")
Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 17
Myostatin/HIF2α-Mediated Ferroptosis is Involved in Skeletal Muscle Dysfunction in Chronic Obstructive Pulmonary Disease
Authors Zhang L , Li D, Chang C, Sun Y
Received 3 June 2022
Accepted for publication 12 September 2022
Published 24 September 2022 Volume 2022:17 Pages 2383—2399
DOI https://doi.org/10.2147/COPD.S377226
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Richard Russell
Lijiao Zhang, Danyang Li, Chun Chang, Yongchang Sun
Department of Respiratory and Critical Care Medicine, Peking University Third Hospital, Beijing, 100191, People’s Republic of China
Correspondence: Yongchang Sun, Email [email protected]
Objective: Skeletal muscle dysfunction is an important comorbidity in patients with chronic obstructive pulmonary disease (COPD), and is associated with poor quality of life and reduced survival, but the mechanisms involved remain elusive. Ferroptosis is a newly discovered type of cell death resulting from iron‐dependent lipid peroxide accumulation. The purpose of this study was to examine whether ferroptosis is involved in COPD-associated skeletal muscle dysfunction.
Methods: A mouse model of COPD was established after 24 weeks of cigarette smoke (CS) exposure, and mRNA sequencing, hematoxylin-eosin (H&E) staining, immunostaining (IF), RT-PCR, and Western blot were utilized to identify the changes in gastrocnemius muscles. In vitro, C2C12 myotubes were treated with CS extract (CSE) and evaluated for ferroptosis-related molecules. The pathways regulating ferroptosis were then explored in CSE-stimulated myotubes.
Results: Compared with controls, COPD mice showed an enriched ferroptosis pathway. Gpx4 was decreased, while hypoxia-inducible factor (Hif) 2α was increased, at gene and protein levels. A reduced level of GSH, but increased cell death, Fe2+, lipid ROS, LPO, and 4-HNE were observed in COPD mice or in CSE-stimulated C2C12 myotubes, which could be ameliorated by ferroptosis inhibitors. The expression of myostatin (MSTN) was enhanced in COPD mice and CSE-stimulated myotubes. MSTN up-regulated HIF2α expression and led to ferroptosis in myotubes, whereas inhibition of MSTN binding to its receptor or inhibition/knockdown of HIF2α resulted in decreased cell death, and partially restored GPX4 and GSH.
Conclusion: CS exposure induced ferroptosis in vivo and in vitro. Mechanistically, CS-exposure upregulated MSTN which further induced ferroptosis through HIF2α in skeletal muscles, which may contribute to muscle dysfunction through impairing metabolic capacity and decreasing muscle fiber numbers, revealing a potential novel therapeutic target for COPD-related skeletal muscle dysfunction.
Keywords: chronic obstructive pulmonary disease, skeletal muscle dysfunction, ferroptosis, myostatin, hypoxia-inducible factor 2α
Introduction
Chronic obstructive pulmonary disease (COPD) is a globally prevalent chronic airway disease, and is the third leading cause of death worldwide.1 As a highly heterogeneous disease mostly associated with cigarette smoking and aging, COPD is considered to be a systemic disease with multiple intra- and extra-pulmonary comorbidities, some of which occur independently of COPD, but some of which may be causally related to COPD.1 Sarcopenia/skeletal muscle dysfunction is one of the important comorbidities of COPD, and has a significant impact on patients’ quality of life, disease severity, hospitalization, and mortality.2–4 The prevalence of skeletal muscle dysfunction in patients with stable COPD was from 14.5%2 to 55%.5 Skeletal muscle dysfunction is more prevalent in individuals with emphysema, and more significant in lower extremities, which impairs the ambulatory capacity of the patients, leading to disused muscle atrophy, creating a vicious cycle.4 Unlike irreversible destructive lung lesions, the inherent adaptability of skeletal muscles offers therapeutic opportunities to tackle skeletal muscle dysfunction in COPD patients.6 However, the mechanisms of COPD-related skeletal muscle dysfunction have not been fully elucidated.
A number of signaling pathways may be involved in or contribute to COPD-related skeletal muscle dysfunction, such as proteasome-mediated ubiquitin-dependent protein catabolic pathway (ubiquitin-proteasome pathway),7 autophagy-lysosome pathway,8 NF-κB and inflammatory cytokines,9 and the myostatin (MSTN)-Smad3 pathway,7 among which the ubiquitin-proteasome pathway predominated in muscle proteolysis. As important muscle-specific E3-ubiquitin ligases, muscle-specific Ring finger 1 protein (MuRF1, also known as TRIM63) and Atrogin1 (also known as FBXO32) mediate muscle proteolysis, and loss of either can alleviate muscle atrophy.10 In addition, slow- to fast-twitch fiber-type transformation was found in peripheral muscles of COPD patients, resulting in deterioration of skeletal muscle anti-fatigue ability and decreased aerobic metabolism.4
Ferroptosis is a form of iron-dependent and regulated cell death characterized by accumulation of reactive oxygen species (ROS) and massive lipid peroxidation (LPO)-mediated cell membrane damage.11 Glutathione (GSH)-glutathione peroxidase 4 (GPX4) axis plays a protective role in ferroptosis which can be triggered by molecules that inhibit the biosynthesis of GSH or GSH-dependent antioxidant enzymes.11 GSH, the most abundant endogenous antioxidant in mammalian cells, is generated from cysteine which can be obtained in its oxidized form cystine through the amino antiporter-system Xc− (a transmembrane protein complex consisting of light-chain subunit SLC7A11 and heavy-chain subunit SLC3A2), or in its reduced form cysteine from methionine through the transsulfuration pathway.12 When GSH is insufficient, excess peroxide leads to LPO. Unrestrained LPO is the hallmark of ferroptosis, in which iron is essential. Skeletal muscle contains 10–15% of the total iron content of the human body. It has been confirmed that excess iron accumulation could disrupt normal cellular function through oxidative stress and the catalysis of toxic hydroxyl radicals.13
Cellular senescence is currently considered to be an important mechanism underlying the pathogenesis of COPD, and cigarette smoke (CS) may act as an inducer of senescence.14 Mounting evidence has demonstrated that ferroptosis is implicated in the development and/or progression of many age-related diseases,15 and that age-related iron accumulation has been found in the skeletal muscles from both animal models of sarcopenia and aged patients.16
Hence, based on this evidence, we speculate that ferroptosis may be involved in the pathogenesis of COPD-related skeletal muscle dysfunction. Therefore, we investigated ferroptosis and the potential molecules involved in its regulation in skeletal muscles from COPD mice induced by CS exposure and in an in vitro model. We found that CS exposure induced ferroptosis in skeletal muscles from mice and in C2C12 myotubes. CS exposure up-regulated expression of MSTN, which induced ferroptosis through HIF2α in skeletal muscle cells. These findings revealed a novel mechanistic link between COPD and skeletal muscle dysfunction.
Materials and Methods
Cigarette-Smoke Induced Mouse Model of COPD
Aged-matched C57BL/6 mice (6–8 weeks old) were purchased from Beijing Vital River Laboratory or Charles Rivers Laboratories and kept in ventilated specific pathogen-free grade animal facility. All animal handling and procedures were approved by the Animal Care Committee of Peking University (No. LA2021545) according to China’s Guidelines on Welfare and Ethical Review for Laboratory Animals. All mice were housed on a 12 h:12 h light/dark cycle with ad libitum access to food and water. The mouse model of COPD was established using a nose-only cigarette smoking exposure method as reported previously.17 Briefly, mice were simultaneously exposed to cigarettes with a filter (tar 10 mg, nicotine 0.9 mg, CO 12 mg) twice/day for 50 minutes each time, with 20-minute smoke-free intervals, 5 days a week, for 24 weeks using a nose-only smoke exposure system (SG-300; SIBATA, Tokyo, Japan). Cigarettes were actively burned and smoke was generated by a computer-controlled suction, with 20 mL smoke per 8 seconds into the exposure chamber. The optimal smoke: air ratio of 1:9 was obtained. The control mice were exposed to filtered air. After 24 weeks of CS exposure, the mice underwent lung function tests and morphometric assessment of lung sections for verifying the successful modeling of COPD.
Pulmonary Function Test
The pulmonary function of mice was performed by animal breathing devices (AniRes2005, Bolan Technology, Beijing, China). Briefly, mice were anesthetized by intraperitoneal injection of 3% pentobarbital sodium (0.6 mg/10 g). A tracheotomy was conducted and a cannula was inserted into the laryngeal trachea. The mice were ventilated with a tidal volume of 10 mL/kg, a respiration rate of 500 breaths/min, and a positive end-expiratory pressure of 2 cmH2O. Forced vital capacity (FVC), forced expiratory volume in 0.1 s (FEV0.1), forced expiratory flow at 25% of FVC (FEF25%), FEF75%, FEF25-75%, lung compliance, and the resistance of lung (RL) were measured.18
Grip Force Test
The grip force test was performed to measure the muscle strength of four limbs of the mice using a grip strength meter (unibiolab, Beijing, China). The mouse was grasping the mesh bar connected to a force transducer with all four limbs. Then, the mouse was pulled back by the tail at a constant speed until its grip was released. The peak force (g) was automatically recorded by the transducer. Each mouse was tested 4 times, with a 2-minute rest interval between each test. The mean of four measurements was recorded as grip strength.
Histology Analysis of the Mouse Lungs and Skeletal Muscles
After the pulmonary function test, the bronchoalveolar lavage fluid (BALF) was collected for fluorescent-activated cell sorting (FACS). Then, the mice were sacrificed by cervical dislocation. Lung tissues and gastrocnemius muscles of hind limbs were fixed for 24 h in 4% paraformaldehyde, and were then embedded in paraffin. The lung tissue was sectioned at a thickness of 5 μm for H&E staining. Then, the mean linear intercept (MLI) and destructive index (DI) of lung tissue were evaluated for the degree of emphysema, following our previous study.19 Specifically, MLI was measured using a grid of 100×100 μm randomly passing through the alveoli and calculated the total length of each line of the grid divided by the number of alveolar intercepts. The measurement of DI was performed by a grid with 42 points located in the center of hairline crosses superimposed on the lung field. Structures lying under these points were marked as normal (N) or destroyed (D) alveolar and/or duct spaces. Points falling on the other structures, such as duct walls and alveolar walls, did not enter into the calculations. DI was calculated as D/(D+N). The 10 μm thick cross-sections were cut from the paraffin-embedded gastrocnemius muscle by a cryostat (Leica, Solms, Germany) and were stained with H&E.
Immunofluorescence (IF) Stainings
Skeletal muscle tissues embedded in paraffin were sectioned to a thickness of 5 µm. Then, the paraffin slides were deparaffinized and rehydrated through a series of graded alcohols until water was used. The slides were subjected to antigen retrieval in a thermostatic water bath, at 95°C for 15 min, and they were blocked with 5% goat serum in TBST at room temperature for 30 min. The samples were then incubated with 1000-fold diluted primary antibodies in blocking buffer at 4°C overnight, followed by washes with 1×PBS for 3 times, incubation with 1000-fold diluted secondary antibody (goat anti-rabbit IgG (H+L) highly cross-adsorbed secondary antibody-Alexa Fluor 647) and DAPI in blocking buffer at room temperature for 1 h, and washed with 1×PBS for 3 times. The primary antibodies included anti-slow skeletal myosin heavy chain (MyHC) primary antibody (GB111858, Servicebio, Wuhan, China), anti-4 hydroxynonenal (4-HNE) primary antibody (ab46545, Abcam, Cambridge, UK), anti-GPX4 primary antibody (AF7020, Beyotime, Shanghai, China) and DAPI (Beyotime, Shanghai, China). The expression of slow skeletal MyHC, 4-HNE, and GPX4 in skeletal muscles was imaged under a fluorescence microscope.
Cigarette Smoke Extract Preparation
Cigarette smoke extract (CSE) was prepared as reported previously.20 CSE was prepared by bubbling the smoke from 5 cigarettes (tar 10 mg, nicotine 0.9 mg, CO 12 mg) through 10 mL of serum-free cell culture medium. The 5 cigarettes were smoked at constant airflow, lasting 5 minutes for each cigarette. Then, the obtained extract was adjusted the PH to 7.4 and filtrated through a 0.22 μm pore filter (Merk Millipore, Massachusetts, Germany). The concentration of 100% was calculated with UV absorbance 4.0 at 320 nm. CSE was freshly prepared and utilized within 30 minutes.
Cell Culture and Myotubes Formation
The mouse C2C12 myoblasts (ATCC, Manassas, USA) were cultured at 37°C with 5% CO2 in DMEM (ThermoFisher, Massachusetts, USA), 10% FBS (ThermoFisher, Massachusetts, USA), 2 mM L-glutamine, 100 IU/mL penicillin, and 100 μg/mL streptomycin (ThermoFisher Scientific). When the C2C12 myoblasts grew to 70–80% confluence, the culture medium was switched to a differentiation medium which consisted of DMEM, 2% horse serum (Thermofisher, Massachusetts, USA), 100 IU/mL penicillin, and 100 μg/mL streptomycin. After 5 days, most of the myoblasts differentiated into myotubes. Our previous studies had shown that the concentration of CSE below 3% had no significant effects on the viability of C2C12 myotubes,17 so subsequent experiments took 3% as the optimal effective dose. In the cell culture, C2C12 myotubes were also treated with indicated agents, including transforming growth factor (TGF)-β receptor activing-like kinase (ALK) 4/5 blocker TEW-7197 (S7530, Selleck, Shanghai, China), ferroptosis inhibitor UAMC-3203 (S8792, Selleck, Shanghai, China), hypoxia-inducible factor (HIF) 2α (400087, Merk Millipore), recombinant MSTN (120–00-10, Dogesce, Beijing, China) and vehicle (dimethylsulfoxide, DMSO).
Cell Transfection with shRNA Hif2α Lentiviral Particles
Mouse negative control-short hairpin RNA (shRNA) and shRNA Epas1/Hif2α were purchased from Santa Cruz Biotechnology (Texas, USA). C2C12 myotubes were transfected with shRNA (1 nM) using in vitro transfection reagent (Yeasen, Shanghai, China) according to the manufacturer’s instructions.
RNA Extraction, Reverse Transcription, and Quantitative PCR (RT-qPCR)
Total RNA was extracted from the gastrocnemius muscle of mice and the C2C12 myotubes using Trizol (Invitrogen, California, USA) following the manufacturer’s instruction. Then, the extracted RNA was reverse transcribed into cDNA with PrimeScript RT Master Mix (TAKARA biotechnology, Dalian, China). cDNA was quantified by SYBR green real-time PCR with 500 nM primers using a QuantStudio™ 5 (Thermo Fisher). Gapdh was used as reference genes. The primer sequences used for RT-qPCR as follow: Gapdh: F: TCAACGACCACTTTGTCAAGCTCA, R: GCTGGTGGTCCAGGGGTCTTACT; Gpx4: F: GATGGAGCCCATTCCTGAACC, R: CCCTGTACTTATCCAGGCAGA; Ncoa4: F: GAACCATCAGGACACATGGAAA, R: AGGAGCCATAGCCTTGGGT; Slc7a11: F: GGCACCGTCATCGGATCAG, R: CTCCACAGGCAGACCAGAAAA; Slc3a2: F: CGTCCTGCAACCAAGAACTC, R: TCGTACAGCAGCGGAAGT; Tfr1: F: TCGTACAGCAGCGGAAGT, R: TCTCCACGAGCGGAATACAG; Acls4: F: CTCACCATTATATTGCTGCCTGT, R: TCTCTTTGCCATAGCGTTTTTCT; Lpcat3: F: GACGGGGACATGGGAGAGA, R: GTAAAACAGAGCCAACGGGTAG. Relative gene expression to control was calculated using the formula: 2−ΔΔCT=2sample−ΔCT/2Ctrl−ΔCT.
Western Blotting
Gastrocnemius muscles and C2C12 myotubes were homogenized in radioimmunoprecipitation assay (RIPA) lysis buffer (Solarbio, Beijing, China) with protease and phosphatase inhibitors (Macgene, Beijing, China) for 30 min on ice and then centrifuged at 10,000 g for 10 min at 4°C. The protein in the supernatant was measured using a BCA assay. Approximately 30 μg protein of each sample was separated by 10% SDS-PAGE and transferred onto a PVDF membrane (Merk Millipore, Germany). The membranes were then sequentially incubated with primary and secondary antibodies. Protein signals were detected using Immobilon Western Chemiluminescent HRP Substrate (Merk Millipore). The antibodies included GAPDH (sc-47742, Santa Cruz Biotechnology, Texas, USA) (1:1000), fast skeletal myosin heavy chains (MyHC) (GB112130, Servicebio, Wuhan, China) (1:1000), slow skeletal MyHC (GB111857, Servicebio, Wuhan, China) (1:1000), MuRF1 (bs-2539R, Bioss, Beijing, China) (1:1000), Atrogin1 (bsm-54451R, Bioss, Beijing, China) (1:1000), GPX4 (AF7020, Beyotime, Shanghai, China) (1:1000), GDF8/myostatin (ab203076, Abcam, Cambridge, UK) (1:1000), and hypoxia-inducible factor (HIF) 2α (57921, Cell Signaling Technology, MA, USA) (1:1000).
Measurement of Cell Death by 7-AAD Staining
7-amino actinomycin D (7-AAD) is frequently used to stain and exclude dead cells in flow cytometry.21 C2C12 myotubes were harvested and stained with 7-AAD (BioLegend, California, USA) for 30 min at 4°C, and then washed twice with pre-cooled PBS. The C2C12 myotubes were analyzed immediately using a CytoFlex LX flow cytometer (Beckman Coulter, CA, USA) and analyzed with FlowJo (Tree Star, San Carlos, California, USA).
Ferrous Iron (Fe2+) Detection
Cellular Fe2+ was detected by FerroOrange (MX4559, Maokangbio, Shanghai, China). FerrOrange is a fluorescent probe that specifically detects Fe2+ which is mainly located in the endoplasmic reticulum. Once FerroOrange reacts with Fe2+, it can generate an orange fluorescent product (Absmax=542 nm, FLmax=572 nm) irreversibly. The C2C12 myotubes were washed 3 times in a serum-free medium and then 1 μM of FerrOrange was added to the medium for 30 min at 37°C. The level of Fe2+ was detected using a CytoFlex LX flow cytometer.
Measurement of GSH
The GSH and oxidized glutathione (GSSG) levels were measured using a GSH and GSSG Assay Kit (S0053, Beyotime, Shanghai, China) according to the manufacturer’s instructions. GSSG is reduced to GSH by glutathione reductase, which can react with the chromogenic substrate DTNB to produce yellow TNB and GSSG. The amount of total glutathione, which was calculated by spectrophotometer A412, determined the amount of yellow TNB. The content of GSSG was then determined using reagents to remove GSH in samples. The level of GSH was calculated by subtracting GSSG from the total glutathione. The values of GSH were normalized by protein concentration.
Detection of Lipid Reactive Oxygen Species (ROS)
The level of lipid ROS in C2C12 myotubes was detected by the live cell analysis reagent C11-BODIPY581/591 (C10445, Thermofisher, Massachusetts, USA). Briefly, after treatment with CSE and/or drugs, the C2C12 myotubes were harvested and then labeled with C11-BODIBY581/591 (10 μM), and incubated for 30 min at 37°C in the dark. Then, C2C12 myotubes were washed 3 times with 1×PBS, centrifuged, and resuspended in 500 μL PBS for detection by flow cytometer and laser confocal microscopy.
Lipid Peroxidation Measurements
The lipid peroxide (LPO) level in lysates of skeletal muscles and C2C12 myotubes was detected using a lipid peroxidation assay kit (E-BC-K176, Elabscience, Wuhan, China) according to the manufacturer’s protocol. In acid conditions, LPO reacted with chromogenic reagents to generate the stable chromogenic group at 45°C for 60 min, followed by centrifugation at 1100 g for 10 min at room temperature. The level of the stable chromogenic group in supernatants was quantified colorimetrically at OD586. The value of LPO was normalized by protein concentration detected by the BCA method.
RNA-Seq and Bioinformatics Analysis
mRNA-sequencing was performed with skeletal muscles from mice (GEO: GSE197463) and humans (GEO: GSE100281). Gene expression analysis was performed using R packages ggplot2 (v3.3.5). Correlation analysis was performed using the M function in corrplot (v0.90).
Statistical Analysis
Data were shown as mean ± S.D. Unpaired Student’s t-test (two groups) or one-way analysis of variance (ANOVA) (more than two groups) was performed to evaluate statistical differences between groups with SPSS 20.0 software (IBM Corporation, Armonk, NY, USA). p<0.05 was considered a statistically significant difference.
Results
Long-Term CS Exposure Led to Emphysema and Skeletal Muscle Dysfunction
After being exposed to CS for 24 weeks, the mice showed typical changes consistent with COPD. Lung function measurements showed a significant increase in airway resistance (RL) (Figure 1A) but a significant decrease in lung compliance in CS-exposed mice (Figure 1B). Compared with controls, CS-exposed mice also showed an increase in FVC (Figure 1C), but significant decreases in FEV0.1, FEV0.1/FVC, and FEF25-75% (Figure 1D–F). Lung histological analysis showed damaged alveolar walls and enlargement of airway spaces (Figure 1G), as demonstrated by increased MLI and DI in CS-exposed mice (Figure 1H and I). In addition, there was also a marked inflammatory infiltration in the lung parenchyma and interstitial spaces (Figure 1G). Neutrophils and eosinophils were also increased in BALF from CS-exposed mice (Figure 1J). We evaluated the muscle strength of the four limbs of the mice, and the results showed a marked decline in grip strength (Figure 1K), accompanied by a narrower cross-sectional area of myofibers and looser texture of gastrocnemius muscles (Figure 1L) in CS-exposed mice as compared with the control group. As a potent negative regulator of muscle mass, the myokine MSTN was up-regulated in skeletal muscles from mice17 and patients with COPD,22 and is believed to play a vital role in COPD-related skeletal muscle dysfunction.23 Here, we confirmed that the expressions of MSTN, MuRF1, and Atrogin1 were enhanced in the muscles of CS-exposed mice (Figure 1M). The COPD mice also showed a decreased MyHC of slow-twitch fiber-type but an increased MyHC of fast-twitch fiber-type (Figure 1M and N), which was consistent with findings from COPD patients.4
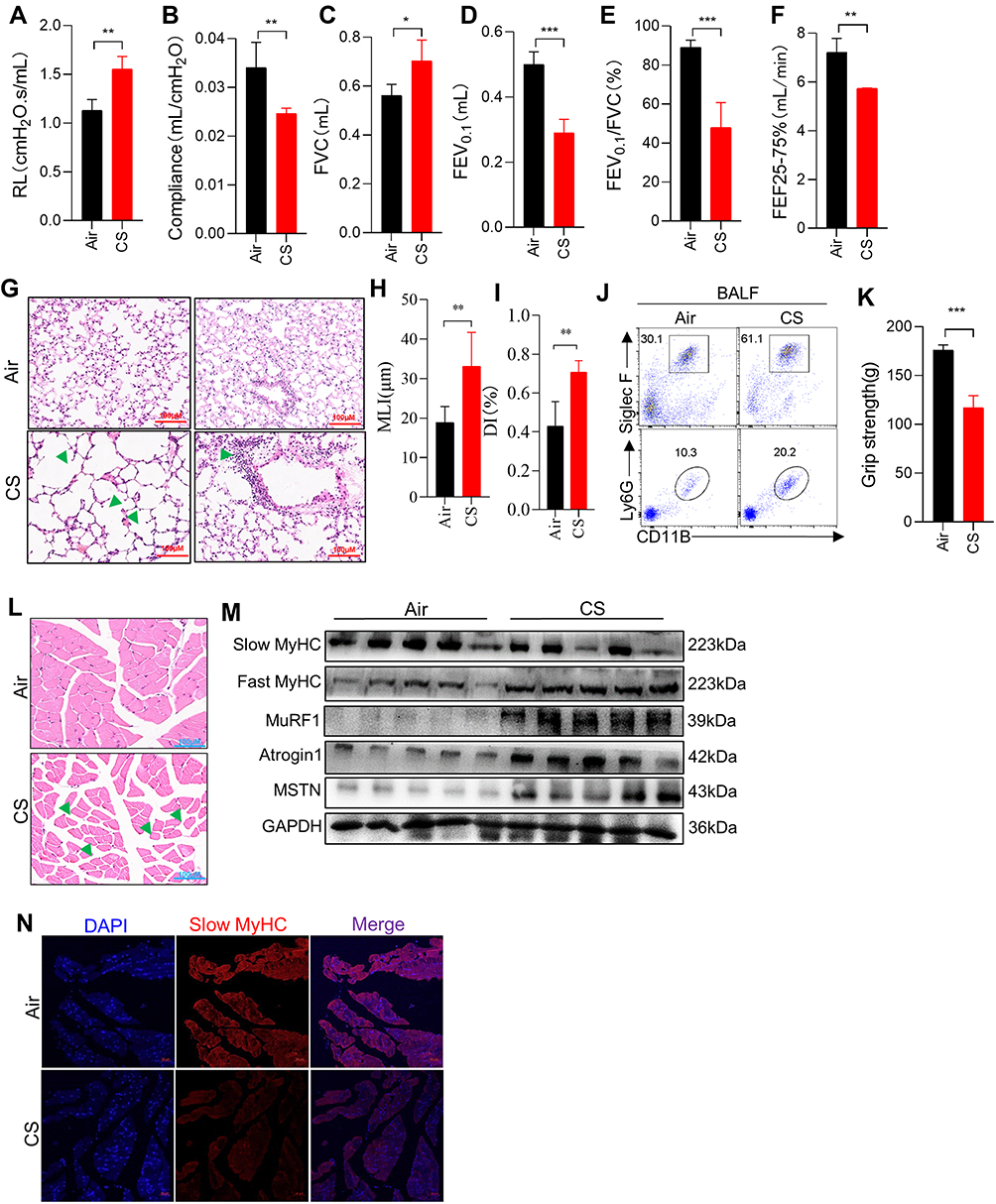 |
Figure 1 Long-term CS exposure led to emphysema and skeletal muscle dysfunction. (A–F) Airway resistance (RL), lung compliance, FVC, FEV0.1, FEV0.1/FVC, and FEF25-75% were measured in mice; (G) H&E staining of lung tissues from mice exposed to air and CS; Arrows (left) indicate destruction of the alveolar wall; Arrow (right) indicate Inflammatory cells infiltrates in the lung parenchyma and interstitial spaces; The scale in each image is 100 μm; (H and I) histogram of MLI and DI; (J) FACS showed the neutrophils (CD11B+Ly6G+) and eosinophils (CD11B+SiglecF+) in BALF; (K) histogram of grip strength; (L) H&E staining of muscles from mice exposed to air and CS; Arrows indicate changes in muscle fibers and widening of gaps; The scale in each image is 100 μm; (M) WB showed the protein expression of Slow MyHC, Fast MyHC, MuRF1, Atrogin1 and MSTN in muscles of air and CS-exposed mice; (N) the expression of Slow MyHC in muscles from mice. n = 5, *P < 0.05, **P < 0.01, ***P < 0.001. |
Chronic CS Exposure Induced Ferroptosis in Skeletal Muscles from Mice
Cell death is one of the important mechanisms affecting organ or tissue function, so we explored the death patterns of skeletal muscle cells in COPD. Firstly, RNA-sequencing performed on skeletal muscles from mice (GEO: GSE197463) and COPD patients24 (GEO: GSE100281) was analyzed. The results showed that the ferroptosis pathway was significantly enriched in COPD patients (Figure 2A), and that Gpx4 and nuclear receptor coactivator (Ncoa4) which can inhibit and promote ferroptosis, respectively, were dysregulated in CS-exposed mice (Figure 2B and C). Therefore, we next examined some important ferroptosis-related molecules in skeletal muscles from our COPD mice. We found that the expression of GPX4 was reduced in muscles from CS-exposed mice as compared with the control ones (Figure 2D–F). In addition, the mRNA levels of the cystine transporter Slc7a11, transferrin receptor 1 (Tfr1), and Ncoa4 were upregulated in CS-exposed mice (Figure 2D).
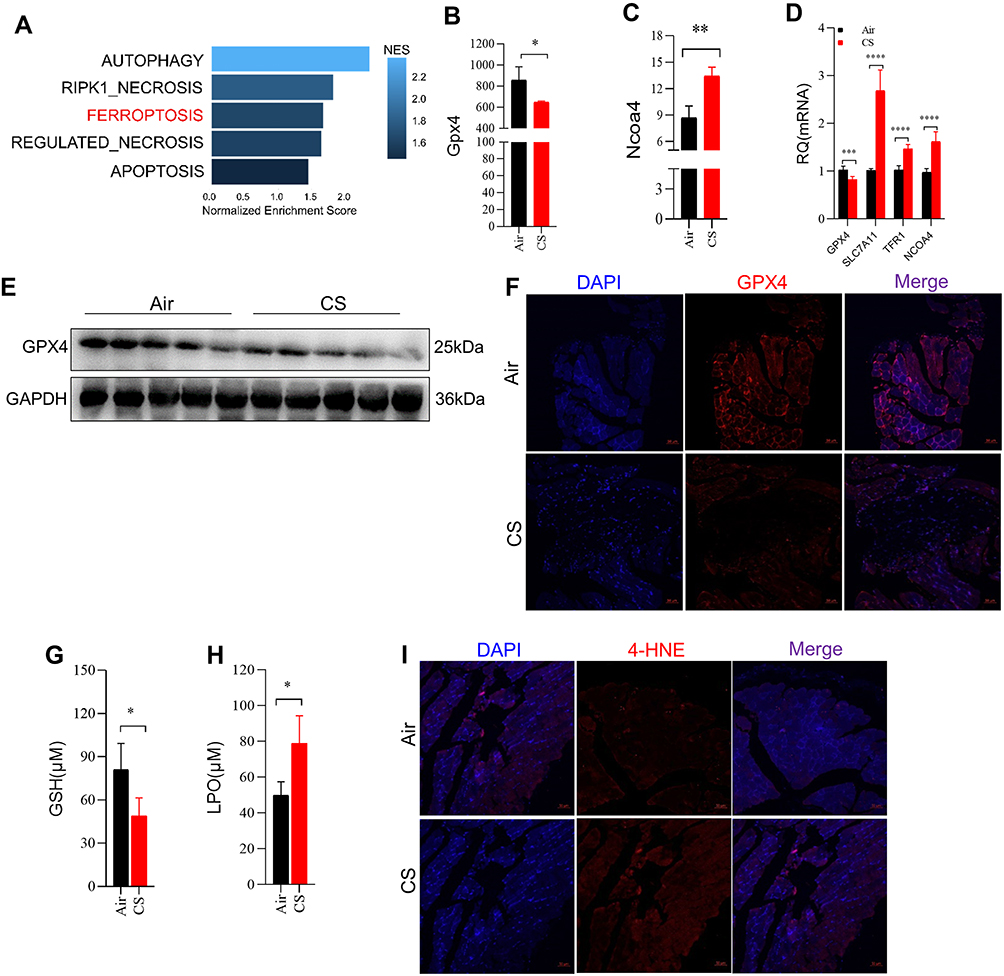 |
Figure 2 Chronic CS exposure caused ferroptosis in skeletal muscles from mice. (A) Cell death was enriched in skeletal muscles from COPD patients and healthy control by GSEA. (B and C) Gene expression of Gpx4 and Ncoa4 in skeletal muscles from mice by RNA-sequencing; (D) RT-qPCR examined the expression of Gpx4, Slc7a11, Tfr1, and Ncoa4 in muscles; (E) WB showed the protein expression of GPX4 in muscles; (F) IF showed the expression of GPX4 in muscles from mice exposed to air and CS; The scale in each image is 50 μm; (G) the content of GSH in muscles; (H) the level of LPO in muscles determined by colorimetry; (I) IF showed the expression of 4-HNE in muscles from mice exposed to air and CS; The scale in each image is 50 μm. n = 5, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. |
Ferroptosis is caused by decreased antioxidant capacity and lethal accumulation of LPO, and consistently, we found decreased GSH but increased LPO in CS-exposed mice as compared with the control ones (Figure 2G and H). Also, 4-hydroxynonenal (4-HNE), a major marker of LPO, was accumulated in muscle tissues of CS-exposed mice (Figure 2I). Taken together, these findings confirmed that CS exposure induced skeletal muscle ferroptosis, which may be involved in skeletal muscle dysfunction in COPD mice.
CSE Stimulation Induced Ferroptosis in C2C12 Myotubes
After C2C12 myoblasts were induced with a medium containing 2% horse serum for 5 days, the majority of myoblasts fused to form C2C12 myotubes (Figure 3A), which were utilized for subsequent studies in vitro. To determine the direct effect of CSE on myotubes, 7AAD was used to determine whether CSE caused myotubes death. We found that the death proportion of C2C12 myotubes was increased in CSE-stimulated cells (Figure 3B). Compared with the blank controls, the CSE-stimulated myotubes showed an increased level of Slc7a11, but reduced levels of Gpx4, Slc3a2, Tfr1, Ncoa4, acyl-CoA synthetase long-chain family member 4 (Acls4), and lysophosphatidylcholine acyltransferase 3 (Lpcat3) (Figure 3C). GSH, one of the most important antioxidants capable of preventing damage to important cellular components caused by ROS, can be regulated by GPX4 which converts GSH into GSSG and reduces the cytotoxic lipid peroxides (L-OOH) to the corresponding alcohols (L-OH). Here, we found that GPX4 expression was decreased in CSE-treated C2C12 myotubes (Figure 3D), which was consistent with the results in muscles from CS-exposed mice. Of note, we also found that the expressions of MuRF1, Atrogin1, and MSTN were enhanced in CSE-stimulated myotubes (Figure 3D).
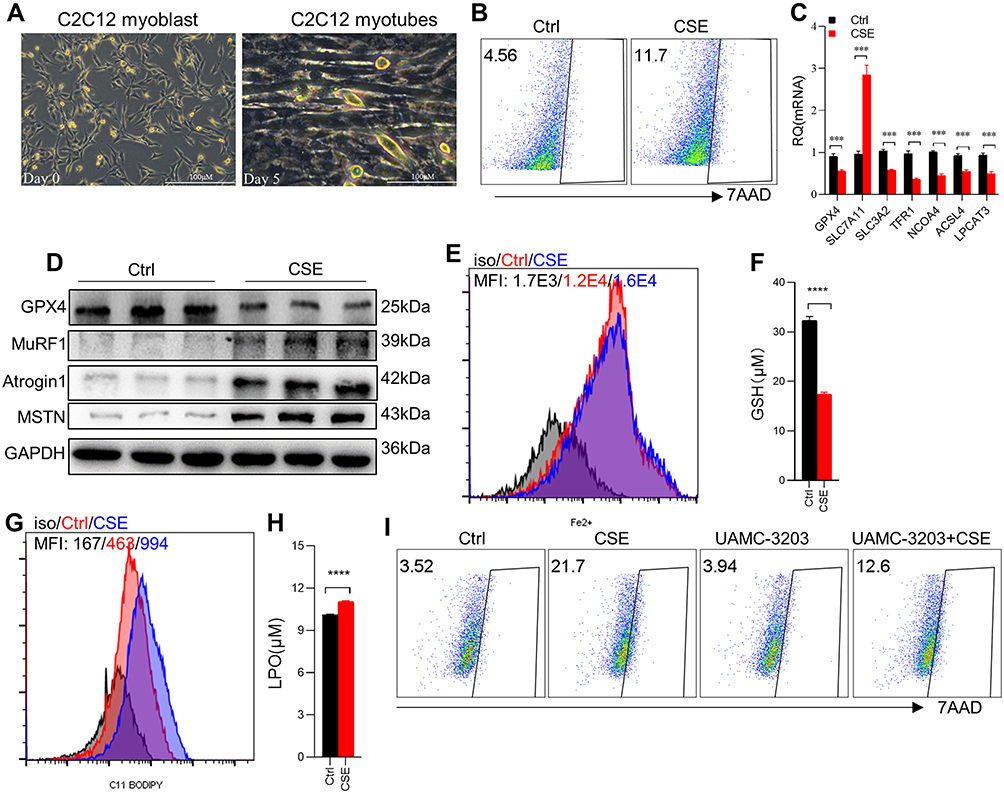 |
Figure 3 CSE stimulation caused ferroptosis in C2C12 myotubes. (A) Cellular morphology of C2C12 myoblasts and C2C12 myotubes. The scale in each image is 100 μm; (B) the proportion of cell death was detected by flow cytometry with 7AAD staining in Ctrl and 3% CSE-stimulated myotubes; (C) RT-qPCR examined the expression of Gpx4, Slc7a11, Slc3a2, Tfr1, Ncoa4, Acls4 and Lpcat3 in C2C12 myotubes; (D) WB showed the protein expression of GPX4, MuRF1, Atrogin1, and MSTN in C2C12 myotubes; (E) the level of Fe2+ was detected by flow cytometry with the fluorescent probe; (F) the content of GSH in C2C12 myotubes; (G) lipid ROS was detected by flow cytometry with C11 BODIPY; (H) the level of LPO in C2C12 myotubes. (I) The proportion of cell death was detected by flow cytometry with 7AAD staining. UAMC-3203, a ferroptosis inhibitor. ***P < 0.001, ****P < 0.0001. |
Excessive accumulation of intracellular Fe2+ can directly catalyze the production of lipid ROS through the Fenton reaction, eventually causing ferroptosis. Therefore, we detected Fe2+ in myotubes by Fe2+ fluorescent probes, and found a significantly increased level of Fe2+ in C2C12 myotubes treated with CSE (Figure 3E). Furthermore, a significant decrease in GSH (Figure 3F) but a marked increase in lipid ROS (Figure 3G) and LPO (Figure 3H) were observed in CSE-stimulated C2C12 myotubes. To further confirm the occurrence of ferroptosis, we found that the death proportion of C2C12 myotubes induced by CSE was significantly alleviated after intervention with the ferroptosis inhibitor UAMC-3203 (Figure 3I).
CSE Induced Ferroptosis by Up-Regulating the Expression of MSTN
Given that CSE induced upregulation of MSTN, we asked whether CSE caused ferroptosis through MSTN. To test this hypothesis, we used a MSTN blocker TEW-7197, which could inhibit the binding of MSTN to its receptor ALK4/5, in our myotubes model. We found that GPX4 expression was restored (Figure 4A), and that lipid ROS labeled with C11-BODIPY was significantly decreased in CSE-treated C2C12 myotubes after TEW-7197 treatment (Figure 4B). Consistently, exogenous recombinant MSTN induced myotubes death (Figure 4C). Furthermore, MSTN down-regulated GPX4 expression (Figure 4D), decreased the level of GSH (Figure 4E), and increased the level of Fe2+ (Figure 4F), lipid ROS (Figure 4G) and LPO (Figure 4H). In addition, the ferroptosis inhibitor UAMC-3203 was shown to down-regulate MSTN-induced ferroptosis, as manifested by decreased cell death (Figure 5A), recovered GPX4 and GSH (Figure 5B and C), and reduction of Fe2+, lipid ROS, LPO (Figure 5D–F), and lipid ROS labeled by C11-BODIPY (Figure 5G).
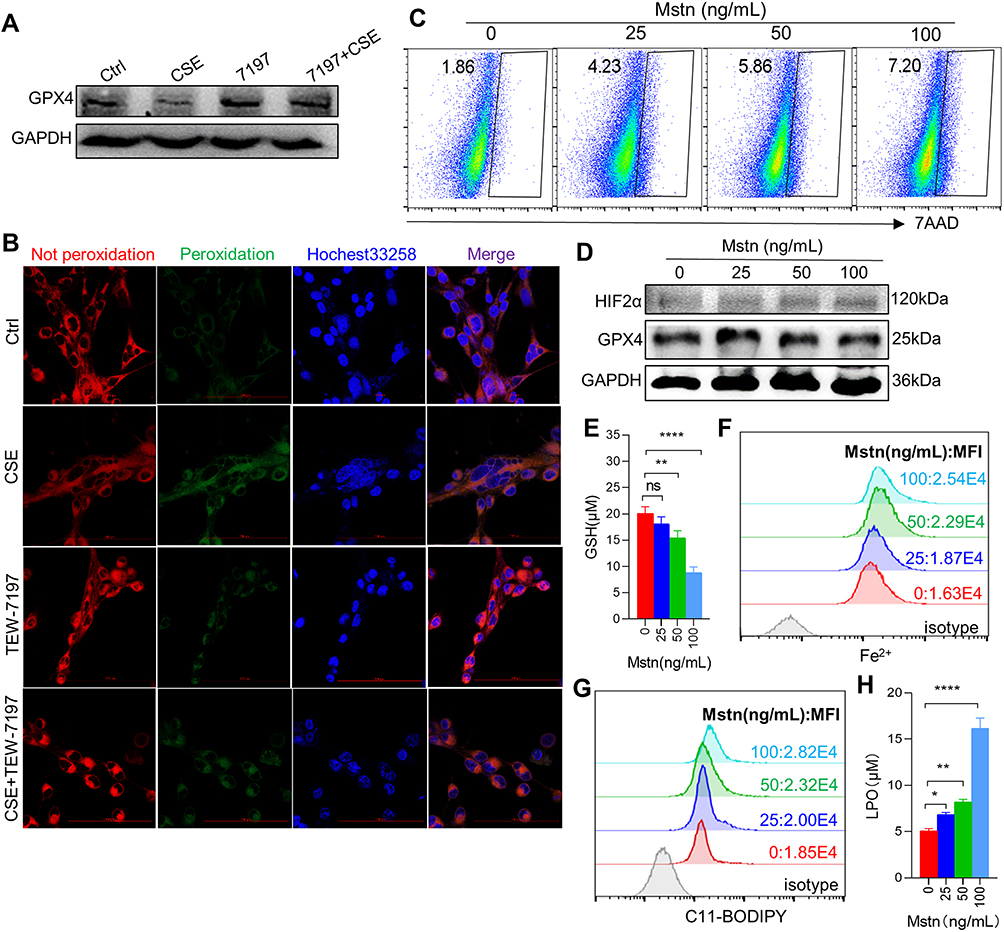 |
Figure 4 CSE induced ferroptosis by up-regulating the expression of MSTN. (A) WB showed the expression of GPX4 in myotubes; (B) the expression of lipid ROS in myotubes by confocal microscopy with C11 BODIPY; The scale in each image is 100 μm; (C) the proportion of cell death was detected by flow cytometry with 7AAD staining in different concentration of MSTN treated myotubes; (D) WB showed the protein expression of GPX4 and HIF2α; (E) histogram of the content of GSH in myotubes; (F and G) flow cytometry detected the levels of Fe2+ and lipid ROS in different concentration of MSTN treated myotubes; (H) histogram of the content of LPO in myotubes. *P < 0.05,**P < 0.01,****P < 0.0001. Abbreviation: ns, not significant. |
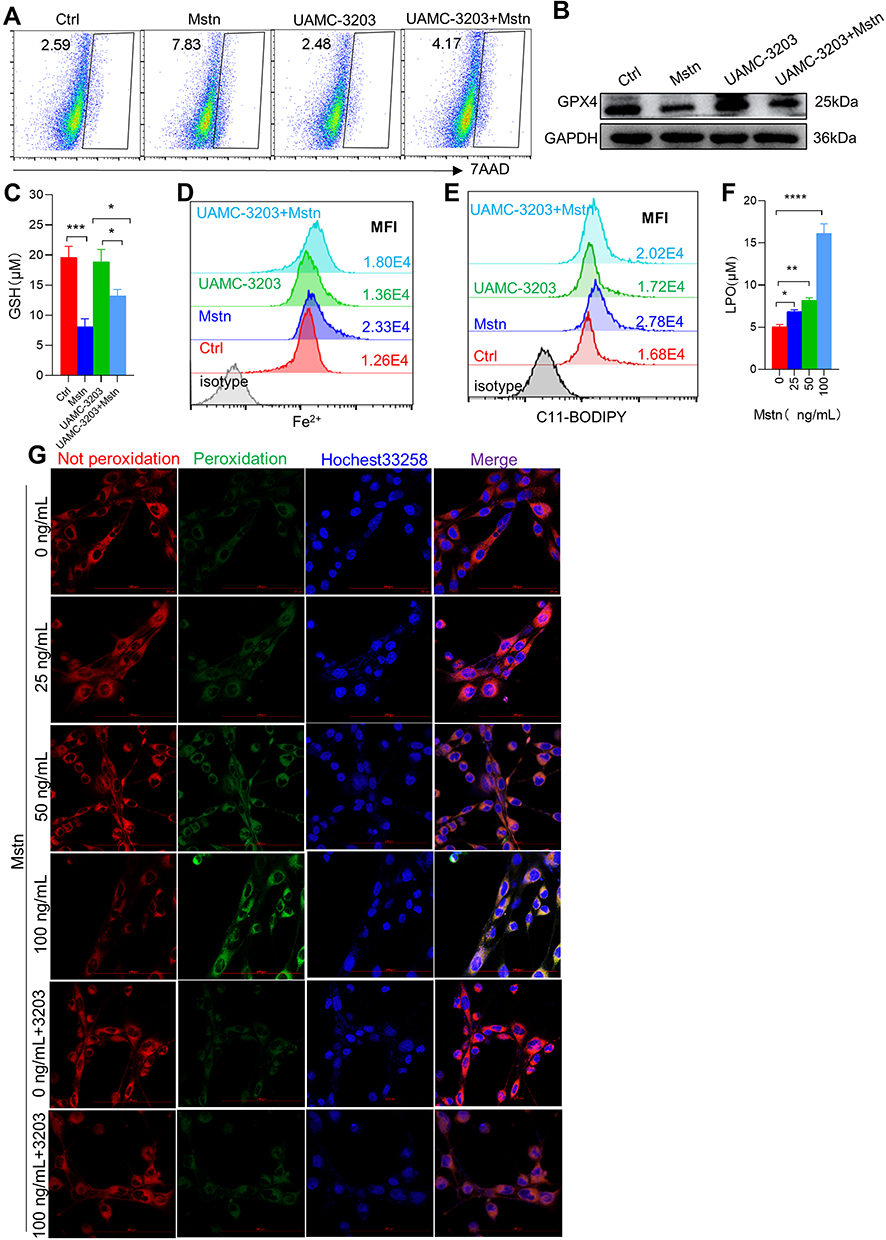 |
Figure 5 Ferroptosis inhibitor intervention alleviated ferroptosis-related indicators caused by MSTN. (A) Cell death was detected by flow cytometry; (B) the protein expression of GPX4; (C) Histogram of the content of GSH; (D and E) flow cytometry detected the levels of Fe2+ and lipid ROS; (F) histogram of the content of LPO; (G) the expression of lipid ROS in myotubes using confocal microscopy; The scale in each image is 100 μm. *P < 0.05,**P < 0.01,***P < 0.001, ****P < 0.0001. |
CSE Induced Ferroptosis Through the HIF2α Pathway
To explore the pathways underlying CSE-induced ferroptosis, we firstly analyzed the mRNA sequencing of skeletal muscles from mice, and found that Hif1α, Epas1 (the gene encoding HIF2α), and Hif3α were negatively correlated with Gpx4, among which Epas1 had the strongest correlation with Gpx4 (r=−0.51) (Figure 6A). There were differences in Epas1 and Hif3α mRNA levels in the skeletal muscles of air-exposed and CS-exposed mice, but the most significant difference was noted in Epas1 (P<0.01) (Figure 6B). Therefore, we speculated that HIF2α may be involved in ferroptosis in skeletal muscles. We found that HIF2α was significantly enhanced in gastrocnemius muscles from CS-exposed mice and in CSE-treated C2C12 myotubes (Figure 6C). To verify the role of HIF2α in ferroptosis, we detected ferroptosis-related markers after intervening C2C12 myotubes with a HIF2α inhibitor. The results showed that the death proportion of C2C12 myotubes was reduced markedly upon stimulation with a CSE+HIF2α inhibitor (Figure 6D), followed by the rescued GPX4 (Figure 6E and F). The HIF2α inhibitor decreased Fe2+ level (Figure 6G), lipid ROS (Figure 6H) and LPO (Figure 6I) in CSE-treated C2C12 myotubes, but also increased GSH (Figure 6J).
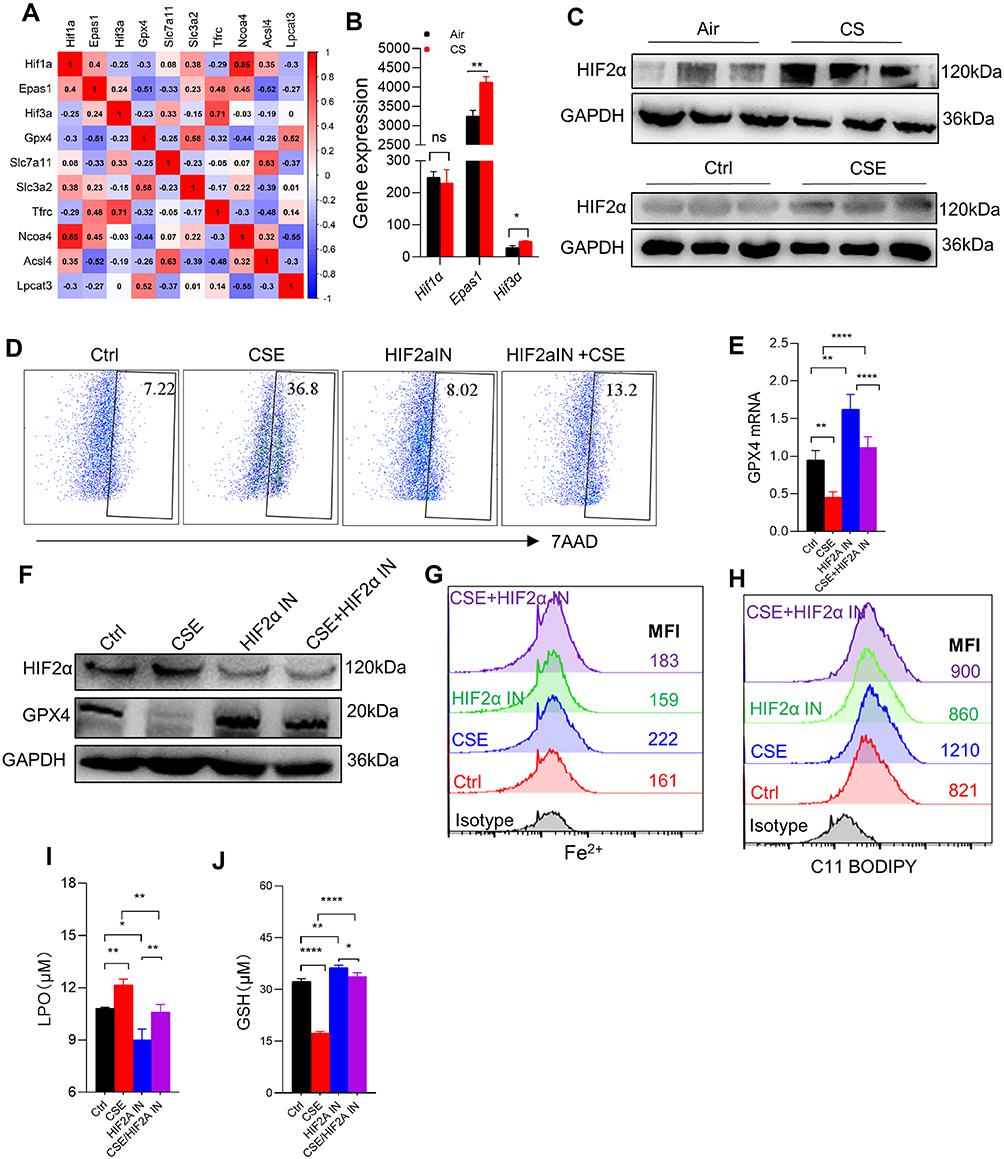 |
Figure 6 HIF2α inhibition alleviated ferroptosis-related indicators caused by CSE. (A) Corplot showed the correlation between Hif1α, Epas1, Hif3α, and Gpx4. (B) Gene expression of Hif1α, Epas1, and Hif3α in skeletal muscles from mice by RNA-sequencing; (C) WB showed the protein expression of HIF2α in muscles (up) and C2C12 myotubes (down); (D) the proportion of cell death was detected by flow cytometry with 7AAD staining in myotubes; (E) RT-qPCR detected the expression of Gpx4 in myotubes; (F) the expression of HIF2α and GPX4 were detected by WB; (G and H) the levels of Fe2+ and lipid ROS were detected by flow cytometry; (I and J) histogram of the content of LPO and GSH in C2C12 myotubes. HIF2α IN, an inhibitor of HIF2α. *P < 0.05,**P < 0.01,****P < 0.0001. Abbreviation: ns, not significant. |
To further explore the role of HIF2α in skeletal muscle ferroptosis, we transfected C2C12 myotubes with shRNA Hif2α lentiviral particles to knock-down Hif2α and confirmed that HIF2α was down-regulated after shRNA Hif2α transfected (Figure 7A). As expected, the loss of Hif2α alleviated ferroptosis-related markers in CSE-stimulated C2C12 myotubes, as demonstrated by reduced cell death (Figure 7B), increased GPX4 (Figure 7A) and GSH (Figure 7C), accompanied by lower Fe2+ (Figure 7D), lipid ROS (Figure 7E) and LPO (Figure 7F), indicating that CSE induced ferroptosis in C2C12 myotubes through HIF2α.
 |
Figure 7 HIF2α knocking down alleviated ferroptosis-related indicators caused by CSE. (A) WB showed the protein expression of GPX4 and HIF2α; (B) the proportion of cell death was detected by flow cytometry with 7AAD staining in myotubes; (C) histogram of the content of GSH in myotubes; (D and E) flow cytometry detected the levels of Fe2+ and lipid ROS in myotubes; (F) histogram of the content of LPO in myotubes. *P < 0.05,**P < 0.01,***P < 0.001. Abbreviations: NC-shRNA, negative control shRNA; shRNA Hif2α, shRNA Hif2α lentiviral particles; ns, not significant. |
Discussion
Skeletal muscle dysfunction/sarcopenia is one of the major comorbidities of COPD, but the mechanisms linking the lung and the skeletal muscles remain to be elucidated. In the current study, for the first time to our knowledge, we found that chronic CS exposure induced ferroptosis in skeletal muscles from COPD mice with comorbidity limb weakness. The findings were recapitulated in an in vitro model of C2C12 myotubes exposed to CSE. Mechanistically, CS exposure upregulated the myokine-MSTN, which induced ferroptosis through HIF2α in skeletal muscles. These novel results provide insights into understanding the potential mechanisms of COPD-related skeletal muscle dysfunction and may reveal potential targets of intervention.
Ferroptosis is a newly identified type of cell death resulting from iron‐dependent lipid peroxide accumulation. Although iron is an essential trace element for cell survival, inappropriate iron accumulation can also damage the structure and function of cells through catalysis of highly toxic hydroxyl radicals and oxidative stress,13 and then contributes to pathogenesis by damaging DNAs, lipids, and proteins.25 In recent years, a few studies have suggested that ferroptosis may be associated with skeletal muscle dysfunction. For example, specific deletion of Tfr1 in satellite cells could activate ferroptosis and impaired muscle regeneration in skeletal muscles of aging mice.26 Another study with a sarcopenic mouse model suggested that iron overload upregulated p53 expression, thereby inhibiting the expression of SLC7A11, and ultimately leading to ferroptosis through LPO accumulation in skeletal muscle cells.16 Ferroptosis has also been found to be involved in rhabdomyolysis caused by exertional heat stroke27 and in atorvastatin-induced myopathy.28 In our study, mRNA sequencing performed on skeletal muscles showed that the ferroptosis pathway was markedly enriched in COPD, accompanied by abnormal expression of Gpx4 and Ncoa4. Our study further demonstrated a decrease in GPX4 and GSH, but an increase in 4-HNE and LPO in skeletal muscles from CS-induced COPD mice, which was consistent with the in vitro results observed in C2C12 myotubes stimulated by CSE, as demonstrated by an increase in cell death, Fe2+ and lipid ROS. GSH, a tripeptide antioxidant containing a thiol group, is synthesized from glutamate, cysteine, and glycine, in which the thiol on the cysteine is the active group of GSH.29 GSH serves as a direct antioxidant as well as an important substrate for antioxidant GPX4 to prevent lipid ROS accumulation. GPX4 contains a selenocysteine unit that can use GSH as a cofactor to detoxify LPO, thereby inhibiting ferroptosis.30
In our study, we also detected the elevated levels of Fe2+,which is essential for ferroptosis. On the one hand, iron is indispensable for the metabolic enzymes that are associated with phospholipid peroxidation and cellular ROS production. On the other hand, an unenzymatic, iron-dependent Fenton chain reaction is essential for ferroptosis.12 These canonical changes were demonstrated in CS-exposed mice and in CSE-treated C2C12 myotubes, suggesting that the intracellular glutathione antioxidant system is impaired and that polyunsaturated fatty acids (PUFAs) are peroxidized into toxic LPO. After intervention with ferroptosis inhibitor UAMC-3203, the cell death of C2C12 myotubes was attenuated in CSE-treated C2C12 myotubes, suggesting that ferroptosis did occur in COPD-associated skeletal muscle dysfunction.
We also examined the mRNA levels of Slc7a11, Slc3a2, Tfr1, Ncoa4, Acsl4, and Lpcat3, which were implicated in the process of ferroptosis. SLC7A11 and SLC3A2 together formed the cell surface heterodimeric amino antiporter-system Xc−, which can exchange cystine and glutamate in a 1:1 ratio for intracellular GSH synthesis.31 Here, we found Slc7a11 enhancement in muscles from CS-exposed mice and in CSE-treated C2C12 myotubes, suggesting that elevated Slc7a11 may be the compensatory changes caused by decreased intracellular GSH. However, the trends of Tfr1 and Ncoa4 in muscles and C2C12 myotubes were not completely consistent. TFR1 plays a significant role in regulating iron homeostasis and is responsible for internalizing transferrin-bound iron which is then released inside cells and stored in ferritin with a non-toxic form.32 Previous studies demonstrated that autophagy increased intracellular Fe2+ by degrading ferritin and inducing TFR1 expression in mouse lung primary fibroblasts, thereby inducing iron-dependent ferroptosis.33 However, studies also showed that specific deletion of Tfr1 in satellite cells activated ferroptosis and impaired muscle regeneration.26 TFR1 played opposite roles in these two studies, indicating that the mechanism of involvement of TFR1 in ferroptosis was complex and may vary in different cell types. NCOA4 is an autophagosome component involved in ferritinophagy, and the arginine located on the surface of ferritin heavy chain 1 can specifically bind to the carbon-terminal element of NCOA4 and fuse with lysosome via newly formed autophagosome, thereby promoting ferroptosis.34 We found an opposite trend of NCOA4 in muscles from CS-exposed mice and in CSE-treated C2C12 myotubes, suggesting that intermolecular interactions associated with ferroptosis were complicated, and only some of the signaling pathways such as GSH-GPX4 are activated in COPD-related skeletal muscle dysfunction.
Previous studies have confirmed that MSTN is involved in skeletal muscle dysfunction in COPD, mainly by affecting the balance of muscle proteolysis and protein synthesis. In this study, we investigated whether MSTN mediated CS exposure-induced ferroptosis. It is interesting to note that exogenous MSTN induced C2C12 myotube ferroptosis, which was alleviated after inhibiting the binding of MSTN to its receptor or intervening with a ferroptosis inhibitor. This finding raises an intriguing hypothesis that MSTN induces skeletal muscle dysfunction and/or sarcopenia via ferroptosis, which warrants further investigation in appropriate disease models.
HIFs are heterodimers composed of the regulated HIF-α protein subunits (HIF1α, HIF2α, or HIF3α) expressed only during hypoxia and the constitutively expressed HIF-β subunit.35 Studies demonstrated that activation of HIF2α up-regulated lipid and iron regulatory genes in murine colorectal cancer cells and colon tumors, and put cells into a ferroptosis-sensitive state and promoted cell death.36 However, whether HIF mediates ferroptosis in skeletal muscle cells has not been reported. Because skeletal muscle mRNA sequencing showed enhanced Hif2α expression which was correlated negatively with Gpx4 in CS-exposed mice, we hypothesized that HIF2α may be implicated in ferroptosis of skeletal muscles in COPD mice. Here, we observed elevated HIF2α protein expression in skeletal muscle from COPD mice and CSE-treated C2C12 myotubes. After pharmacological inhibition or genetic deletion of HIF2α in C2C12 myotubes, CSE-induced ferroptosis was attenuated, suggesting a critical role of HIF2α in skeletal muscle ferroptosis. Studies have reported that HIF2α could keep satellite cells in a quiescent state to block myogenic differentiation, but to promote self-renewal, thereby maintaining the stemness and long-term stability. HIF2α ablation caused satellite cells to deplete, leading to long-term regeneration failure of skeletal muscles, while transient pharmacological inhibition of HIF2α accelerated muscle regeneration by increasing the proliferation and differentiation of satellite cells.37 Therefore, as an upstream molecule regulating ferroptosis, HIF2α inhibition may be a potential target for treating COPD-related skeletal muscle dysfunction. Moreover, we also found that MSTN enhanced HIF2α production. To sum up, MSTN/HIF2α-mediated ferroptosis may contribute to skeletal muscle dysfunction in COPD by increasing muscle cell death and breaking the oxidative and anti-oxidative balance in the cells.
Our present study has several limitations. Firstly, the major focus of our current investigation is on the classic pathway of ferroptosis, ie, the GSH-GPX4 axis. Whether other signaling pathways of ferroptosis are involved in COPD-related skeletal muscle dysfunction requires further exploration. Secondly, we did not investigate whether targeting ferroptosis is beneficial for skeletal muscle dysfunction in CS exposure-induced COPD in mice, which is warranted in future studies.
Conclusion
As one of the major comorbidities of COPD, skeletal muscle dysfunction is associated with poor prognosis of the patients, but the potential mechanisms remain to be investigated. Our study demonstrated that CS exposure-induced ferroptosis, as manifested by decreased GPX4 and GSH, but increased cell death, Fe2+, lipid ROS, 4-HNE and LPO, was involved in the pathogenesis of COPD-related skeletal muscle dysfunction. Skeletal muscle ferroptosis was mediated by the classical myokine-MSTN via the HIF2α pathway. These novel findings shed light on the pathogenesis of COPD-associated skeletal muscle dysfunction, and provide evidence for developing newer modalities by targeting ferroptosis in the management of this important comorbidity of COPD.
Data Sharing Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The animal study was reviewed and approved by the Animal Care Committee of Peking University (No. LA2021545) according to China’s Guidelines on Welfare and Ethical Review for Laboratory Animals.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the National Nature Science Foundation of China (No. 82170048, 82090014, and 81770040).
Disclosure
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Vogelmeier CAA, Anzueto A, Barnes P, Bourbeau J, Criner G. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease. In: 2021 Report. Global Initiat Chronic Obstruct Lung Dis. http://www.goldcopd.org.
2. Jones SE, Maddocks M, Kon SS, et al. Sarcopenia in COPD: prevalence, clinical correlates and response to pulmonary rehabilitation. Thorax. 2015;70(3):213–218. doi:10.1136/thoraxjnl-2014-206440
3. Sepúlveda-Loyola W, Osadnik C, Phu S, Morita AA, Duque G, Probst VS. Diagnosis, prevalence, and clinical impact of sarcopenia in COPD: a systematic review and meta-analysis. J Cachexia Sarcopenia Muscle. 2020;11(5):1164–1176. doi:10.1002/jcsm.12600
4. Jaitovich A, Barreiro E. Skeletal muscle dysfunction in chronic obstructive pulmonary disease what we know and can do for our patients. Am J Respir Crit Care Med. 2018;198(2):175–186. doi:10.1164/rccm.201710-2140CI
5. Cebron Lipovec N, Schols AM, van den Borst B, et al. Sarcopenia in advanced COPD affects cardiometabolic risk reduction by short-term high-intensity pulmonary rehabilitation. J Am Med Dir Assoc. 2016;17(9):814–820. doi:10.1016/j.jamda.2016.05.002
6. Passey SL, Hansen MJ, Bozinovski S, McDonald CF, Holland AE, Vlahos R. Emerging therapies for the treatment of skeletal muscle wasting in chronic obstructive pulmonary disease. Pharmacol Ther. 2016;166:56–70. doi:10.1016/j.pharmthera.2016.06.013
7. Plant PJ, Brooks D, Faughnan M, et al. Cellular markers of muscle atrophy in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. 2010;42(4):461–471. doi:10.1165/rcmb.2008-0382OC
8. Guo Y, Gosker HR, Schols AM, et al. Autophagy in locomotor muscles of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2013;188(11):1313–1320. doi:10.1164/rccm.201304-0732OC
9. Agusti A, Morla M, Sauleda J, Saus C, Busquets X. NF-kappaB activation and iNOS upregulation in skeletal muscle of patients with COPD and low body weight. Thorax. 2004;59(6):483–487. doi:10.1136/thx.2003.017640
10. Cohen S, Nathan JA, Goldberg AL. Muscle wasting in disease: molecular mechanisms and promising therapies. Nat Rev Drug Discov. 2015;14(1):58–74. doi:10.1038/nrd4467
11. Tang D, Chen X, Kang R, Kroemer G. Ferroptosis: molecular mechanisms and health implications. Cell Res. 2021;31(2):107–125. doi:10.1038/s41422-020-00441-1
12. Jiang X, Stockwell BR, Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol. 2021;22(4):266–282. doi:10.1038/s41580-020-00324-8
13. Ikeda Y, Imao M, Satoh A, et al. Iron-induced skeletal muscle atrophy involves an Akt-forkhead box O3-E3 ubiquitin ligase-dependent pathway. J Trace Elem Med Biol. 2016;35:66–76. doi:10.1016/j.jtemb.2016.01.011
14. Barnes PJ, Baker J, Donnelly LE. Cellular senescence as a mechanism and target in chronic lung diseases. Am J Respir Crit Care Med. 2019;200(5):556–564. doi:10.1164/rccm.201810-1975TR
15. Zhou RP, Chen Y, Wei X, et al. Novel insights into ferroptosis: implications for age-related diseases. Theranostics. 2020;10(26):11976–11997. doi:10.7150/thno.50663
16. Huang Y, Wu B, Shen D, Chen J, Yu Z, Chen C. Ferroptosis in a sarcopenia model of senescence accelerated mouse prone 8 (SAMP8). Int J Biol Sci. 2021;17(1):151–162. doi:10.7150/ijbs.53126
17. Xiong J, Le Y, Rao Y, et al. RANKL mediates muscle atrophy and dysfunction in a cigarette smoke-induced model of COPD. Am J Respir Cell Mol Biol. 2021;64(5):617–628. doi:10.1165/rcmb.2020-0449OC
18. Vanoirbeek J, Devos F, Maaske A, et al. Lung function measurements in mouse models of lung disease: what to expect from FEV0.1? Eur Res J. 2016;48(suppl60):PA4131. doi:10.1183/13993003.congress-2016.PA4131
19. Zhou L, Le Y, Tian J, et al. Cigarette smoke-induced RANKL expression enhances MMP-9 production by alveolar macrophages. Int J Chron Obstruct Pulmon Dis. 2019;14:81–91. doi:10.2147/COPD.S190023
20. Gellner CA, Reynaga DD, Leslie FM. Cigarette smoke extract: a preclinical model of tobacco dependence. Curr Protoc Neurosci. 2016;77:954. doi:10.1002/cpns.14
21. Zembruski NC, Stache V, Haefeli WE, Weiss J. 7-Aminoactinomycin D for apoptosis staining in flow cytometry. Anal Biochem. 2012;429(1):79–81. doi:10.1016/j.ab.2012.07.005
22. Sancho-Muñoz A, Guitart M, Rodríguez DA, Gea J, Martínez-Llorens J, Barreiro E. Deficient muscle regeneration potential in sarcopenic COPD patients: role of satellite cells. J Cell Physiol. 2021;236(4):3083–3098. doi:10.1002/jcp.30073
23. Polkey MI, Praestgaard J, Berwick A, et al. Activin Type II receptor blockade for treatment of muscle depletion in chronic obstructive pulmonary disease. A randomized trial. Am J Respir Crit Care Med. 2019;199(3):313–320. doi:10.1164/rccm.201802-0286OC
24. Willis-Owen SAG, Thompson A, Kemp PR, et al. COPD is accompanied by coordinated transcriptional perturbation in the quadriceps affecting the mitochondria and extracellular matrix. Sci Rep. 2018;8. doi:10.1038/s41598-018-29789-6
25. Zhao G. Is iron accumulation a possible risk factor for sarcopenia? Biol Trace Elem Res. 2018;186(2):379–383. doi:10.1007/s12011-018-1332-z
26. Ding HR, Chen SJ, Pan XH, et al. Transferrin receptor 1 ablation in satellite cells impedes skeletal muscle regeneration through activation of ferroptosis. J Cachexia Sarcopeni. 2021;12(3):746–768. doi:10.1002/jcsm.12700
27. He S, Li R, Peng Y, et al. ACSL4 contributes to ferroptosis-mediated rhabdomyolysis in exertional heat stroke. J Cachexia Sarcopenia Muscle. 2022;13(3):1717–1730. doi:10.1002/jcsm.12953
28. Zhang Q, Qu H, Chen Y, et al. Atorvastatin induces mitochondria-dependent ferroptosis via the modulation of Nrf2-xCT/GPx4 Axis. Front Cell Dev Biol. 2022;10:806081. doi:10.3389/fcell.2022.806081
29. Zhang Y, Swanda RV, Nie L, et al. mTORC1 couples cyst(e)ine availability with GPX4 protein synthesis and ferroptosis regulation. Nat Commun. 2021;12(1):1589. doi:10.1038/s41467-021-21841-w
30. Ingold I, Berndt C, Schmitt S, et al. Selenium Utilization by GPX4 Is required to prevent hydroperoxide-induced ferroptosis. Cell. 2018;172(3):409. doi:10.1016/j.cell.2017.11.048
31. Xu W, Deng H, Hu S, et al. Role of ferroptosis in lung diseases. J Inflamm Res. 2021;14:2079–2090. doi:10.2147/JIR.S307081
32. Dixon SJ, Stockwell BR. The role of iron and reactive oxygen species in cell death. Nat Chem Biol. 2014;10(1):9–17. doi:10.1038/nchembio.1416
33. Park E, Chung SW. ROS-mediated autophagy increases intracellular iron levels and ferroptosis by ferritin and transferrin receptor regulation. Cell Death Dis. 2019;10(11):822. doi:10.1038/s41419-019-2064-5
34. Mou Y, Wu J, Zhang Y, Abdihamid O, Duan C, Li B. Low expression of ferritinophagy-related NCOA4 gene in relation to unfavorable outcome and defective immune cells infiltration in clear cell renal carcinoma. BMC Cancer. 2021;21(1):18. doi:10.1186/s12885-020-07726-z
35. Lee P, Chandel NS, Simon MC. Cellular adaptation to hypoxia through hypoxia inducible factors and beyond. Nat Rev Mol Cell Bio. 2020;21(5):268–283. doi:10.1038/s41580-020-0227-y
36. Singhal R, Mitta SR, Das NK, et al. HIF-2alpha activation potentiates oxidative cell death in colorectal cancers by increasing cellular iron. J Clin Invest. 2021;131(12). doi:10.1172/JCI143691
37. Xie L, Yin A, Nichenko AS, Beedle AM, Call JA, Yin H. Transient HIF2A inhibition promotes satellite cell proliferation and muscle regeneration. J Clin Invest. 2018;128(6):2339–2355. doi:10.1172/JCI96208
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.