")
Back to Journals » Journal of Hepatocellular Carcinoma » Volume 8
NAFLD-Associated HCC: Progress and Opportunities
Authors Geh D , Anstee QM, Reeves HL
Received 18 January 2021
Accepted for publication 8 March 2021
Published 8 April 2021 Volume 2021:8 Pages 223—239
DOI https://doi.org/10.2147/JHC.S272213
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Ahmed Kaseb
Daniel Geh,1 Quentin M Anstee,2,3 Helen L Reeves2– 4
1Biosciences Institute, Faculty of Medical Sciences, Newcastle University, Newcastle upon Tyne, UK; 2Translational and Clinical Research Institute, Faculty of Medical Sciences, Newcastle University, Newcastle upon Tyne, UK; 3The Liver Unit, Newcastle Upon Tyne Hospitals NHS Foundation Trust, Freeman Hospital, Newcastle upon Tyne, UK; 4Hepatopancreatobiliary Multidisciplinary Team, Newcastle Upon Tyne Hospitals NHS Foundation Trust, Freeman Hospital, Newcastle upon Tyne, UK
Correspondence: Helen L Reeves
Translational and Clinical Research Institute, Faculty of Medical Sciences, Newcastle University, Newcastle upon Tyne, NE2 4HH, UK
Tel +44 191 208 4423
Fax +44 191 208 4301
Email [email protected]
Abstract: Due to an increase in the obesity-associated metabolic syndrome of epidemic proportions, nonalcoholic fatty liver disease (NAFLD) is becoming a leading cause of hepatocellular carcinoma (HCC) in western countries. This presents added challenges, as NAFLD-associated HCC tends to present at an advanced stage in older patients with co-morbidities. Their prognosis is generally poor with the benefits of standard therapies less certain. The pathogenesis of NAFLD-associated HCC is multifactorial and not well understood, although the risk of HCC developing undoubtedly increases as NAFLD progresses to steatohepatitis and cirrhosis. Recent advances in our understanding of the drivers of NAFLD and HCC will hopefully lead to the development of clinically relevant biomarkers, tools and strategies to aid the identification of high-risk patients, inform preventive measures, and introduction of better tolerated targeted therapies. Lifestyle modification and chemoprevention with drugs such as anti-platelets, statins and anti-diabetics are being evaluated for HCC prevention. The landmark IMBrave150 study introducing the combination of atezolizumab and bevacizumab has recently transformed the landscape of systemic therapies in HCC, with follow-up analyses and real-world data for patients with NAFLD-associated HCC eagerly anticipated. While responses may vary in ways not yet appreciated, the rate of discovery and progress suggests imminent change and opportunities.
Keywords: hepatocellular carcinoma, HCC, nonalcoholic fatty liver disease, NAFLD, liver cirrhosis, biomarkers, prevention, systemic therapy, immunotherapy
Introduction
Primary liver cancer is the sixth most common incident cancer and the fourth leading cause of cancer-related mortality worldwide.1 Hepatocellular carcinoma (HCC) accounts for 90% of primary liver cancers and usually develops on the background of liver cirrhosis.2 Historically, the most common underlying etiologies of cirrhosis associated with HCC have been chronic hepatitis B virus (HBV) and hepatitis C virus (HCV) infection.3 In recent decades, the implementation of HBV and HCV eradication programmes combined with the modern epidemic of lifestyle-related diseases such as obesity, hypertension, dyslipidemia and type 2 diabetes mellitus (T2DM) has led to nonalcoholic fatty liver disease (NAFLD) becoming a leading cause of liver cirrhosis and HCC in many western countries.4,5 Survival rates in HCC are dependent on stage at presentation, but poor overall, with an average 5-year survival of 5–15%.6–8 Worryingly the mortality from HCC is increasing compared to other cancers, in which mortality is generally decreasing.9,10 This growing clinical problem has led to a considerable focus of research into the pathogenesis, diagnosis and management of NAFLD-HCC, often highlighting differences compared to HCC of other etiologies. This review will summarize recent progress in our understanding of NAFLD-HCC, whilst highlighting areas of unmet or ongoing research with the greatest potential.
Search Criteria
PubMed and MEDLINE databases were searched using the keywords NAFLD, nonalcoholic steatohepatitis (NASH) and HCC. Recent studies investigating pathogenesis, biomarkers, prevention and treatment of NAFLD-HCC were included in this narrative review.
NAFLD-HCC: A Growing Problem
Nonalcoholic fatty liver disease (NAFLD) encompasses a wide spectrum of liver disease ranging from simple steatosis to nonalcoholic steatohepatitis (NASH) to liver fibrosis, cirrhosis, end-stage liver disease and the development of HCC. NAFLD is strongly associated with components of the metabolic syndrome such as hypertension, type 2 diabetes mellitus (T2DM) and cardiovascular disease.11 The relationship between NAFLD and the metabolic syndrome is complex and bidirectional.12 NAFLD has become the most common cause of chronic liver disease worldwide with an estimated 25% of the world population being affected.11,13 The natural history of NAFLD is highly variable with serial biopsy studies demonstrating dynamic progression and regression between stages, with the majority not progressing to end-stage liver disease.14–16 The huge patient population, combined with the lack of inexpensive, non-invasive and reliable biomarkers, makes identifying NAFLD patients with progressive disease and those at most risk of developing HCC extremely difficult.
In keeping with the rise of NAFLD, the prevalence of NAFLD-HCC is also increasing. Between 2004 and 2009 the incidence of NAFLD-HCC in the United States increased by 9% annually.17 This was imitated in the liver transplant programme in the United States, with NAFLD-HCC being the fastest growing indication for liver transplantation, increasing from 2.1% to 16.2% between 2000 and 2016.5 Between 2000 and 2010 the north of England saw an over tenfold increase in HCC related mortality from NAFLD and in 2010 NAFLD-HCC accounted for 34.8% of all HCC.4 Between 25–50% of NAFLD-HCC develops in the absence of cirrhosis.4,18–20 Although HCC can develop in other etiologies of chronic liver disease in the absence of cirrhosis, this appears to be more frequent in NAFLD.18 This presents a huge challenge, given that current evidence, summarized in clinical guidelines, does not support abdominal ultrasound surveillance for early-stage HCC detection in patients without cirrhosis or at least advanced fibrosis.21–24 Published series indicate that patients with NAFLD-HCC generally have a worse prognosis compared to HCC of other etiologies.4,19,25,26 Contributory factors include NAFLD-HCC being diagnosed at a more advanced stage of disease, hand-in-hand with either ineffective or absent surveillance, as well as NAFLD-HCC patients often being older with more co-morbidities, limiting the use of curative treatments such as liver resection and liver transplantation.4,19,25–27 Current understanding of the pathogenesis underlying the development of HCC, especially in the absence of cirrhosis is poorly understood. It is hoped that by further understanding this, reliable non-invasive biomarkers will be developed which will allow effective screening and early diagnosis of HCC, particularly in the non-cirrhotic NAFLD population. It is also hoped that by understanding the pathogenesis better strategies to prevent the development of HCC from NAFLD may be fostered. More effective and better tolerated systemic therapies are also desperately needed, given the majority of patients with NAFLD-HCC present at an advanced stage.
In summary, NAFLD-HCC is becoming a common cause of HCC worldwide. Not only are the numbers of patients increasing, patients are older, with more co-morbidities, with cancers detected at a more advanced stage. These features adversely affect prognosis and the likelihood of treatment benefit. Paradoxically, despite these features, liver function is better preserved in those patients without cirrhosis, which also impacts prognosis and treatment choice. The reality is that patients with NAFLD-HCC, either in the presence or absence of cirrhosis, are not well represented in the studies on which clinical practice guidelines have been based. Understanding the disease pathogenesis of NAFLD-HCC is an essential need, in the search for biomarkers to guide surveillance, diagnosis and prognosis, as well as targets for prevention and systemic therapies. In addition, however, we need to consider carefully the published and emerging data from ongoing clinical trials, looking for evidence of benefit in elderly patients with NAFLD-HCC.
Understanding of the Pathogenesis of NAFLD-HCC
The development of HCC in NAFLD is complex, multifactorial and not fully understood. Systemically both obesity and insulin resistance contribute to HCC development through systemic inflammation and promotion of oncogenic pathways.28 Increased hepatic lipid storage leads to lipotoxicity, endoplasmic reticulum stress and reactive oxygen species-mediated DNA damage which can further drive oncogenesis.29 The influence of certain genetic polymorphisms, gender and an altered gut microbiome are also becoming apparent.29–31 Here we will focus on recent developments in the understanding of NAFLD-HCC and highlight areas of ongoing research with the most potential. These are summarized in Figure 1.
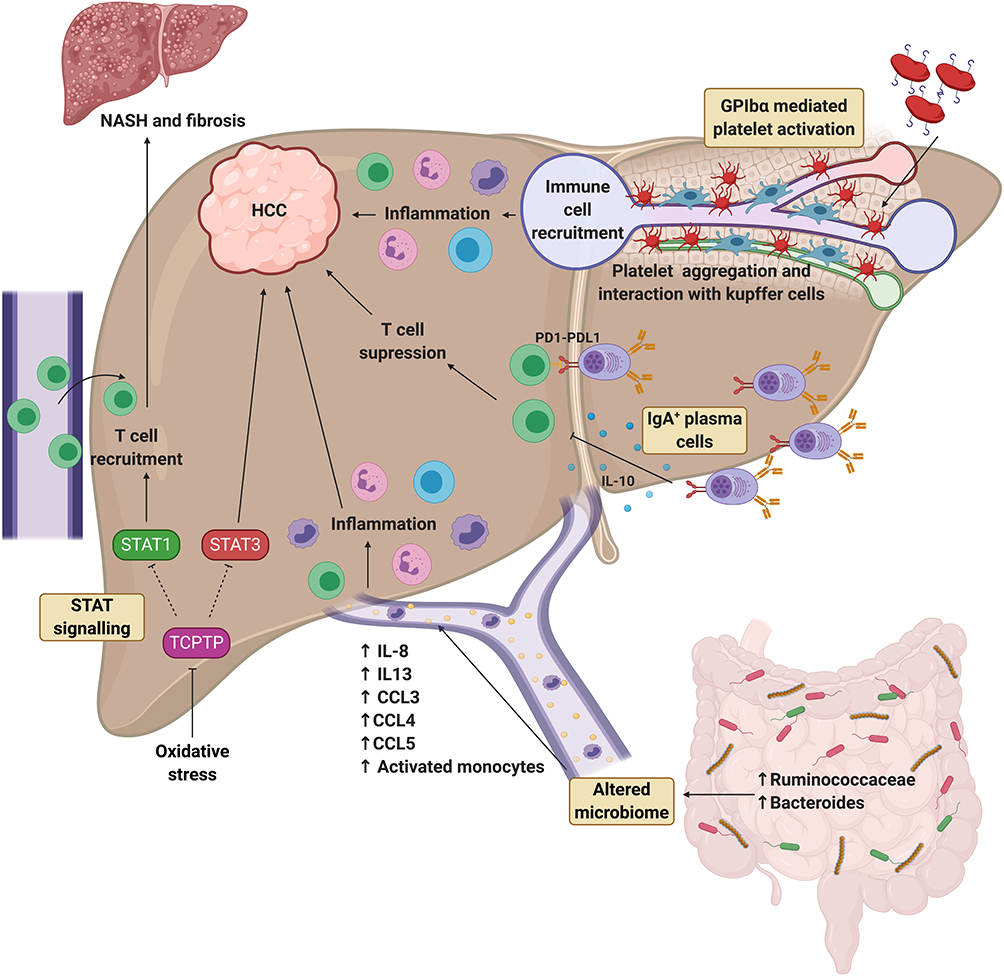 |
Figure 1 Recent progress in understanding the pathogenesis of NAFLD-associated HCC. Differential role of STAT1 and STAT3 signalling. Oxidative stress induces inactivation of TCPTP, which promotes STAT1 and STAT3 signalling. STAT1 signalling is responsible for T cell recruitment and progression to NASH and fibrosis, whereas STAT3 signalling is responsible for the development of HCC independent to NASH and fibrosis. Role of the altered microbiome. Cirrhotic patients with NAFLD-HCC have been found to have increased Bacteroides and Ruminococcaceae bacteria compared to patients with NAFLD cirrhosis without HCC. This profile is associated with high systemic levels of IL-8, IL-13, CCL3, CCL4, CCL5 and more activated monocytes in the circulation, which may be driving hepatic inflammation and subsequent HCC development. Role of IgA+ plasma cells. IgA+ plasma cells have been found to accumulate in NASH fibrosis and suppress CD8+ T cells via PD-L1 and IL-10 expression resulting in HCC development. Role of GPIbα platelet activation. Platelet activation and aggregation via GPIbα is elevated in NASH compared to steatosis and drives hepatic inflammation and HCC development via interaction with Kupffer cells and promotion of immune cell recruitment. Created with BioRender.com. Abbreviations: CCL, chemokine (C-C motif) ligand; GPIbα, glycoprotein Ibα; HCC, hepatocellular carcinoma; IL, interleukin; NAFLD, nonalcoholic fatty liver disease; NASH, nonalcoholic steatohepatitis; PD-L1, programme cell death ligand 1; STAT, signal transducer and activator of transcription; TCPTP, T cell protein tyrosine phosphate. |
Sexual Dimorphism in NAFLD-HCC
There is evidence of sexual dimorphism in the prevalence of NAFLD and HCC.31,32 Epidemiological studies indicate that the prevalence of NAFLD may be higher in men compared to pre-menopausal women.32,33 Gender-specific biological factors such as body fat composition, lipid metabolism and sex hormones likely contribute to this dimorphism in NAFLD.33 Similarly, the prevalence of HCC is higher in men compared to women with the proportional difference varying according to the underlying etiology.34 A combination of biological factors such as MyD88-dependent interleukin (IL) 6 production, sex hormones, adiponectin production and sex-related co-morbidities as well as behavioural factors such as smoking likely all contribute.31,35–38 Importantly this sexual dimorphism gives us a naturally occurring disease model which identifies important underlying biological mechanisms driving HCC development in NAFLD. Further work however is needed to translate these findings into strategies to prevent or treat NAFLD-HCC.
Genetic and Epigenetic Drivers of NAFLD-HCC
A genetic predisposition to HCC has long been recognised and we have long anticipated that identifying genetic drivers will aid early detection and preventive strategies. To date, these goals have proved elusive. A number of single nucleotide polymorphisms (SNPs) and epigenetic mechanisms have been identified as having roles in the pathogenesis of NAFLD-HCC, although no single factor has changed clinical practice.
Single Nucleotide Polymorphisms
The SNPs presently identified as predisposing to HCC development in NAFLD, essentially increase the level of fat and the severity of NAFLD. Patatin-like phospholipase domain containing 3 (PNPLA3) hydrolyses triglycerides and retinyl esters, with the PNPLA3 (rs738409 c.444 C>G, p.I148M) SNP encoding a methionine substitution in the protein which delays its proteasomal degradation and hampers lipid mobilisation. The rs58542926 c.449 C>T, p.Glu167Lys SNP in the gene encoding Transmembrane 6 superfamily member 2 (TM6SF2) accelerates degradation of the protein which regulates lipoprotein export. SNPs in these two genes, in addition to one in the phospholipid remodelling gene, membrane bound 0-acyltransferase domain containing 7 (MBOAT7; rs641738), have been linked to NAFLD progression and development of HCC.39–48 PNPLA3 rs738409 confers an increased risk of steatohepatitis and fibrosis in both European and Japanese populations and is an independent risk factor for the development of HCC.41,44,45 MBOAT7 rs641738 confers risk of fibrotic progression in a number of etiologies of chronic liver disease, including NAFLD and has been shown to be independently associated with the development of HCC even in the absence of cirrhosis.39,40,43,47 TM6SF2 rs58542926 is again associated with NAFLD progression to fibrosis but has conflicting reports of its independent association with HCC.42 Although a recent meta-analysis of 5 studies including a total of 6873 patients has confirmed an independent association,46 the requirement for such a large number of patients to confirm the risk does draw attention to some of the limitations of the predictive role of individual SNPs. These SNPs do not predispose to a distinct monogenic condition. They are instead modifiers of a complex, polygenic disease trait - that is subject both to other unknown genetic modifiers as well as fundamental environmental influences. The clinical value of any modifying risk factor needs to be considered in terms of its sensitivity and specificity, often expressed as the positive and negative predictive values. The positive predictive value reports the probability that subjects with the risk allele truly have HCC. As there are so many individuals with the risk alleles who do not have HCC, the positive predictive value of these single SNPs is insufficient to be of any clinical use. The negative predictive value reports the probability that subjects without the risk allele truly do not have HCC. For PNPLA3 rs738409, the negative predictive value has been reported as over 80% for patients with cirrhosis and high as 97% in patients with NAFLD-HCC in the absence of cirrhosis.49 Negative predictive values may have value identifying those who do not need surveillance, rather than those who do. Going forward, both sensitivity and specificity may be improved using combinations of SNPs and Bianco et al have recently reported the development of a polygenic risk score, combining SNP data from PNPLA3, TM6SF2, and MBOAT7, as well from glucokinase regulator (GCKR) predicting an elevated risk, with that from another gene called 17β-hydroxysteroid dehydrogenase type 13 (HSD17B13), which suppresses risk.50
Epigenetics – Non-Coding RNAs
Non-coding RNAs (ncRNAs) are an important group of epigenetic regulators. Their main function is to regulate gene expression, which is achieved by interactions with DNA and RNA causing alterations in gene transcription and translation.51 ncRNAs are classified by their size into three main groups; long ncRNA (lncRNAs), small ncRNAs and micro ncRNAs (miRNAs).52 The involvement of lncRNAs and miRNAs in NAFLD-HCC pathogenesis has recently become apparent and is a rapidly developing area of research, with potential diagnostic and therapeutic applications. Levels of miR-122, miR-192 and miR-194 miRNAs are all reported decreased in both NAFLD and HCC.53–58 Mechanistically these miRNAs are thought to alter hepatic lipid metabolism and enhance free fatty acid oxidation contributing to NAFLD progression, while also interfering with the c-Myc and epithelial–mesenchymal transition (EMT) pathways that promote HCC.59 Decreased expression of these miRNAs is associated with a poorer prognosis in HCC and quantification of their levels in tissues have been proposed as both diagnostic and prognostic biomarkers.54,55,58 miR-21, miR-34a, miR-16, miR-375, miR-10b, miR-155 and miR-15b have also been linked to both the pathogenesis of NAFLD and HCC.60 Importantly these miRNAs can be found in the blood of patients in the form of cell-free RNA (cfRNA) with many of these being differentially expressed in NAFLD compared to HCC making these promising diagnostic biomarkers.60,61 Furthermore, many of these circulating miRNAs have been shown to accurately select for HCC when combined with other clinical or biochemical parameters as well as correlate with disease stage in various retrospective studies.61
Increased tissue expression of the highly up-regulated in liver cancer (HULC) lncRNA and the metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) lncRNA have been observed in both NAFLD and HCC.62–67 HULC is thought to contribute to NAFLD progression through its effect on mitogen-activated protein kinase (MAPK) signalling and promote HCC proliferation and metastasis through the miR-200a-3p/ZEB1 EMT pathway.63,65,68 MALAT1 promotes NAFLD fibrosis via the silent information regulator 1 (SIRT1), transforming growth factor beta 1 (TGF-β1) pathways in hepatic stellate cells, while promoting HCC growth via upregulation of serine/arginine-rich splicing factor 1 (SRSF1) and the activation of Wnt signalling.64,67 MALAT1 appears to be a sensitive marker of human HCC with overexpression corresponding to recurrence after liver transplantation.69,70 Similar to miRNAs circulating lncRNAs are also found in HCC patients and have the potential to be used as diagnostic biomarkers.61 Circulating levels of IncRNA-CTBP, LINC00151, RP11-160H22.5, XLOC014171 and LINC00974 have all been found to differentiate between HCC and chronic liver disease.61
Epigenetics – DNA Methylation
Dysregulation in DNA methylation has been increasingly recognised as an important driver of carcinogenesis.71 The streptozotocin + high-fat diet (STAM) model of NASH mimics NASH-associated HCC in mice using a combination of streptozotocin-induced type 1 diabetes and high-fat diet-induced steatosis giving rise to HCC at around 16 weeks of age.72 A recent study using this model revealed that increasing hypomethylation and overexpression of the tubulin beta 2B class IIB (Tubb2b) gene occurred during the development and progression of HCC.73 Tubb2b overexpression was also found in the human HepG2 cell line suggesting this could be a potential biomarker or therapeutic target.73 DNA methylation may also be an important biomarker for identifying NAFLD progression. DNA methylation patterns from liver biopsies are known to correlate to NAFLD severity.74,75 Importantly the methylation of circulating cell-free DNA (cfDNA) can be analysed. Hardy et al demonstrated that plasma DNA methylation of peroxisome proliferation-activated receptor gamma (PPARγ) corresponds to NAFLD stage and changes in hepatocellular DNA methylation.76 Although this requires further validation this identifies circulating DNA methylation as a potential biomarker to identify patients with advanced NAFLD. Although this study did not specifically address NAFLD-HCC risk, cases with advanced fibrosis may also be at greater risk of developing HCC. Further studies with greater longitudinal follow-up will be required to address this.
Genetics and Epigenetics – Future Opportunities
Many of these genetic and epigenetic factors associate with both NAFLD progression and the presence of NAFLD-HCC, identifying lipid handling, cell proliferation, fibrogenesis and DNA methylation mechanisms as being involved in HCC development, with the potential for exploitation as clinically relevant biomarkers or therapeutic strategies. The precise interplay between these factors and the pathogenesis of NAFLD-HCC remains unclear and further research is needed to uncover the complex underlying biology.
Many of these epigenetic markers are specific to HCC, have prognostic value and can be isolated from peripheral blood giving these the potential to be used as biomarkers or “liquid biopsies” for surveillance, diagnosis and prognosis.59,77,78 Multiple ncRNAs can be found in the blood of NAFLD and HCC patients and have been shown to accurately select for HCC in retrospective studies, especially when combined with other clinical or biochemical parameters.61 Further larger prospective studies are needed to validate these as useful biomarkers for either surveillance and/or diagnosis of HCC.
From a therapeutic perspective, several preclinical studies have demonstrated that restoration of miRNAs in HCC can suppress tumour progression and improve chemosensitivity.79,80 A Phase I clinical trial of a miRNA mimic showed evidence of efficacy in a subset of HCC patients, but was stopped due to serious immune-related adverse events.81 Further work is needed to develop targeted genetic and epigenetic-based therapies which are both effective against HCC and have an acceptable safety profile.
Role of the Microbiome in NAFLD-HCC
Microbiome dysbiosis has been well described in obesity, T2DM, NAFLD and alcoholic liver disease.82–85 Increased gut permeability, translocation of lipopolysaccharides, immune activation and altered bile acid signalling likely all contribute to liver inflammation and fibrosis in NAFLD.29 An association between microbiome dysbiosis and NAFLD-HCC has also recently been reported.86 Within their gut microbiome, cirrhotic NAFLD-HCC patients have increased Bacteroides and Ruminococcaceae populations compared to patients with NALFD cirrhosis and no HCC.86 The microbiota profile found in NAFLD-HCC patients correlated with higher levels of interleukin (IL) 8, IL-13, chemokine (C-C motif) ligand (CCL) 3, CCL4 and CCL5, as well as activated circulating monocytes, in blood. While these HCC-associated changes in microbiota may be bystanders or consequences of the cancer, it is also possible that they precede the development of HCC, exacerbating inflammation and driving hepatocarcinogenesis (Figure 1).86 As the reported study was relatively small and was restricted to patients with similar ethnicity, diet and body mass index, larger and more heterogeneous cohort studies will be needed to confirm associations and explore the microbiome signatures as biomarkers of risk, diagnosis and prognosis in HCC.86 Studies using pre-clinical models to explore the causal relationship and mechanism linking microbiota to HCC are also needed. If a causal relationship is confirmed it is hoped that this may pave the way for future therapeutic approaches aimed at modifying the microbiome for the prevention or treatment of NAFLD-HCC.
Signalling Pathways – The Role of STAT-3 in NAFLD-HCC
The interleukin-6 (IL-6), janus-activated kinase (JAK), and signal transducer and activator of transcription (STAT) signalling pathways are established as drivers in the development and progression of HCC.87–89 Recently, it has been suggested that there are differences in obesity-related STAT signalling driving NASH or HCC.90 Using high-fat diet (HFD) and choline-deficient high-fat diet (CD-HFD) models of NAFLD, it was shown that the oxidative hepatic environment in obesity increased STAT-1 and STAT-3 signalling, via the inactivation of STAT-1 and STAT-3 phosphatase T cell protein tyrosine phosphate (TCPTP), promoting hepatic T cell recruitment, NASH, fibrosis and HCC. Using a series of deletion and enhancement experiments, STAT-1 signalling was identified as responsible for T cell recruitment and the development of NASH and fibrosis but not HCC, while STAT-3 signalling was responsible for the development of HCC independent of NASH and fibrosis (Figure 1).90 The identification of a pathway central to the development of HCC but not the fibrotic healing response makes it attractive to explore as a therapeutic target. The androgen receptor-driven oncogene, cell cycle-related kinase (CCRK) has likewise been identified as a driver of HCC in a NAFLD model by its induction of STAT-3 and the mTORC1/4E-BP1/S6K/SREBP1 pathway.91 Interestingly, CCRK knockdown resulted in protection against the development of both NASH and HCC although STAT-1 signalling was not investigated.91 While confirmatory studies in human NAFLD are needed to confirm the importance of STAT-1 and STAT-3 inhibition in NASH, fibrosis and HCC, respectively, a phase I clinical trial of an oral STAT-3 inhibitor (OPB-111077) in patients with advanced HCC after failing sorafenib has demonstrated that the drug is reasonably well tolerated.92 While the focus in these trials is on anti-tumour activity, the recent mechanistic work suggesting a role for STAT-3 in the development of HCC suggests that its inhibition may be of value in the early or pre-malignant stages of HCC.
The Role of the Immune Response in the Development of NAFLD-HCC
The current knowledge of the roles of the immune response in NAFLD and HCC has been comprehensively summarized elsewhere.29 In brief, chronic inflammation - in part driven by damage-associated molecular patterns (DAMPs) and pathogen-associated molecular patterns (PAMPs) - appear to be a significant driver of hepatocarcinogenesis.93,94 Immune escape from anti-tumour responses is also vital, with populations of T regulatory cells (Tregs) and myeloid-derived suppressor cells (MDSCs) exerting immunosuppressive effects on cytotoxic CD8+ T cells and natural killer (NK) cells.29 The recruitment of these immune cells into the liver is an important and potentially targetable step in the pathogenesis of NASH and HCC, with a recent study identifying platelets as playing a key role worthy of special mention.95 Malehmir et al demonstrated that platelet number, activation and aggregation in the liver were increased in NASH but not steatosis, with antiplatelet therapies limiting immune cell infiltration and preventing the development of NASH and HCC in the CD-HFD model of NASH (Figure 1). Kupffer cells and platelet glycoprotein Ibα (GPIbα) were shown to be key mediators, while platelet inhibitors (aspirin and ticagrelor) were proposed as treatments to suppress NASH and prevent the development of HCC. A small pilot study in patients starting anti-platelet therapy at the time of stenting for coronary artery disease, confirmed a significant reduction in liver fat mass and liver volume after 6 months using imaging techniques, compared to baseline and compared to patients not treated with anti-platelet therapy.95 While encouraging, larger studies are needed to confirm the potential of anti-platelet therapy in preventing NAFLD progression and HCC.
The role of adaptive immunity in the development of NAFLD-HCC has also become apparent. Shalapour et al demonstrated that a population of IgA+ plasma cells accumulates in NASH fibrosis and contributes to the development of NAFLD-HCC by suppressing CD8+ T cells via programmed cell death ligand 1 (PD-L1) and interleukin 10 (IL-10) expression (Figure 1).96 Given that IgA regulates mucosal immunity these immunosuppressive IgA+ plasma cells may be a key effector in the gut microbiome – liver axis that contributes to hepatocarcinogenesis. Further work is needed to confirm this in humans.
Biomarkers for Surveillance, Diagnosis and Prognosis
Effective HCC surveillance strategies and early diagnostic tools represent huge areas of unmet need. Surveillance with 6-monthly abdominal ultrasound has benefit in patients with advanced fibrosis and cirrhosis,21–24 but despite this still has multiple limitations and questionable cost-effectiveness.97 Identifying NAFLD patients with fibrosis remains challenging due to the large size of the at-risk population and the variable natural history of NAFLD progression.14 Therefore, better biomarkers are needed to identify NAFLD patients with advanced fibrosis who would benefit from HCC surveillance, and to detect early-stage HCC. These biomarkers should be inexpensive, quick, easily accessible and have superior predictive values to current biomarkers. Here we will highlight the most promising recent advances in biomarker development. These are also summarized in Figure 2.
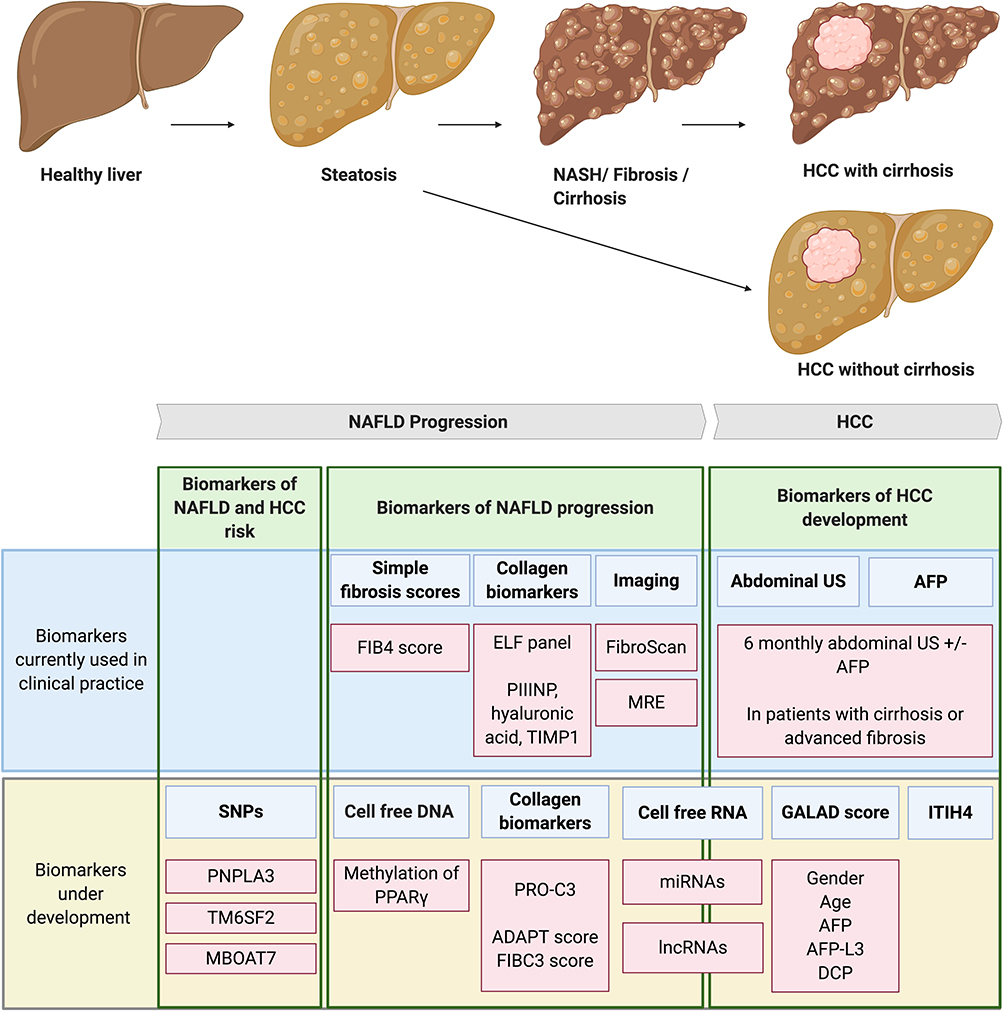 |
Figure 2 Biomarkers of NAFLD progression and HCC development. NAFLD is a spectrum of liver disease ranging from steatosis to NASH, fibrosis and eventually cirrhosis. HCC mainly develops on the background of liver cirrhosis however between 25–50% of HCC develops in the absence of cirrhosis. Biomarkers of NAFLD and HCC risk. SNPs in PNPLA3, TM6SF2 and MBOAT7 genes are associated with both NAFLD progression and the development of NAFLD-HCC. TM6SF2 in particular is an independent risk factor for HCC development in the absence of cirrhosis. The predictive values of these SNPs are insufficient to be used in clinical practice on their own, however risk stratification tools may be developed using combinations of SNPs. Biomarkers of NAFLD progression. Biomarkers currently used in clinical practice to detect NAFLD fibrotic progression include; simple fibrosis scores such as the FIB4 score, panels using collagen biomarkers such as the ELF panel and imaging techniques such as FibroScan and MRE. Biomarkers currently under development include; PPARγ methylation of cell-free DNA, scoring systems using the PRO-C3 collagen neo-epitope (ADAPT and FIBC3) and circulating cell-free RNA. Biomarkers of HCC development. Current clinical guidelines recommend screening for HCC development with 6-monthly abdominal US with or without AFP measurements in patients with cirrhosis or advanced fibrosis. Biomarkers being developed to better detect HCC include; the GALAD score, measurement of serum ITIH4 and detection of cell-free RNA. Created with BioRender.com. Abbreviations: AFP, alpha-fetoprotein; AFP-L3, alpha-fetoprotein L3; DCP, des-gamma-carboxy-prothrombin; ELF, enhanced liver fibrosis; FIB4, fibrosis-4; HCC, hepatocellular carcinoma; ITIH4, inter-alpha-trypsin inhibitor heavy chain 4; lncRNAs, long non-coding RNAs; MBOAT7, membrane-bound 0-acyltransferase domain containing 7; miRNAs, micro non-coding RNAs; MRE, magnetic resonance elastography; NAFLD, nonalcoholic fatty liver disease; NASH, nonalcoholic steatohepatitis; PNPLA3, patatin-like phospholipase domain containing 3; PIIINP, type III procollagen peptide; PPARγ, peroxisome proliferation-activated receptor gamma; PRO-C3, N-terminal type III collagen propeptide; SNPs, single nucleotide polymorphisms; TIMP1, tissue inhibitor of metalloproteinases 1; TM6SF2, transmembrane 6 superfamily member 2; US, ultrasound. |
Biomarkers of NAFLD Progression
Current biomarkers used in routine clinical practice to identify fibrotic progression in NAFLD include; simple scores such as the fibrosis-4 (FIB4) index, specific fibrosis panels such as the enhanced liver fibrosis (ELF) panel and imaging biomarkers such as FibroScan.98 Liver biopsy remains the gold standard for identifying fibrotic progression however is limited by its invasiveness and sample variability.98 Advanced imaging techniques such as magnetic resonance elastography (MRE) have superior performance but are limited to clinical trials due to high costs and limited availability.98
Collagen Neo-Epitope PRO‐C3 in the FIBC3 and ADAPT Scores
N-terminal type III collagen propeptide (PRO-C3), a marker of type III collagen formation, is thought to correspond to hepatic fibrogenesis and can be measured using an enzyme-linked immunosorbent assay on peripheral blood. Two recent studies have incorporated PRO-C3 into composite scores to assess for advanced fibrosis in large NAFLD cohorts with extensive clinical and liver biopsy data. The ADAPT score, based on age, presence of diabetes, PRO-C3 and platelet count, correlated with fibrosis stage, exhibiting an area under the receiver operating curve (AUROC) of 0.86 in the derivation and 0.87 in the validation cohorts, respectively.99 The FIBC3 score, based on age, body mass index, presence of diabetes, platelet count and PRO-C3, correlated with fibrosis stage and was associated with the presence of advanced fibrosis with a weighted AUROC of 0.89 and 0.83 in independent derivation and validation cohorts, respectively.100 Both scores were superior to current ‘simple scores’, such as FIB-4.99,100 While further independent validation of both scores are required, the FIBC3 and ADAPT scores offer the potential to more easily identify NAFLD patients with advanced fibrosis and enter them into the HCC surveillance programme. Comparisons with imaging-based biomarkers, as well exploring its usefulness in high-risk populations such as patients with T2DM, would also be valuable.
Experimental Biomarkers of NAFLD Progression
As discussed earlier the role of epigenetic factors such as ncRNAs and DNA methylation in the pathogenesis of NAFLD has recently become apparent and has identified circulating cfDNA and cfRNA as potential biomarkers of NAFLD progression. For example levels of circulating miRNA-122 and methylation of PPARγ in circulating cfDNA have been correlated to NAFLD progression and offer promise as biomarkers.53,76 Further prospective studies evaluating these biomarkers are awaited.
HCC Biomarkers
Serum Inter-Alpha-Trypsin Inhibitor Heavy Chain 4 (ITIH4)
Serum inter-alpha-trypsin inhibitor heavy chain 4 (ITIH4) is another potential biomarker of both NAFLD progression and HCC development.101 A preclinical study using a pig model of NAFLD demonstrated that ITIH4 expression was increased in both hepatocytes and HCC cells in NAFLD and that serum ITIH4 levels correlated to both NAFLD progression and HCC development. Serum ITIH4 was also elevated in patients with NAFLD-HCC compared to NASH and steatosis in a small patient cohort.101 As for ADAPT, further validation of ITIH4 as a biomarker guiding HCC surveillance is required.
The GALAD Score
The GALAD score is based on gender, age, alpha-fetoprotein (AFP), AFP-L3 and Des-gamma-carboxy-prothrombin (DCP) and has shown promise as a sensitive biomarker for HCC.102,103 In a case-control study, the GALAD score detected HCC with an AUROC of 0.96.103 Importantly in a small prospective study, the GALAD score was higher in patients with NASH who developed HCC compared to those who did not develop HCC up to 1.5 years before HCC diagnosis.103 Although large prospective validation studies using the GALAD score as a surveillance tool are awaited, the cumulative data are compelling and in March 2020 the Food and Drug Administration (FDA) granted breakthrough device designation to the GALAD Score for the early diagnosis of HCC, with its marketing by Roche anticipated.104
HCC Arising in the Absence of Cirrhosis
HCC occurring in the absence of cirrhosis is a significant challenge. A reported 25–50% of NAFLD-HCC develop in the absence of cirrhosis, meaning few are diagnosed early.4,18–20 While these individuals with better liver reserve may be more likely to be offered curative resection, their late stage at presentation contributes to poorer prognosis overall.4,18–20 HCC surveillance in NAFLD patients without advanced fibrosis or cirrhosis is currently not recommended. The incidence of around 0.01% in this population is so small, surveillance - despite the increased chance of surgical cure with early detection, is far from cost-effective.97 It is hoped that by better understanding the pathogenesis of non-cirrhotic NAFLD-HCC biomarkers to identify those at risk will be identified. As discussed above there has been extensive work investigating the genetic and epigenetic drivers of HCC in NAFLD. Their use as biomarkers promises potential as a risk stratification tool to identify those at high risk of developing non-cirrhotic HCC whom will benefit from regular surveillance. With advances in the understanding of disease pathogenesis, in combination with risk calculation tools based on genotypes combined with clinical variables, there is hope that cost-effective approaches will emerge.97,105
Prevention of NAFLD-HCC
Reducing the risk of HCC development in the growing NAFLD patient population needs to be a priority. As obesity and T2DM are risk factors for NAFLD progression as well as independent risk factors for HCC development, lifestyle interventions to combat these should be at the centre of preventative strategies.106–108 Here we will discuss both lifestyle and chemoprevention strategies, which are also summarized in Table 1.
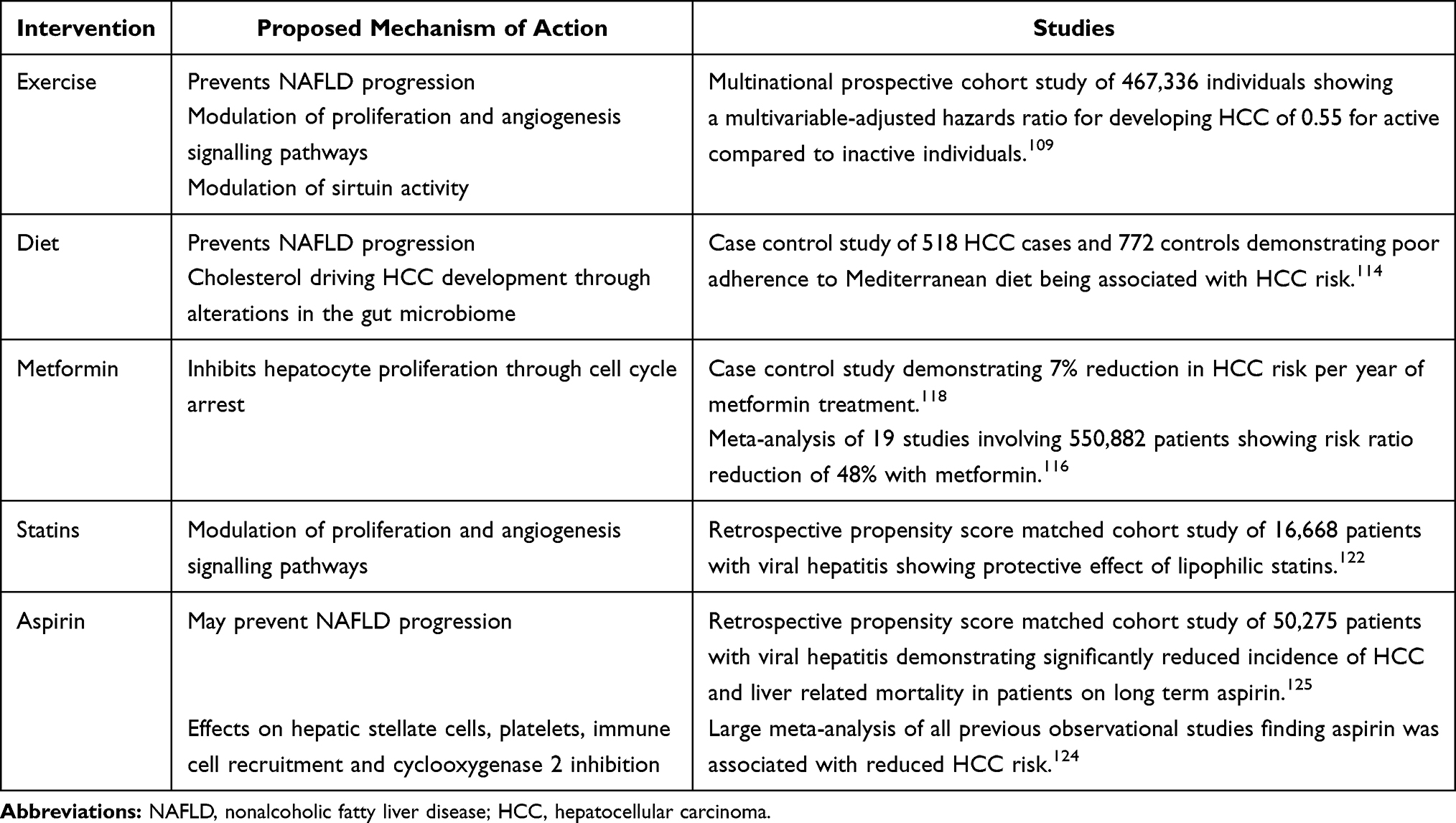 |
Table 1 Interventions That May Prevent the Development of NAFLD-Associated HCC |
Lifestyle Intervention
Exercise
A recently published large multinational cohort study has demonstrated that physical activity can reduce the risk of developing HCC.109 The investigators identified 275 HCC cases developing in a cohort of 467,336 individuals in the European Prospective Investigation into Cancer and Nutrition cohort (EPIC), followed up for a median of 14.9 years. The multivariable-adjusted hazard ratio of HCC was 0.55 (95% CI 0.38–0.80) comparing active and inactive individuals and 0.50 (95% CI 0.33–0.76) for those reporting >2 hours/week vigorous activity compared to those with no vigorous activity. This reduction was found to be independent of other liver cancer risk factors, as well as age, gender, body weight, alcohol consumption and smoking. Preclinical studies suggest exercise attenuates HCC progression by altering key signalling pathways in proliferation and angiogenesis.110,111 Bianchi et al have recently taken this forward in an exercise study using nuclear factor kappa B subunit 1 knockout (nfkb1-/-) mice, which develop inflammation, steatosis and cancer with age.112 They found that 3 months of moderate aerobic exercise was sufficient to suppress inflammatory liver disease and cancer.112 The exercise intervention reversed steatosis within the liver and reduced inflammation, oxidative damage and cellular senescence. Changes in the activity of sirtuin, an enzyme that deacetylates transcription factors that contribute to cellular regulation, were proposed as underpinning the changes seen.112
Dietary Prevention
Together with exercise, a balanced and healthy diet is the cornerstone of NAFLD management. In particular, the Mediterranean diet which is rich in fibre, unsaturated fats, vitamins and natural anti-oxidants and low in cholesterol has been reported to be beneficial in NAFLD.113 Studies also support the Mediterranean diet as being protective in HCC.114 In a case–control study of 518 HCC cases and 772 controls poor adherence to the Mediterranean diet was associated with HCC risk with this risk being amplified in cases with underlying chronic HBV or HCV infection.114 A recent study using the high-fat/high cholesterol (HFHC) diet model of NAFLD-associated HCC in mice demonstrated that cholesterol drives HCC development by altering the gut microbiota.115 They found that only mice fed with the high cholesterol diet developed HCC on top of NAFLD and that this was associated with a defined change in the gut microbiota and metabolites. They showed that the change in gut microbiota and metabolites may be driving HCC by transplanting the fecal microbiota from HFHC fed mice into germ-free mice fed with normal chow who then developed NAFLD and increased hepatocyte proliferation.115
Chemoprevention
Anti-Diabetic Drugs
The chemopreventive properties of metformin have been demonstrated in a number of large studies.116,117 Metformin may well have chemoprotective properties beyond its effect on diabetic control, as protection against HCC development is not conferred by other hypoglycemic drugs such as sulphonylureas and insulin.117 In a large case–control study a 7% reduction in HCC risk for each year a diabetic patient was on metformin was observed.118 Furthermore, a large meta-analysis of 19 studies involving 550,882 patients found that metformin reduced the risk ratio of HCC development by 48%.116 Mechanistically metformin is thought to have direct anti-tumour effects. In vitro studies demonstrate that metformin inhibits hepatocyte proliferation by inducing cell cycle arrest at G0/G1 through p21/Cip1 and p27/Kip1 upregulation and cyclin D1 downregulation.118 It is worth also noting that metformin use may be associated with poorer responses to sorafenib as an anti-cancer treatment.119 It is proposed that both metformin and sorafenib exert anti-tumour effects via inhibition of the mammalian target of rapamycin (mTOR), with metformin users who develop HCC, developing tumours with inherent mTOR resistance.119
Statins
Statins have also been shown to reduce cancer incidence, attributed to their tumour-suppressive actions on a number of proliferation and angiogenic pathways.120–123 A large propensity-score matched cohort study of patients with chronic HBV and HCV infection has suggested that not all statins are equal, as HCC protection was observed from lipophilic statins such as atorvastatin and simvastatin, rather than from hydrophilic statins such as pravastatin and rosuvastatin.122 The primary goal of these drugs is to reduce cardiovascular risk and as yet, there is no prospective evidence supporting the use of lipophilic statins over hydrophilic statins in patients with NAFLD.
Aspirin
NAFLD patients commonly take aspirin for either primary or secondary prevention of cardiovascular disease, however recent studies reveal that aspirin may also be beneficial in the treatment of NAFLD and prevention of HCC.124–126 Aspirin is thought to ameliorate NAFLD progression and prevent HCC development through effects on hepatic stellate cells, cyclooxygenase 2 inhibition and immune cell recruitment.95,127–129 A prospective cohort study of 361 biopsy-proven NAFLD patients showed a significantly lower odds ratio of progressing to NASH or fibrosis (0.68 and 0.54, respectively) in patients taking aspirin compared to those not taking aspirin.126 A large propensity score-matched cohort study of 50,275 patients with chronic HBV or HCV infection demonstrated a significantly lower incidence of HCC and liver-related mortality in patients taking aspirin compared to patients not taking aspirin.125 A subsequent meta-analysis confirmed this protective effect.124 Although these studies are encouraging NAFLD patients are generally underrepresented in these primary prevention studies, therefore care is needed when extrapolating the results. Further studies powered to detect the effect on HCC risk in NAFLD specifically are awaited.
Combination Preventive Strategies
While further data with metformin, anti-platelet drugs and lipophilic statins are awaited, to assess benefits and risks, lifestyle interventions, tight glycaemic control and statins – which are all central to the management of NAFLD – should be adhered to as a matter of priority for HCC prevention.130 Research into methods to better implement and ensure lasting adherence to lifestyle interventions in NAFLD may be very worthwhile.
Systemic Therapies for NAFLD-HCC
The Barcelona Clinic Liver Cancer (BCLC) algorithm is widely used to guide treatments for patients with HCC. Patients with early localized disease, good liver function and good performance status (BCLC 0 and A) may be offered curative treatments, such as liver resection, transplantation or ablation.21,22 Similarly, fit patients with more advanced disease confined to their liver may be offered locoregional therapies, such as transarterial chemoembolization (TACE).21,22 NAFLD-HCC patients, however, tend to present with advanced disease and/or have poor performance status meaning the majority are staged as BCLC C or D.4,19 For BCLC C, systemic therapies are advised first line – although the evidence these benefit patients with NAFLD-HCC is limited. If patients have small but growing tumours, with preserved liver function, ablation or TACE might be considered. Realistically though, these patients may live for longer receiving best supportive care.4,19,21,22,26
The Changing Landscape and Immuno-Oncology Therapies
Sorafenib has been the first-line systemic therapy for HCC for the last decade, with Lenvatinib coming on to the scene as an equally effective alternative – with a different side effect profile, over the last 2 years.131,132 Although the efficacy of sorafenib in NAFLD-HCC is reportedly less than that seen in patients with HCV-HCC,133 it is has been tolerated in patients with NAFLD-HCC in real-life practice, with some successes.134 Elderly patients with NAFLD were not represented in the Lenvatinib study,131 but its use has been adopted in some centres, with real-world data anticipated. However, changes in the immune-oncology field with relevance to patients with HCC are having a major impact. Both the programmed cell death protein (PD-1) checkpoint inhibitors, nivolumab and pembrolizumab, are licensed in the USA for second-line therapy for advanced HCC, based on data from non-randomized clinical trials.135,136 As a first line therapy, the randomized study evaluating nivolumab versus sorafenib in patients with unresectable HCC did not achieve statistical significance for its primary endpoint of overall survival.137,138 However, the combination of the vascular endothelial growth factor (VEGF) monoclonal antibody bevacizumab with the programmed death ligand 1 (PD-L1) antibody atezolizumab has proved to be a game changer, showing superiority over sorafenib as a first line treatment.139 In this landmark study called IMbrave150, there was improved overall survival, with progression-free survival 2.5 months longer, in patients treated with the combination therapy compared to sorafenib, whilst showing a comparable side effect profile.139 IMbrave150 study patients were largely those with viral hepatitis, with less than one-third being of “non-viral” etiology.139 Subgroup analysis suggests that atezolizumab plus bevacizumab may be less effective in non-viral HCC, so whether the combination is superior to sorafenib in patients with NAFLD-HCC, especially those elderly or classed as performance status 2, is not yet known. These data are likely to emerge in the coming year or two, as will results from other trials of systemic therapies, either alone or in combination for first line and second line, that are ongoing and likely to change practice in the not too distant future.140,141
Summary of Progress and Opportunities
Due to the increasing prevalence of the metabolic syndrome, NAFLD is becoming a leading cause of HCC. Steady progress in basic and translational research is improving our understanding of the disease pathogenesis, aiding the identification and exploration of biomarker candidates, as well as strategies to prevent HCC development, some of which have been summarized here. The positive impact of exercise is perhaps one of the key take home messages, highlighting the need to try and address levels of activity in our sedentary NAFLD population. The landmark IMbrave150 study is also a highlight of 2020. Although the implications for our patients with NAFLD may not truly emerge for some time, the recent increase in numbers of clinical trials of medical therapies with positive outcomes has been highly encouraging, with the prospect of greater opportunities for life-prolonging treatments drawing closer.
Disclosure
Dr Daniel Geh is funded by the Newcastle CRUK Clinical Academic Training Programme. Professor Quentin M Anstee reports research grant funding from Abbvie, Allergan/Tobira, AstraZeneca, GlaxoSmithKline, Glympse Bio, Novartis Pharma AG, Pfizer Ltd., Vertex. He is also involved in active research collaborations (including research collaborations supported through the EU IMI2 LITMUS Consortium*) with Abbvie, Antaros Medical*, Allergan/Tobira*, AstraZeneca*, BMS*, Boehringer Ingelheim International GMBH*, Echosens*, Ellegaard Gottingen Minipigs AS*, Eli Lilly & Company Ltd.*, Exalenz Bioscience Ltd.*, Genfit SA*, Glympse Bio, GlaxoSmithKline, HistoIndex*, Intercept Pharma Europe Ltd.*, iXscient Ltd.*, Nordic Bioscience*, Novartis Pharma AG*, Novo Nordisk A/S*, One Way Liver Genomics SL*, Perspectum Diagnostics*, Pfizer Ltd.*, Resoundant*, Sanofi-Aventis Deutschland GMBH*, SomaLogic Inc.*, Takeda Pharmaceuticals International SA*; consultancy for 89Bio, Abbott Laboratories, Acuitas Medical, Allergan/Tobira, Altimmune, AstraZeneca, Axcella, Blade, BMS, BNN Cardio, Celgene, Cirius, CymaBay, EcoR1, E3Bio, Eli Lilly & Company Ltd., Galmed, Genentech, Genfit SA, Gilead, Grunthal, HistoIndex, Indalo, Imperial Innovations, Intercept Pharma Europe Ltd., Inventiva, IQVIA, Janssen, Madrigal, MedImmune, Metacrine, NewGene, NGMBio, North Sea Therapeutics, Novartis, Novo Nordisk A/S, PathAI, Pfizer Ltd., Poxel, ProSciento, Raptor Pharma, Servier, Terns, Viking Therapeutics. Speaker Abbott Laboratories, Allergan/Tobira, BMS, Clinical Care Options, Falk, Fishawack, Genfit SA, Gilead, Integritas Communications, Kenes, Medscape; royalties from Elsevier Ltd (Davidson’s Principles & Practice of Medicine textbook). Professor Quentin M Anstee and Professor Helen L Reeves receive support from the CRUK HUNTER Accelerator Award (C9380/A26813). Professor Helen L Reeves also reports speaker honoraria from Bayer and Eisai. The authors report no other conflicts of interest in this work.
References
1. Akinyemiju T, Abera S, Ahmed M, et al. The Burden of Primary Liver Cancer and underlying etiologies from 1990 to 2015 at the Global, Regional, and National Level: results from the Global Burden of Disease Study 2015. JAMA Oncol. 2017;3(12):1683–1691. doi:10.1001/jamaoncol.2017.3055
2. El-Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology. 2007;132(7):2557–2576. doi:10.1053/j.gastro.2007.04.061
3. Yang JD, Roberts LR. Hepatocellular carcinoma: a global view. Nat Rev Gastroenterol Hepatol. 2010;7(8):448–458. doi:10.1038/nrgastro.2010.100
4. Dyson J, Jaques B, Chattopadyhay D, et al. Hepatocellular cancer: the impact of obesity, type 2 diabetes and a multidisciplinary team. J Hepatol. 2014;60(1):110–117. doi:10.1016/j.jhep.2013.08.011
5. Younossi Z, Stepanova M, Ong JP, et al. Nonalcoholic steatohepatitis is the fastest growing cause of hepatocellular carcinoma in liver transplant candidates. Clin Gastroenterol Hepatol. 2019;17(4):748–755.e743. doi:10.1016/j.cgh.2018.05.057
6. Caselitz M, Masche N, Flemming P, et al. [Prognosis of hepatocellular carcinoma according to new staging classifications]. Dtsch Med Wochenschr. 2004;129(33):1725–1730. doi:10.1055/s-2004-829023. German.
7. Llovet JM, Beaugrand M. Hepatocellular carcinoma: present status and future prospects. J Hepatol. 2003;38(Suppl 1):S136–S149. doi:10.1016/S0168-8278(02)00432-4
8. Sarveazad A, Agah S, Babahajian A, Amini N, Bahardoust M. Predictors of 5 year survival rate in hepatocellular carcinoma patients. J Res Med Sci. 2019;24(1):86. doi:10.4103/jrms.JRMS_1017_18
9. El-Serag HB, Davila JA, Petersen NJ, McGlynn KA. The continuing increase in the incidence of hepatocellular carcinoma in the United States: an update. Ann Intern Med. 2003;139(10):817–823. doi:10.7326/0003-4819-139-10-200311180-00009
10. Ryerson AB, Eheman CR, Altekruse SF, et al. Annual Report to the Nation on the Status of Cancer, 1975–2012, featuring the increasing incidence of liver cancer. Cancer. 2016;122(9):1312–1337. doi:10.1002/cncr.29936
11. Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology. 2016;64(1):73–84. doi:10.1002/hep.28431
12. Anstee QM, Targher G, Day CP. Progression of NAFLD to diabetes mellitus, cardiovascular disease or cirrhosis. Nat Rev Gastroenterol Hepatol. 2013;10(6):330–344.
13. Younossi Z, Anstee QM, Marietti M, et al. Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol. 2018;15(1):11–20. doi:10.1038/nrgastro.2017.109
14. Hardy T, Oakley F, Anstee QM, Day CP. Nonalcoholic fatty liver disease: pathogenesis and disease spectrum. Annu Rev Pathol. 2016;11:451–496. doi:10.1146/annurev-pathol-012615-044224
15. McPherson S, Hardy T, Henderson E, Burt AD, Day CP, Anstee QM. Evidence of NAFLD progression from steatosis to fibrosing-steatohepatitis using paired biopsies: implications for prognosis and clinical management. J Hepatol. 2015;62(5):1148–1155. doi:10.1016/j.jhep.2014.11.034
16. Singh S, Allen AM, Wang Z, Prokop LJ, Murad MH, Loomba R. Fibrosis progression in nonalcoholic fatty liver vs nonalcoholic steatohepatitis: a systematic review and meta-analysis of paired-biopsy studies. Clin Gastroenterol Hepatol. 2015;13(4):643–654 e649. doi:10.1016/j.cgh.2014.04.014
17. Younossi ZM, Otgonsuren M, Henry L, et al. Association of nonalcoholic fatty liver disease (NAFLD) with hepatocellular carcinoma (HCC) in the United States from 2004 to 2009. Hepatology. 2015;62(6):1723–1730. doi:10.1002/hep.28123
18. Mittal S, El-Serag HB, Sada YH, et al. Hepatocellular carcinoma in the absence of cirrhosis in United States veterans is associated with nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol. 2016;14(1):124–131.e121. doi:10.1016/j.cgh.2015.07.019
19. Piscaglia F, Svegliati-Baroni G, Barchetti A, et al. Clinical patterns of hepatocellular carcinoma in nonalcoholic fatty liver disease: a multicenter prospective study. Hepatology. 2016;63(3):827–838. doi:10.1002/hep.28368
20. Stine JG, Wentworth BJ, Zimmet A, et al. Systematic review with meta-analysis: risk of hepatocellular carcinoma in non-alcoholic steatohepatitis without cirrhosis compared to other liver diseases. Aliment Pharmacol Ther. 2018;48(7):696–703. doi:10.1111/apt.14937
21. European Association for the Study of the Liver. EASL clinical practice guidelines: management of hepatocellular carcinoma. J Hepatol. 2018;69(1):182–236.
22. Heimbach JK, Kulik LM, Finn RS, et al. AASLD guidelines for the treatment of hepatocellular carcinoma. Hepatology. 2018;67(1):358–380. doi:10.1002/hep.29086
23. Omata M, Cheng AL, Kokudo N, et al. Asia-Pacific clinical practice guidelines on the management of hepatocellular carcinoma: a 2017 update. Hepatol Int. 2017;11(4):317–370.
24. Vogel A, Cervantes A, Chau I, et al. Hepatocellular carcinoma: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2019;30(5):871–873. doi:10.1093/annonc/mdy510
25. Mittal S, Sada YH, El-Serag HB, et al. Temporal trends of nonalcoholic fatty liver disease-related hepatocellular carcinoma in the veteran affairs population. Clin Gastroenterol Hepatol. 2015;13(3):594–601.e591. doi:10.1016/j.cgh.2014.08.013
26. Weinmann A, Alt Y, Koch S, et al. Treatment and survival of non-alcoholic steatohepatitis associated hepatocellular carcinoma. BMC Cancer. 2015;15(1):210. doi:10.1186/s12885-015-1197-x
27. Than NN, Ghazanfar A, Hodson J, et al. Comparing clinical presentations, treatments and outcomes of hepatocellular carcinoma due to hepatitis C and non-alcoholic fatty liver disease. Qjm. 2017;110(2):73–81. doi:10.1093/qjmed/hcw151
28. Margini C, Dufour JF. The story of HCC in NAFLD: from epidemiology, across pathogenesis, to prevention and treatment. Liver Int. 2016;36(3):317–324. doi:10.1111/liv.13031
29. Anstee QM, Reeves HL, Kotsiliti E, Govaere O, Heikenwalder M. From NASH to HCC: current concepts and future challenges. Nat Rev Gastroenterol Hepatol. 2019;16(7):411–428.
30. Rattan P, Minacapelli CD, Rustgi V. The microbiome and hepatocellular carcinoma. Liver Transpl. 2020;26(10):1316–1327. doi:10.1002/lt.25828
31. Lonardo A, Ballestri S, Chow PKH, Suzuki A. Sex disparity in hepatocellular carcinoma owing to NAFLD and non-NAFLD etiology: epidemiological findings and pathobiological mechanisms. Hepatoma Res. 2020;6:83.
32. Lonardo A, Nascimbeni F, Ballestri S, et al. Sex differences in nonalcoholic fatty liver disease: state of the art and identification of research gaps. Hepatology. 2019;70(4):1457–1469. doi:10.1002/hep.30626
33. Lonardo A, Suzuki A. Sexual dimorphism of NAFLD in adults. Focus on clinical aspects and implications for practice and translational research. Clin Med. 2020;9(5):1278. doi:10.3390/jcm9051278
34. Petrick JL, Braunlin M, Laversanne M, Valery PC, Bray F, McGlynn KA. International trends in liver cancer incidence, overall and by histologic subtype, 1978–2007. Int J Cancer. 2016;139(7):1534–1545. doi:10.1002/ijc.30211
35. Manieri E, Herrera-Melle L, Mora A, et al. Adiponectin accounts for gender differences in hepatocellular carcinoma incidence. J Exp Med. 2019;216(5):1108–1119. doi:10.1084/jem.20181288
36. Naugler WE, Sakurai T, Kim S, et al. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science. 2007;317(5834):121–124. doi:10.1126/science.1140485
37. Wu EM, Wong LL, Hernandez BY, et al. Gender differences in hepatocellular cancer: disparities in nonalcoholic fatty liver disease/steatohepatitis and liver transplantation. Hepatoma Res. 2018;4(10):66. doi:10.20517/2394-5079.2018.87
38. Yeh SH, Chen PJ. Gender disparity of hepatocellular carcinoma: the roles of sex hormones. Oncology. 2010;78(Suppl 1):172–179. doi:10.1159/000315247
39. Buch S, Stickel F, Trépo E, et al. A genome-wide association study confirms PNPLA3 and identifies TM6SF2 and MBOAT7 as risk loci for alcohol-related cirrhosis. Nat Genet. 2015;47(12):1443–1448. doi:10.1038/ng.3417
40. Donati B, Dongiovanni P, Romeo S, et al. MBOAT7 rs641738 variant and hepatocellular carcinoma in non-cirrhotic individuals. Sci Rep. 2017;7(1):4492. doi:10.1038/s41598-017-04991-0
41. Liu YL, Patman GL, Leathart JB, et al. Carriage of the PNPLA3 rs738409 C >G polymorphism confers an increased risk of non-alcoholic fatty liver disease associated hepatocellular carcinoma. J Hepatol. 2014;61(1):75–81. doi:10.1016/j.jhep.2014.02.030
42. Liu YL, Reeves HL, Burt AD, et al. TM6SF2 rs58542926 influences hepatic fibrosis progression in patients with non-alcoholic fatty liver disease. Nat Commun. 2014;5(1):4309. doi:10.1038/ncomms5309
43. Mancina RM, Dongiovanni P, Petta S, et al. The MBOAT7-TMC4 variant rs641738 increases risk of nonalcoholic fatty liver disease in individuals of European descent. Gastroenterology. 2016;150(5):1219–1230.e1216. doi:10.1053/j.gastro.2016.01.032
44. Seko Y, Sumida Y, Tanaka S, et al. Development of hepatocellular carcinoma in Japanese patients with biopsy-proven non-alcoholic fatty liver disease: association between PNPLA3 genotype and hepatocarcinogenesis/fibrosis progression. Hepatol Res. 2017;47(11):1083–1092. doi:10.1111/hepr.12840
45. Singal AG, Manjunath H, Yopp AC, et al. The effect of PNPLA3 on fibrosis progression and development of hepatocellular carcinoma: a meta-analysis. Am J Gastroenterol. 2014;109(3):325–334. doi:10.1038/ajg.2013.476
46. Tang S, Zhang J, Mei TT, et al. Association of TM6SF2 rs58542926 T/C gene polymorphism with hepatocellular carcinoma: a meta-analysis. BMC Cancer. 2019;19(1):1128. doi:10.1186/s12885-019-6173-4
47. Thabet K, Asimakopoulos A, Shojaei M, et al. MBOAT7 rs641738 increases risk of liver inflammation and transition to fibrosis in chronic hepatitis C. Nat Commun. 2016;7(1):12757. doi:10.1038/ncomms12757
48. Anstee QM, Darlay R, Cockell S, et al. Genome-wide association study of non-alcoholic fatty liver and steatohepatitis in a histologically characterised cohort. J Hepatol. 2020;73(3):505–515. doi:10.1016/j.jhep.2020.04.003
49. Anstee QM, Liu YL, Day CP, Reeves HL. Reply to: HCC and liver disease risk in homozygous PNPLA3 p.I148M carriers approach monogenic inheritance. J Hepatol. 2015;62(4):982–983. doi:10.1016/j.jhep.2014.12.028
50. Bianco C, Jamialahmadi O, Pelusi S, et al. Non-invasive stratification of hepatocellular carcinoma risk in non-alcoholic fatty liver using polygenic risk scores. J Hepatol. 2020. doi:10.1016/j.jhep.2020.11.024.
51. Geisler S, Coller J. RNA in unexpected places: long non-coding RNA functions in diverse cellular contexts. Nat Rev Mol Cell Biol. 2013;14(11):699–712. doi:10.1038/nrm3679
52. St Laurent G, Wahlestedt C, Kapranov P. The Landscape of long noncoding RNA classification. Trends Genet. 2015;31(5):239–251. doi:10.1016/j.tig.2015.03.007
53. Cheung O, Puri P, Eicken C, et al. Nonalcoholic steatohepatitis is associated with altered hepatic MicroRNA expression. Hepatology. 2008;48(6):1810–1820. doi:10.1002/hep.22569
54. Coulouarn C, Factor VM, Andersen JB, Durkin ME, Thorgeirsson SS. Loss of miR-122 expression in liver cancer correlates with suppression of the hepatic phenotype and gain of metastatic properties. Oncogene. 2009;28(40):3526–3536. doi:10.1038/onc.2009.211
55. Lian J, Jing Y, Dong Q, et al. miR-192, a prognostic indicator, targets the SLC39A6/SNAIL pathway to reduce tumor metastasis in human hepatocellular carcinoma. Oncotarget. 2016;7(3):2672–2683. doi:10.18632/oncotarget.6603
56. Liu XL, Cao HX, Wang BC, et al. miR-192-5p regulates lipid synthesis in non-alcoholic fatty liver disease through SCD-1. World J Gastroenterol. 2017;23(46):8140–8151. doi:10.3748/wjg.v23.i46.8140
57. Zarrinpar A, Gupta S, Maurya MR, Subramaniam S, Loomba R. Serum microRNAs explain discordance of non-alcoholic fatty liver disease in monozygotic and dizygotic twins: a prospective study. Gut. 2016;65(9):1546–1554. doi:10.1136/gutjnl-2015-309456
58. Zhao Y, Li F, Zhang X, et al. MicroRNA-194 acts as a prognostic marker and inhibits proliferation in hepatocellular carcinoma by targeting MAP4K4. Int J Clin Exp Pathol. 2015;8(10):12446–12454.
59. Heo MJ, Yun J, Kim SG. Role of non-coding RNAs in liver disease progression to hepatocellular carcinoma. Arch Pharm Res. 2019;42(1):48–62. doi:10.1007/s12272-018-01104-x
60. Afonso M, Rodrigues P, Simão A, Castro R. Circulating microRNAs as potential biomarkers in non-alcoholic fatty liver disease and hepatocellular carcinoma. J Clin Med. 2016;5(3):30. doi:10.3390/jcm5030030
61. Mocan T, Simão AL, Castro RE, et al. Liquid biopsies in hepatocellular carcinoma: are we winning? Clin Med. 2020;9(5):1541. doi:10.3390/jcm9051541
62. Hou Z, Xu X, Fu X, et al. HBx-related long non-coding RNA MALAT1 promotes cell metastasis via up-regulating LTBP3 in hepatocellular carcinoma. Am J Cancer Res. 2017;7(4):845–856.
63. Li SP, Xu HX, Yu Y, et al. LncRNA HULC enhances epithelial-mesenchymal transition to promote tumorigenesis and metastasis of hepatocellular carcinoma via the miR-200a-3p/ZEB1 signaling pathway. Oncotarget. 2016;7(27):42431–42446. doi:10.18632/oncotarget.9883
64. Malakar P, Shilo A, Mogilevsky A, et al. Long Noncoding RNA MALAT1 promotes hepatocellular carcinoma development by SRSF1 upregulation and mTOR activation. Cancer Res. 2017;77(5):1155–1167. doi:10.1158/0008-5472.CAN-16-1508
65. Shen X, Guo H, Xu J, Wang J. Inhibition of lncRNA HULC improves hepatic fibrosis and hepatocyte apoptosis by inhibiting the MAPK signaling pathway in rats with nonalcoholic fatty liver disease. J Cell Physiol. 2019;234(10):18169–18179. doi:10.1002/jcp.28450
66. Sookoian S, Flichman D, Garaycoechea ME, San Martino J, Castaño GO, Pirola CJ. Metastasis-associated lung adenocarcinoma transcript 1 as a common molecular driver in the pathogenesis of nonalcoholic steatohepatitis and chronic immune-mediated liver damage. Hepatol Commun. 2018;2(6):654–665. doi:10.1002/hep4.1184
67. Wu Y, Liu X, Zhou Q, et al. Silent information regulator 1 (SIRT1) ameliorates liver fibrosis via promoting activated stellate cell apoptosis and reversion. Toxicol Appl Pharmacol. 2015;289(2):163–176. doi:10.1016/j.taap.2015.09.028
68. Zhang Y, Li Z, Zhang Y, Zhong Q, Chen Q, Zhang L. Molecular mechanism of HEIH and HULC in the proliferation and invasion of hepatoma cells. Int J Clin Exp Med. 2015;8(8):12956–12962.
69. Lin R, Maeda S, Liu C, Karin M, Edgington TS. A large noncoding RNA is a marker for murine hepatocellular carcinomas and a spectrum of human carcinomas. Oncogene. 2007;26(6):851–858. doi:10.1038/sj.onc.1209846
70. Lai MC, Yang Z, Zhou L, et al. Long non-coding RNA MALAT-1 overexpression predicts tumor recurrence of hepatocellular carcinoma after liver transplantation. Med Oncol. 2012;29(3):1810–1816. doi:10.1007/s12032-011-0004-z
71. Rakyan VK, Down TA, Balding DJ, Beck S. Epigenome-wide association studies for common human diseases. Nat Rev Genet. 2011;12(8):529–541. doi:10.1038/nrg3000
72. Fujii M, Shibazaki Y, Wakamatsu K, et al. A murine model for non-alcoholic steatohepatitis showing evidence of association between diabetes and hepatocellular carcinoma. Med Mol Morphol. 2013;46(3):141–152. doi:10.1007/s00795-013-0016-1
73. Dreval K, Tryndyak V, de Conti A, Beland FA, Pogribny IP. Gene expression and DNA methylation alterations during non-alcoholic steatohepatitis-associated liver carcinogenesis. Front Genet. 2019;10:486. doi:10.3389/fgene.2019.00486
74. Murphy SK, Yang H, Moylan CA, et al. Relationship between methylome and transcriptome in patients with nonalcoholic fatty liver disease. Gastroenterology. 2013;145(5):1076–1087. doi:10.1053/j.gastro.2013.07.047
75. Zeybel M, Hardy T, Robinson SM, et al. Differential DNA methylation of genes involved in fibrosis progression in non-alcoholic fatty liver disease and alcoholic liver disease. Clin Epigenetics. 2015;7(1). doi:10.1186/s13148-015-0056-6.
76. Hardy T, Zeybel M, Day CP, et al. Plasma DNA methylation: a potential biomarker for stratification of liver fibrosis in non-alcoholic fatty liver disease. Gut. 2017;66(7):1321–1328. doi:10.1136/gutjnl-2016-311526
77. Li C, Chen J, Zhang K, Feng B, Wang R, Chen L. Progress and Prospects of Long Noncoding RNAs (lncRNAs) in Hepatocellular Carcinoma. Cell Physiol Biochem. 2015;36(2):423–434. doi:10.1159/000430109
78. Xu RH, Wei W, Krawczyk M, et al. Circulating tumour DNA methylation markers for diagnosis and prognosis of hepatocellular carcinoma. Nat Mater. 2017;16(11):1155–1161. doi:10.1038/nmat4997
79. Lou G, Song X, Yang F, et al. Exosomes derived from miR-122-modified adipose tissue-derived MSCs increase chemosensitivity of hepatocellular carcinoma. J Hematol Oncol. 2015;8(1):122. doi:10.1186/s13045-015-0220-7
80. Tsai WC, Hsu SD, Hsu CS, et al. MicroRNA-122 plays a critical role in liver homeostasis and hepatocarcinogenesis. J Clin Invest. 2012;122(8):2884–2897. doi:10.1172/JCI63455
81. Hong DS, Kang YK, Borad M, et al. Phase 1 study of MRX34, a liposomal miR-34a mimic, in patients with advanced solid tumours. Br J Cancer. 2020;122(11):1630–1637. doi:10.1038/s41416-020-0802-1
82. Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature. 2006;444(7122):1022–1023. doi:10.1038/4441022a
83. Llopis M, Cassard AM, Wrzosek L, et al. Intestinal microbiota contributes to individual susceptibility to alcoholic liver disease. Gut. 2016;65(5):830–839. doi:10.1136/gutjnl-2015-310585
84. Mouzaki M, Wang AY, Bandsma R, et al. Bile acids and dysbiosis in non-alcoholic fatty liver disease. PLoS One. 2016;11(5):e0151829. doi:10.1371/journal.pone.0151829
85. Qin J, Li Y, Cai Z, et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature. 2012;490(7418):55–60. doi:10.1038/nature11450
86. Ponziani FR, Bhoori S, Castelli C, et al. Hepatocellular carcinoma is associated with gut microbiota profile and inflammation in nonalcoholic fatty liver disease. Hepatology. 2019;69(1):107–120. doi:10.1002/hep.30036
87. Calvisi DF, Ladu S, Gorden A, et al. Ubiquitous activation of Ras and Jak/Stat pathways in human HCC. Gastroenterology. 2006;130(4):1117–1128. doi:10.1053/j.gastro.2006.01.006
88. He G, Dhar D, Nakagawa H, et al. Identification of liver cancer progenitors whose malignant progression depends on autocrine IL-6 signaling. Cell. 2013;155(2):384–396. doi:10.1016/j.cell.2013.09.031
89. He G, Yu GY, Temkin V, et al. Hepatocyte IKKbeta/NF-kappaB inhibits tumor promotion and progression by preventing oxidative stress-driven STAT3 activation. Cancer Cell. 2010;17(3):286–297. doi:10.1016/j.ccr.2009.12.048
90. Grohmann M, Wiede F, Dodd GT, et al. Obesity Drives STAT-1-Dependent NASH and STAT-3-Dependent HCC. Cell. 2018;175(5):1289–1306.e1220. doi:10.1016/j.cell.2018.09.053
91. Sun H, Yang W, Tian Y, et al. An inflammatory-CCRK circuitry drives mTORC1-dependent metabolic and immunosuppressive reprogramming in obesity-associated hepatocellular carcinoma. Nat Commun. 2018;9(1):5214. doi:10.1038/s41467-018-07402-8
92. Yoo C, Kang J, Lim HY, et al. Phase I Dose-Finding Study of OPB-111077, a Novel STAT3 inhibitor, in patients with advanced hepatocellular carcinoma. Cancer Res Treat. 2019;51(2):510–518. doi:10.4143/crt.2018.226
93. Arrese M, Cabrera D, Kalergis AM, Feldstein AE. Innate Immunity and Inflammation in NAFLD/NASH. Dig Dis Sci. 2016;61(5):1294–1303. doi:10.1007/s10620-016-4049-x
94. Park EJ, Lee JH, Yu GY, et al. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell. 2010;140(2):197–208. doi:10.1016/j.cell.2009.12.052
95. Malehmir M, Pfister D, Gallage S, et al. Platelet GPIbα is a mediator and potential interventional target for NASH and subsequent liver cancer. Nat Med. 2019;25(4):641–655. doi:10.1038/s41591-019-0379-5
96. Shalapour S, Lin X-J, Bastian IN, et al. Inflammation-induced IgA+ cells dismantle anti-liver cancer immunity. Nature. 2017;551(7680):340–345. doi:10.1038/nature24302
97. Geh D, Rana FA, Reeves HL. Weighing the benefits of hepatocellular carcinoma surveillance against potential harms. J Hepatocell Carcinoma. 2019;6:23–30. doi:10.2147/JHC.S159581
98. Wong VW-S, Adams LA, De Lédinghen V, Wong GL-H, Sookoian S. Noninvasive biomarkers in NAFLD and NASH — current progress and future promise. Nat Rev Gastroenterol Hepatol. 2018;15(8):461–478. doi:10.1038/s41575-018-0014-9
99. Daniels SJ, Leeming DJ, Eslam M, et al. ADAPT: an algorithm incorporating PRO-C3 accurately identifies patients with NAFLD and advanced fibrosis. Hepatology. 2019;69(3):1075–1086. doi:10.1002/hep.30163
100. Boyle M, Tiniakos D, Schattenberg JM, et al. Performance of the PRO-C3 collagen neo-epitope biomarker in non-alcoholic fatty liver disease. JHEP Rep. 2019;1(3):188–198. doi:10.1016/j.jhepr.2019.06.004
101. Nakamura N, Hatano E, Iguchi K, et al. Elevated levels of circulating ITIH4 are associated with hepatocellular carcinoma with nonalcoholic fatty liver disease: from pig model to human study. BMC Cancer. 2019;19(1). doi:10.1186/s12885-019-5825-8.
102. Yang JD, Addissie BD, Mara KC, et al. GALAD score for hepatocellular carcinoma detection in comparison with liver ultrasound and proposal of GALADUS score. Cancer Epidemiol Biomarkers Prev. 2019;28(3):531–538. doi:10.1158/1055-9965.EPI-18-0281
103. Best J, Bechmann LP, Sowa JP, et al. GALAD score detects early hepatocellular carcinoma in an International cohort of patients with nonalcoholic steatohepatitis. Clin Gastroenterol Hepatol. 2020;18(3):728–735.e724. doi:10.1016/j.cgh.2019.11.012
104. Roche. FDA grants breakthrough device designation for Roche’s Elecsys GALAD score to support earlier diagnosis of hepatocellular carcinoma (news release); 2020. Available from: https://www.roche.com/media/releases/med-cor-2020-03-04.htm.
105. Hassan MM, Kaseb A, Etzel CJ, et al. Genetic variation in the PNPLA3 gene and hepatocellular carcinoma in USA: risk and prognosis prediction. Mol Carcinog. 2013;52 Suppl 1:E139–E147. doi:10.1002/mc.22057
106. Nair S, Mason A, Eason J, Loss G, Perrillo RP. Is obesity an independent risk factor for hepatocellular carcinoma in cirrhosis? Hepatology. 2002;36(1):150–155. doi:10.1053/jhep.2002.33713
107. Yang JD, Ahmed F, Mara KC, et al. Diabetes is associated with increased risk of hepatocellular carcinoma in patients with cirrhosis from nonalcoholic fatty liver disease. Hepatology. 2020;71(3):907–916. doi:10.1002/hep.30858
108. El-Serag HB, Hampel H, Javadi F. The association between diabetes and hepatocellular carcinoma: a systematic review of epidemiologic evidence. Clin Gastroenterol Hepatol. 2006;4(3):369–380. doi:10.1016/j.cgh.2005.12.007
109. Baumeister SE, Schlesinger S, Aleksandrova K, et al. Association between physical activity and risk of hepatobiliary cancers: a multinational cohort study. J Hepatol. 2019;70(5):885–892. doi:10.1016/j.jhep.2018.12.014
110. Piguet AC, Saran U, Simillion C, et al. Regular exercise decreases liver tumors development in hepatocyte-specific PTEN-deficient mice independently of steatosis. J Hepatol. 2015;62(6):1296–1303. doi:10.1016/j.jhep.2015.01.017
111. Saran U, Guarino M, Rodríguez S, et al. Anti-tumoral effects of exercise on hepatocellular carcinoma growth. Hepatol Commun. 2018;2(5):607–620. doi:10.1002/hep4.1159
112. Bianchi A, Marchetti L, Hall Z, et al. Moderate exercise inhibits age-related inflammation, liver steatosis, senescence, and tumorigenesis. J Immunol. 2021;206(4):ji2001022. doi:10.4049/jimmunol.2001022
113. Anania C, Perla FM, Olivero F, Pacifico L, Chiesa C. Mediterranean diet and nonalcoholic fatty liver disease. World J Gastroenterol. 2018;24(19):2083–2094. doi:10.3748/wjg.v24.i19.2083
114. Turati F, Trichopoulos D, Polesel J, et al. Mediterranean diet and hepatocellular carcinoma. J Hepatol. 2014;60(3):606–611. doi:10.1016/j.jhep.2013.10.034
115. Zhang X, Coker OO, Chu ES, et al. Dietary cholesterol drives fatty liver-associated liver cancer by modulating gut microbiota and metabolites. Gut. 2020:
116. Ma S, Zheng Y, Xiao Y, Zhou P, Tan H. Meta-analysis of studies using metformin as a reducer for liver cancer risk in diabetic patients. Medicine (Baltimore). 2017;96(19):e6888. doi:10.1097/MD.0000000000006888
117. Zhou YY, Zhu GQ, Liu T, et al. Systematic review with network meta-analysis: antidiabetic medication and risk of hepatocellular carcinoma. Sci Rep. 2016;6:33743. doi:10.1038/srep33743
118. Chen HP, Shieh JJ, Chang CC, et al. Metformin decreases hepatocellular carcinoma risk in a dose-dependent manner: population-based and in vitro studies. Gut. 2013;62(4):606–615. doi:10.1136/gutjnl-2011-301708
119. Casadei Gardini A, Faloppi L, De Matteis S, et al. Metformin and insulin impact on clinical outcome in patients with advanced hepatocellular carcinoma receiving sorafenib: validation study and biological rationale. Eur J Cancer. 2017;86:106–114. doi:10.1016/j.ejca.2017.09.003
120. Mansourian PG, Yoneda M, Krishna Rao M, Martinez FJ, Thomas E, Schiff ER. Effects of statins on the risk of hepatocellular carcinoma. Gastroenterol Hepatol (N Y). 2014;10(7):417–426.
121. Kim G, Jang SY, Nam CM, Kang ES. Statin use and the risk of hepatocellular carcinoma in patients at high risk: a nationwide nested case-control study. J Hepatol. 2018;68(3):476–484. doi:10.1016/j.jhep.2017.10.018
122. Simon TG, Duberg AS, Aleman S, et al. Lipophilic statins and risk for hepatocellular carcinoma and death in patients with chronic viral hepatitis: results from a Nationwide Swedish Population. Ann Intern Med. 2019;171(5):318–327. doi:10.7326/M18-2753
123. Singh S, Singh PP, Singh AG, Murad MH, Sanchez W. Statins are associated with a reduced risk of hepatocellular cancer: a systematic review and meta-analysis. Gastroenterology. 2013;144(2):323–332. doi:10.1053/j.gastro.2012.10.005
124. Memel ZN, Arvind A, Moninuola O, et al. Aspirin use is associated with a reduced incidence of hepatocellular carcinoma: a systematic review and meta‐analysis. Hepatol Commun. 2020. doi:10.1002/hep4.1640.
125. Simon TG, Duberg A-S, Aleman S, Chung RT, Chan AT, Ludvigsson JF. Association of aspirin with hepatocellular carcinoma and liver-related mortality. N Engl J Med. 2020;382(11):1018–1028. doi:10.1056/NEJMoa1912035
126. Simon TG, Henson J, Osganian S, et al. Daily aspirin use associated with reduced risk for fibrosis progression in patients with nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol. 2019;17(13):2776–2784.e2774. doi:10.1016/j.cgh.2019.04.061
127. Paik YH, Kim JK, Lee JI, et al. Celecoxib induces hepatic stellate cell apoptosis through inhibition of Akt activation and suppresses hepatic fibrosis in rats. Gut. 2009;58(11):1517–1527. doi:10.1136/gut.2008.157420
128. Yoshida S, Ikenaga N, Liu SB, et al. Extrahepatic platelet-derived growth factor-β, delivered by platelets, promotes activation of hepatic stellate cells and biliary fibrosis in mice. Gastroenterology. 2014;147(6):1378–1392. doi:10.1053/j.gastro.2014.08.038
129. Foderà D, D’Alessandro N, Cusimano A, et al. Induction of apoptosis and inhibition of cell growth in human hepatocellular carcinoma cells by COX-2 inhibitors. Ann N Y Acad Sci. 2004;1028:440–449. doi:10.1196/annals.1322.052
130. European Association for the Study of the Liver (EASL); European Association for the Study of Diabetes (EASD); European Association for the Study of Obesity (EASO). EASL-EASD-EASO clinical practice guidelines for the management of non-alcoholic fatty liver disease. J Hepatol. 2016;64(6):1388–1402. doi:10.1016/j.jhep.2015.11.004
131. Kudo M, Finn RS, Qin S, et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised Phase 3 non-inferiority trial. Lancet. 2018;391(10126):1163–1173. doi:10.1016/S0140-6736(18)30207-1
132. Llovet JM, Ricci S, Mazzaferro V, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359(4):378–390. doi:10.1056/NEJMoa0708857
133. Bruix J, Cheng AL, Meinhardt G, Nakajima K, De Sanctis Y, Llovet J. Prognostic factors and predictors of sorafenib benefit in patients with hepatocellular carcinoma: analysis of two Phase III studies. J Hepatol. 2017;67(5):999–1008. doi:10.1016/j.jhep.2017.06.026
134. King J, Palmer DH, Johnson P, et al. Sorafenib for the treatment of advanced hepatocellular cancer – a UK audit. Clin Oncol. 2017;29(4):256–262. doi:10.1016/j.clon.2016.11.012
135. El-Khoueiry AB, Sangro B, Yau T, et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet. 2017;389(10088):2492–2502. doi:10.1016/S0140-6736(17)31046-2
136. Zhu AX, Finn RS, Edeline J, et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): a non-randomised, open-label Phase 2 trial. Lancet Oncol. 2018;19(7):940–952. doi:10.1016/S1470-2045(18)30351-6
137. Yau T, Park J, Finn R, Cheng A, Mathurin P, Edeline J. CheckMate 459: a randomized, multi-center phase 3 study of nivolumab vs sorafenib as first-line treatment in patients with advanced hepatocellular carcinoma. Ann Oncol. 2019;30(suppl5):v851. doi:10.1093/annonc/mdz394.029
138. Sangro B, Park J, Finn R, et al. LBA-3 CheckMate 459: long-term (minimum follow-up 33.6 months) survival outcomes with nivolumab versus sorafenib as first-line treatment in patients with advanced hepatocellular carcinoma. Ann Oncol. 2020;31:S241–S242. doi:10.1016/j.annonc.2020.04.078
139. Finn RS, Qin S, Ikeda M, et al. Atezolizumab plus bevacizumab in unresectable hepatocellular carcinoma. N Engl J Med. 2020;382(20):1894–1905. doi:10.1056/NEJMoa1915745
140. von Felden J. New systemic agents for hepatocellular carcinoma: an update 2020. Curr Opin Gastroenterol. 2020;36(3):177–183. doi:10.1097/MOG.0000000000000626
141. Kudo M. Recent advances in systemic therapy for hepatocellular carcinoma in an Aging Society: 2020 update. Liver Cancer. 2020;9(6):640–662. doi:10.1159/000511001
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.