")
Back to Journals » Drug Design, Development and Therapy » Volume 15
Nα-1, 3-Benzenedicarbonyl-Bis-(Amino Acid) and Dipeptide Candidates: Synthesis, Cytotoxic, Antimicrobial and Molecular Docking Investigation
Authors Naglah AM , Moustafa GO , Elhenawy AA , Mounier MM , El-Sayed H , Al-Omar MA , Almehizia AA , Bhat MA
Received 14 August 2020
Accepted for publication 23 January 2021
Published 25 March 2021 Volume 2021:15 Pages 1315—1332
DOI https://doi.org/10.2147/DDDT.S276504
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Anastasios Lymperopoulos
Ahmed M Naglah,1,2 Gaber O Moustafa,2 Ahmed A Elhenawy,3,4 Marwa M Mounier,5 Heba El-Sayed,6 Mohamed A Al-Omar,1 Abdulrahman A Almehizia,1,7 Mashooq A Bhat7
1Department of Pharmaceutical Chemistry, Drug Exploration & Development Chair (DEDC), College of Pharmacy, King Saud University, Riyadh, 11451, Saudi Arabia; 2Peptide Chemistry Department, Chemical Industries Research Division, National Research Centre, Cairo, Egypt; 3Chemistry Department, Faculty of Science, Al-Azhar University (Boys’Branch), Cairo, Egypt; 4Chemistry Department, Faculty of Science, Albaha University, Al Baha, Saudi Arabia; 5Pharmacognosy Department, Pharmaceutical and Drug Industries Research Division, National Research Centre, Giza, Egypt; 6Botany and Microbiology Department, Faculty of Science, Helwan University, Helwan, Egypt; 7Department of Pharmaceutical Chemistry, College of Pharmacy, King Saud University, Riyadh, 11451, Saudi Arabia
Correspondence: Gaber O Moustafa; Ahmed Elhenawy Tel +201003123355
; +966599044526
Email [email protected]; [email protected]
Purpose: The objective of our work was to prepare a potent and safe antimicrobial and anticancer agents, through synthesis of several peptides and examine their biological activities, namely as, cytotoxically potent and antimicrobial and antifungal agents.
Introduction: Multidrug-resistant microbial strains have arisen against all antibiotics in clinical use. Infections caused by these bacteria threaten global public health and are associated with high mortality rates.
Methods: The main backbone structure for the novel synthesized linear peptide is Nα-1, 3-benzenedicarbonyl-bis-(Amino acids)-X, ( 3– 11). A computational docking study against DNA gyrase was performed to formulate a mode of action of the small compounds as antimicrobial agents.
Results: The peptide-bearing methionine-ester ( 4) exhibited potent antimicrobial activity compared to the other synthesized compounds, while, peptide ( 8), which had methionine-hydrazide fragment was the most potent as antifungal agent against Aspergillus niger with 100% inhibition percent. Compounds ( 6 and 7) showed the highest potency against breast human tumor cell line “MCF-7” with 95.1% and 79.8% of cell inhibition, respectively. The nine compounds possessed weak to moderate antiproliferative effect over colon tumor cell line. The docking results suggest good fitting through different hydrogen bond interactions with the protein residues. In silico ADMET study also evaluated and suggested that these compounds had promising oral bioavailability features.
Conclusion: The tested compounds need further modification to have significant antimicrobial and antitumor efficacy compared to the reference drugs.
Keywords: anticancer, antimicrobial activity, linear-peptides, Nα-1, 3-benzenedicarbonyl-bis-peptides, molecular docking
Introduction
The application of peptide-based drugs for the treatment of different diseases has been widely developed during the last few decades. Peptides and peptide derivatives are inherently active against infectious diseases within biological systems. Moreover, antimicrobial peptides (AMPs) have been used as broad-spectrum antibiotics against several microorganisms including bacteria, fungi, and viruses.1 The emergence of multi-resistance pathogens is still potentially problematic for the treatment of infectious diseases. Furthermore, weakening of the immune system or inflammation associated with chronic infection makes patients more vulnerable to the emergence of cancer.2 Such an association between cancer and infection urges the identification of a new class of molecules that are dually-active as broad-spectrum antimicrobial agents and as anticancer agents.3 Promising new molecules, defined as antimicrobial peptides with anticancer properties, should present activities against broad multi-resistance pathogens and the ability to interfere with tumor growth and invasion towards healthy tissues. Physicochemical properties of peptide sequences exhibit dual-activity against pathogens and cancer. For example, the presence of positively charged or bulky hydrophobic amino acids in such sequences electrostatically interacts with negatively charged groups in the bacterial membranes. Furthermore, these amino acids provide the required characteristics to interact with tampered cancer cell membrane, through which the activity of AMPs toward cancer cells is promoted through the interactions of negatively charged phospholipids exposed in the membrane of cancer cells or by interacting with the cell membrane potential and fluidity.4 Despite the versatility of peptide design and synthesis to achieve such dual activities, their metabolic and conformational instabilities represent the main obstacles for the successful implementation of peptide-based drugs. This drawback urged us to overcome such obstacles by the design and synthesis of peptide derivatives with rigid conformations and specific amino acid sequences that impart, simultaneously, anticancer and antimicrobial activities. Our research group has published several studies indicating that peptide candidates are promising as potential anticancer drugs,5–15 anti-inflammatory and analgesic drugs,16 and as antibacterial drugs.17–28 The incidence of microbial infections in cancer patients increases due to the low circulating neutrophil levels in these patients in addition to the disruption of the microbial flora. Thus, the objective of the current study is to synthesize a new peptide derivative with multiple biological activities including anticancer, anti-inflammatory, and antibacterial activities to be considered as potential candidates used in the treatment of cancer.
Materials and Methods
Chemistry
These companies; Sigma (Ronkonkoma, NY, USA), Fluka (Buchs, Switzerland), and E. Merck (Hohenbrunn, Germany) were used to import all chemical and related materials. All “melting points; M.P.” uncorrected which gained from “Digital melting point apparatus; model: IA9100”. The “Micro-Analytical center” in Cairo University of Egypt was used for measuring the following spectroscopic analysis; (i) Elemental Micro-analyses for C, N, and H, which gained through a good restriction of the theoretical values (±0.4%). (ii) “Potassium bromide; KBr” disks were handled for preparation tested samples to implement into the “Fourier transforms infrared spectrophotometer; IR Shimadzu; Model: with 1S affinity”. (iii) Mass spectra measured by “Shimadzu gas chromatograph–spectrometer, Model: QP2010ultra; Kyoto, Japan”. NMR-spectra in ”DMSO-d6” as proton and carbon were obtained from “JöEL500 MHz instrument”.
Synthesis of Nα-1, 3-Benzenedicarbonyl-Bis-[Amino Acid Methyl Ester]
First Route: The Acid Chloride Method
A DCM of compound 1 (1 mmol, 50 mL) was added dropwise while stirring to a cold (−20°C) dichloromethane solution (50 mL) of free L-alanine and L-methionine ester (2 mmol), obtained by the addition of two equivalent amounts of triethylamine (2 mmol) to the amino acid methyl ester hydrochloride and cold (−20°C) dichloromethane, 50mL, while stirring. The obtained mixture was stirred for an additional 3 hours at (−20°C), and then for 24 hours at room temperature, followed by washes with distilled water, 1 N sodium bicarbonate, 1 N potassium hydrogen sulphate, and distilled water, and then for dried for 24 hours at 0°C over anhydrous sodium sulphate. The volatile materials were evaporated until dry and were then triturated with petroleum ether (boiling point B.P. 40–60°C) to obtain residual material. The obtained precipitate was collected and then recrystallized from methanol (MeOH) to obtain compounds 3 and 4.
Second Route: The Mixed Anhydride Method
Ethyl chloroformate (2.9 mL, 30.1 mmol), a DCM solution (50 mL) of isophthalic acid (5 g, 30.1 mmol) and triethylamine (6.6 mL, 60.2 mmol), was stirred at −20°C. The reaction mixture was stirred for an additional 30 minutes, and then the DCM solution (50 mL) free of L-alanine and L- methionine ester (8.4 g; 60.2 mmol, −20°C) was added to the dichloromethane (50 mL) under continuous stirring for 7 hours at −20°C, and subsequently for 24 hours at room temperature. The crude product was recrystallized from methanol to obtain pure compounds 3 and 4; as determined by the melting point and TLC “thin layer chromatography“ compared to the original sample prepared according to Method A.
Nα-1, 4-Benzenedicarbonyl-Bis-[L-Ala-OMe], (3)
Yield: [A]: 90; [B]: 80; m.p. 108–110°C.Rf x100 (solvent system) 55(S2). [α]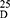 : - 12.04 (C, 0.02, MeOH). IR in (cm−1; KBr): 3400 (NH-stretching), 3066 (CH-aromatic), 2950 (CH-aliphatic), 1738 (C=O, ester), 1644 and 1534 (C=O, amide I + II).1H-NMR (500 MHz, DMSO-d6) δ:9.04, 8.97 (s, 2H, CONH, D2O, exchangeable), 8.41 (s, 1H, aromatic H, C2), 8.15–8.04 (d, 2H, aromatic H, C4, 6), 7.65–7.60 (t, 1H, aromatic H, C5), 4.54, 4.50 (q,2H, CHNH), 3.66 (s, 6H, OCH3), 1.42 (d, 6H, 2CH3, CHCH3). 13C-NMR (125 MHz, δ, ppm, DMSO-d6): 173.57 (2C, CO, ester), 166.33 (2C, CONH), 134.38 (2C, isophthaloyl, C1, 5), 130.77 (2C, isophthaloyl, C2, 4), 128.81 (1C, isophthaloyl, C3), 127.27 (1C, isophthaloyl, C6), 52.35 (2C, OCH3, L-ala), 48.85 (2C, CHCH3, L-ala), 17.15 (2C, CHCH3, L-ala). MS (70-eV;-EI):-m/z-(%) =337 (M+-+1, 14.75%), 336 (M+, 1.2%), 277 (87.52%), 234 (100%), 50 (7.13%). Molecular formula (M.wt.), C16H20N2O6 – (336.3), calculated-analysis; C, 57.14; H, 5.99; N, 8.33, found; –C 57.00, –H 6.02, –N 8.30.
: - 12.04 (C, 0.02, MeOH). IR in (cm−1; KBr): 3400 (NH-stretching), 3066 (CH-aromatic), 2950 (CH-aliphatic), 1738 (C=O, ester), 1644 and 1534 (C=O, amide I + II).1H-NMR (500 MHz, DMSO-d6) δ:9.04, 8.97 (s, 2H, CONH, D2O, exchangeable), 8.41 (s, 1H, aromatic H, C2), 8.15–8.04 (d, 2H, aromatic H, C4, 6), 7.65–7.60 (t, 1H, aromatic H, C5), 4.54, 4.50 (q,2H, CHNH), 3.66 (s, 6H, OCH3), 1.42 (d, 6H, 2CH3, CHCH3). 13C-NMR (125 MHz, δ, ppm, DMSO-d6): 173.57 (2C, CO, ester), 166.33 (2C, CONH), 134.38 (2C, isophthaloyl, C1, 5), 130.77 (2C, isophthaloyl, C2, 4), 128.81 (1C, isophthaloyl, C3), 127.27 (1C, isophthaloyl, C6), 52.35 (2C, OCH3, L-ala), 48.85 (2C, CHCH3, L-ala), 17.15 (2C, CHCH3, L-ala). MS (70-eV;-EI):-m/z-(%) =337 (M+-+1, 14.75%), 336 (M+, 1.2%), 277 (87.52%), 234 (100%), 50 (7.13%). Molecular formula (M.wt.), C16H20N2O6 – (336.3), calculated-analysis; C, 57.14; H, 5.99; N, 8.33, found; –C 57.00, –H 6.02, –N 8.30.
Nα-1, 4-Benzenedicarbonyl-Bis-[L-Met-OMe], (4)
Yield: [A]: 85; [B]: 70; m.p. 130–132 °C. Rf x100 (solvent system) 65 (S2). [α]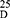 : - 24.21 (C, 0.02, DMSO). IR in (cm−1; KBr): 3421 (NH-stretching), 3015 (CH-aromatic), 2959 (CH-aliphatic), 1725 (C=O-ester), 1608 and 1523 (C=O-amideI+II). 1H-NMR (500 MHz, DMSO-d6) δ: 8.46 (s, 2H, CONH, D2O, exchangeable), 8.20–7.69 (4H, aromatic H), 3.90 (q 2H, CHNH), 3.34 (s, 6H, OCH3), 2.51 (t, 8H, 4CH2, CH2CH2), 2.18 (s, 6H, SCH3). 13C-NMR (125 MHz, δ, ppm, DMSO-d6): 165.84 (2C, CO, ester), 162.00 (2C, CONH), 134.04 (2C, isophthaloyl, C1, 5), 130.61–130.01 (3C, isophthaloyl, C2, 3, 4, 6), 52.90–52.00 (4C, 2OCH3, 2CHCH2, L-met), 40.19–39.35 (4C, 2CHCH2CH2, L-met), 17.00 (2C, SCH3, L-met). MS (70-eV;-EI):-m/z-(%) =457 (M+-+1, 52.94%), 456 (M+, 63.73%), 442 (100%), 272 (90.20%), 233 (55.88%), 51 (66.67%). Molecular formula (M.wt.), C20H28N2O6S2 (456.6), calculated analysis; C, 52.61; H, 6.18; N, 6.14; S, 14.05, found; C 52.56, H 6.10, N 6.09; S, 14.00.
: - 24.21 (C, 0.02, DMSO). IR in (cm−1; KBr): 3421 (NH-stretching), 3015 (CH-aromatic), 2959 (CH-aliphatic), 1725 (C=O-ester), 1608 and 1523 (C=O-amideI+II). 1H-NMR (500 MHz, DMSO-d6) δ: 8.46 (s, 2H, CONH, D2O, exchangeable), 8.20–7.69 (4H, aromatic H), 3.90 (q 2H, CHNH), 3.34 (s, 6H, OCH3), 2.51 (t, 8H, 4CH2, CH2CH2), 2.18 (s, 6H, SCH3). 13C-NMR (125 MHz, δ, ppm, DMSO-d6): 165.84 (2C, CO, ester), 162.00 (2C, CONH), 134.04 (2C, isophthaloyl, C1, 5), 130.61–130.01 (3C, isophthaloyl, C2, 3, 4, 6), 52.90–52.00 (4C, 2OCH3, 2CHCH2, L-met), 40.19–39.35 (4C, 2CHCH2CH2, L-met), 17.00 (2C, SCH3, L-met). MS (70-eV;-EI):-m/z-(%) =457 (M+-+1, 52.94%), 456 (M+, 63.73%), 442 (100%), 272 (90.20%), 233 (55.88%), 51 (66.67%). Molecular formula (M.wt.), C20H28N2O6S2 (456.6), calculated analysis; C, 52.61; H, 6.18; N, 6.14; S, 14.05, found; C 52.56, H 6.10, N 6.09; S, 14.00.
Synthesis of Nα-1, 4-Benzenedicarbonyl-Bis-[Amino Acid]
To a stirred solution (−5°C, 20 mL) containing methyl esters of methanol (3, 4) (2 g, 5.94 mmol), sodium hydroxide (NaOH) (1 N, 35 mL) was added dropwise and then stirred for 3 hours at −5°C, followed by stirring at room temperature for an additional 24 hours. The volatile materials were evaporated under reduced pressure and the aqueous solution was acidified (−5°C, 1 N hydrochloric acid [HCL], pH=3). The residual material was obtained and then recrystallized in an ethanol/water solution to obtain the acids 5, 6.
Nα-1, 4-Benzenedicarbonyl-Bis-[L-Ala], (5)
Yield: 75; m.p. 184–186 0C. Rf x100 (solvent system) 44 (S1). [α]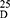 : - 55.15 (C, 0.06, DMSO). IR in (cm−1; KBr): 3318 (NH-stretching), 3131 (CH, aromatic), 2997 (CH, aliphatic), 1715 (C=O, acid), 1643 and 1530 (C=O, amide I and amide II). 1H-NMR (500 MHz, DMSO-d6) δ: 8.93, 8.91 (s, 2H, OH), 8.82, 8.80 (s, 2H, CONH, D2O exchangeable), 8.38 (s, 1H, aromatic H, C2), 8.18–8.02 (d, 2H, aromatic H, C4, 6), 7. 66–7.57 (t, 1H, aromatic H, C5), 4.48, 4.44 (q, 2H, CHNH), 3.66 (s, 6H, OCH3, disappeared), 1.43, 1.41 (d, 6H, 2CH3, CHCH3). 13C-NMR (125 MHz, δ, ppm, DMSO-d6): 174.62 (2C, CO, acid), 167.38, 167.10 (2C, CONH), 166.31, 165.94 (2C, isophthaloyl, C1, 5), 134.76, 134, 61 (2C, isophthaloyl, C2, 4), 133.85 (1C, isophthaloyl, C3), 130.64 (1C, isophthaloyl, C6), 52.35 (2C, OCH3, L-ala, disappeared), 48.41 (2C, CHCH3, L-ala), 18.25 (2C, CHCH3, L-ala). MS (EI, 70 eV): m/z (%) =309 (M++1, 7.38%), 308 (M+, 3.37%), 263 (23.82%), 76 (100%), 50 (41.95%). Molecular formula (M.wt.), C14H16N2O6 (308.3), calculated analysis; C, 54.54; H, 5.23; N, 9.09, found; C 54.51, H 5.20, N 9.02.
: - 55.15 (C, 0.06, DMSO). IR in (cm−1; KBr): 3318 (NH-stretching), 3131 (CH, aromatic), 2997 (CH, aliphatic), 1715 (C=O, acid), 1643 and 1530 (C=O, amide I and amide II). 1H-NMR (500 MHz, DMSO-d6) δ: 8.93, 8.91 (s, 2H, OH), 8.82, 8.80 (s, 2H, CONH, D2O exchangeable), 8.38 (s, 1H, aromatic H, C2), 8.18–8.02 (d, 2H, aromatic H, C4, 6), 7. 66–7.57 (t, 1H, aromatic H, C5), 4.48, 4.44 (q, 2H, CHNH), 3.66 (s, 6H, OCH3, disappeared), 1.43, 1.41 (d, 6H, 2CH3, CHCH3). 13C-NMR (125 MHz, δ, ppm, DMSO-d6): 174.62 (2C, CO, acid), 167.38, 167.10 (2C, CONH), 166.31, 165.94 (2C, isophthaloyl, C1, 5), 134.76, 134, 61 (2C, isophthaloyl, C2, 4), 133.85 (1C, isophthaloyl, C3), 130.64 (1C, isophthaloyl, C6), 52.35 (2C, OCH3, L-ala, disappeared), 48.41 (2C, CHCH3, L-ala), 18.25 (2C, CHCH3, L-ala). MS (EI, 70 eV): m/z (%) =309 (M++1, 7.38%), 308 (M+, 3.37%), 263 (23.82%), 76 (100%), 50 (41.95%). Molecular formula (M.wt.), C14H16N2O6 (308.3), calculated analysis; C, 54.54; H, 5.23; N, 9.09, found; C 54.51, H 5.20, N 9.02.
Nα-1, 4-Benzenedicarbonyl-Bis-[L-Met], (6)
Yield: 70; m.p. 210–212 0C. Rf x100 (solvent system) 50 (S1). [α]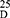 : - 38.75 (C, 0.05, DMSO). IR in (cm−1; KBr): 3435 (NH-stretching), 3086 (CH-arom.), 3006 (CH-aliphatic), 1687 (C=O, acid), 1612 and 1580 (C=O, amide I and amide II). 1H-NMR (500 MHz, DMSO-d6) δ: 13.00 (br, 2H, OH), 8.48 (s, 2H, CONH, D2O exchangeable), 8.15–7.60 (4H, aromatic H), 3.75 (q 2H, CHNH), 3.34 (s, 6H, OCH3, disappeared), 2.50 (t, 8H, 4CH2, CH2CH2), 2.16 (s, 6H, SCH3). 13C-NMR (125 MHz, δ, ppm, DMSO-d6): 167.03 (2C, CO, acid), 165.00 (2C, CONH), 133.81 (2C, isophthaloyl, C1, 5), 131.67 (2C, isophthaloyl, C2, 4), 130.43 (1C, isophthaloyl, C3), 129.55 (1C, isophthaloyl, C6), 52.50 (2C, 2CHCH2, L-met), 40.47–39.52 (4C, 2CHCH2CH2, L-met), 15.20 (2C, SCH3, L-met). MS (EI, 70 eV): m/z (%) = 429 (M++1, 0.80%), 428 (M+, 0.85%), 76 (19.17%), 65 (100%), 50 (48.76%). Molecular formula (M.wt.), C18H24N2O6S2 (428.5), calculated analysis; C, 50.45; H, 5.65; N, 6.54; S, 14.97, found; C 50.39, H 5.66, N 6.50; S, 14.95.
: - 38.75 (C, 0.05, DMSO). IR in (cm−1; KBr): 3435 (NH-stretching), 3086 (CH-arom.), 3006 (CH-aliphatic), 1687 (C=O, acid), 1612 and 1580 (C=O, amide I and amide II). 1H-NMR (500 MHz, DMSO-d6) δ: 13.00 (br, 2H, OH), 8.48 (s, 2H, CONH, D2O exchangeable), 8.15–7.60 (4H, aromatic H), 3.75 (q 2H, CHNH), 3.34 (s, 6H, OCH3, disappeared), 2.50 (t, 8H, 4CH2, CH2CH2), 2.16 (s, 6H, SCH3). 13C-NMR (125 MHz, δ, ppm, DMSO-d6): 167.03 (2C, CO, acid), 165.00 (2C, CONH), 133.81 (2C, isophthaloyl, C1, 5), 131.67 (2C, isophthaloyl, C2, 4), 130.43 (1C, isophthaloyl, C3), 129.55 (1C, isophthaloyl, C6), 52.50 (2C, 2CHCH2, L-met), 40.47–39.52 (4C, 2CHCH2CH2, L-met), 15.20 (2C, SCH3, L-met). MS (EI, 70 eV): m/z (%) = 429 (M++1, 0.80%), 428 (M+, 0.85%), 76 (19.17%), 65 (100%), 50 (48.76%). Molecular formula (M.wt.), C18H24N2O6S2 (428.5), calculated analysis; C, 50.45; H, 5.65; N, 6.54; S, 14.97, found; C 50.39, H 5.66, N 6.50; S, 14.95.
Synthesis of Nα-1, 3-Benzenedicarbonyl-Bis-[Amino Acid Hydrazides]
To a stirred solution (50 mL methanol) of esters (3 and 4; 1 mmol), hydrazine hydrate (0.35 mL, 10 mmol) was added. The mixture was refluxed for 3 hours; and the volatile materials were evaporated under reduced pressure. The obtained residue was triturated with ether and recrystallized from mixture of methanol/ether solution to produce the hydrazides (7 and 8).
Nα-1, 4-Benzenedicarbonyl-Bis-[L-Ala-NHNH2], (7)
Yield: 65; m.p. 269–271 0C. Rf x100 (solvent system) 35 (S). [α]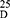 : - 112.00 (C, 0.02, DMSO). IR in (cm−1; KBr): 3298 (NH-stretching), 3100 (CH-arom), 2900 (CH-aliphatic.), 1643 (C=O, hydrazide), 1610 and 1531 (C=O- amide I+ II). 1H-NMR (500 MHz, DMSO-d6) δ: 8.75 (s, 2H, CONHNH2, D2O exchangeable), 8.40, 8.34 (s, 2H, CONHCH, D2O exchangeable), 8.02–7.52 (s, 4H, aromatic H),4.49–4.47 (q, 2H, CHNH), 4.19 (s, 4H, CONHNH2), 3.66 (s, 6H, OCH3, disappeared), 1.35, 1.33 (d, 6H, 2CH3, CHCH3). 13C-NMR (125 MHz, δ, ppm, DMSO-d6): 172.27 (2C, CO, hydrazide), 166.27 (2C, CONH), 165.00 (2C, isophthaloyl, C1, 5), 134.51 (2C, isophthaloyl, C2, 4),130.78 (1C, isophthaloyl, C3),128.67 (1C, isophthaloyl, C6), 52.35 (2C, OCH3, L-ala, disappeared), 48.74 (2C, CHCH3, L-ala), 17.33, 17.25 (2C, CHCH3, L-ala). MS (EI, 70 eV): m/z (%) =337 (M++1, 1.65%), 336 (M+, 0.76%), 273 (100%), 76 (29.37%), 50 (7.46%). Molecular formula (M.wt.), C16H20N2O6 (336.3), calculated analysis; C, 57.14; H, 5.99; N, 8.33, found; C 57.11, H 5.90, N 8.31.
: - 112.00 (C, 0.02, DMSO). IR in (cm−1; KBr): 3298 (NH-stretching), 3100 (CH-arom), 2900 (CH-aliphatic.), 1643 (C=O, hydrazide), 1610 and 1531 (C=O- amide I+ II). 1H-NMR (500 MHz, DMSO-d6) δ: 8.75 (s, 2H, CONHNH2, D2O exchangeable), 8.40, 8.34 (s, 2H, CONHCH, D2O exchangeable), 8.02–7.52 (s, 4H, aromatic H),4.49–4.47 (q, 2H, CHNH), 4.19 (s, 4H, CONHNH2), 3.66 (s, 6H, OCH3, disappeared), 1.35, 1.33 (d, 6H, 2CH3, CHCH3). 13C-NMR (125 MHz, δ, ppm, DMSO-d6): 172.27 (2C, CO, hydrazide), 166.27 (2C, CONH), 165.00 (2C, isophthaloyl, C1, 5), 134.51 (2C, isophthaloyl, C2, 4),130.78 (1C, isophthaloyl, C3),128.67 (1C, isophthaloyl, C6), 52.35 (2C, OCH3, L-ala, disappeared), 48.74 (2C, CHCH3, L-ala), 17.33, 17.25 (2C, CHCH3, L-ala). MS (EI, 70 eV): m/z (%) =337 (M++1, 1.65%), 336 (M+, 0.76%), 273 (100%), 76 (29.37%), 50 (7.46%). Molecular formula (M.wt.), C16H20N2O6 (336.3), calculated analysis; C, 57.14; H, 5.99; N, 8.33, found; C 57.11, H 5.90, N 8.31.
Nα-1, 4-Benzenedicarbonyl-Bis-[L-Met-NHNH2], (8)
Yield: 70; m.p. 292–294 0C. Rf x100 (solvent system) 40 (S). [α]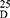 : - 86.25 (C, 0.03, DMSO). IR in (cm−1; KBr): 3288 (NH-stretching), 3155 (CH-aromatic), 3053 (CH-Aliphatic), 1661 (C=O, hydrazide), 1625 and 1525 (C=O, amide I and amide II). 1H-NMR (500 MHz, DMSO-d6)δ: 9.85 (br, 2H, CONHNH2, D2O exchangeable), 8.28 (s, 2H, CONH, D2O exchangeable), 7.94–7.51 (4H, aromatic H), 4.00 (br, 2H, CONHNH2), 3.17 (q, 2H, CHNH), 3.34 (s, 6H, OCH3, disappeared), 2.51 (t, 8H, 4CH2, CH2CH2), 1.75 (s, 6H, SCH3). 13C-NMR (125 MHz, δ, ppm, DMSO-d6): 165.91 (2C, CO, hydrazide), 160.00 (2C, CONH), 133.55 (2C, isophthaloyl, C1, 5), 129.88 (2C, isophthaloyl, C2, 4), 128.97 (1C, isophthaloyl, C3), 126.45 (1C, isophthaloyl, C6), 50.45 (2C, 2CHCH2, L-met), 40.18–39.34 (4C, 2CHCH2CH2, L-met), 16.25 (2C, SCH3, L-met). MS (EI, 70 eV): m/z (%) =456 (M+, 3.42%), 265 (5%), 76 (100%), 64 (10.92%), 50 (81.49%). Molecular formula (M.wt.), C20H28N2O6S2 (456.6), calculated analysis; C, 52.61; H, 6.18; N, 6.14; S, 14.05, found; C– 52.55, H– 6.15, N- 6.11; S– 14.00.
: - 86.25 (C, 0.03, DMSO). IR in (cm−1; KBr): 3288 (NH-stretching), 3155 (CH-aromatic), 3053 (CH-Aliphatic), 1661 (C=O, hydrazide), 1625 and 1525 (C=O, amide I and amide II). 1H-NMR (500 MHz, DMSO-d6)δ: 9.85 (br, 2H, CONHNH2, D2O exchangeable), 8.28 (s, 2H, CONH, D2O exchangeable), 7.94–7.51 (4H, aromatic H), 4.00 (br, 2H, CONHNH2), 3.17 (q, 2H, CHNH), 3.34 (s, 6H, OCH3, disappeared), 2.51 (t, 8H, 4CH2, CH2CH2), 1.75 (s, 6H, SCH3). 13C-NMR (125 MHz, δ, ppm, DMSO-d6): 165.91 (2C, CO, hydrazide), 160.00 (2C, CONH), 133.55 (2C, isophthaloyl, C1, 5), 129.88 (2C, isophthaloyl, C2, 4), 128.97 (1C, isophthaloyl, C3), 126.45 (1C, isophthaloyl, C6), 50.45 (2C, 2CHCH2, L-met), 40.18–39.34 (4C, 2CHCH2CH2, L-met), 16.25 (2C, SCH3, L-met). MS (EI, 70 eV): m/z (%) =456 (M+, 3.42%), 265 (5%), 76 (100%), 64 (10.92%), 50 (81.49%). Molecular formula (M.wt.), C20H28N2O6S2 (456.6), calculated analysis; C, 52.61; H, 6.18; N, 6.14; S, 14.05, found; C– 52.55, H– 6.15, N- 6.11; S– 14.00.
Synthesis of Nα-1, 3-Benzenedicarbonyl-Bis-[L-Ala-L-Phe Methyl Esters], (9)
Ethyl chloroformate (2.9 mL, 30.1 mmol) was added to solution of 1, 3-benzenedicarbonyl-bis-[L-Alanine], 5, (−20 °C, 15.05mmol) and triethylamine (3.3 mL, 30.1mmol) in dichloromethane (50 mL). The reaction mixture was stirred for additional 30 minutes, then the two equivalents amount of free amino acid ester (L-Phe) (30.1mmol)(−20°C) in dichloromethane (50mL) was added. Then, the same procedure, mentioned in the synthesis of compound 3 using the mixed anhydride method was completed. The crude product was collected, and then purified by recrystallization from ethanol to get the corresponding ester 9.
Yield: 85; m.p. 86–88 0C. Rf x100 (solvent system) 70 (S2); [α]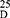 : - 22.10 (C, 0.02, MeOH). IR in (cm−1; KBr): 3422 (NH-stretching) 3140 (CH, aromatic) 2950 (CH, aliphatic) 1732 (C=O, ester) 1648, 1530, 1441 and 1382 (C=O, amide I, amide II, amide III and amide IV). 1H-NMR (500 MHz, DMSO-d6)δ: 9.09–9.01 (s, 4H, CONH, D2O exchangeable), 8.66 (s, 1H, isophthaloyl, aromatic H, C2), 8.48–8.40 (d, 2H, aromatic H, isophthaloyl, C4, 6), 7.65 (t, 1H, isophthaloyl, aromatic H, C5), 7.50–7.21 (10H, aromatic H, L-phe), 4.56–4.51 (q, 2H, CHNH, L-phe), 4.29–4.23 (t,2H, CHNH, L-ala), 3.64–3.59 (s, 6H, OCH3), 3.19–2.88 (d, 4H, CH2, L-phe), 1.41–1.03 (d, 6H, 2CH3, CHCH3, L-ala). 13C-NMR (125 MHz, δ, ppm, DMSO-d6): 173.00 (2C, CO, ester), 169.00–170.25 (4C, CONH), 135.50 (1C, L-Phe, C1), 133.20 (2C, isophthaloyl, C1, 5), 131.50 (2C, isophthaloyl, C2, 4), 128.80–124-25 (10C, the remaining aromatic carbon), 53.25 (2C, CHCH2, L-phe), 51.50 (2C, OCH3, L-ala), 50.00 (2C, CHCH3, L-ala), 38.20 (2C, CH2, L-phe), 18.00 (2C, CHCH3, L-ala). MS (EI, 70 eV): m/z (%) =631 (M++1, 18.77%), 630 (M+, 23.60%), 611 (100%), 74 (2.15%), 55 (1.33%). Molecular formula (M.wt.), C34H38N4O8 (630.7), calculated analysis; C, 64.75; H, 6.07; N, 8.88, found; C 64.70, H 6.04, N 8.90.
: - 22.10 (C, 0.02, MeOH). IR in (cm−1; KBr): 3422 (NH-stretching) 3140 (CH, aromatic) 2950 (CH, aliphatic) 1732 (C=O, ester) 1648, 1530, 1441 and 1382 (C=O, amide I, amide II, amide III and amide IV). 1H-NMR (500 MHz, DMSO-d6)δ: 9.09–9.01 (s, 4H, CONH, D2O exchangeable), 8.66 (s, 1H, isophthaloyl, aromatic H, C2), 8.48–8.40 (d, 2H, aromatic H, isophthaloyl, C4, 6), 7.65 (t, 1H, isophthaloyl, aromatic H, C5), 7.50–7.21 (10H, aromatic H, L-phe), 4.56–4.51 (q, 2H, CHNH, L-phe), 4.29–4.23 (t,2H, CHNH, L-ala), 3.64–3.59 (s, 6H, OCH3), 3.19–2.88 (d, 4H, CH2, L-phe), 1.41–1.03 (d, 6H, 2CH3, CHCH3, L-ala). 13C-NMR (125 MHz, δ, ppm, DMSO-d6): 173.00 (2C, CO, ester), 169.00–170.25 (4C, CONH), 135.50 (1C, L-Phe, C1), 133.20 (2C, isophthaloyl, C1, 5), 131.50 (2C, isophthaloyl, C2, 4), 128.80–124-25 (10C, the remaining aromatic carbon), 53.25 (2C, CHCH2, L-phe), 51.50 (2C, OCH3, L-ala), 50.00 (2C, CHCH3, L-ala), 38.20 (2C, CH2, L-phe), 18.00 (2C, CHCH3, L-ala). MS (EI, 70 eV): m/z (%) =631 (M++1, 18.77%), 630 (M+, 23.60%), 611 (100%), 74 (2.15%), 55 (1.33%). Molecular formula (M.wt.), C34H38N4O8 (630.7), calculated analysis; C, 64.75; H, 6.07; N, 8.88, found; C 64.70, H 6.04, N 8.90.
Synthesis of Nα-1, 3-Benzenedicarbonyl-Bis-[L-Ala-L-Phe], (10)
To a stirred solution (−5 °C, 20 mL methanol) of ester (9; 2 mmol), NaOH (1N, 25 mL) was added dropwise, then stirring was continuous for additional three 3 hours at −5 °C. The reaction mixture was left over night at room temperature. Then complete the same procedure which was mentioned in the synthesis of compounds 5 and 6. The precipitated crude product was filtered off and recrystallized from ethanol to get the corresponding acid 10.
Yield: 75; m.p. 169–171 0C. Rf x100 (solvent system) 60 (S1); [α]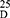 : −33.41 (C, 0.02, MeOH). IR in (cm−1; KBr): 3430 (NH-stretching) 3175 (CH, aromatic) 2928 (CH, aliphatic) 1643 (C=O, acid) 1529, 1480, 1421 and 1386 (C=O, amide I, amide II, amide III and amide IV). 1H-NMR (500 MHz, DMSO-d6)δ: 12.55 (br, 2H, OH), 9.19–9.09 (s, 4H, CONH, D2O exchangeable), 8.70 (s, 1H, isophthaloyl, aromatic H, C2), 8.40–8.33 (d, 2H, aromatic H, isophthaloyl, C4, 6), 7.77 (t, 1H, isophthaloyl, aromatic H, C5), 7.47–7.18 (10H, aromatic H, L-phe), 4.61–4.49 (q, 2H, CHNH, L-phe), 4.33–4.20 (t, 2H, CHNH, L-ala), 3.64–3.59 (s, 6H, OCH3, disappeared), 3.25–2.75 (d, 4H, CH2, L-phe), 1.55–1.25 (d, 6H, 2CH3, CHCH3, L-ala). 13C-NMR (125 MHz, δ, ppm, DMSO-d6): 174.50 (2C, CO, acid), 170.15–172.00 (4C, CONH), 139.20 (1C, L-Phe, C1), 136.00 (2C, isophthaloyl, C1, 5), 134.20 (2C, isophthaloyl, C2, 4), 130.50–125-00 (10C, the remaining aromatic carbon), 54.50 (2C, CHCH2, L-phe), 51.50 (2C, OCH3, L-ala, disappeared), 48.50 (2C, CHCH3, L-ala), 39.70 (2C, CH2, L-phe), 17.50 (2C, CHCH3, L-ala). MS (EI, 70 eV): m/z (%) =603 (M++1, 66.92%), 602 (M+, 79.23%), 484 (100%), 367 (72.31%), 159 (79.23%), 53 (64.62%). Molecular formula (M.wt.), C32H34N4O8 (602.6), calculated analysis; C, 63.78; H, 5.69; N, 9.30, found; C 63.70, H 5.70, N 9.32.
: −33.41 (C, 0.02, MeOH). IR in (cm−1; KBr): 3430 (NH-stretching) 3175 (CH, aromatic) 2928 (CH, aliphatic) 1643 (C=O, acid) 1529, 1480, 1421 and 1386 (C=O, amide I, amide II, amide III and amide IV). 1H-NMR (500 MHz, DMSO-d6)δ: 12.55 (br, 2H, OH), 9.19–9.09 (s, 4H, CONH, D2O exchangeable), 8.70 (s, 1H, isophthaloyl, aromatic H, C2), 8.40–8.33 (d, 2H, aromatic H, isophthaloyl, C4, 6), 7.77 (t, 1H, isophthaloyl, aromatic H, C5), 7.47–7.18 (10H, aromatic H, L-phe), 4.61–4.49 (q, 2H, CHNH, L-phe), 4.33–4.20 (t, 2H, CHNH, L-ala), 3.64–3.59 (s, 6H, OCH3, disappeared), 3.25–2.75 (d, 4H, CH2, L-phe), 1.55–1.25 (d, 6H, 2CH3, CHCH3, L-ala). 13C-NMR (125 MHz, δ, ppm, DMSO-d6): 174.50 (2C, CO, acid), 170.15–172.00 (4C, CONH), 139.20 (1C, L-Phe, C1), 136.00 (2C, isophthaloyl, C1, 5), 134.20 (2C, isophthaloyl, C2, 4), 130.50–125-00 (10C, the remaining aromatic carbon), 54.50 (2C, CHCH2, L-phe), 51.50 (2C, OCH3, L-ala, disappeared), 48.50 (2C, CHCH3, L-ala), 39.70 (2C, CH2, L-phe), 17.50 (2C, CHCH3, L-ala). MS (EI, 70 eV): m/z (%) =603 (M++1, 66.92%), 602 (M+, 79.23%), 484 (100%), 367 (72.31%), 159 (79.23%), 53 (64.62%). Molecular formula (M.wt.), C32H34N4O8 (602.6), calculated analysis; C, 63.78; H, 5.69; N, 9.30, found; C 63.70, H 5.70, N 9.32.
Synthesis of Nα-1, 3-Benzenedicarbonyl-Bis-[L-Ala-L-Phe-Hydrazide], (11)
The ester (9; 1 mmol) in 50 mL methanol was stirred with hydrazine hydrate (0.35 mL, 10 mmol) for 3 hours; the volatile materials were evaporated under reduced pressure. The residual product diluted with ether and recrystallized by mixture of methanol/ether to afford the corresponding hydrazide 11.
Yield: 65; m.p. 235–237 0C. Rf x100 (solvent system) 45 (S); [α] : −133.80 (C, 0.04, DMSO). IR in (cm−1; KBr): 3424 (NH-stretching), 3273 (CH, aromatic) 3066 (CH, aliphatic) 1647 (C=O, hydrazide) 1538, 1449, 1400 and 1382 (C=O, amide I, amide II, amide III and amide IV). 1H-NMR (500 MHz, DMSO-d6)δ: 9.92 (br, 2H, CONHNH2, 9.23–9.15 (s, 4H, CONH, D2O exchangeable), 8.82 (s, 1H, isophthaloyl, aromatic H, C2), 8.35–8.24 (d, 2H, aromatic H, isophthaloyl, C4, 6), 7.86 (t, 1H, isophthaloyl, aromatic H, C5), 7.62–7.38 (10H, aromatic H, L-phe), 4.74–4.60 (q, 2H, CHNH, L-phe), 4.44–4.30 (t, 2H, CHNH, L-ala), 4.00 (br, 2H, CONHNH2), 3.64–3.59 (s, 6H, OCH3, disappeared), 3.40–2.80 (d, 4H, CH2, L-phe), 1.90–1.60 (d, 6H, 2CH3, CHCH3, L-ala). 13C-NMR (125 MHz, δ, ppm, DMSO-d6): 176.60 (2C, CO, hydrazide), 171.25–172.50 (4C, CONH), 138.00 (1C, L-Phe, C1), 135.20 (2C, isophthaloyl, C1, 5), 133.00 (2C, isophthaloyl, C2, 4), 128.00–123-50 (10C, the remaining aromatic carbon), 55.75 (2C, CHCH2, L-phe), 51.50 (2C, OCH3, L-ala, disappeared), 46.00 (2C, CHCH3, L-ala), 36.00 (2C, CH2, L-phe), 16.20 (2C, CHCH3, L-ala). MS (EI, 70 eV): m/z (%) =631 (M++1, 41.67%), 630 (M+, 69.70%), 403 (74.24%), 91 (100%), 71 (47.73%), 52 (92.42%). Molecular formula (M.wt.), C32H38N8O6 (630.7), calculated analysis; C, 60.94; H, 6.07; N, 17.77, found; C 60.92, H 6.02, N 17.72.
: −133.80 (C, 0.04, DMSO). IR in (cm−1; KBr): 3424 (NH-stretching), 3273 (CH, aromatic) 3066 (CH, aliphatic) 1647 (C=O, hydrazide) 1538, 1449, 1400 and 1382 (C=O, amide I, amide II, amide III and amide IV). 1H-NMR (500 MHz, DMSO-d6)δ: 9.92 (br, 2H, CONHNH2, 9.23–9.15 (s, 4H, CONH, D2O exchangeable), 8.82 (s, 1H, isophthaloyl, aromatic H, C2), 8.35–8.24 (d, 2H, aromatic H, isophthaloyl, C4, 6), 7.86 (t, 1H, isophthaloyl, aromatic H, C5), 7.62–7.38 (10H, aromatic H, L-phe), 4.74–4.60 (q, 2H, CHNH, L-phe), 4.44–4.30 (t, 2H, CHNH, L-ala), 4.00 (br, 2H, CONHNH2), 3.64–3.59 (s, 6H, OCH3, disappeared), 3.40–2.80 (d, 4H, CH2, L-phe), 1.90–1.60 (d, 6H, 2CH3, CHCH3, L-ala). 13C-NMR (125 MHz, δ, ppm, DMSO-d6): 176.60 (2C, CO, hydrazide), 171.25–172.50 (4C, CONH), 138.00 (1C, L-Phe, C1), 135.20 (2C, isophthaloyl, C1, 5), 133.00 (2C, isophthaloyl, C2, 4), 128.00–123-50 (10C, the remaining aromatic carbon), 55.75 (2C, CHCH2, L-phe), 51.50 (2C, OCH3, L-ala, disappeared), 46.00 (2C, CHCH3, L-ala), 36.00 (2C, CH2, L-phe), 16.20 (2C, CHCH3, L-ala). MS (EI, 70 eV): m/z (%) =631 (M++1, 41.67%), 630 (M+, 69.70%), 403 (74.24%), 91 (100%), 71 (47.73%), 52 (92.42%). Molecular formula (M.wt.), C32H38N8O6 (630.7), calculated analysis; C, 60.94; H, 6.07; N, 17.77, found; C 60.92, H 6.02, N 17.72.
Biological Evaluation
In vitro Antimicrobial Activity
Antimicrobial efficiency for the investigated compounds (3–11) was evaluated using “well diffusion method” against pathogenic Gram-positive bacteria including Streptococcus pneumonia, Staphylococcus aureus ATCC25923, and Micrococcus luteus. Subsequentially, these compounds were tested against Gram-negative bacteria “Escherichia coli (ATCC25922), Pseudomonas aeruginosa (ATCC7853) and Proteus mirabilis, and Candida albicans (ATCC1031)” as the human yeast pathogen. While antifungal activity was tested against different plant pathogens (Rhizocotonia solani, Fusarium oxysporum, and Fusarium solani) and human pathogen Aspergillus niger in terms of the percent inhibition of mycelial growth. All tested bacteria and yeast were incubated at 37°C for 24 hours by inoculation into nutrient broth. The culture suspensions were prepared and adjusted at absorbance 0.5 by spectrophotometer.
Well Diffusion Method
Petri dishes with nutrient agar were inoculated with a 100 μL suspension of each bacterial or yeast culture. Wells were generated using the agar surface around (5 mm) below the cork-borer. The tested compounds (100 µL) were poured into the well using a sterile syringe. The bacteria seeded plate was placed in the refrigerator for 8 hours at 4°C. Next, the plates were incubated at 37±2°C for 24 hours. The inhibition zone formed around the well in the plates was measured as the diameter of the inhibition zone around the well (in mm) through the well diameter. DMSO was used to dissolve the investigated compounds (3 to 11; 1 mg/mL) as well as for the dilution of the compounds to be tested.23 The average values for the three different trials (measurements were taken in three different fixed directions) are tabulated in Table 1.
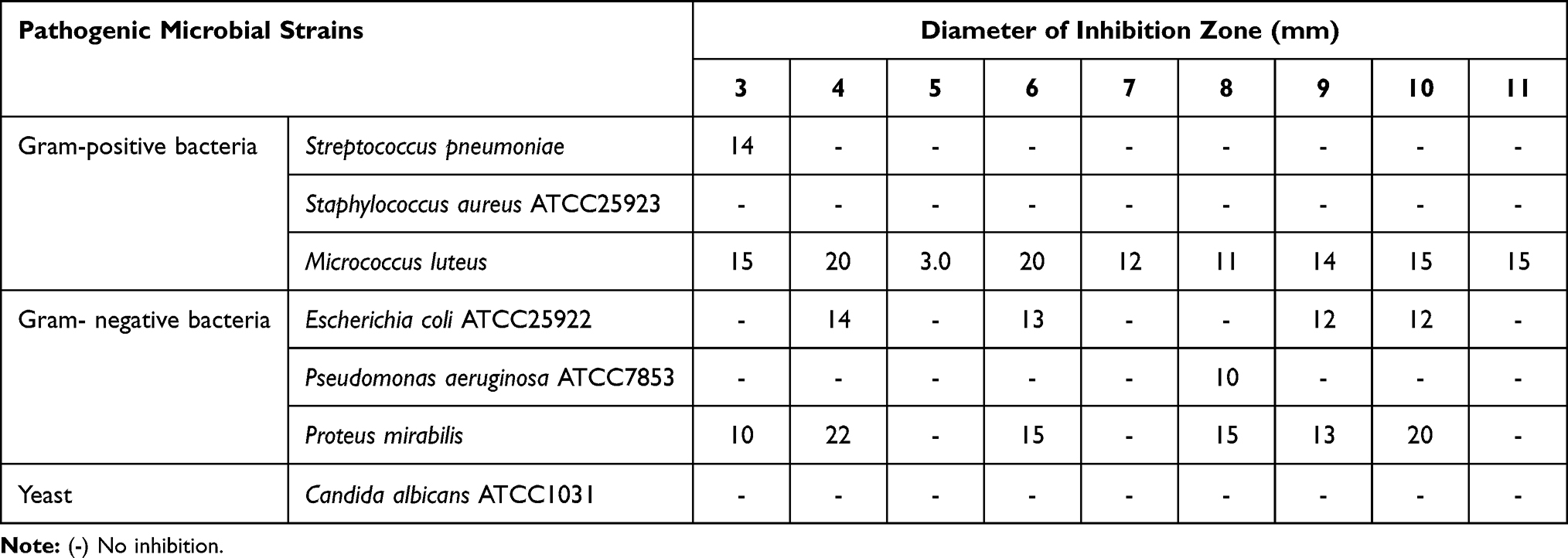 |
Table 1 Antimicrobial Activity of the Synthesized Compounds by Well Diffusion Assay Method |
Antifungal Activity
The antifungal potency for compounds 3 to 11 was evaluated as the inhibition percent for expansion mycelial(IPEM).29 The test compounds 3 to 11(100 μL) were added to potato dextrose agar medium (PDA) plates at a concentration of 1 mg/mL. Agar discs (5 mm) were taken from 7 day-old cultures of 4 pathogenic fungi (Aspergillus niger, Rhizoctonia solani, Fusarium oxysporum, and Fusarium solani) and were placed separately in the center of the Petri plates. For the control condition, agar discs of the same size as the tested pathogen were positioned similarly on a fresh PDA plate. The incubation period for all paired cultures was performed at 25°C for 6 days. Inhibitory efficiency was measured by determining the expansion radius of the mycelium on the treated media (R2) versus the control (R1). The IPEM were obtained using the following formula: IPEM= {(R1 − R2)/R1} × 100.
Antioxidant Activity
Scavenging efficiency for free radicals was measured as the change the color of DPPH• radical from strong violet to yellow color as previously reported.30
Anticancer Activity
Human cancer cell lines used in this study, included MCF7 (Breast carcinoma cell line), Hep-G2 (Liver carcinoma cell line), and HCT116 (Colon carcinoma cell line). They were obtained frozen in liquid nitrogen (−180°C) from Vacsera (Giza, Egypt). The tumor cell lines were maintained by serial sub-culturing. Cytotoxicity was determined using the MTT assay.
Cell Viability Assay
For the assessment of the cytotoxic activities of compounds 3 to 11, cells were seeded in 96-well flat-bottomed microtiter plates at a density of approximately (10x103/well for MCF-7 and HepG2, while in HCT-116 20x103 cells/well), in complete RPMI-1640Medium. Cells were incubated in a water jacketed CO2 incubator at 37°C for 24 hours. The remaining procedures, including the MTT assays, were performed as previously reported.31,32
BCL-2 Measurement
Based on previous reports,32 the effects on BCL-2 levels for the compounds under examination and the standards were tested. A contaminated biotin with antibody was added and was then streptavidin HRP. The termination of the reaction takes place by addition of acid, and the absorbance was determined at 450 nm.
LC50 Determination
LC50 values were calculated using SPSS Statistics Base v.9 for the active extracts which showed promising values in the analysis.
Enzyme-Linked Immunosorbent Assay for Tubulin Beta (TUBb)
A total of 1.2–1.8×10,000 cells/well of MCF-7 cells were seeded in DMEM (supplemented with 10% FBS and 1% penicillin-streptomycin). Before assessment of tubulin levels, the compound under investigation was added for 18–24 hours. Next, Avidin combined with HRP was added to each microplate well and incubated. The remaining steps were performed as previously reported.33
Human P53 Estimation
ELISA was used to measure the amount of human p53 bound to antibodies in the sample or to standards adsorbed onto the microwells. A solution containing biotin was added. The contaminated biotin with antibodies was supplemented. Then, Human p53 was measured as previously reported.34
Computational Study
Preparation of Ligands and Protein
The 3-dimensional structure protein files were downloaded from the RCSB protein data bank for DNA Gyrase B (ID: 4uro),35 14α-sterol demethylase Candida albicans (ID:1EA1),3 and transforming growth factor beta receptor type I (TGFBR1) (ID: 1VJY).37 For mining of protein data, we used the BLAST P tool for analysis of the FASTA protein sequence. Then, the CLUSTALW46 package was utilized for the alignment of multiple of amino acid sequences.38–44
The Docking computations were completed using the MOE 2015 package.45 Compounds 3 to 11 were built then energy minimized based on “Semi-empirical” PM3 then DFT ”discrete Fourier transform” based on B3LYP/6-311G. The error correction for the structure of the catalytic sites was performed by supplementing hydrogens and partial charges using (Amber12: EHT), then minimized by utilized the same force field with RMSD= 0.100. The catalytic site was identified and analyzed using the Site Finder program. This method is based on alpha spheres as well as an energy model.46 The catalytic zone was predicted by the MOE-Site finder.47
Stepwise Docking Experiment
Water and inhibiting molecules were eliminated, then H-atoms were supplemented to the obtained crystal structure. The charges were designed using MMFF94x force field. The alpha-site spheres were added based on the findings of the on-site-finder module. The final energy, generated structures, and final scores were measured using procedures described previously.48
ADMET Predictions
The in silico ADMET profile was simulated using MOE and admetSAR Lipinski criteria and ADMET properties (absorption, distribution, metabolism, excretion, and toxicity).49
Results and Discussion
Chemistry
The Nα-1, 3-benzenedicarbonyl-bis-(L-Alanine and L- Methionine methyl esters (3, 4, respectively), were obtained using two synthesis routes. The first involved acylation of L-alanine and L-methionine methyl esters with 1, 3-benzenedicarbonyl dichloride 2, which was obtained from the reaction of 1, 3-benzenedicarboxlaic acid 1 with thionyl chloride. Compound 2 was then coupled with the L-alanine and L-methionine esters at a low temperature in the presence of trimethylamine. In the second synthesis route: the bis-esters (3, 4) were prepared starting from Nα-1,3-benzenedicarboxlaic acid 1, with L-alanine and L-methionine esters, in the presence of ethyl chloroformate, as a mixed anhydride partner. Hydrolysis of (3, 4) with methanolic NaOH resulted in the corresponding Nα-1,3-benzenedicarbonyl-bis-(L-alanine and L-methionine; 5, 6). The hydrazinolysis of 3, 4 with alcoholic hydrazine hydrate, generated the corresponding Nα-1,3-benzenedicarbonyl-bis-(L-alanine and L-methionine; 7, 8) (Scheme 1).
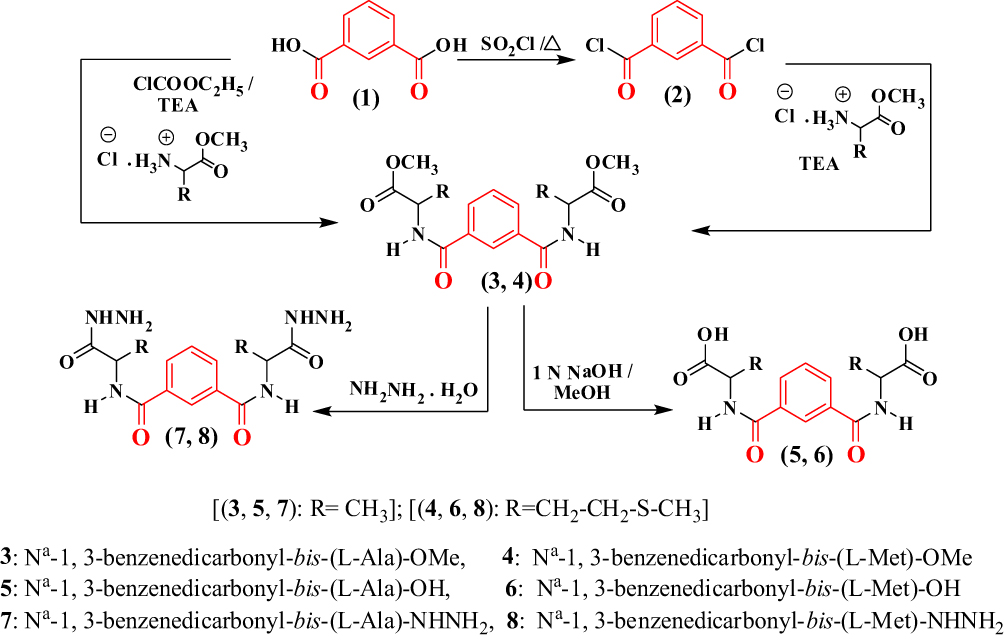 |
Scheme 1 |
Treatment of 5 with free L-phenyl alanine ester generated the corresponding Nα-1,3-benzenedicarbonyl-bis-(dipeptide)-ester (9). This compound was converted to the corresponding Nα-1,3-benzenedicarbonyl-bis-(dipeptide; 10) under hydrolysis conditions by ethanolic sodium hydroxide. Likewise, hydrazinolysis of the dipeptide methyl ester (10) with methanolic hydrazine hydrate generated the corresponding dipeptidehydrazide (11) (Scheme 2).
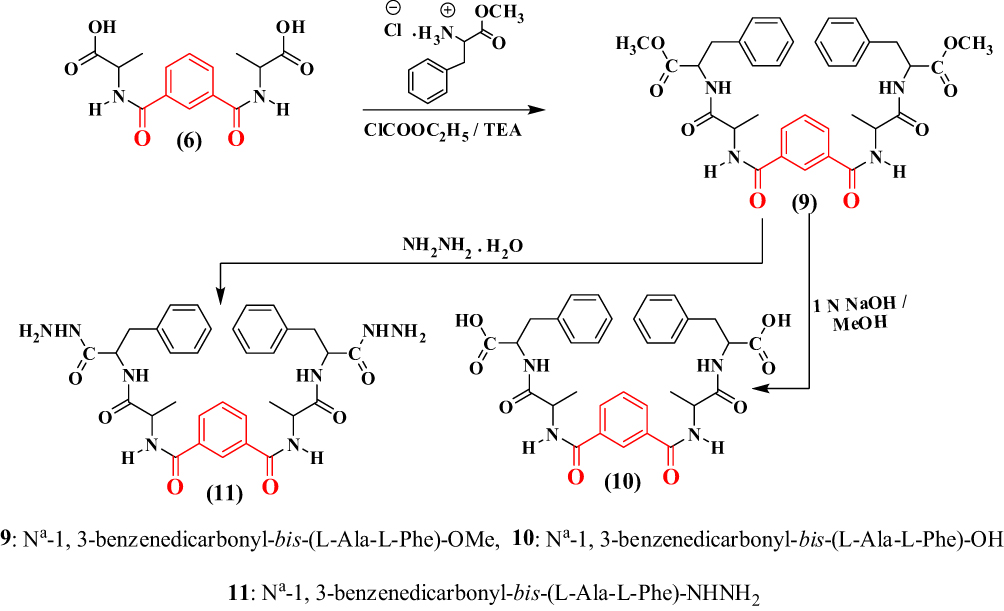 |
Scheme 2 |
Biological Evaluations
Antimicrobial Activity
The antimicrobial activity of the synthesized compounds 3 to 11 against pathogenic bacterial and fungal strains is shown in Tables 1 and 2. The tested compounds showed different degrees of potency against pathogenic bacteria. Micrococcus luteus was the most sensitive bacteria against compound 6, which showed the highest activity. Compounds 5, 7 produced the lowest inhibitory activity and showed resistance towards all tested pathogenic microorganisms tested with the exception of Micrococcus luteus.
 |
Table 2 Antifungal Activity of the Synthesized Compounds by the Percent Inhibition of Mycelial Growth |
None of the tested compounds exhibited any activity against Staphylococcus aureus and yeast. The pathogen Proteus sp. was inhibited by most synthesized peptides except for compounds 6 and 11, while Streptococcus pneumonia pathogen was only resistant to compound 3, among all members of all synthesized compounds.
Most synthesized compounds showed the most significant antifungal activity against Aspergillus niger, except for compounds 4, 7, and 9. Exposure to compounds 3, 5, 6, and 10 changed the black color of Aspergillus niger spores to grey. Compound 8 was the most potent antifungal agent against Aspergillus niger, which produced 100% inhibition, while compound 11 inhibited the formation of the respective fungal spores. The inhibitory mechanism of these compounds may be attributed to a fungicidal inhibition of the THN-reductase enzyme, involved in melanin synthesis via the DHN-melanin pathway.50 Compounds 4, 9, and 11 showed a moderate inhibition against Rhizocotonia solani, while compounds 5 and 9 showed a relatively low degree of inhibition (11%) against Fusarium oxysporum. Most synthesized compounds (ie, 3, 4, 6, 7, 8, and 10) showed no antifungal activity against Fusarium solani.
Antioxidant Activity
Nine compounds were screened to investigate their antioxidant potency using the free radical scavenging activity DPPH assay, and all nine compounds showed weak free radical scavenging activity (Table 3).
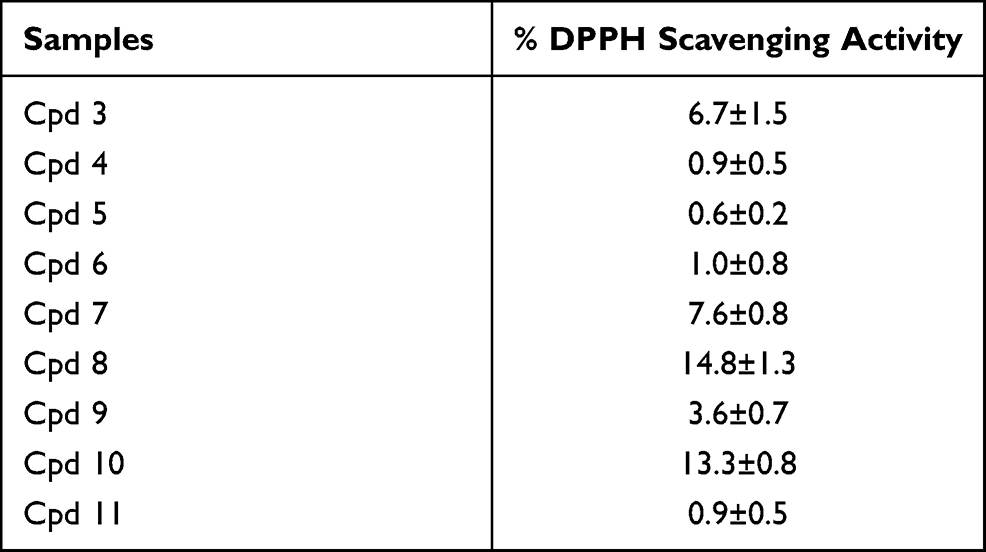 |
Table 3 Free Radical Scavenging Activity of the Synthesized Compounds on Free Radical DPPH at 50ug/mL |
Anticancer Activity
Nine samples (3 to 11) were preliminary screening for their antiproliferative activity against the following human carcinoma cell lines, breast MCF-7 (Figure 1), colon HCT-116 (Figure 2), and hepatocellular carcinoma HepG2 (Figure 3) cell lines. The nine compounds possessed weak to moderate antiproliferative effects in the HCT-116 cell line at concentration 10 ppm or 10µg/mL, with cytotoxicity ranging from 14% to 60.7%. While compound 9 possessed weak cytotoxic activity in the liver human tumor cell line HepG2, with cytotoxicity ranging from 2.2% to 9%. All the cytotoxicity results of the studied compounds were compared with the results of the doxorubicin reference drug, which ranged from 80% to 95% cytotoxicity for all three cell lines tested.
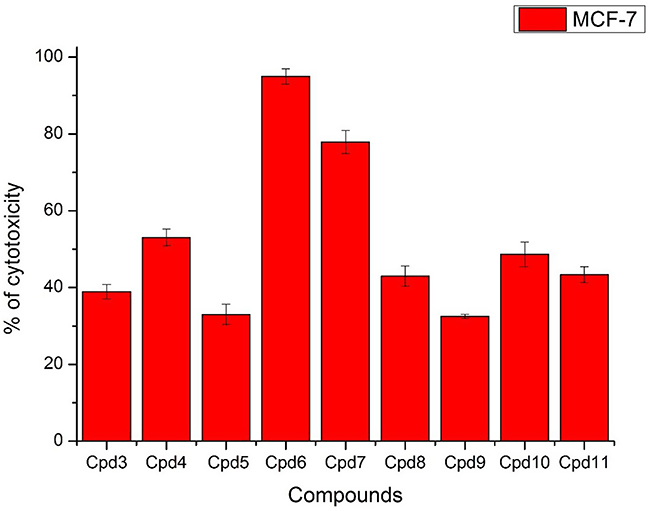 |
Figure 1 In vitro cytotoxicity screening for nine tested compounds against colon carcinoma (MCF-7). |
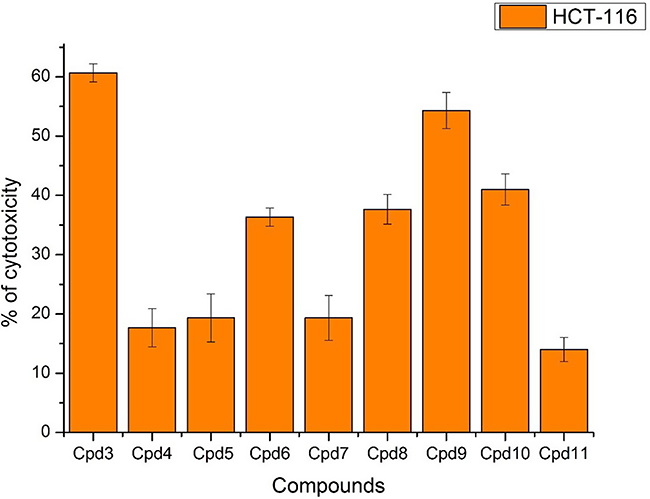 |
Figure 2 In vitro cytotoxicity screening for nine tested compounds via against breast carcinoma (HCT-116). |
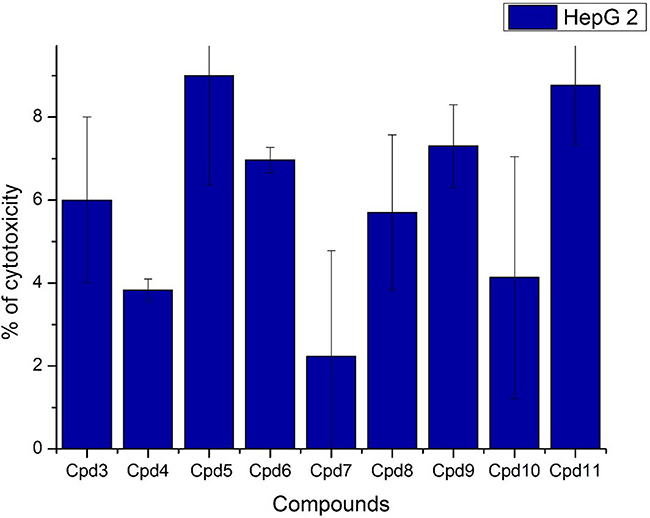 |
Figure 3 In vitro cytotoxicity screening for nine tested compounds via liver carcinoma (HepG 2). |
Conversely, compounds 6 and 7, showed a strong cytotoxic effect on the breast cancer cell line MCF-7 with 95.1% and 79.8% of cell inhibition, respectively. Based on the promising results of compound 6 in the breast cancer cell line, it was selected for further investigation of additional anticancer activity.
The proto-oncogene coding the B-cell lymphoma 2 (Bcl-2) is known as apoptosis regulator by maintaining the equilibrium between cell death and proliferation. Low levels of Bcl-2 are associated with cell death through apoptosis.
MCF-7 cells treated with compound 6 produced BCL2 levels of 103.3 pg/mL, which lies near the borderline calibration curve of BCL2, ranging from 78.1 to 2500 pg/mL. Weak expression of Bcl2 was observed in comparison to the reference drug doxorubicin, indicates compound 6 induced apoptosis of MCF-7cells.
P53 is a tumor suppressor that is triggered in response to stress. Studies demonstrate that p53 is critical for the prevention of tumorigenesis and may potentially be involved in at least half of all known cancers. MCF-7 treated with compound 6 showed a slight downregulation of the protein levels of p53 to 70.15 pg/mL, which suggests that compound 6, induces apoptosis via another pathway rather than acting via p53 protein.
Microtubules are a major component of the cytoskeleton. They play a crucial role in cell division. Tubulin-binding agents are amongst the most widely used chemotherapeutic drugs in cancer treatment. We investigated the effects of compound 6 at a concentration of 10 µg/mL on TubBin MCF-7 cells. TubBlevels were 0.19 pg/mL which indicated that compound 6 had no significant effect on tubulin polymerization as compared with reference drug colchicine.
Docking Studies
The docking study targeted (ID: 4uro) DNA Gyrase B, (ID:1EA1) Candida albicans lanosterol14-α-demethylase, and (ID: 1VJY) transforming growth factor (TGF) for β-receptor type I (β-RI), to examine the potential mode of action of the small compounds as antimicrobial and antitumor agents. The ligand–protein interactions were estimated based on the docking score using the functions implemented in the Molecular Operating Environment MOE 2015.10 package. All calculations of the docking experiments against crystal structures (PDB: 4uro, 1EA1 and 1VJy) are shown in Tables 4 and 5. Bacterial DNA-gyrase plays a vital role in the activity of antibacterial agents and acts by breaking double-stranded DNA by catalyzing negative supercoiling which is essential for DNA replication, transcription, and recombination.51 Analysis of the co-crystallized DNA-gyrase cleavage complex with novobiocin, which is an effective antibacterial agent that acts by cleaving DNA and restricting the ATPase binding site located on the vital peptidoglycan of the bacterial cell wall (Ser 55, Ala64, Asn.65, Asp89, Thr164, Thr173, and Val79). The inhibitory effect may be a result of the distinct structure of cell wall that characterizes Gram-negative and Gram-positive bacteria. The cell wall of Gram-negative bacteria is composed of a thin peptidoglycan layer (7–8 nm) with an additional an outer membrane. The Gram-positive bacteria contain a thick peptidoglycan layer (20–80 nm) outside of the cell wall with no outer membrane. The peptidoglycan is a mesh-like polymer consisting of sugars and amino acids. A peptidoglycan layer protects microorganisms against antibacterial agents such as antibiotics, toxins, chemicals, and degradative enzymes.52,53 Furthermore, a docking study evaluated the lanosterol 14-α-demethylase-fluconazolecomplex (PDB ID: 1EA1), extracted from Mycobacterium tuberculosis and showed a high similarity with the lanosterol-14-α-demethylase obtained from Candida species, for which it is difficult to obtain X-ray crystal structure for bound membrane enzyme. In addition, the crystal structure ofTGF of β-RI bound with naphthyridine (ligand 460) was obtained as PDB file (ID: 1VJY) with 2.0Å resolution.
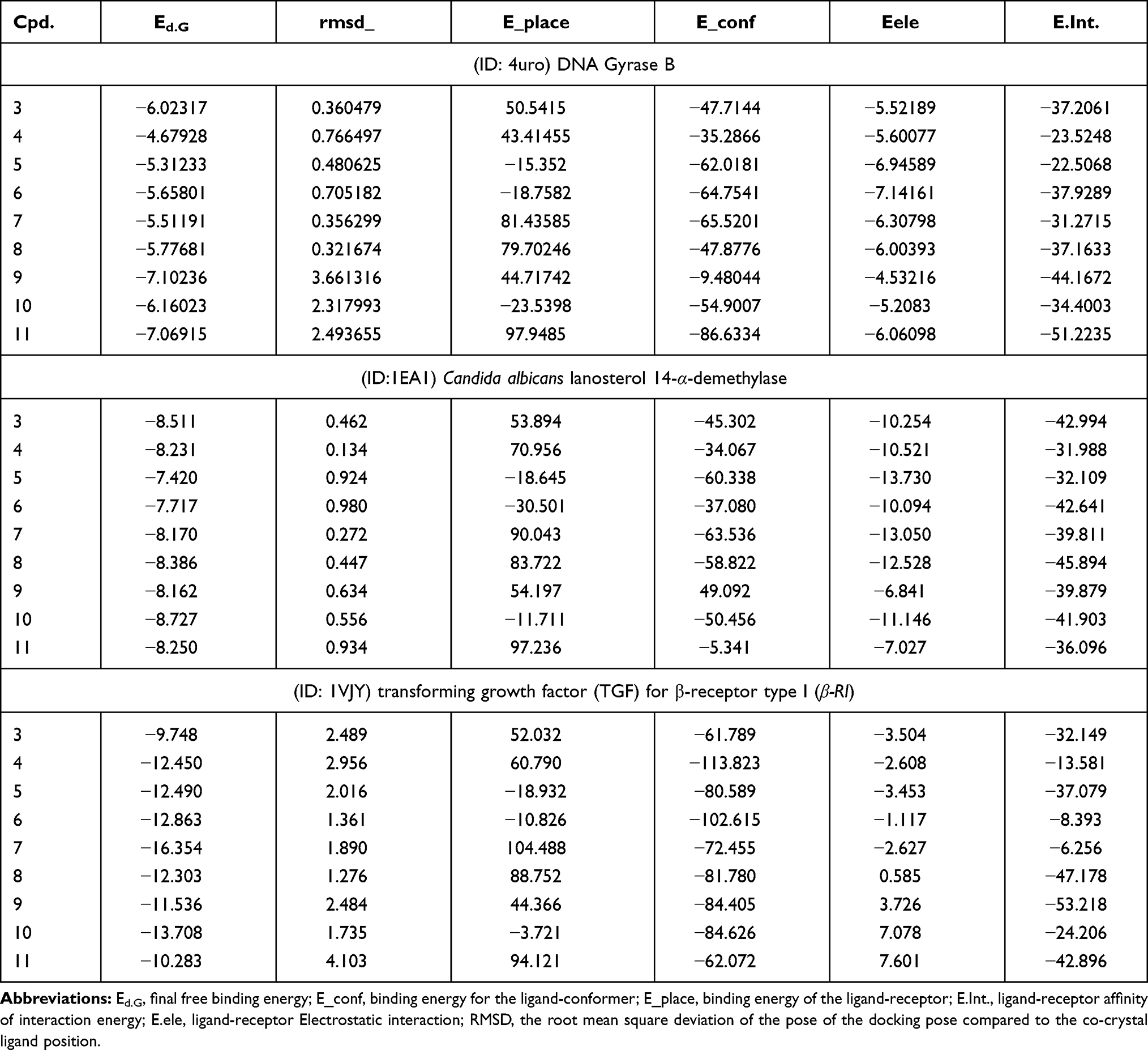 |
Table 4 Simulated Molecular Docking Energy in (kcal/mol) for the Tested Ligands |
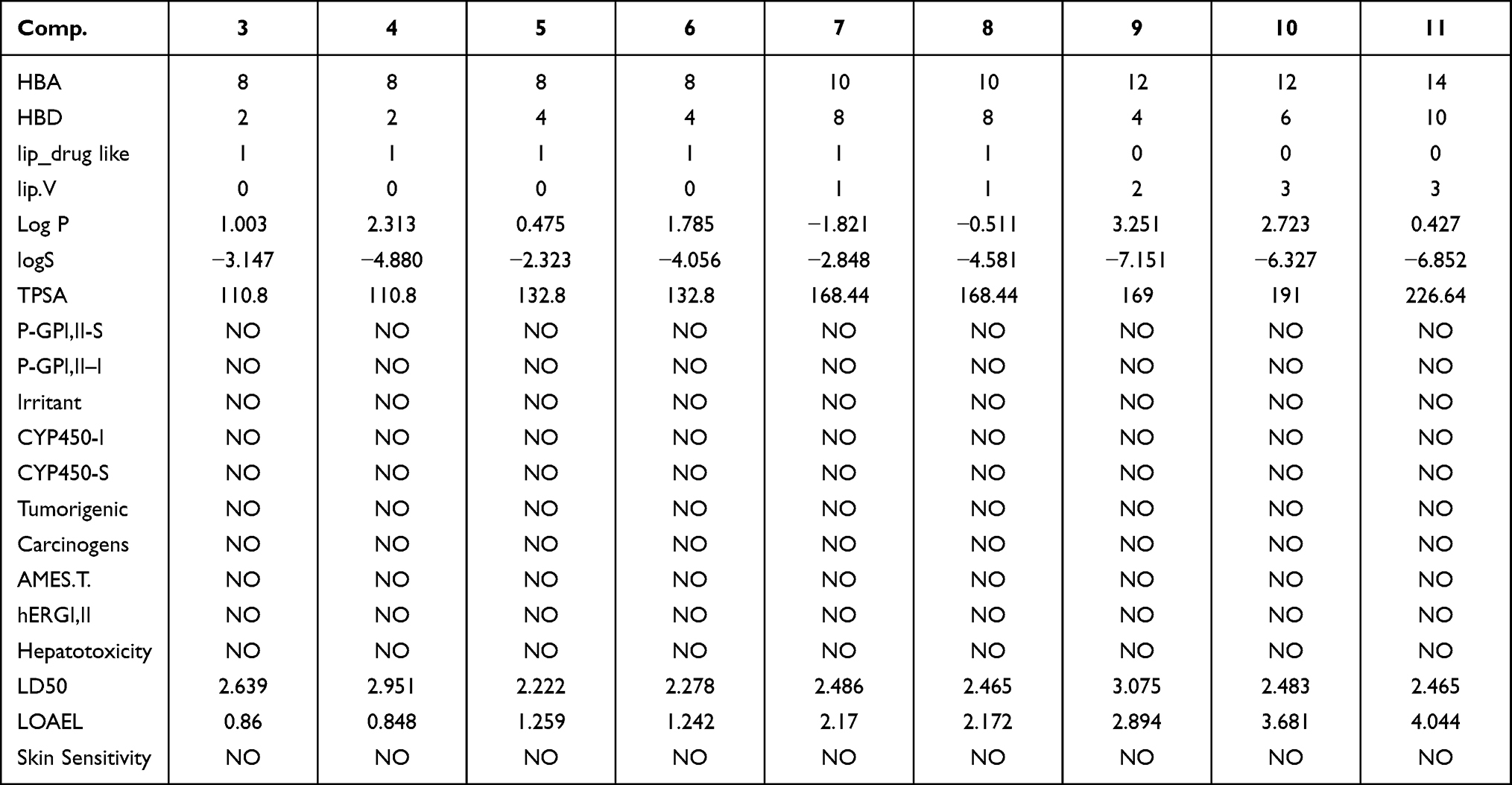 |
Table 5 Pharmacokinetic Features for Compounds (3–11) |
All compounds were redocked into active sites in the absence of the reference inhibitor. The ligands were successfully complexed with active fsites of enzymes. The extracted docked poses of ligands were energy-minimized with a molecular mechanics (Amber12: EHT) force field, up to 0.05 kcal/mol for the gradient convergence. The poses were initially selected among those with a docking score of E.dG<-7 kcal/mol). Next, poses were filtered based on lowest docking MOE score with lowest root means quart deviation (RMSD) against reference drugs. Finally, the highest MOE scoring function for the tested compounds was applied to evaluate the binding affinities of the tested compounds (Table).
(ID: 4uro) DNA Gyrase B: The bispeptides 9,10, and 11 exhibited the highest binding affinity and RMSD over other compounds for DNA gyrase (−7.1,-6.1, and −7.0 kcal/mol.), respectively, (summarized in Table 4). The other compounds showed considerable binding affinity (RMSD < 1), which was attributed to the higher stability of these compounds in the binding site (Table 4). The binding scores for compounds 3 to 8 could be arranged as 3> 8> 6 >7 > 5> 4 with a trend for RMSD between ~0.3–0.7 (Table 4). All compounds allowed H-bond formation with important amino acid residues (Ala64, Asn.65, and Thr164) at the DNA gyrase active site (Table S1, Supplementary materials). All compounds were arranged in parallel with Asn65 and Thr164, the hydrophobic portion of these compounds was considered a stabilization key in the receptor conformation. Furthermore, compounds 3, 4, and 6 were linked via an H2O molecule which bound to three important backbone amino acids Val79, Ser 55, and Thr173 (Figure 4). The different ligand conformations indicated the interactions with hydrophilic amino acid backbone at the 4uro binding site (Table S1, Supplementary materials). From (Figure 4), we can postulate that the hydrophobicity fragment generated in the synthesized compounds (Ala and Met) is an important pharmabiotic characteristic for binding to the DNA-gyrase pocket. Furthermore, the inhibitory efficiency of the synthesized compounds against bacterial growth may be explained by their ability to attack the naked peptidoglycan of the cell wall.54,55 As a result, the antibacterial mechanism of the synthesized compounds involves the possibility of attacking the bacterial membrane and changing its permeability. Hence, the synthesized particles may leak through sugars and proteins to deactivate hydrogen respiratory chain enzymes, and subsequently, they produce pits and gaps in the bacterial membrane (peptidoglycan layer), to induce irregular fragmentation of the bacterial cells.56,57 We therefore concluded that these indicators suggested that the compounds tested would induce suitable biological functions.
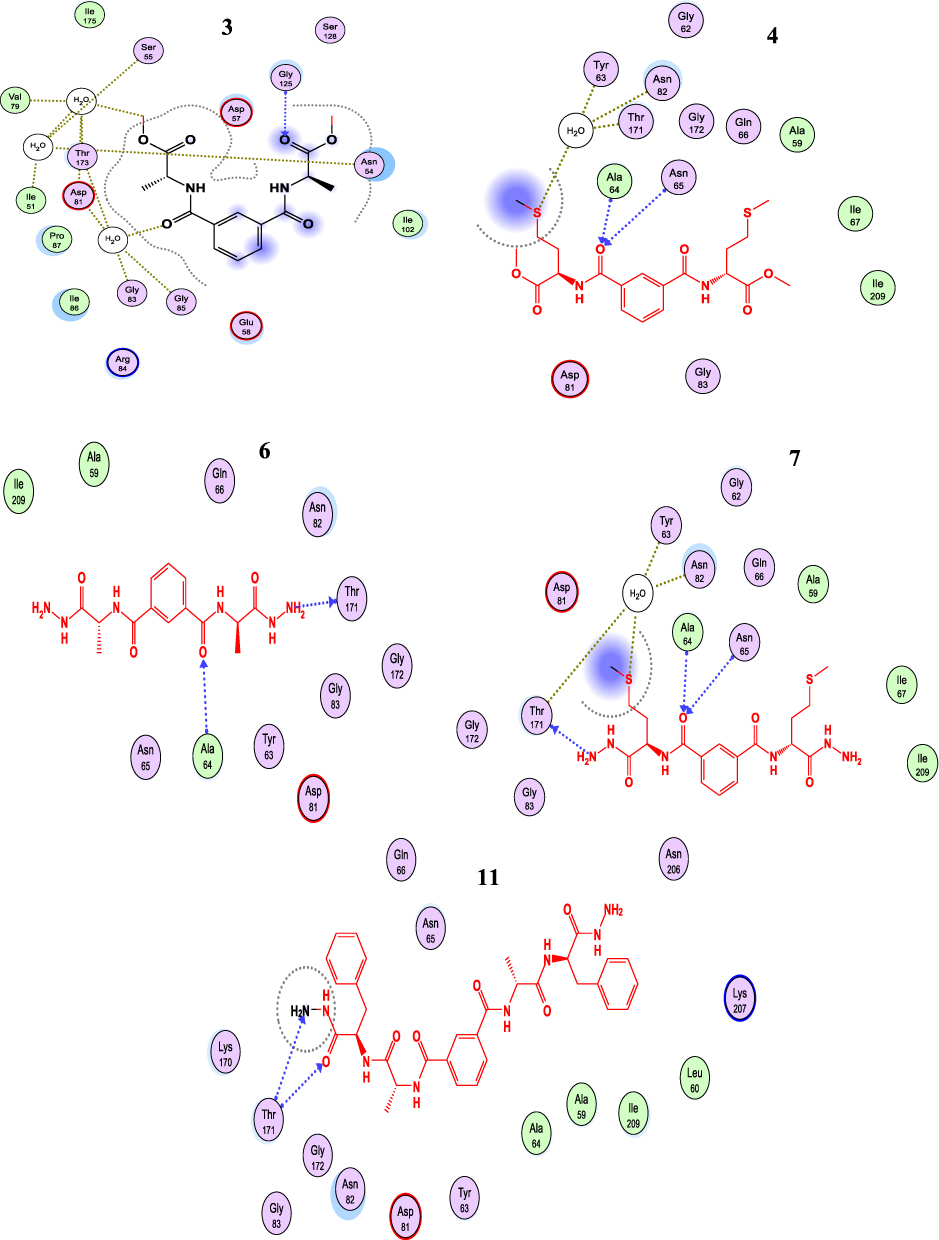 |
Figure 4 The 2D binding mode of 3,4,7,8 and 11 into the active site of DNA Gyrase, H-bond represented as blue dashed color. |
(ID:1EA1) Candida albicans Lanosterol 14-α-Demethylase
The tested compounds 3–11 showed good binding affinity against 1EA1 (Table 4, Figure S1, 2; Supplementary material). The binding pockets of 1EA1 included amino acid residues Arg324, Arg324, Gln70, Tyr74, Arg93, Hip390, Gln70, and Arg94, which produce H-, π-π and hydrophobic interactions with the tested compounds (Table S2, Supplementary Materials).
(ID: 1VJY) Transforming Growth Factor (TGF) for β-Receptor Type I (β-RI)
All synthesized compounds showed higher binding affinity (Table 4) than the reference ligand 460 (Ed.G= −4.81 kcal/mol). The Ala-hydrazide derivative 7 showed the highest binding score (Ed.G= −16.354 kcal/mol). Compound 7 interacted with binding site by forming five H-bonds with the following amino acids Lys232, Ser280, and Asp290, respectively. While at the same time it formed H-bonds with H2O which interacted with backbone amino acids Lys232 and Glu245 (Figure S3; Supplementary material). The binding affinity for peptide derivatives (4 to 6 and 8) showed almost equally binding affinity (Ed.G=~ −12 kcal/mol). This compound occupied the binding site through similar interactions with important amino acids of the binding sites for compound 3 with Gln70, Ala254, Leu319, andArg324; for compound 5 with Arg94; for 6 with Glu255, Leu319, and Phe385; and for 8 with Ala254, Thr258, and Cys392. The bis-peptide 9 to 11 derivatives showed lower binding affinity than the amino acid derivatives 3 to 8 (Table 4). Bis-peptide 9 formed two H-bondswithArg93 and two π- π interactions with Arg94. Compound 10 formed the following interactions: H-bonds withHip390, Arg93, and π- π bonds with Cys392. While compound 11 produced two bonds with Val393 and Arg389 (Figure S3, S4; Table S3, Supplementary Material).
ADMET Profile in silico
For any bioactive molecule to be considered a potential drug should possess low toxicity with good pharmacokinetic properties. ADME/T has interfered with several agents from reaching the clinical trials. Hence, the ADME/T profile for compounds 3 to 11 was predicted using MOE and admet-SAR.[52]The pharmacokinetic descriptors in Table 5 indicate that compounds 3 to 8 fulfilled the Lipinski rules, while the other compounds 9 to 11 showed more than one violation when the Lipinski restrictions were applied. The low lipophilicity values of compound 5 (Log P<5 mol/L) and generally for all compounds (3–11) predicted good permeability and absorption across the cell membrane.57 The topological polar surface area (TPSA) is another important pharmacokinetic parameter that predicts the permeability of the membrane by polar atoms.36 The passive absorption value is TPSA>140Å2. Compounds (7 to 11) were predicted to be passively absorbed with values ranging from ~168 to −226 Å2. The tested compounds did not act as inhibitors or substrates against the P-glycoprotein isoforms P-GPI, II–I and P-GPI, II-S, thus, it can be safely presumed that these compounds would not exert any relevant activity.58,59 Based on the above findings, synthesized compounds 3 to 11 may present good absorption properties.
As shown in Table 5, (i) our compounds did not show any inhibition or activation of any Cytochrome P450 (CYP450) isoform. CYP450 is an important detoxification enzyme in the body and is mainly located in liver. It oxidizes xenobiotics to facilities their excretion. CYP450 can activate or deactivate many drugs. It is therefore important to evaluate the potential inhibitory effects against CYP450 enzymes (ie, CYP1A2, CYP2C19, CYP2C9, CYP2D6, CYP3A4) or activation of CYP2D6 or CYP3A4 for the synthesized compounds against 18,000 compounds known to inhibit or mimic these isoforms. (ii) There were no observed carcinogenic or tumorigenic effects induced by the synthesized compounds, comparing 981 known carcinogenic modules obtained from the Carcinogenic Potency Database (CPDB).60 (iii) The chemical structure of each of the investigated compounds was compared with 10,000 models that have been previously tested in rats to predict the lethal dose value for these compounds. Our findings showed low values of LD50 (~2.2–3.07 mg/kg) for the synthesized compounds. (iv) The LOAE was predicted by comparing the tested compounds with 576 known agents. This test is applied in chronic evaluation studies for the identification of the lowest doses of chemicals tolerable over a long period. The predictor simulated that no LOAE was detectable for any of the investigated compounds. (v) The synthesized compounds exerted no mutagenic effect on DNA of microorganisms when tested in the Ames test with over 8000 compounds. The Ames test is a widely used examination of mutagenic effects of bioactive molecules against microorganism DNA.61,62 (vi) A weak inhibitory action was determined against the human Ether-a-gogo-Related Gene (hERG), which can lead to long QT syndrome, for the tested compounds.63,64 (viii) The tested compounds did not exhibit any skin sensitivity, when tested utilizing 254 compounds able to induce skin sensitization, or hepatoxic effects when evaluated against 531 compounds which have liver associated side effects in human. (vii) Based on the above characteristics, we can conclude that the synthesized compounds may have a good oral bioavailability without any potential for marked health adverse effects via rodent toxicity profiles.
This study introduces novel peptide derivatives based on two amino acids: alanine and methionine. Based on the activity results and docking studies, the Ala, Met, and Ala-Phe, at Nα-1, 3-benzenedicarbonyl do not induce any meaningful differences in antimicrobial or antioxidant activities. The docking study showed that a binding affinity ranging, −5 to −7 kcal/mol) against DNA gyrase as the model of anti-microbial activity, and against antifungal model these compounds showed higher binding score than antibacterial results (−7 to −8 kcal/mol.). In addition, all the compounds were evaluated as anticancer agents in three cell lines MCF-7, HCT-116, and HepG-2. In case of MCF-7, the presence of Met.OMe (7) and Ala.hydrazide (8) in the parent compound increased the activity against all similar members of the tested compounds. Furthermore, the parent compound that carried other fragments did not present any significant variation in cytotoxic activity. In the HCT-116 cell line, introducing Ala (3) and Ala.Phe (9) fragments into the parent compound showed highest activity against HCT116, with inhibition potency percentage (56% and 61%), respectively. These compounds showed binding scores −9.7 and −13.7 kcal/mol, respectively for compounds 3 and 9. The presence of Met.OMe (6) and its derivatives from the hydrazide (8), in addition to the presence of Ala.Phe.OMe (10) showed moderate activity. The other compounds showed the lowest activity among the compounds. In the HepG-2 cell line, all compounds showed a low toxic potency in the range of 2–10%.
Conclusion
In the present study, nine novel peptide derivatives, characterized by the general stretcher, Nα-1, 3-benzenedicarbonyl-bis-(amino acid) and dipeptide candidates were synthesized. All synthesized compounds showed potent antimicrobial activity of different degrees against different pathogenic bacteria and fungi, and only compounds 5 and 7 had the lowest inhibitory activity. These compounds showed good antimicrobial activity against important pathogens so they can be considered potent therapeutic drugs that exert biological control, warranting further investigations. Furthermore, three human cancer cell lines (MCF-7, HCT-116, and HepG-2) were used to evaluate the anticancer potency of the synthesized compounds. The novel compounds (3–11) showed a strong cytotoxic effect in the MCF-7 human breast cancer cell line with 95.1% and 79.8% cell inhibition, respectively. The docking results suggest good fitting through different hydrogen bond interactions with protein residues allowing eliciting anticancer activity. The ADME/T profile in silico suggested compounds 3–11 may have a good absorption property.
Sample Availability
Samples of the compounds (1–11) are available from Dr Gaber O. Moustafa.
Funding
This work was funded by the Deanship of Scientific Research, king Saud University through vice Deanship of Scientific Research Chairs.
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Hancock RE, Haney EF, Gill EE. The immunology of host defence peptides: beyond antimicrobial activity. Nat Rev Immunol. 2016;16:321–334. doi:10.1038/nri.2016.29
2. Rolston KV. The spectrum of pulmonary infections in cancer patients. Curr Opin Oncol. 2001;13:218–223. doi:10.1097/00001622-200107000-00002
3. Gaspar D, Veiga AS, Castanho MA. From antimicrobial to anticancer peptides. Review Front Microbiol. 2013;4:294. doi:10.3389/fmicb.2013.00294
4. Felício MR, Silva ON, Gonçalves S, Santos NC, Franco OL. Peptides with dual antimicrobial and anticancer activities. Front Chem. 2017;5:1–9. doi:10.3389/fchem.2017.00005
5. Abo-Ghalia MH, Moustafa GO, Alwasidi AS, Naglah AM. Cytotoxic Investigation of Isophthaloyl Cyclopenta peptides. Lat Am J Pharm. 2017;36:1957–1962.
6. Moustafa GO, El-Sawy AA, Abo-Ghalia MH. Synthesis of novel cyclopeptide candidates: i-cyclo-[Nα-isophthaloyl-bis-(Glycine-amino acid)-L-lysine] derivatives with expected anticancer activity. Egypt J Chem. 2013;5:473–494.
7. Hassan AS, Moustafa GO, Awad HM. Synthesis and in vitro anticancer activity of pyrazolo [1, 5-a] pyrimidines and pyrazolo [3, 4-d][1, 2, 3] triazines. Synth Commun. 2017;47(21):1963–1972. doi:10.1080/00397911.2017.1358368
8. Amr AE, Abo-Ghalia MH, Moustafa GO, Al-Omar MA, Nossier ES, Elsayed EA. Design, synthesis and docking studies of novel macrocyclic pentapeptides as anticancer multi-targeted kinase inhibitors. Molecules. 2018;23(10):10. doi:10.3390/molecules23102416
9. Moustafa GO, Younis A, Al-Yousef SA, Mahmoud SY. Design, synthesis of novel cyclic pentapeptide derivatives based on 1, 2-benzenedicarbonyl chloride with expected anticancer activity. J Comput Theor Nanosci. 2019;16(5–6):1733–1739. doi:10.1166/jctn.2019.8114
10. Kassem AF, Moustafa GO, Nossier ES, et al. In vitro anticancer potentiality and molecular modelling study of novel amino acid derivatives based on N 1, N 3-bis-(1-hydrazinyl-1-oxopropan-2-yl) isophthalamide. J Enzyme Inhib Med Chem. 2019;34(1):1247–1258. doi:10.1080/14756366.2019.1613390
11. Mohamed FH, Shalaby AM, Soliman HA, et al. Design, synthesis and molecular docking studies of novel cyclic pentapeptides based on phthaloyl chloride with expected anticancer activity. Egypt J Chem. 2020;63(5):1723–1736. doi:10.21608/EJCHEM.2019.18452.2137
12. Abo-Ghalia MH, Moustafa GO, Amr AE, Naglah AM, Elsayed EA, Bakheit AH. Anticancer activities and 3D-QSAR studies of some new synthesized macrocyclic heptapeptide derivatives. Molecules. 2020;25(5):1253. doi:10.3390/molecules25051253
13. Kalmouch A, Radwan MAA, Omran MM, Sharaky M, Moustafa GO. Synthesis of novel 2, 3ʹ-bipyrrole derivatives from chalcone and amino acids as antitumor agents. Egypt J Chem. 2020;63(11):4409–4421.
14. Moustafa GO, Al-Wasidi AS, Naglah AM, Refat MS. Isolation and synthesis of dibenzofuran derivatives possessing anticancer activities: a review. Egyptian J Chem. 2020;63(6):2355–2367. doi:10.21608/ejchem.2020.21937.2310
15. Moustafa GO. Therapeutic potentials of cyclic peptides as promising anticancer drugs. Egyptian J Chem. 2021;64(4):2160–2172.
16. Elhenawy AA, Al-Harbi LM, Moustafa GO, El-Gazzar MA, Abdel-Rahman RF, Salim AE. Synthesis, comparative docking, and pharmacological activity of naproxen amino acid derivatives as possible anti-inflammatory and analgesic agents. Drug Des Devel Ther. 2019;13:1773. doi:10.2147/DDDT.S196276
17. Moustafa GO, Khalaf H, Naglah A, et al. Synthesis, molecular docking studies, in vitro antimicrobial and antifungal activities of novel dipeptide derivatives based on N-(2-(2-hydrazinyl-2-oxoethylamino)-2-oxoethyl)-Nicotinamide. Molecules. 2018;23:761. doi:10.3390/molecules23040761
18. Naglah AM, Moustafa GO, Al-Omar MA, Al-Salem HAS, Hozzein WN. Synthesis, characterization and in vitro antimicrobial investigation of novel amino acids and dipeptides based on dibenzofuran-2-sulfonyl-chloride. J Comput Theor Nanosci. 2017;14:3183–3190. doi:10.1166/jctn.2017.6613
19. Al-Salem HAS, Naglah AM, Moustafa GO, Mahmoud AZ, Al-Omar MA. Synthesis of novel tripeptides based on Dibenzofuran-2-Sulfonyl-[Aromatic and Hydroxy Aromatic Residues]: towards Antimicrobial and Antifungal Agents. J Comput Theor Nanosci. 2017;14:3958–3966. doi:10.1166/jctn.2017.6702
20. Hassan AS, Moustafa GO, Askar AA, Naglah AM, Al-Omar MA. Synthesis and antibacterial evaluation of fused pyrazoles and Schiff bases. Synth Commun. 2018;48(21):2761–2772. doi:10.1080/00397911.2018.1524492
21. Hassan AS, Askar AA, Nossier ES, Naglah AM, Moustafa GO, Al-Omar MA. Antibacterial evaluation, in silico characters and molecular docking of schiff bases derived from 5-aminopyrazoles. Molecules. 2019;24(17):3130. doi:10.3390/molecules24173130
22. Hasanin MS, Moustafa GO. New potential green, bioactive and antimicrobial nanocomposites based on cellulose and amino acid. Int J Biol Macromol. 2019;144:441–448. doi:10.1016/j.ijbiomac.2019.12.133
23. Elsherif MA, Hassan AS, Moustafa GO, Awad HM, Morsy NM. Antimicrobial evaluation and molecular properties prediction of pyrazolines incorporating benzofuran and pyrazole moieties. Bull Chem Soc Ethiop. 2020;10(02):037–043. doi:10.7324/JAPS.2020.102006
24. Hassan AS, Moustafa GO, Morsy NM, Abdou AM, Hafez TS. Design, synthesis and antibacterial activity of N-aryl-3-(arylamino)-5-(((5-substituted furan-2-yl)methylene)amino)-1H-pyrazole-4-carboxamide as Nitrofurantoin® analogues. Egypt J Chem. 2020;63(11):4485–4497.
25. Khalaf HS, Naglah AM, Al-Omar MA, Awad HM, Bakheit AH, Bakheit AH. Synthesis, docking, computational studies, and antimicrobial evaluations of new dipeptide derivatives based on nicotinoylglycylglycine hydrazide. Molecules. 2020;25(16):3589. doi:10.3390/molecules25163589
26. Al-Wasidi AS, Naglah AM, Kalmouch A, Adam AMA, Refat MS, Moustafa GO. Preparation of Cr2O3, MnO2, Fe2O3, NiO, CuO, and ZnO oxides using their glycine complexes as precursors for in situ thermal decomposition. Egyptian J Chem. 2020;63(3):1109–1118.
27. Al-Wasidi AS, Naglah AM, Refat MS, El-Megharbel SM, Kalmouch A, Moustafa GO. Synthesis, spectroscopic characterization and antimicrobial studies of Mn(II), Co(II), Ni(II), Cr(III) and Fe(III) melatonin drug complexes. Egyptian J Chem. 2020;63(4):1469–1481.
28. Al-Wasidi AS, Wafeek M, Abd El-Ghaffar HA., Naglah A.M., Kalmouch A., Hamed M., Moustafa G.O. Effect of Density on Growth Hormone and Some Physiological Parameters and its Relation to Growth Performance. Egyptian J Chem. 2020;63(4):1575–1584.
29. Imtiaj A, Jayasinghe C, Lee GW, Lee TS. Antibacterial and antifungal activities of stereumostrea, an inedible wild mushroom. Mycobiology. 2007;35:210–214. doi:10.4489/MYCO.2007.35.4.210
30. El-Feky AM, Elbatanony MM, Mounier MM. Anti-cancer potential of the lipoidal and flavonoidal compounds from Pisum sativum and Viciafaba peels. Egypt J Basic Applied Sci. 2018;5(4):258–264. doi:10.1016/j.ejbas.2018.11.001
31. Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Meth. 1983;65(1):55–63. doi:10.1016/0022-1759(83)90303-4
32. Barbareschi M, Caffo O, Veronese S, et al. Bcl-2 and p53 expression in node-negative breast carcinoma: a study with long-term follow-up. Hum Pathol. 1996;27:1149–1155. doi:10.1016/S0046-8177(96)90307-X
33. Liliom K, Lehotzky A, Molnar A, Ovadi J. Characterization of tubulin-alkaloid interactions by enzyme-linked immunosorbent assay. Anal Biochem. 1995;228(1):18–26. doi:10.1006/abio.1995.1309
34. Thomas MD, McIntosh GG, Anderson JJ, et al. Novel quantitative immunoassay system for p53 using antibodies selected for optimum designation of p53 status. J Clin Pathol. 1997;50(2):143–147. doi:10.1136/jcp.50.2.143
35. Gellibert F, Woolven J, Fouchet MH, Mathews N, Goodland H, Lovegrove V. Identification of 1, 5-naphthyridine derivatives as a novel series of potent and selective TGF-β type I receptor inhibitors. J Med Chem. 2004;47(18):4494–4506. doi:10.1021/jm0400247
36. Podust LM, Poulos TL, Waterman MR. Crystal structure of cytochrome P450 14α-sterol demethylase (CYP51) from Mycobacterium tuberculosis in complex with azole inhibitors. Proc Natl Acad Sci, 2001;98(6):3068–3073.
37. Chatzou M, Magis C, Chang J, et al. Multiple sequence alignment modeling: methods and applications. Briefings in Bioinformatics. 2016;17(6):1009–1023. doi:10.1093/bib/bbv099
38. Kumar A, Sasmal D, Jadav SS, Sharma N. Mechanism of immunoprotective effects of curcumin in DLM-induced thymic apoptosis and altered immune function: an in silico and in vitro study. Immunopharmacol Immunotoxicol. 2015;37(6):488–498. doi:10.3109/08923973.2015.1091004
39. Rana R, Sharma R, Kumar A. Repurposing of Fluvastatin against Candida albicans CYP450 lanosterol 14 α-demethylase, a target enzyme for antifungal therapy: an In silico and In vitro study. Curr Mol Med. 2019;19(7):506–524. doi:10.2174/1566524019666190520094644
40. Jacob KS, Ganguly S, Kumar P, Poddar R, Kumar A. Homology model, molecular dynamics simulation and novel pyrazole analogs design of Candida albicans CYP450 lanosterol 14 α-demethylase, a target enzyme for antifungal therapy. J Biomol Struct Dyn. 2017;35(7):1446–1463. doi:10.1080/07391102.2016.1185380
41. Kant K, Lal UR, Kumar A, Ghosh M. A merged molecular docking, ADME-T and dynamics approaches towards the genus of Arisaema as herpes simplex virus type 1 and type 2 inhibitors. Comput Biol Chem. 2019;78:217–226. doi:10.1016/j.compbiolchem.2018.12.005
42. Navyashree V, Kant K, Kumar A. Natural chemical entities from Arisaema Genus might be a promising break-through against Japanese encephalitis virus infection: a molecular docking and dynamics approach. J Biomol Struct Dyn. 2020;1–13.
43. Gupta M, Sharma R, Kumar A. Comparative potential of simvastatin, Rosuvastatin and Fluvastatin against bacterial infection: an in silico and in vitro study. Orient Pharm Exp Med. 2019;19(3):259–275. doi:10.1007/s13596-019-00359-z
44. Pontius J, Richelle J, Wodak SJ. Deviations from standard atomic volumes as a quality measure for protein crystal structures. 1996.
45. Chemical Computing Group Inc. Molecular Operating Environment (MOE). Version 2015.10. Montreal, Quebec, Canada: Chemical Computing Group Inc;2016
46. Dale GE, Kostrewa D, Gsell B, Steiger M, D’Arcy A. Crystal engineering: deletion mutagenesis of the 24 kDa fragment of the DNA gyrase B subunit from Staphylococcus aureus. Acta Crystallographica D Biol Crystallography. 1999;55(9):1626–1629. doi:10.1107/S0907444999008227
47. Frisch MJ, Trucks GW, Schlegel HB, et al. Gaussian 09, Revision D. 01, Gaussian, Inc. Wellingford CT. Gaussian. Inc, Wellingford CT Google Scholar; 2013.
48. Elhenawy AA, Al-Harbi L, El-Gazzar M, Khowdiary MM, Moustfa A. Synthesis, molecular properties and comparative docking and QSAR of new 2-(7-hydroxy-2-oxo-2H-chromen-4-yl) acetic acid derivatives as possible anticancer agents. J Spectrochimica Acta Part A. 2019;218:248–262. doi:10.1016/j.saa.2019.02.074
49. Lipinski CA. Lead-and drug-like compounds: the rule-of-five revolution. Drug Discov Today Technol. 2004;1(4):337–341. doi:10.1016/j.ddtec.2004.11.007
50. Tsai HF, Chang YC, Washburn RG, Wheeler MH, Kwon-Chung KJ. The developmentally regulated alb1 gene of Aspergillus fumigatus: its role in modulation of conidial morphology and virulence. J Bacteriol. 1998;180:3031–3038. doi:10.1128/JB.180.12.3031-3038.1998
51. Aldred KJ, McPherson SA, Turnbough CL, Kerns RJ, Osheroff N. Topoisomerase IV-quinolone interactions are mediated through a water-metal ion bridge: mechanistic basis of quinolone resistance. Nucleic Acids Res. 2013;41(8):4628–4639. doi:10.1093/nar/gkt124
52. Jung WK, Koo HC, Kim KW, Shin S, Kim SH, Park YH. Antibacterial activity and mechanism of action of the silver ion in Staphylococcus aureus and Escherichia coli. Appl Environ Microbiol. 2008;74(7):2171–2178. doi:10.1128/AEM.02001-07
53. Feng QL, Wu J, Chen GQ, Cui FZ, Kim TN, Kim JO. A mechanistic study of the antibacterial effect of silver ions on Escherichia coli and Staphylococcus aureus. J Biomed Mater Res. 2000;52(4):662–668. doi:10.1002/1097-4636(20001215)52:4<662::AID-JBM10>3.0.CO;2-3
54. Amro NA, Kotra LP, Wadu-Mesthrige K, Bulychev A, Mobashery S, Liu GY. High-resolution atomic force microscopy studies of the Escherichia coli outer membrane: structural basis for permeability. Langmuir. 2000;16(6):2789–2796. doi:10.1021/la991013x
55. Pellieux C, Dewilde A, Pierlot C, Aubry JM. [18] Bactericidal and virucidal activities of singlet oxygen generated by thermolysis of naphthalene endoperoxides. Methods Enzymol. 2000;319:197–207.
56. Yang H, Lou C, Sun L, et al. Admet SAR 2.0: web-service for prediction and optimization of chemical ADMET properties. Bioinformatics. 2019;35:1067–1069. doi:10.1093/bioinformatics/bty707
57. Clark DE, Pickett SD. Computational methods for the prediction of ‘drug-likeness‘. Drug Discov Today. 2000;5(2):49–58. doi:10.1016/S1359-6446(99)01451-8
58. Amin ML. P-glycoprotein Inhibition for Optimal Drug Delivery. Drug Target Insights. 2013;7:27–34. doi:10.4137/DTI.S12519
59. Bento A, Gaulton A, Hersey A, Bellis L, Chambers J, Davies M. The ChEMBL bioactivity database: an update. Nucleic Acids Res. 2014;42(D1):D1083–D1090. doi:10.1093/nar/gkt1031
60. Ames BN, McCann J, Yamasaki E. Methods for detecting carcinogens and mutagens with the Salmonella/mammalian-microsome mutagenicity test. Mutat Res. 1975;31(6):347–364. doi:10.1016/0165-1161(75)90046-1
61. Mortelmans K, Zeiger E. The Ames Salmonella/microsome mutagenicity assay”. Mutat Res. 2000;455(1–2):29–60. doi:10.1016/S0027-5107(00)00064-6
62. Witchel HJ, Hancox JC. Familial and acquired long QT syndrome and the cardiac rapid delayed rectifier potassium current. Clin Exp Pharmacol Physiol. 2000;27(10):753–766. doi:10.1046/j.1440-1681.2000.03337.x
63. Balakumar R, Sivaprakasam E, Kavitha D, Sridhar S, Kumar JS. Antibacterial and antifungal activity of fruit bodies of Phellinus mushroom extract. Int J Biosci. 2011;1:72–77.
64. Batran RZ, Kassem AF, Abbas EM, Elseginy SA, Mounier MM. Design, synthesis and molecular modeling of new 4-phenylcoumarin derivatives as tubulin polymerization inhibitors targeting MCF-7 breast cancer cells. Bioorg Med Chem. 2018;26(12):3474–3490. doi:10.1016/j.bmc.2018.05.022
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.