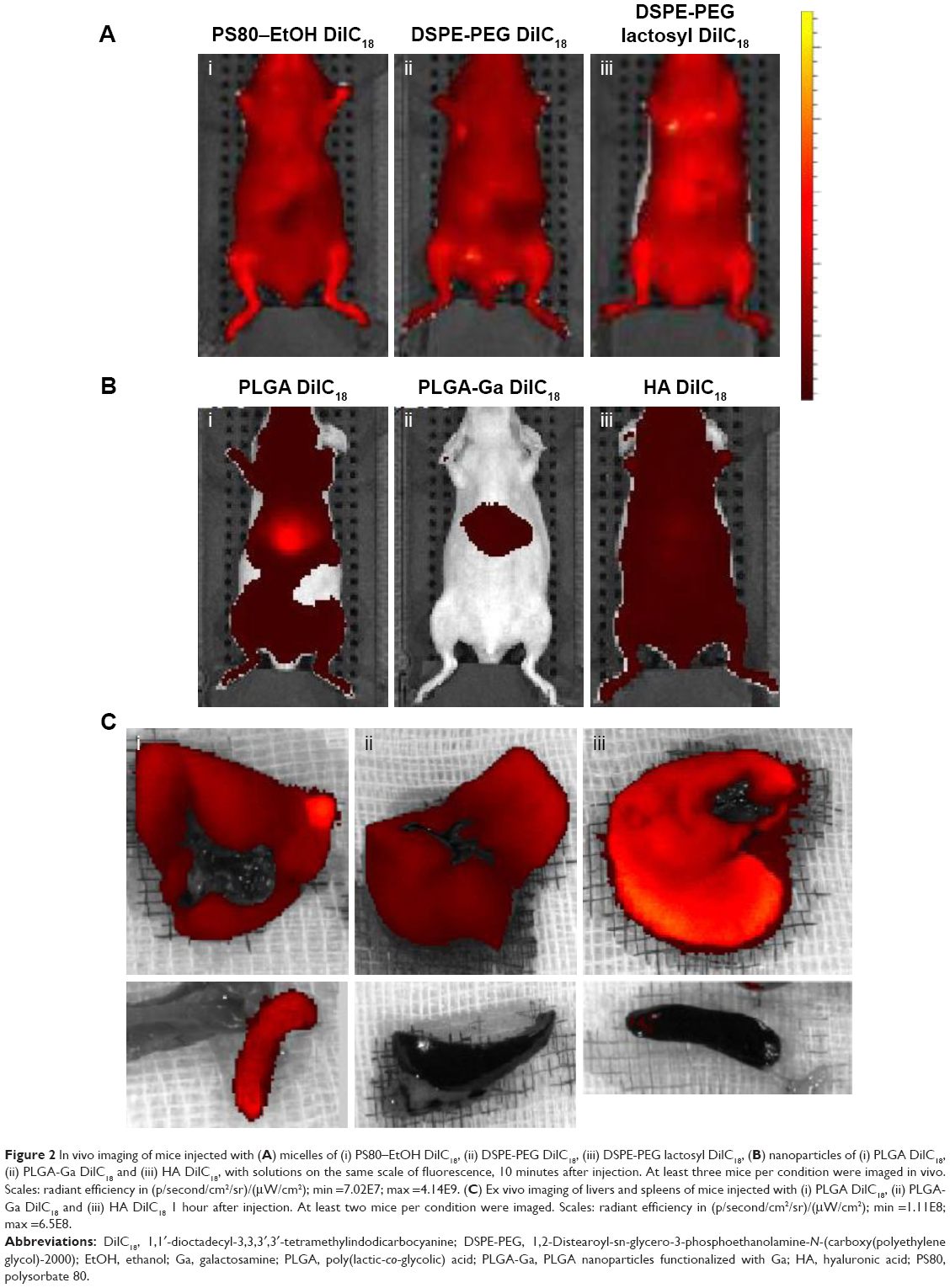 | Figure 2 In vivo imaging of mice injected with (A) micelles of (i) PS80–EtOH DilC18, (ii) DSPE-PEG DilC18, (iii) DSPE-PEG lactosyl DilC18, (B) nanoparticles of (i) PLGA DilC18, (ii) PLGA-Ga DilC18 and (iii) HA DilC18, with solutions on the same scale of fluorescence, 10 minutes after injection. At least three mice per condition were imaged in vivo. Scales: radiant efficiency in (p/second/cm2/sr)/(μW/cm2); min =7.02E7; max =4.14E9. (C) Ex vivo imaging of livers and spleens of mice injected with (i) PLGA DilC18, (ii) PLGA-Ga DilC18 and (iii) HA DilC18 1 hour after injection. At least two mice per condition were imaged. Scales: radiant efficiency in (p/second/cm2/sr)/(μW/cm2); min =1.11E8; max =6.5E8. |
Next, we evaluated the ability of the nanocarriers to target hepatocytes at the cellular level. Mice were injected with nanocarriers loaded with DilC18, at iso-concentration of polymer or surfactant.
As observed in the IVIS biodistribution study, PS80–EtOH micelles poorly accumulated in hepatocytes, with only 3.6% of hepatocytes marked with DilC18, 1 hour after injection, whereas hepatocytes were highly targeted by PLGA nanoparticles, especially PLGA-Ga nanoparticles, which showed >80% of marked hepatocytes (Figure 3A). A reduction in the marked population of hepatocytes after 20 hours is believed to be due to the release of DilC18 from PLGA nanoparticles and subsequent clearance from hepatocytes. On the opposite, as observed in the in vivo imaging distribution study, HA nanoparticles accumulate progressively in the liver, ~40% of hepatocytes are marked after 1 hour and 30% remain marked after 20 hours: there is no important difference in marked population between the two time points for HA nanoparticles (Figure 3B).
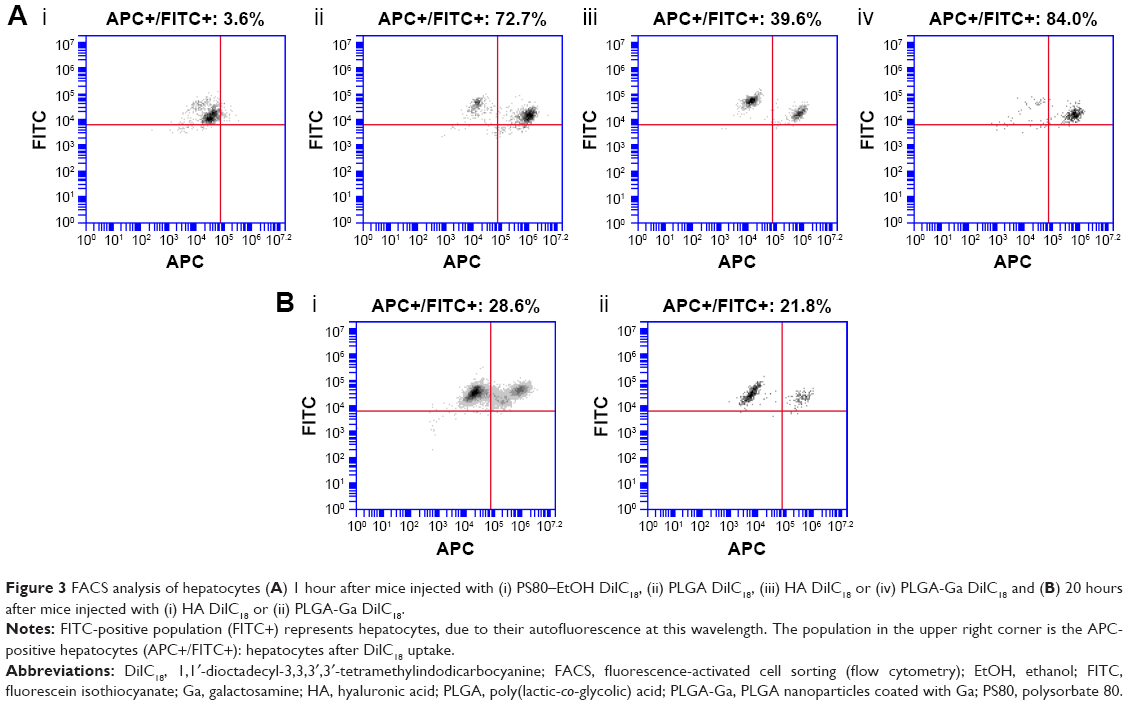 | Figure 3 FACS analysis of hepatocytes (A) 1 hour after mice injected with (i) PS80–EtOH DilC18, (ii) PLGA DilC18, (iii) HA DilC18 or (iv) PLGA-Ga DilC18 and (B) 20 hours after mice injected with (i) HA DilC18 or (ii) PLGA-Ga DilC18. |
Considering their advantageous biodistribution, specifically in the liver and their ability to target hepatocytes, PLGA-Ga BM nanoparticles were used to deliver BM. Mice xenografted with MDA-MB-231 were treated with docetaxel 20 mg/kg, without pretreatment (group 3), with pretreatment of PS80–EtOH BM micelles (BM =5 mg/kg; group 4) or PLGA-Ga BM nanoparticles (BM =0.097 mg/kg; group 5; Table S1; Figure 4).
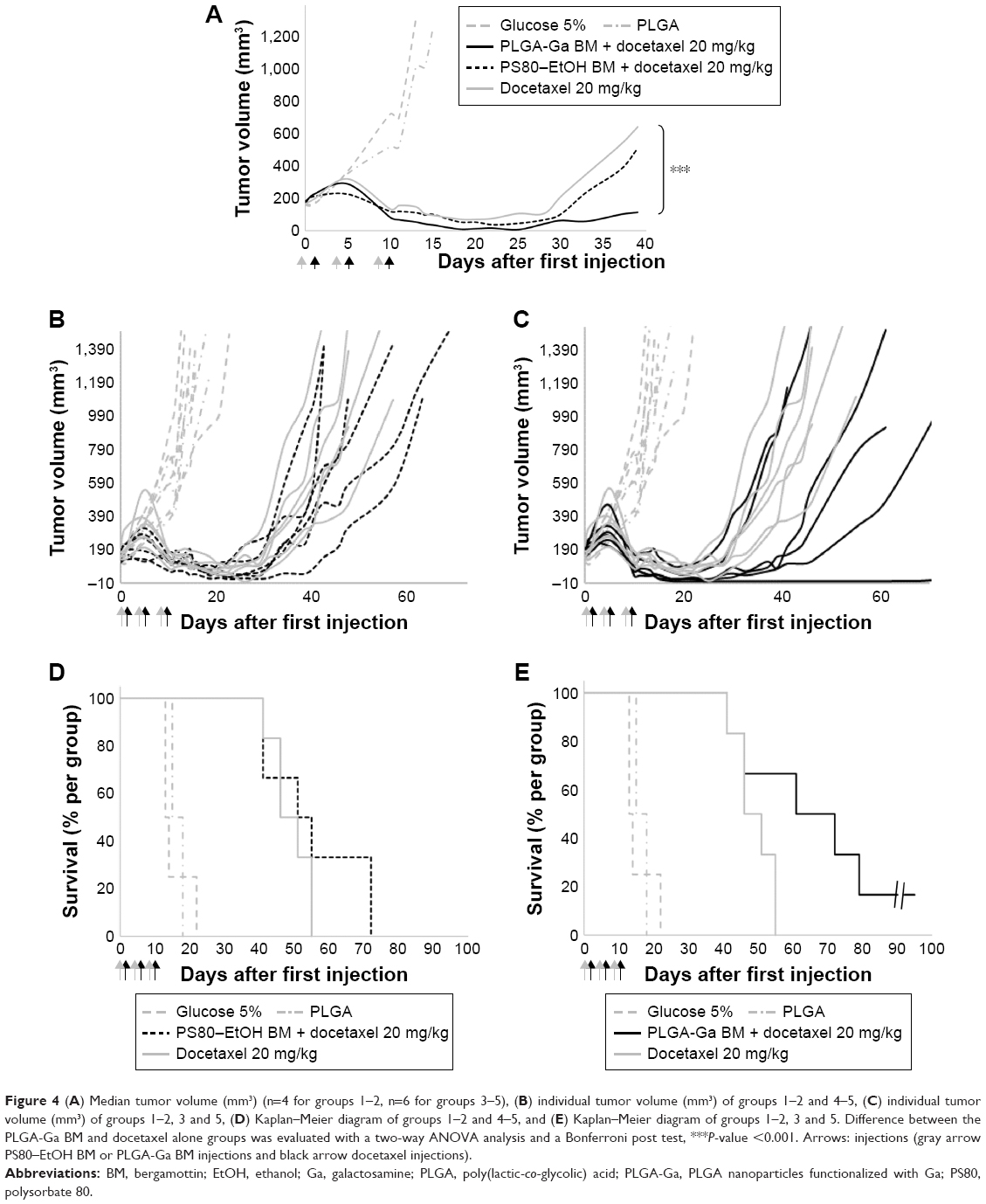 | Figure 4 (A) Median tumor volume (mm3) (n=4 for groups 1–2, n=6 for groups 3–5), (B) individual tumor volume (mm3) of groups 1–2 and 4–5, (C) individual tumor volume (mm3) of groups 1–2, 3 and 5, (D) Kaplan–Meier diagram of groups 1–2 and 4–5, and (E) Kaplan–Meier diagram of groups 1–2, 3 and 5. Difference between the PLGA-Ga BM and docetaxel alone groups was evaluated with a two-way ANOVA analysis and a Bonferroni post test, ***P-value <0.001. Arrows: injections (gray arrow PS80–EtOH BM or PLGA-Ga BM injections and black arrow docetaxel injections). |
BM was shown not to sensitize MDA-MB-231 cells to docetaxel in vitro (Figure S8). Good tolerance to docetaxel treatment was observed for all groups with individual weight loss <10% (Figure S9). As a control, PLGA-Ga BM nanoparticles alone did not show a significant antitumoral effect in vivo (group 2; Figure 4). The optimal %T/C was, respectively, 62% for PS80–EtOH BM-treated group versus docetaxel alone and 32% for PLGA-Ga BM-treated group versus docetaxel alone, on day 18. The %T/C stays inferior to 40% for 10 consecutive days for the PLGA-Ga BM-treated group. Using a two-way ANOVA and a Bonferroni posttest, the difference in tumor growth delay between the two groups, docetaxel alone and PLGA-Ga BM + docetaxel, was statistically different (P-value <0.001). The median survival was 66 days for PLGA-Ga BM-treated group, compared to 48 days for the docetaxel alone group and the overall survival rate was 67% versus 0% at day 55.
Discussion
Different nanocarriers were designed to encapsulate the hydrophobic BM, inhibitor of CYP3A4, to make it injectable and to deliver it to hepatocytes. First, micelles made of the surfactant PS80 were included as a benchmark of small nanocarrier encapsulating BM. Such micelles are known to be relatively unstable in plasma,29,30 although they were injected at a final concentration in plasma above their CMC. Micelles made of the lipid DSPE-PEG were also tested because of their small size (~20 nm) and their relatively more important stability compared to PS80–EtOH micelles. However, even with a lactosyl surface functionalization, they did not accumulate in the liver within 1 hour, either due to the micelle instability, leaking or prolonged blood circulation. Indeed, their PEGylated surface and low charge may favor a stealth behavior. Moreover, the homogeneous biodistribution of micelles encapsulating the fluorescent DilC18 probe, different from the biodistribution of the “free” probe injected in EtOH-H2O (1:1 v/v), tends to discard the leaking hypothesis in <1 hour.
BM was also encapsulated into a soft nanoparticle made of a biocompatible and biodegradable polymer: PLGA.31 Nanoparticles had hydrodynamic diameters <70 nm and were negatively charged, allowing them to extravasate and be able to cross the space of Disse to reach hepatocytes. PLGA nanoparticles were coated with Ga to test whether this functionalization would provide an active targeting. The Ga surface functionalization reduces the absolute value of the PLGA nanoparticles’ charge and is usually used as a molecular targeting agent to hepatocytes.32–34
We also designed DilC18-loaded HA nanoparticles. HA nanoparticles were larger: 96 nm, although still <150 nm. Their surface charge laid between the surface charges of PLGA and PLGA-Ga nanoparticles. Their different behavior in vivo may come from their larger size or difference in functionalization and degradability. Indeed, as a polysaccharide, HA is reported to confer stealth properties to the nanoparticles they functionalize.35,36
To test the CYP3A4 inhibition of the BM in the nanocarriers, their IC50 was evaluated on the hepatocyte cell line, HepaRG, induced to express active CYP3A4. IC50 was relatively close for nonencapsulated BM in ethanol, PS80–EtOH, DSPE-PEG and DSPE-PEG (lactosyl) micelles, ranging from 1.0 μM to 2.9 μM. IC50 of BM in PLGA nanoparticles was lower: 0.26 μM for PLGA and 0.56 μM for PLGA-Ga. Such a difference may result from a favored uptake of nanoparticles by HepaRG cells. Although it is generally considered that a fivefold difference of IC50 is considered acceptable between two CYP450 activity assays, difference among nanocarriers may not be significant.18
HepaRG cells do express ASGPR, a specific receptor at the surface of hepatocytes, receptor of Ga and lactosyl.37 A differential uptake of functionalized versus unfunctionalized micelles and nanoparticles was qualitatively observed in vitro by confocal imaging. To do so, nanocarriers were loaded with the fluorescent probe DilC18 instead of BM. Qualitatively, a lactosyl or Ga functionalization increases the uptake of nanocarriers in ASGPR-expressing cells in vitro, after 3 hours of incubation.
The mouse model was chosen for in vivo studies. Mouse and human express homologous cytochromes P450 and mouse was recognized as relevant for CYP450 inhibition studies.38–40 A limit to the mouse model was the size of the space of Disse fenestrae, which was 280 nm, slightly wider than in human.41
To test the hypothesis that the physicochemical properties of the PLGA, as well as its Ga functionalization, would make these nanoparticles accumulate rapidly in the liver and target specifically hepatocytes in vivo, we injected such nanocarriers, loaded with the fluorescent probe DilC18. When “free” DilC18 accumulated in the lungs, the PS80–EtOH and DSPE-PEG micelles were biodistributed evenly in the body, indicating they may circulate, whereas PLGA nanoparticles reached the liver specifically within seconds. Uncoated PLGA nanoparticles were trapped in the spleen, showing that they do not avoid effectively the MPS, contrary to PLGA-Ga nanoparticles. HA nanoparticles also accumulate in the liver, although part of them circulate and take a longer time to be trapped in the liver. Like PLGA-Ga nanoparticles, they do not accumulate in the spleen.
At a cellular scale, beyond their ability to reach the liver, PLGA, PLGA-Ga and HA nanoparticles were uptaken by hepatocytes in vivo. After isolating mice hepatocytes, we showed that 1 hour after injection, PLGA nanoparticles were uptaken in ~70% of hepatocytes, and PLGA-Ga nanoparticles in >80% of hepatocytes. HA nanoparticles were uptaken by only 40% of hepatocytes. Then, 20 hours after PLGA-Ga injection, only 20% of hepatocytes still fluoresced at DilC18 wavelength and 30% after HA injection. Most probably, in the meantime, DilC18 was released from nanoparticles and cleared. HA nanoparticles circulating longer before being trapped in the liver, more hepatocytes were positively marked 20 hours after HA injection than after PLGA-Ga injection.
Since PLGA-Ga reach 80% of hepatocytes very rapidly and are not observed in the spleen, we selected PLGA-Ga nanoparticles to be injected 24 hours prior to docetaxel treatment. Even though PLGA-Ga reach the hepatocytes very rapidly, BM is a “suicide” inhibitor of CYP3A4: its inhibition lasts until the neosynthesis of CYP3A4 proteins, so for at least 24 hours.42 The 24-hour time lapse is more ethical regarding volumes injected to mice and allows BM to exit from PLGA nanoparticles and to bind to CYP3A4.
Our strategy is to inhibit CYP3A4 activity in hepatocytes, decrease docetaxel’s metabolism in the liver and increase its concentration in the tumor. The antitumor efficacy of docetaxel was indeed enhanced with a preinjection of PS80–EtOH BM at 5 mg/kg of BM injected, with an optimal %T/C of 62% on day 20. The efficacy of docetaxel was even more boosted with a pre-injection of PLGA-Ga BM, at 0.097 mg/kg of BM injected, with an optimal %T/C of 32% on day 20, indicating that this pretreatment is active. Even though BM concentration injected in mice was ~50 times lower for PLGA-Ga BM-treated mice than for the PS80–EtOH BM ones, docetaxel’s efficiency was enhanced by two in terms of %T/C. Besides, a marked improvement of overall survival was observed with 67% of animals alive at day 55 with pretreatment with PLGA-Ga BM nanoparticles against 0% without pretreatment and an increase in median survival of 18 days. No clinical sign of toxicity was observed in treated groups. The use of PLGA-GA BM formulation allows injecting 50 times less BM and 50 times less polymer/surfactant, which limits the potential toxicity or immunogenicity.
Intra-tumor expression of CYP450 may lower the dose of medication, when the drug is a substrate of the CYP450 isotype expressed. Our strategy could be developed in the indication of hepatocellular carcinoma (HCC). When HCC is inferior to 3 cm in diameter, it is most frequently composed of well-differentiated to moderately differentiated hepatocytes,43 meaning that they still express CYP450 enzymes.44 Inhibiting CYP3A4 in particular, by which docetaxel is metabolized, would allow increasing the concentration of docetaxel in HCCs and avoid resistance mechanisms. HCC may also express the efflux pump P-glycoprotein, which is known to be at the origin of drug resistance in many tumor types. The impact of PLGA-Ga BM nanoparticles on P-glycoprotein activity should be evaluated in vivo.11
Other natural inhibitors of hepatic cytochromes P450 could be used in place of, or together with, furanocoumarins, into hepatocyte-specific nanocarriers for optimal inhibition of drug metabolism. Indeed, most of drug metabolism results from the activity of multiple cytochromes P450. Furanocoumarins inhibit several CYP450 (CYP3A4, CYP1A2, CYP1B1, CYP2C9, CYP2C19 and CYP2D6),11,12 but other CYP450 are involved in common drug metabolism. The taxane paclitaxel for instance is metabolized by both CYP3A4 and CYP2C8.45 A strategy to bring patients’ drug metabolism to a baseline could be to combine a furanocoumarin with a flavonoid, such as quercetin.18,46
Conclusion
In this study, we designed a nanocarrier to encapsulate a CYP450 3A4 inhibitor, using the mechanism of nanoparticle clearance by the liver, to target hepatocytes. Our aim was to increase the efficiency of the drug docetaxel. We showed an effective targeting and CYP3A4 inhibition following intravenous injection of PLGA-Ga BM nanoparticles. For a given dose of docetaxel intravenously administered to mice bearing MDA-MB-231 tumors, a significant delay of tumor growth and improvement in overall survival were observed without increasing docetaxel clinical signs of toxicity, when compared to docetaxel alone. These results open the opportunity to decrease the dose of docetaxel for the same efficacy and reduced toxicity.
Acknowledgments
We are grateful to the Imaging and Cytometry Platform (UMS 23/3655, Gustave Roussy Cancer Campus, University of Paris Saclay, Villejuif, France), especially to Valérie Rouffiac for the image acquisitions and processing and to the ImagoSeine platform (Institut Jacques Monod, University Paris, Diderot-Paris 7) for providing the confocal microscope. FEG-SEM instrumentation was facilitated by the IMPC FR2482 (Institut des Matériaux de Paris Centre) financially supported by the C’Nano projects of the Région Ile-de-France. We also acknowledge Hugo Lenormand and Laura Fabre who helped in the characterizations of nanoparticles. Funding support from the ANRT (Association nationale de la recherche et de la technologie) is gratefully received.
Disclosure
MP, LP, CB, MG, AD, MB, AP and LL are employees of Nanobiotix, Paris, which is conducting the study presented in the paper. EV, LP, CB, MG, AD, MB, AP and LL have financial involvement with Nanobiotix. MP, LP, CB, MG, AP and LL are coinventors of patent applications related to the study presented in the paper. The authors report no other conflicts of interest in this work.
References
Cheetham P, Petrylak DP. Tubulin-targeted agents including docetaxel and cabazitaxel. Cancer J. 2013;19(1):59–65. | ||
van Oosterom AT, Schrijvers D, Schriivers D [corrected to Schrijvers D]. Docetaxel (Taxotere), a review of preclinical and clinical experience. Part II: clinical experience. Anticancer Drugs. 1995;6(3):356–368. | ||
Marre F, Sanderink GJ, de Sousa G, Gaillard C, Martinet M, Rahmani R. Hepatic biotransformation of docetaxel (Taxotere) in vitro: involvement of the CYP3A subfamily in humans. Cancer Res. 1996;56(6):1296–1302. | ||
Bardelmeijer HA, Ouwehand M, Buckle T, et al. Low systemic exposure of oral docetaxel in mice resulting from extensive first-pass metabolism is boosted by ritonavir. Cancer Res. 2002;62(21):6158–6164. | ||
Hirth J, Watkins PB, Strawderman M, Schott A, Baker LH. The effect of an individual’s cytochrome CYP3A4 activity on docetaxel clearance. Clin Cancer Res. 2000;6(4):1255–1258. | ||
Pond GR, Armstrong AJ, Galsky MD, Wood BA, Leopold L, Sonpavde G. Efficacy of docetaxel-based chemotherapy following ketoconazole in metastatic castration-resistant prostate cancer: implications for prior therapy in clinical trials. Urol Oncol. 2013;31(8):1457–1463. | ||
Hendrikx JJ, Lagas JS, Song J-YY, et al. Ritonavir inhibits intratumoral docetaxel metabolism and enhances docetaxel antitumor activity in an immunocompetent mouse breast cancer model. Int J Cancer. 2016;138(3):758–769. | ||
Huisman MT, Smit JW, Wiltshire HR, Beijnen JH, Schinkel AH. Assessing safety and efficacy of directed P-glycoprotein inhibition to improve the pharmacokinetic properties of saquinavir coadministered with ritonavir. J Pharmacol Exp Ther. 2003;304(2):596–602. | ||
Tsoi KM, MacParland SA, Ma X-Z, et al. Mechanism of hard-nanomaterial clearance by the liver. Nat Mater. 2016;15(11):1212–1221. | ||
Braet F, Wisse E. Structural and functional aspects of liver sinusoidal endothelial cell fenestrae: a review. Comp Hepatol. 2002;1(1):1. | ||
Hanley MJ, Cancalon P, Widmer WW, Greenblatt DJ. The effect of grapefruit juice on drug disposition. Expert Opin Drug Metab Toxicol. 2011;7(3):267–286. | ||
Valenzuela B, Rebollo J, Pérez T, Brugarolas A, Pérez-Ruixo JJ. Effect of grapefruit juice on the pharmacokinetics of docetaxel in cancer patients: a case report. Br J Clin Pharmacol. 2011;72(6):978–981. | ||
Bailey DG, Malcolm J, Arnold O, Spence JD. Grapefruit juice-drug interactions. Br J Clin Pharmacol. 1998;46(2):101–110. | ||
Fiske CH, Subbarow Y. The colorimetric determination of phosphorus. J Biol Chem. 1925;66(2):375–400. | ||
Chen PS, Toribara TY, Warner H. Microdetermination of phosphorus. Anal Chem. 1956;28(11):1756–1758. | ||
Chang J, Paillard A, Passirani C, et al. Transferrin adsorption onto PLGA nanoparticles governs their interaction with biological systems from blood circulation to brain cancer cells. Pharm Res. 2012;29(6):1495–1505. | ||
Hu Z, Xia X, Tang L. Process for synthesizing oil and surfactant-free hyaluronic acid nanoparticles and microparticles. 2009. US Patent 20060040892 A1. | ||
Donato MT, Jiménez N, Castell JV, Gómez-Lechón MJ. Fluorescence-based assays for screening nine cytochrome P450 (P450) activities in intact cells expressing individual human P450 enzymes. Drug Metab Dispos. 2004;32(7):699–706. | ||
Price RJ, Surry D, Renwick AB, Meneses-Lorente G, Lake BG, Evans DC. CYP isoform induction screening in 96-well plates: use of 7-benzyloxy-4-trifluoromethylcoumarin as a substrate for studies with rat hepatocytes. Xenobiotica. 2000;30(8):781–795. | ||
Seglen PO. Preparation of isolated rat liver cells. Methods Cell Biol. 1976;13:29–83. | ||
Li W-C, Ralphs KL, Tosh D. Isolation and culture of adult mouse hepatocytes. Methods Mol Biol. 2010;633:185–196. | ||
Langdon SP, Hendriks HR, Braakhuis B-JM, et al. Preclinical phase II studies in human tumor xenografts: a European multicenter follow-up study. Ann Oncol. 1994;5(5):415–422. | ||
Trivedi R, Kompella UB. Nanomicellar formulations for sustained drug delivery: strategies and underlying principles. Nanomedicine (Lond). 2010;5(3):485–505. | ||
Chou DK, Krishnamurthy R, Randolph TW, Carpenter JF, Manning MC. Effects of Tween 20 and Tween 80 on the stability of Albutropin during agitation. J Pharm Sci. 2005;94(6):1368–1381. | ||
Blanco E, Shen H, Ferrari M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat Biotechnol. 2015;33(9):941–951. | ||
Nel AE, Mädler L, Velegol D, et al. Understanding biophysicochemical interactions at the nano-bio interface. Nat Mater. 2009;8(7):543–557. | ||
Kanebratt KP, Andersson TB. Evaluation of HepaRG cells as an in vitro model for human drug metabolism studies. Drug Metab Dispos. 2008;36(7):1444–1452. | ||
Anthérieu S, Chesné C, Li R, et al. Stable expression, activity, and inducibility of cytochromes P450 in differentiated HepaRG cells. Drug Metab Dispos. 2010;38(3):516–525. | ||
Reddy LH, Bazile D. Drug delivery design for intravenous route with integrated physicochemistry, pharmacokinetics and pharmacodynamics: illustration with the case of taxane therapeutics. Adv Drug Deliv Rev. 2014;71:34–57. | ||
van Tellingen O, Beijnen JH, Verweij J, Scherrenburg EJ, Nooijen WJ, Sparreboom A. Rapid esterase-sensitive breakdown of polysorbate 80 and its impact on the plasma pharmacokinetics of docetaxel and metabolites in mice. Clin Cancer Res. 1999;5(10):2918–2924. | ||
Díaz-Moscoso A. Soft versus hard nanoparticles in the delivery of aromatic macrocycles for photodynamic therapy of cancer. Int J Med Biomed Res. 2012;1(1):12–23. | ||
Kang J-H, Toita R, Murata M. Liver cell-targeted delivery of therapeutic molecules. Crit Rev Biotechnol. 2016;36(1):132–143. | ||
D’Souza AA, Devarajan PV. Asialoglycoprotein receptor mediated hepatocyte targeting – strategies and applications. J Control Release. 2015;203:126–139. | ||
Zhang YN, Poon W, Tavares AJ, McGilvray ID, Chan WCW. Nanoparticle-liver interactions: cellular uptake and hepatobiliary elimination. J Control Release. 2016;240:332–348. | ||
Salmaso S, Caliceti P. Stealth properties to improve therapeutic efficacy of drug nanocarriers. J Drug Deliv. 2013;2013:374252. | ||
Hu C-MJ, Fang RH, Luk BT, Zhang L. Polymeric nanotherapeutics: clinical development and advances in stealth functionalization strategies. Nanoscale. 2014;6(1):65–75. | ||
Rennert K, Steinborn S, Gröger M, et al. A microfluidically perfused three dimensional human liver model. Biomaterials. 2015;71:119–131. | ||
Tang C, Prueksaritanont T. Use of in vivo animal models to assess pharmacokinetic drug-drug interactions. Pharm Res. 2010;27(9):1772–1787. | ||
Martignoni M, Groothuis GM, de Kanter R. Species differences between mouse, rat, dog, monkey and human CYP-mediated drug metabolism, inhibition and induction. Expert Opin Drug Metab Toxicol. 2006;2(6):875–894. | ||
Renaud HJ, Cui JY, Khan M, Klaassen CD. Tissue distribution and gender-divergent expression of 78 cytochrome p450 mRNAs in mice. Toxicol Sci. 2011;124(2):261–277. | ||
Sarin H. Physiologic upper limits of pore size of different blood capillary types and another perspective on the dual pore theory of microvascular permeability. J Angiogenes Res. 2010;2(1):1–19. | ||
Zhou S, Chan SY, Goh BC, et al. Mechanism-based inhibition of cytochrome P450 3A4 by therapeutic drugs. Clin Pharmacokinet. 2005;44(3):279–304. | ||
Kōjiro M. Pathology of Hepatocellular Carcinoma. Malden, MA: Blackwell Publishing; 2006. | ||
Tsunedomi R, Iizuka N, Hamamoto Y, et al. Patterns of expression of cytochrome P450 genes in progression of hepatitis C virus-associated hepatocellular carcinoma. Int J Oncol. 2005;27(3):661–667. | ||
Spratlin J, Sawyer MB. Pharmacogenetics of paclitaxel metabolism. Crit Rev Oncol Hematol. 2007;61(3):222–229. | ||
Venkatakrishnan K, Von Moltke LL, Greenblatt DJ. Human drug metabolism and the cytochromes P450: application and relevance of in vitro models. J Clin Pharmacol. 2001;41(11):1149–1179. |
Supplementary materials
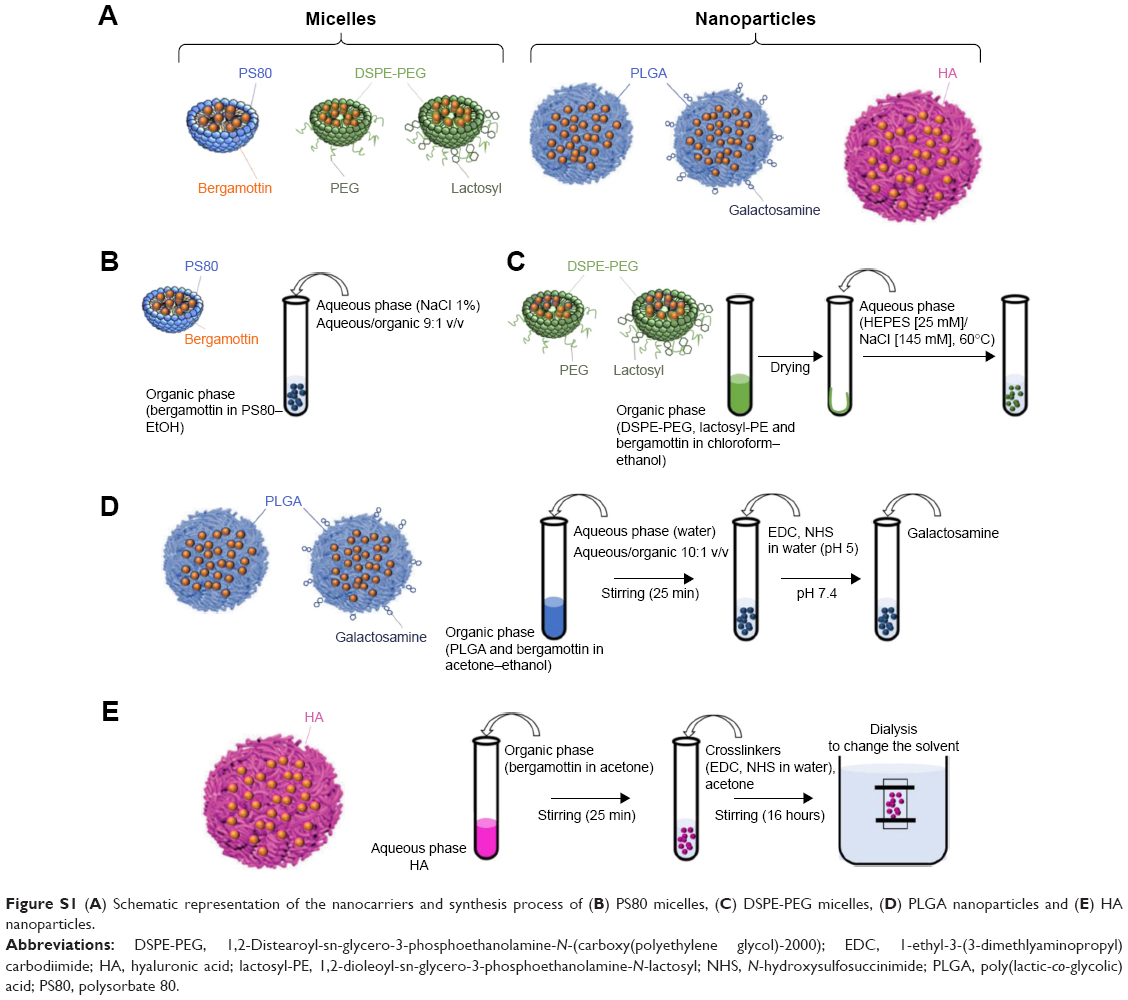 | Figure S1 (A) Schematic representation of the nanocarriers and synthesis process of (B) PS80 micelles, (C) DSPE-PEG micelles, (D) PLGA nanoparticles and (E) HA nanoparticles. |
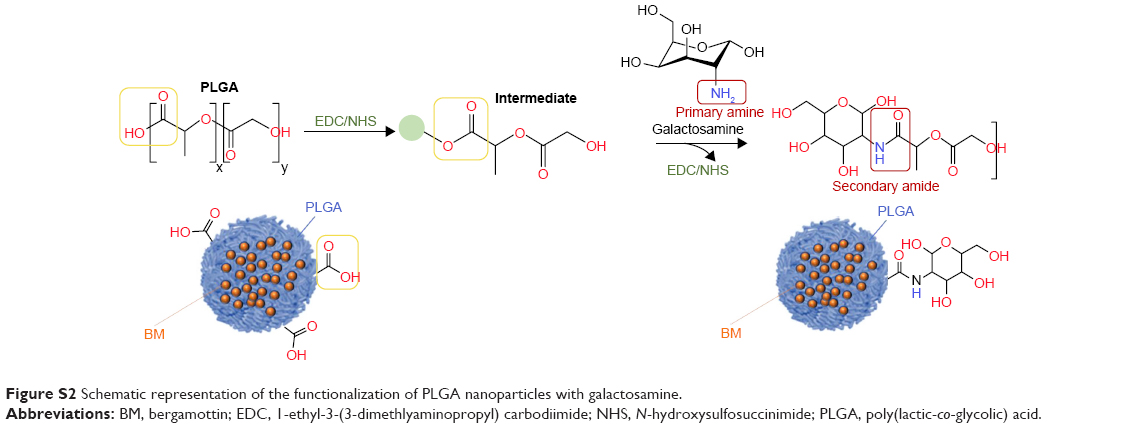 | Figure S2 Schematic representation of the functionalization of PLGA nanoparticles with galactosamine. |
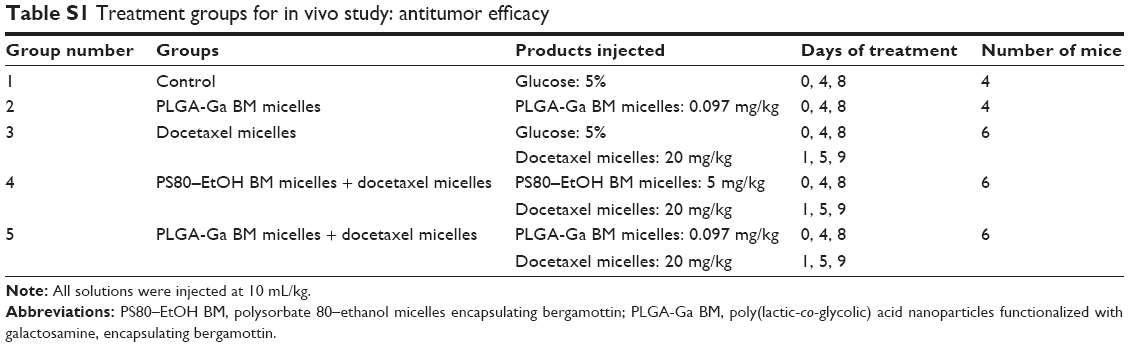 | Table S1 Treatment groups for in vivo study: antitumor efficacy |
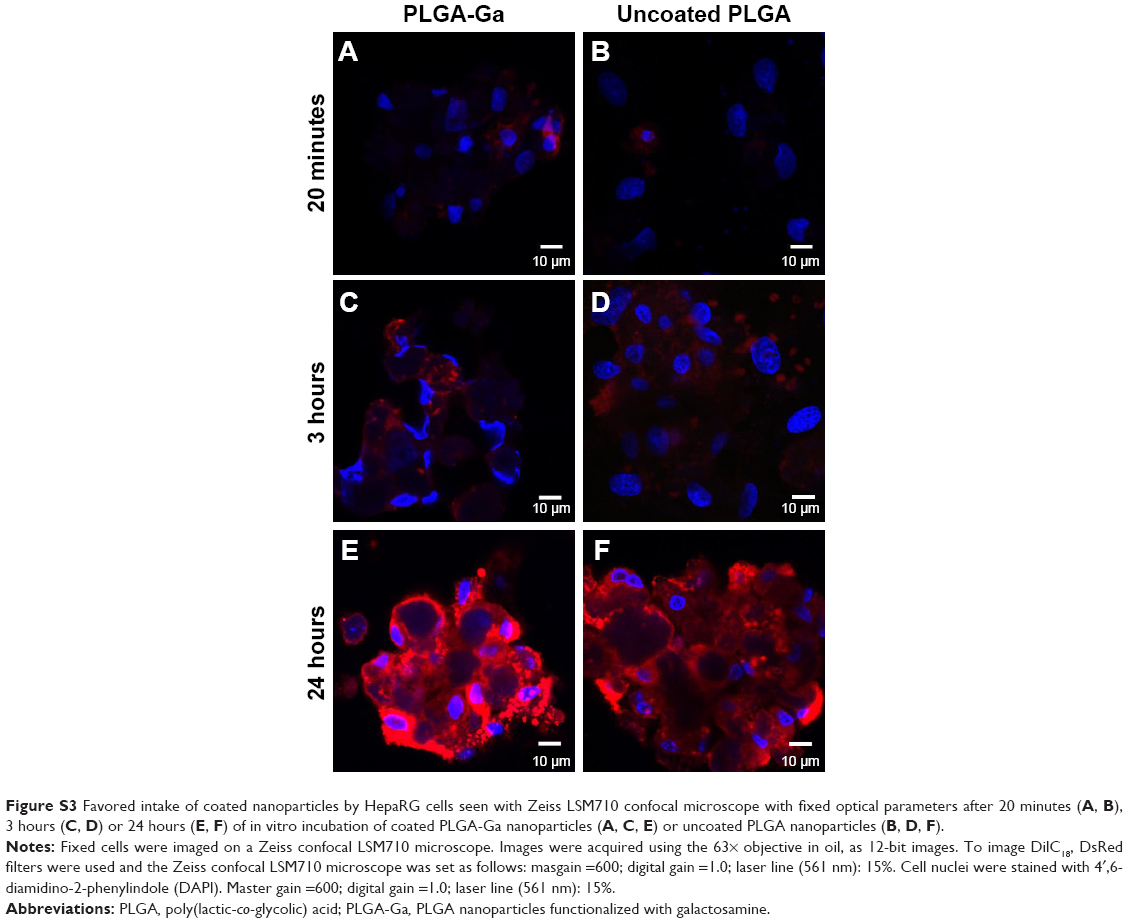 | Figure S3 Favored intake of coated nanoparticles by HepaRG cells seen with Zeiss LSM710 confocal microscope with fixed optical parameters after 20 minutes (A, B), 3 hours (C, D) or 24 hours (E, F) of in vitro incubation of coated PLGA-Ga nanoparticles (A, C, E) or uncoated PLGA nanoparticles (B, D, F). |
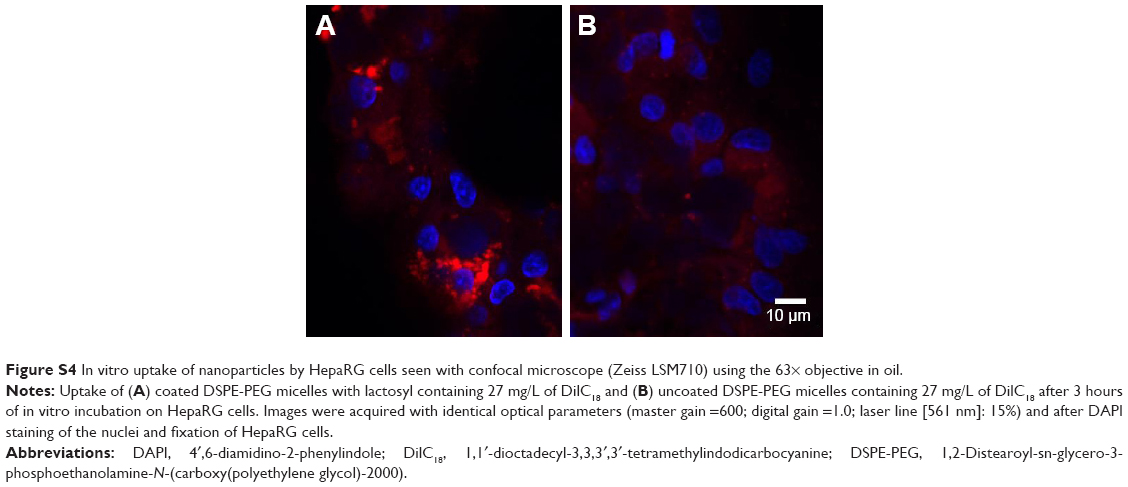 | Figure S4 In vitro uptake of nanoparticles by HepaRG cells seen with confocal microscope (Zeiss LSM710) using the 63× objective in oil. |
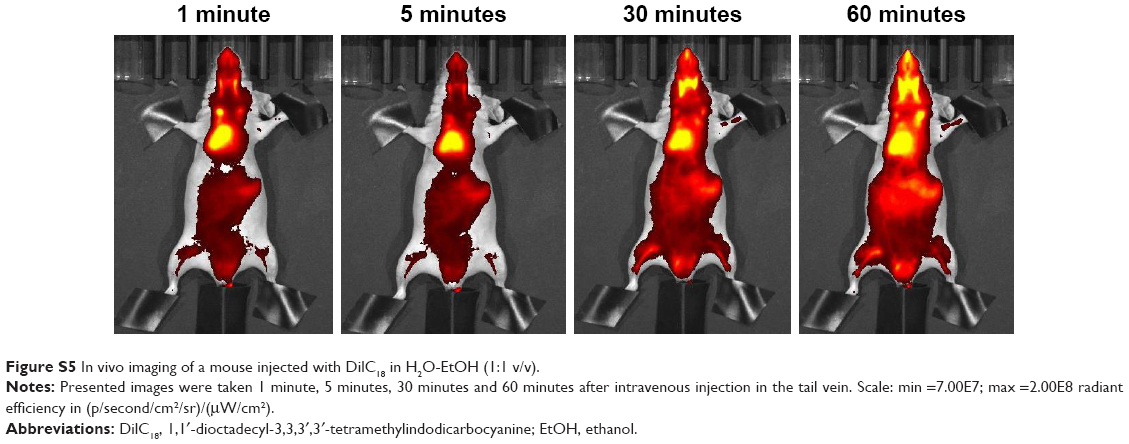 | Figure S5 In vivo imaging of a mouse injected with DilC18 in H2O-EtOH (1:1 v/v). |
 | Figure S6 In vivo imaging of mice injected with (A) PS80–EtOH DilC18, (B) DSPE-PEG DilC18, (C) DSPE-PEG lactosyl DilC18, (D) HA DilC18, (E) PLGA DilC18 and (F) PLGA-Ga DilC18. |
 | Figure S7 In vivo imaging of mice injected with (A) HA DilC18, (B) PLGA DilC18 and (C) PLGA-Ga DilC18, at lowered concentration of nanoparticles (0.25 g/L of polymer). |
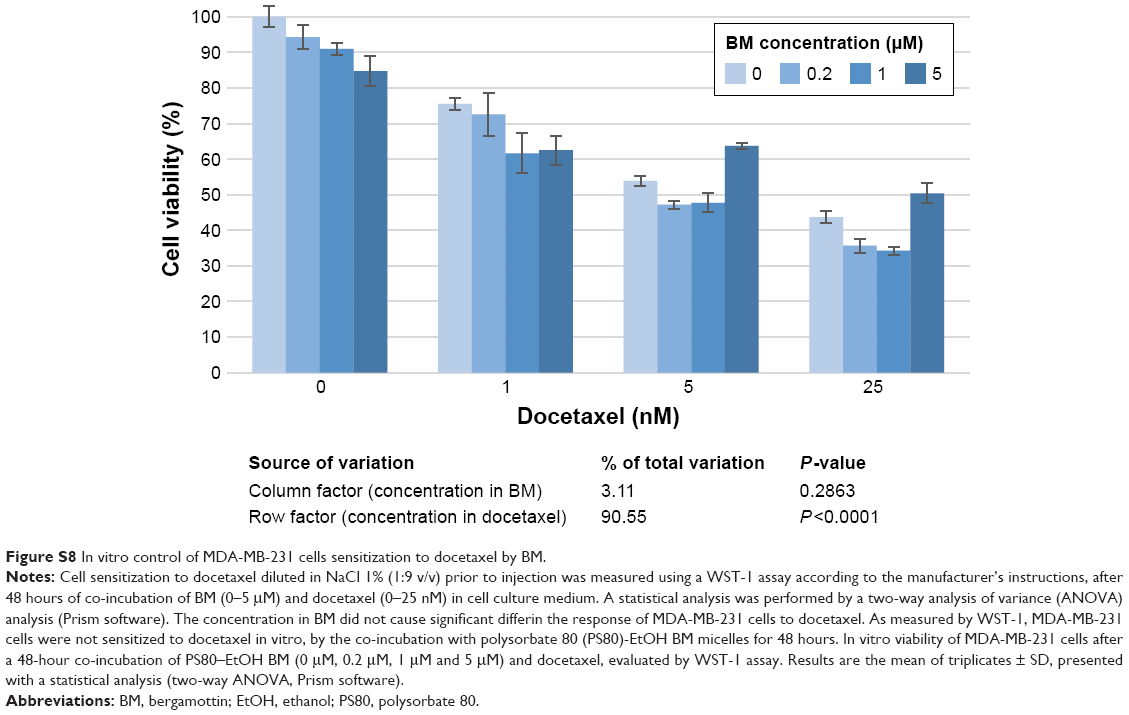 | Figure S8 In vitro control of MDA-MB-231 cells sensitization to docetaxel by BM. |
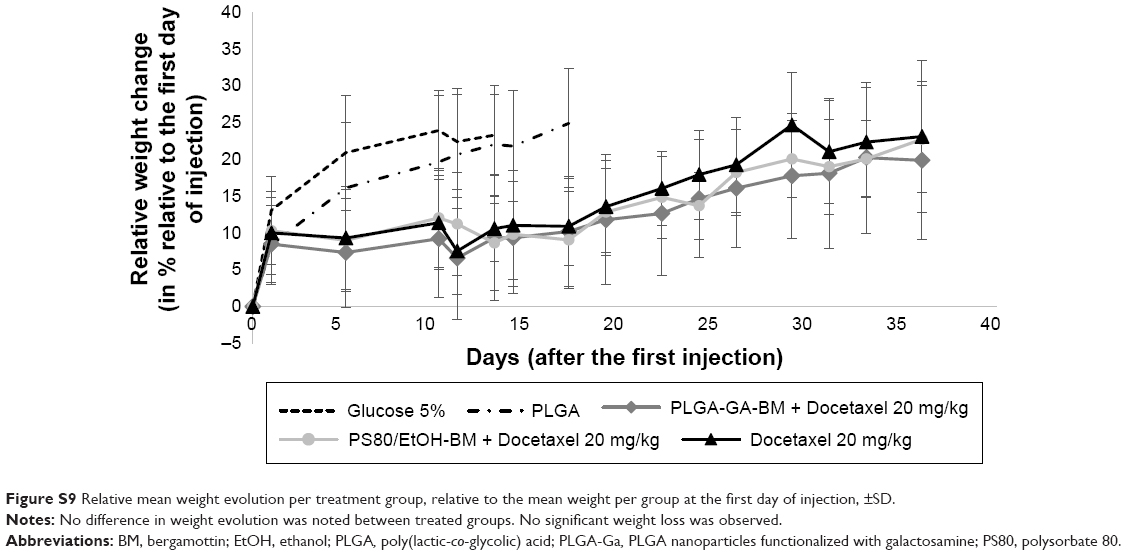 | Figure S9 Relative mean weight evolution per treatment group, relative to the mean weight per group at the first day of injection, ±SD. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.