")
Back to Journals » OncoTargets and Therapy » Volume 9
Myeloid cell signatures in tumor microenvironment predicts therapeutic response in cancer
Received 21 December 2015
Accepted for publication 19 January 2016
Published 1 March 2016 Volume 2016:9 Pages 1047—1055
DOI https://doi.org/10.2147/OTT.S102907
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr William C. Cho
Bhagelu R Achyut, Ali S Arbab
Tumor Angiogenesis Laboratory, Department of Biochemistry and Molecular Biology, Cancer Center, Georgia Regents University, Augusta, GA, USA
Abstract: Tumor microenvironment (TME) consists of several immune and nonimmune cell populations including tumor cells. For many decades, experimental studies have depicted profound contribution of TME toward cancer progression and metastasis development. Several therapeutic strategies have been tested against TME through preclinical studies and clinical trials. Unfortunately, most of them have shown transient effect, and have largely failed due to aggressive tumor growth and without improving survival. Solid tumors are known to have a strong myeloid component (eg, tumor-associated macrophages) in tumor development. Recent data suggest that therapeutic responses in tumor are characterized by alterations in immune cell signatures, including tumor-associated myeloid cells. Polarized tumor-associated myeloid cells (M1–M2) are critical in impairing therapeutic effect and promoting tumor growth. The present review is intended to compile all the literatures related to the emerging contribution of different populations of myeloid cells in the development of tumor and therapeutic failures. Finally, we have discussed targeting of myeloid cell populations as a combination therapy with chemo-, targeted-, or radiation therapies.
Keywords: tumor microenvironment, tumor-associated macrophage, myeloid-derived suppressor cells, therapies, macrophage polarization, radiation, antiangiogenic therapy
Introduction
Tumor microenvironment (TME) has profound contribution toward cancer development and metastasis.1,2 Recent advancements in cancer research have made very clear that tumor is not a single entity, but consists of various host components such as stromal cells, growing blood vessels, and heterogeneous immune cell populations, in addition to the tumor cells.1 Inflammatory cells that are recruited to the tumors from bone marrow significantly contribute to local inflammation.3,4 Depending on the context, infiltrating inflammatory cells in the TME may exert a dual role on tumor growth and progression.5,6 Initially, TME exerts antitumor immune responses by the immune cells that may inhibit tumor cell growth.7 However, at advanced stages, protumoral factors and chemokines secreted by tumor recruit and regulate immune cells to favor tumor growth and progression.6 Among tumor-infiltrating immune cells, heterogeneous populations of myeloid cells (eg, macrophages) are known as distinct critical players in TME to regulate tumor cell migration and metastasis.8–12 The present review is intended to introduce heterogeneous subtypes of myeloid populations and compile the literatures related to the involvement of myeloid cells in the development of tumors and therapeutic failures. Finally, we have discussed targeting of myeloid cell populations as a combination therapy with chemo-, targeted-, or radiation therapies.
Tumor-associated macrophages
Tumor-associated macrophages (TAMs) are part of heterogeneous populations of immunosuppressive myeloid cells that produce chemokines for the activation and maintenance of inflammatory processes in TME.4,9,10,13 TAM recruitment, localization, and phenotypes are regulated by the tumor-secreted factors at the hypoxic areas of the tumor.14,15 Depending on the stimuli, macrophages undergo series of functional reprogramming as described by two different polarization states, known as M1 and M2.15,16 In TME, M1 macrophages are activated by tumor-derived cytokines such as granulocyte monocyte colony-stimulating factor and tumor necrosis factor (TNF). M1 macrophages play an important role as inducer and effector cells in polarized T-helper type 1 cells (Th1) responses. M1 macrophages produce high amount of interleukin (IL)-12 and IL-23, and low IL-10.16 M1 cells also produce reactive oxygen and nitrogen species, and IL-1β, TNF, and IL-6 inflammatory cytokines.17 In addition, M1 macrophages release antitumor chemokines or chemokines that attract Th1 cells such as chemokine (C-X-C motif) ligand (CXCL)-9 and CXCL-10.18–20 Th1 cells drive cellular immunity to eliminate cancerous cells. Studies suggest that recruitment and/or differentiation of M1 macrophages can be inhibited by the T-regulatory cells (Tregs) that promote tumor progression.21 On the other hand, M2 macrophages are induced by IL-4, IL-13, IL-21, and IL-33 cytokines in the TME.22,23 M2 macrophages release high levels of IL-10 and low levels of IL-12 and IL-23 (type 2 cytokines). M2 macrophages also produce chemokine (C-C motif) ligand (CCL)-17, CCL-22, and CCL-24 chemokines that regulate the recruitment of Tregs, Th2, eosinophils, and basophils (type 2 pathway) in tumors.18,20 Th2 response is associated with the anti-inflammatory microenvironment, which promotes tumor growth. In comparison with M1 macrophages, M2 cells are poor antigen presenters, inhibit inflammation, and contribute to tumor progression by angiogenesis and tissue remodeling.16,24,25
TAMs infiltrating to the tumor may participate in local inflammation and may favor tumor progression by acquiring M2-like phenotype.26 Tumor-secreted molecules were associated with the M2-type TAM polarization and tumor growths. For example, secretion of distinct TAM-associated molecules by tumor induces expression of vascular endothelial growth factor (VEGF), mannose receptor-1, arginase-1, IL-10, transforming growth factor-beta (TGF-β), and matrix metallopeptidase 9.27 Overexpression of nodal protein (member of TGF-β superfamily) by tumor contributes to TAM polarization in cancer and contributes tumorigenesis, invasion, and metastasis.28 Nodal protein promotes generation of M2-like macrophages and downregulates expression of IL-12. Interestingly, inhibition of nodal protein reprogrammed TAMs to classically activate M1 macrophages.28 TAMs showed high levels of protumorigenic and hypoxia-associated genes compared with that of splenic myeloid cells.29 Protumoral functions of TAMs are facilitated by inhibiting the antitumoral immune surveillance through participating in the extracellular matrix remodeling and enhancing angiogenesis, cancer cell proliferation, invasion, and metastasis.11 In TME, TAMs are associated with the tumor vasculature development under hypoxic environment. A provascular program is triggered in TAMs by inducing expression of hypoxia-inducible factor-1 and hypoxia-inducible factor-2, and thus, overexpression of tumor-promoting VEGF, basic fibroblast growth factor, IL-8 chemokines, and lymphangiogenic factors.30,31 Surprisingly, tumor cell-derived Sema3A, not VEGF, is responsible for accumulation of TAM into hypoxic niches, which cause TAM to escape antitumor immunity and to promote vasculature development.32 On the other hand, different studies reported that hypoxia is not a major driver for the differentiation of TAM subset found in tumor infiltrate, but rather hypoxia fine-tunes the M2-like macrophage population.33 Moreover, studies suggest that TAM employ antitumoral activities by releasing a wide range of growth factors, cytokines, and chemokines, which activate both the innate and adaptive immune responses.11,21 Other type of myeloid population is known as myeloid-derived suppressor cells (MDSCs), which is critical in regulating TME and thus the tumor progression. We have discussed the characteristics, phenotype, and functions of MDSCs in the next section.
Myeloid-derived suppressor cells
MDSCs are immunosuppressive cells, which are abundant in TME and inhibit T-cell-mediated antitumor immunity.34–36 Myeloid expansion in spleen and peripheral blood are seen in spontaneous and xenograft murine models of cancer.34,37–39 Similar myeloid expansions have been observed in a range of human cancers.40,41 In mice, MDSCs express Gr1+ and CD11b+ myeloid markers, which can be divided into monocytic and granulocytic MDSCs. Monocytic MDSCs express CD11b+Ly6G−/Ly6C+ and granulocytic MDSCs express CD11b+Ly6G+/Ly6Clow markers. On the other hand, human MDSCs express CD11b and CD33 markers. Monocytic MDSCs are characterized by expression of human leukocyte antigen-antigen D related (HLA-DR−), CD11b+, CD33+, and CD14+ markers in humans, whereas mature monocytes express HLA-DR marker. Human granulocytic MDSC are usually characterized by the presence of HLA-DR−, CD11b+, CD33+, and CD15+ markers.35 Gr1 antigen is absent in the human MDSCs. Interestingly, phenotypic characterization of MDSCs through surface markers is heterogeneous and depends on the site of tumor in human cancers.42 Molecular signals that stimulate MDSCs to acquire immunosuppressive properties are signal transducer and activator of transcription (STAT)1, STAT3, and STAT6, and nuclear factor-κB transcription factors.43,44 Arginase 1 (ARG1), NADPH oxidase, inducible nitric oxide synthase, indoleamine 2,3-dioxygenase, and immunosuppressive cytokines that inhibit cytotoxic T-lymphocytes (CTLs), dendritic cells, and natural killer cells are produced by activated MDSCs.36 Surprisingly, expression of CD79a (B-cell receptor component) on immature myeloid cells contributes to their tumor-promoting effects.45 Downregulation of CD40 expression also contribute to accumulation of MDSCs by facilitating MDSC’s resistance to apoptosis.46 In addition, CD4+CD25+FoxP3+ Tregs are expanded due to release of MDSC-secreted factors to generate immunosuppressive TME.47 Overall, it is evident that MDSCs share functional similarities with TAM in TME.
Summary
TME is intricate and consists of heterogeneous subsets of myeloid cells. Growing tumor is capable of modulating antitumor myeloid cells to protumor myeloid cells through secreted factors. M1–M2 polarization of myeloid cells resulted into immunosuppressive and protumor phenotypes. Other category of myeloid cells that exerts protumor function in microenvironment is called MDSCs. All the tumor-promoting myeloid subsets are characterized by the surface markers, secretory factors, and their functions in the microenvironment. Tumor-promoting myeloid cells inhibit antitumor immunity and thus, enhance tumor growth. Next, we have discussed the contribution of key myeloid populations in the therapeutic responses.
Myeloid cell signatures in therapeutic response
Myeloid cells are the key players of microenvironmental regulation of tumor growth and affects therapeutic responses in cancer.48–50 Recently, the role of commensal microbiota on myeloid cell functions and their effect on the response to cancer therapy has been discussed.51 In this section, we have discussed how cellular and molecular myeloid cell signatures are associated with the antiangiogenic therapy (AAT), chemotherapy, and radiotherapy responses.
Myeloid cells in AAT response
Vasculature development is considered one of the major cancer hallmarks in tumor progression, which mediates through VEGF–vascular endothelial growth factor receptor (VEGFR).52,53 AATs have been tried against VEGF–VEGFRs pathways to inhibit the vasculature development in tumor. Surprisingly, most of the treatments resulted into transient decrease in tumor growth followed by enhanced vasculature and tumor growth, which are associated with the presence of MDSCs and TAMs (Table 1). Surprisingly, immune-suppressive myeloid cells mediate tumor resistance to anti-VEGF therapies.54,55 Paracrine signaling network between Th17 and immature myeloid cells or MDSCs induces the expression of granulocyte colony-stimulating factor in the stromal compartment, which in turn attracts MDSCs that drive anti-VEGF-A resistance.56 Recently, we found that myeloid cells mediate escape from AAT in preclinical chimeric mouse model of glioblastoma (GBM).57 AAT through vatalanib, a VEGFR tyrosine kinase inhibitor, was associated with increased bone marrow-derived tumor-associated myeloid cells in GBM.57 Therefore, targeting myeloid cells was proposed using anti-colony-stimulating factor 1 receptor (CSF1R) agents (eg, GW2580) to combat tumor evasion against AATs.58 Depletion of CSF1R+ myeloid cells with GW2580 decreased recruitment of tumor-associated myeloid cells in the tumor and reduced GBM growth. Interestingly, AAT increased expression of CXCL-7 chemokine and CSF1R blockade decreased CXCL-7 in TME. In addition, CXCL-7 expression was correlated with number of tumor-infiltrating bone marrow cells, phosphor-ERK mitogen-activated protein kinase, and proliferation of bone marrow cells in GBM.57
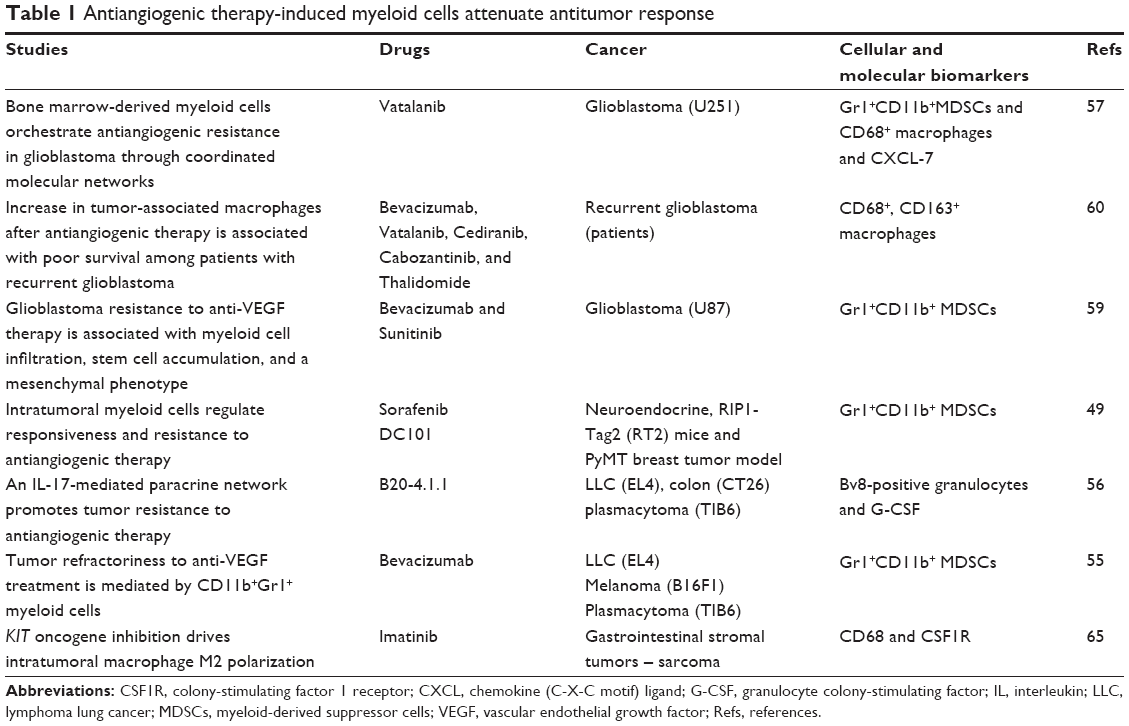 | Table 1 Antiangiogenic therapy-induced myeloid cells attenuate antitumor response |
Adverse effect of AATs has been reported by other study, where bevacizumab (Avastin) and sunitinib initially reduced both infiltration of macrophages and tumor vascularity, and showed sign of improved animal survival. However, multitargeted VEGFR tyrosine kinase inhibitors, but not VEGF inhibitor, rapidly created a vascular gradient in tumor and more rapidly induced hypoxia and reinfiltration of macrophages and CD11b+/Gr1+ myeloid cells. Tumors acquired aggressive mesenchymal features and expressed increased stem cell marker.59 Other group investigated the role of macrophages in patients with recurrent GBM. Specimen from 20 patients with recurrent GBM who received AAT and chemoradiation, and specimen from eight patients who received chemotherapy and/or radiotherapy without AAT or no treatment, was compared.60 Patients who received AAT and had recurrent GBMs showed an increased infiltration of myeloid cells in the tumor bulk and in the infiltrative regions. Higher numbers of CD11b+ cells correlated with poor prognosis of these patients and TAMs may represent a potential biomarker of resistance and a potential therapeutic target in recurrent GBM.60
Similarly, intratumoral myeloid cells are thought to regulate responsiveness and resistance to AAT in other solid cancers.49 The study found that the efficacy of antiangiogenic agents targeting the VEGF–VEGFR axis was dependent on induction of the angiostatic and immune-stimulatory chemokine CXCL-14 in pancreatic neuroendocrine and mammary tumors in mouse.49 Once VEGF–VEGFR axis was blocked, tumor initiated angiogenesis and immune suppression by activating phosphoinositide 3-kinase signaling in all CD11b+ cells, making tumors nonresponsive to VEGF–VEGFR inhibition. Adaptive resistance to AAT was also linked to an increased accumulation of Gr1+CD11b+ cells; however, targeting Gr1+ cells was not sufficient to sensitize antiangiogenic effect. On the other hand, inhibiting phosphoinositide 3-kinase activity in CD11b+ myeloid cells can create an angiostatic and immune-stimulatory environment in the tumor, where AAT can remain efficient. Moreover, studies suggest that AATs, particularly anti-VEGF–VEGFR2, are marked by the overrepresentation of immunosuppressive myeloid cells. Therefore, AAT with the combination of myeloid cell blockade may enhance the therapeutic success in inhibiting tumor growth.
Myeloid cells in chemotherapy response
Studies have reported the involvement of myeloid cell signatures-associated adverse responses with chemotherapeutic modalities (Table 2). Chemotherapy with paclitaxel caused upregulation of chemotactic factors for macrophage CSF1, CCL-8, and IL-34 and increased in CSFR1 expression in TAM in a transgenic mouse model of breast cancer.18,61 Chemotherapy combined with inhibitors of CSFR1 showed enhanced therapeutic activity with decreased metastases, increased T-cells in the tumors, and increased mRNA for various cytotoxic effector molecules such as granzyme A and B and perforin-1.61 When CD8+ CTLs were depleted, the tumor-suppressive effects due to the blockage of macrophage infiltration disappeared, suggesting chemotherapeutic response to be dependent on the depletion of macrophages and activity of CD8+ CTL.61 Recruitment of chemokine (C-C motif) receptor 2 (CCR2)-expressing monocytes occurred following doxorubicin treatment via stroma-derived CCL-2, which contributed to suboptimal treatment response and tumor reemergence in breast cancer model.62 Similarly, chemotherapies of murine breast cancers increased TAM accumulation, which enabled cathepsin protease B- and S-mediated chemoresistance to paclitaxel, etoposide, and doxorubicin.63 Treatment with cyclophosphamide causes the expansion of inflammatory monocytic myeloid cells (CD11b+Ly6ChiCCR2hi), which are immunosuppressive in nature. Ding et al64 showed the initial robust antitumor immune response with adoptive transfer (AT) of tumor-specific CD4+ T-cells following cyclophosphamide treatment (CTX+CD4 AT) in mice with advanced lymphoma, but the combined treatment also resulted in enhanced expansion of monocytic myeloid cells. These therapy-induced monocytic myeloid cells caused failure in long-term tumor control and subsequently caused relapse by mediating functional tolerization of antitumor CD4+ effector cells through the programmed death 1 (PD-1)–programmed death ligand 1 (PD-L1) axis. When PD-1–PD-L1 was blocked after CTX+CD4 AT therapy, there was persistence of CD4+ effector cells and antitumor effects.
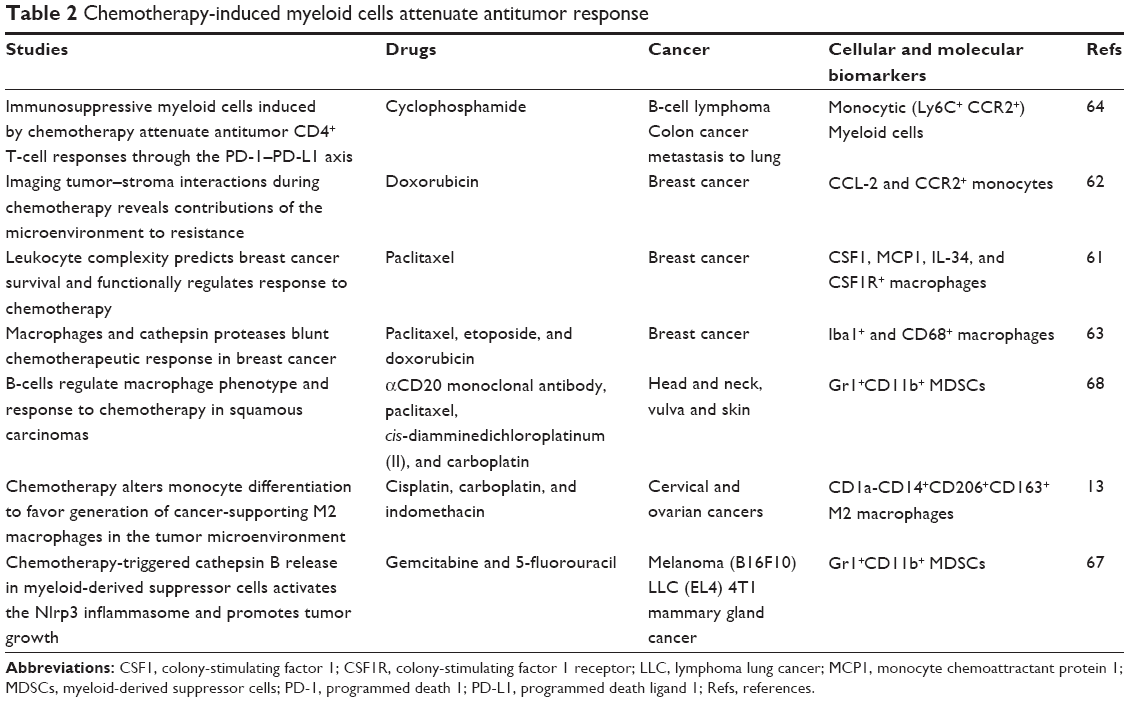 | Table 2 Chemotherapy-induced myeloid cells attenuate antitumor response |
In mice, established gastrointestinal stromal tumors contained M1-like TAMs, which were antitumoral. Imatinib therapy polarized TAMs to become M2-like through the activation of CCAAT/enhancer binding protein (C/EBP)β in this tumor model. Similar findings were observed in human, where TAMs behaved M1-like at baseline and became M2-like after imatinib therapy.65 Macrophages polarized into M2 alternatively activated state of macrophages in response to distinct therapies including platinum-based agents,13 and low-dose irradiation that promotes an inducible nitric oxide synthase-positive M1 phenotype66 that allowed the recruitment of CTL in the tumor and thereby enhanced immunotherapy efficacy in animal models.66 There are some reports that clearly showed that myeloid cells are capable of impairing chemotherapeutic antitumor response. For example, chemotherapeutic agents such as gemcitabine and 5-fluorouracil can activate the NOD-like receptor family (pyrin domain containing-3 protein [Nlrp3]-dependent caspase-1 activation complex [inflammasome]) in MDSCs, leading to production of IL-1β.67 Then IL-1β induced secretion of IL-17 by CD4+ T-cells, which impaired the anticancer efficacy of the chemotherapies.67 In other study, investigators have shown that myeloid-based pathways regulated by humoral immunity limit squamous cell carcinoma responses to chemotherapy not only by fostering tumor angiogenesis but also by impairing CD8+ T-cell infiltration into tumors.68 Here, B-cells educated TAMs toward a tumor-supporting phenotype by the activation of the Fc receptor. Further, B-cell depletion resulted in increased recruitment of CD8+ cells and an enhanced therapeutic response.68 On the other hand, some reports showed that chemotherapy could limit or reverse the expansion of myeloid populations in tumor.69–71 These paradoxical outcomes might be related to the differences in models as well as schedules of the chemotherapy. However, majority of data point toward negative role of tumor-associated myeloid cells (TAMC)s in chemotherapy failures.
Myeloid cells in radiotherapy response
Macrophages accumulated into TME following radiotherapy plays dual roles. Initially accumulated macrophages participate in M1-type responses early in inflammation and then convert to M2 responses at later stages. A series of multiple cytokines cause and sustain the acute phase of radiation-induced inflammation,72 and these cytokine patterns match the status of inflammatory macrophage differentiation in the site of radiation.73 The proinflammatory cascade that is initiated following radiotherapy has been linked to production of cytokines, including the M1 cytokine TNF-α.74,75 At later stages in this inflammatory cascade, the M2 cytokine TGF-β is expressed.76 Studies have shown the mechanisms of radiation-induced inflammation followed by repair and the consequences to adaptive immune responses in the treatment site, and how radiation-induced myeloid cell response may impact immunotherapies designed to improve control of residual cancer cells.77 The impact of radiation-induced myeloid cell response has been reported and discussed.77,78 Peripheral MDSCs together with Treg PD-1-positive cells have shown to predict the response to short-course radiotherapy in rectal cancer patients.79 Treatment with sunitinib increased the efficacy of stereotactic radiotherapy in patients with oligometastases by reversing MDSC and Treg-mediated immune suppression.80 Commonly used nonhypofractionated radiotherapy induced stromal cell-derived factor-1 and caused accumulation of bone marrow-derived myeloid monocytic cells that contributed to vasculogenesis and increased tumor growth.81,82 Similarly, the effect of radiotherapy can be limited due to accumulation of Th2-polarized CD4+ T-cells and macrophages. By depleting macrophages using either a neutralizing monoclonal antibody to CSF1 or a small-molecule inhibitor of the CSF1R (PLX3397) significantly delayed tumor regrowth following radiotherapy in mammary tumor-bearing mice. Delayed tumor growth in this study was thought to be associated with increased accumulation of CD8+ T-cells and reduction of CD4+ T-cells, the main source of the Th2 cytokine IL-4 in mammary tumors.83 Similarly, radiotherapy upregulated CSF1 in prostate cancers and increased myeloid cell numbers and blockade of CSF1R signaling decreased the number of myeloid cells and improved the efficacy of radiotherapy in prostate cancer.84 Radiotherapy could be combined with immunotherapy to improve the antitumor responses.85 On the other hand, one study reported that expansion of peripheral myeloid cells driven by 4T1 murine cancer progression was reversed by radiotherapy.86 Altogether, studies suggested that myeloid cells modulate radiotherapy response and are bonafide target of cancer therapy.
Summary
Majority of tumors are characterized by the overrepresentation of tumor-promoting myeloid subsets. Recent studies suggest that therapies such as AAT, chemotherapy, and radiotherapy against tumors resulted into increased accumulation of myeloid cells. Current evidence suggests that myeloid cells impair antitumor immunity through secreted factors, which constitute immunosuppressive microenvironment. In some experimental studies, targeting myeloid cells by CSF1R inhibitors have improved antitumor immunity by increasing CD8+ T-cells and thus, decreasing tumor growth. Therefore, combining conventional therapies with myeloid inhibitor could enhance the therapeutic efficacy in cancer.
Conclusion and future perspectives
There are many mechanisms known by which cancers can develop resistance to various therapeutic modalities. Therapeutic resistance could be added as an emerging area of interest and cancer hallmark. In addition, biomarkers that can predict response to any type of therapy are urgent to explore.52,87 At this point, we are sure that heterogeneous macrophages are critical players in therapeutic resistance against cancer. Therefore, myeloid cell blockade in addition to AAT or chemotherapy or radiotherapy should provide better antitumor responses. Different myeloid cell blockers have been tested and discussed in literature.50,88,89 Our biggest challenge is the limited understanding of the underlying mechanisms through which therapies modulate distinct subpopulations of macrophages within the TME. Recently, we initiated exploring cytokines, which take part in therapeutic resistance.57 However, we may need complete understanding of the entire TME before and after therapeutic modalities to get clear changes in TME. In addition, we may take advantages of available large-scale therapeutic datasets to rule out gene expression profiles of heterogeneous populations of TAMs using data from whole-tumor samples from patients through bioinformatics approaches.90–92
Acknowledgment
This study was supported by grants from the National Institutes of Health (Nos R01CA160216 and R01CA172048).
Disclosure
The authors report no conflicts of interest in this work.
References
Faurobert E, Bouin AP, Albiges-Rizo C. Microenvironment, tumor cell plasticity, and cancer. Curr Opin Oncol. 2015;27(1):64–70. | ||
Jung HY, Fattet L, Yang J. Molecular pathways: linking tumor microenvironment to epithelial-mesenchymal transition in metastasis. Clin Cancer Res. 2015;21(5):962–968. | ||
Goubran HA, Kotb RR, Stakiw J, Emara ME, Burnouf T. Regulation of tumor growth and metastasis: the role of tumor microenvironment. Cancer Growth Metastasis. 2014;7:9–18. | ||
Gao F, Liang B, Reddy ST, Farias-Eisner R, Su X. Role of inflammation-associated microenvironment in tumorigenesis and metastasis. Curr Cancer Drug Targets. 2014;14(1):30–45. | ||
Whiteside TL. The tumor microenvironment and its role in promoting tumor growth. Oncogene. 2008;27(45):5904–5912. | ||
Zamarron BF, Chen W. Dual roles of immune cells and their factors in cancer development and progression. Int J Biol Sci. 2011;7(5):651–658. | ||
Bissell MJ, Hines WC. Why don’t we get more cancer? A proposed role of the microenvironment in restraining cancer progression. Nat Med. 2011;17(3):320–329. | ||
Sionov RV, Fridlender ZG, Granot Z. The multifaceted roles neutrophils play in the tumor microenvironment. Cancer Microenviron. 2015;8(3):125–158. | ||
Mou W, Xu Y, Ye Y, et al. Expression of Sox2 in breast cancer cells promotes the recruitment of M2 macrophages to tumor microenvironment. Cancer Lett. 2015;358(2):115–123. | ||
Allavena P, Sica A, Solinas G, Porta C, Mantovani A. The inflammatory micro-environment in tumor progression: the role of tumor-associated macrophages. Crit Rev Oncol Hematol. 2008;66(1):1–9. | ||
Condeelis J, Pollard JW. Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell. 2006;124(2):263–266. | ||
Galdiero MR, Bonavita E, Barajon I, Garlanda C, Mantovani A, Jaillon S. Tumor associated macrophages and neutrophils in cancer. Immunobiology. 2013;218(11):1402–1410. | ||
Dijkgraaf EM, Heusinkveld M, Tummers B, et al. Chemotherapy alters monocyte differentiation to favor generation of cancer-supporting M2 macrophages in the tumor microenvironment. Cancer Res. 2013;73(8):2480–2492. | ||
Tripathi C, Tewari BN, Kanchan RK, et al. Macrophages are recruited to hypoxic tumor areas and acquire a pro-angiogenic M2-polarized phenotype via hypoxic cancer cell derived cytokines Oncostatin M and Eotaxin. Oncotarget. 2014;5(14):5350–5368. | ||
Mantovani A, Sozzani S, Locati M, et al. Infiltration of tumours by macrophages and dendritic cells: tumour-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Novartis Found Symp. 2004;256:137–145; discussion 146–148, 259–269. | ||
Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. 2012;122(3):787–795. | ||
Tseng D, Volkmer JP, Willingham SB, et al. Anti-CD47 antibody-mediated phagocytosis of cancer by macrophages primes an effective antitumor T-cell response. Proc Natl Acad Sci U S A. 2013;110(27):11103–11108. | ||
Sica A, Allavena P, Mantovani A. Cancer related inflammation: the macrophage connection. Cancer Lett. 2008;267(2):204–215. | ||
Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454(7203):436–444. | ||
Germano G, Allavena P, Mantovani A. Cytokines as a key component of cancer-related inflammation. Cytokine. 2008;43(3):374–379. | ||
Pommier A, Audemard A, Durand A, et al. Inflammatory monocytes are potent antitumor effectors controlled by regulatory CD4+ T cells. Proc Natl Acad Sci U S A. 2013;110(32):13085–13090. | ||
Pesce J, Kaviratne M, Ramalingam TR, et al. The IL-21 receptor augments Th2 effector function and alternative macrophage activation. J Clin Invest. 2006;116(7):2044–2055. | ||
Kurowska-Stolarska M, Stolarski B, Kewin P, et al. IL-33 amplifies the polarization of alternatively activated macrophages that contribute to airway inflammation. J Immunol. 2009;183(10):6469–6477. | ||
Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol. 2010;11(10):889–896. | ||
Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002;23(11):549–555. | ||
Biswas SK, Gangi L, Paul S, et al. A distinct and unique transcriptional program expressed by tumor-associated macrophages (defective NF-kappaB and enhanced IRF-3/STAT1 activation). Blood. 2006;107(5):2112–2122. | ||
Katara GK, Jaiswal MK, Kulshrestha A, Kolli B, Gilman-Sachs A, Beaman KD. Tumor-associated vacuolar ATPase subunit promotes tumorigenic characteristics in macrophages. Oncogene. 2014;33(49):5649–5654. | ||
Wang XF, Wang HS, Zhang F, et al. Nodal promotes the generation of M2-like macrophages and downregulates the expression of IL-12. Eur J Immunol. 2014;44(1):173–183. | ||
Elpek KG, Cremasco V, Shen H, et al. The tumor microenvironment shapes lineage, transcriptional, and functional diversity of infiltrating myeloid cells. Cancer Immunol Res. 2014;2(7):655–667. | ||
Murdoch C, Giannoudis A, Lewis CE. Mechanisms regulating the recruitment of macrophages into hypoxic areas of tumors and other ischemic tissues. Blood. 2004;104(8):2224–2234. | ||
Zeisberger SM, Odermatt B, Marty C, Zehnder-Fjallman AH, Ballmer-Hofer K, Schwendener RA. Clodronate-liposome-mediated depletion of tumour-associated macrophages: a new and highly effective antiangiogenic therapy approach. Br J Cancer. 2006;95(3):272–281. | ||
Casazza A, Laoui D, Wenes M, et al. Impeding macrophage entry into hypoxic tumor areas by Sema3A/Nrp1 signaling blockade inhibits angiogenesis and restores antitumor immunity. Cancer Cell. 2013;24(6):695–709. | ||
Laoui D, Van Overmeire E, Di Conza G, et al. Tumor hypoxia does not drive differentiation of tumor-associated macrophages but rather fine-tunes the M2-like macrophage population. Cancer Res. 2014;74(1):24–30. | ||
Yan HH, Pickup M, Pang Y, et al. Gr-1+CD11b+ myeloid cells tip the balance of immune protection to tumor promotion in the premetastatic lung. Cancer Res. 2010;70(15):6139–6149. | ||
Achyut BR, Arbab AS. Myeloid derived suppressor cells: fuel the fire. Biochem Physiol. 2014;3:e123. | ||
Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nature Rev Immunol. 2012;12(4):253–268. | ||
Yan HH, Jiang J, Pang Y, et al. CCL9 induced by TGF-beta signaling in myeloid cells enhances tumor cell survival in the premetastatic organ. Cancer Res. 2015;75:5283–5298. | ||
Yang L, DeBusk LM, Fukuda K, et al. Expansion of myeloid immune suppressor Gr+CD11b+ cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell. 2004;6(4):409–421. | ||
Melani C, Sangaletti S, Barazzetta FM, Werb Z, Colombo MP. Amino-biphosphonate-mediated MMP-9 inhibition breaks the tumor-bone marrow axis responsible for myeloid-derived suppressor cell expansion and macrophage infiltration in tumor stroma. Cancer Res. 2007;67(23):11438–11446. | ||
Hoechst B, Ormandy LA, Ballmaier M, et al. A new population of myeloid-derived suppressor cells in hepatocellular carcinoma patients induces CD4(+)CD25(+)Foxp3(+) T cells. Gastroenterology. 2008;135(1):234–243. | ||
Diaz-Montero CM, Salem ML, Nishimura MI, Garrett-Mayer E, Cole DJ, Montero AJ. Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin-cyclophosphamide chemotherapy. Cancer Immunol Immunother. 2009;58(1):49–59. | ||
Solito S, Marigo I, Pinton L, Damuzzo V, Mandruzzato S, Bronte V. Myeloid-derived suppressor cell heterogeneity in human cancers. Ann N Y Acad Sci. 2014;1319:47–65. | ||
Corzo CA, Cotter MJ, Cheng P, et al. Mechanism regulating reactive oxygen species in tumor-induced myeloid-derived suppressor cells. J Immunol. 2009;182(9):5693–5701. | ||
Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9(3):162–174. | ||
Luger D, Yang YA, Raviv A, et al. Expression of the B-cell receptor component CD79a on immature myeloid cells contributes to their tumor promoting effects. PLoS One. 2013;8(10):e76115. | ||
Shen J, Chen X, Wang Z, Zhang G, Chen W. Downregulation of CD40 expression contributes to the accumulation of myeloid-derived suppressor cells in gastric tumors. Oncol Lett. 2014;8(2):775–780. | ||
Almand B, Clark JI, Nikitina E, et al. Increased production of immature myeloid cells in cancer patients: a mechanism of immunosuppression in cancer. J Immunol. 2001;166(1):678–689. | ||
De Palma M, Lewis CE. Macrophage regulation of tumor responses to anticancer therapies. Cancer Cell. 2013;23(3):277–286. | ||
Rivera LB, Meyronet D, Hervieu V, Frederick MJ, Bergsland E, Bergers G. Intratumoral myeloid cells regulate responsiveness and resistance to antiangiogenic therapy. Cell Rep. 2015;11(4):577–591. | ||
Klemm F, Joyce JA. Microenvironmental regulation of therapeutic response in cancer. Trends Cell Biol. 2015;25(4):198–213. | ||
Goldszmid RS, Dzutsev A, Viaud S, Zitvogel L, Restifo NP, Trinchieri G. Microbiota modulation of myeloid cells in cancer therapy. Cancer Immunol Res. 2015;3(2):103–109. | ||
Hainaut P, Plymoth A. Targeting the hallmarks of cancer: towards a rational approach to next-generation cancer therapy. Curr Opin Oncol. 2013;25(1):50–51. | ||
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. | ||
Shojaei F, Wu X, Qu X, et al. G-CSF-initiated myeloid cell mobilization and angiogenesis mediate tumor refractoriness to anti-VEGF therapy in mouse models. Proc Natl Acad Sci U S A. 2009;106(16):6742–6747. | ||
Shojaei F, Wu X, Malik AK, et al. Tumor refractoriness to anti-VEGF treatment is mediated by CD11b+Gr1+ myeloid cells. Nat Biotechnol. 2007;25(8):911–920. | ||
Chung AS, Wu X, Zhuang G, et al. An interleukin-17-mediated paracrine network promotes tumor resistance to anti-angiogenic therapy. Nat Med. 2013;19(9):1114–1123. | ||
Achyut BR, Shankar A, Iskander A, et al. Bone marrow derived myeloid cells orchestrate antiangiogenic resistance in glioblastoma through coordinated molecular networks. Cancer Lett. 2015;369(2):416–426. | ||
Priceman SJ, Sung JL, Shaposhnik Z, et al. Targeting distinct tumor-infiltrating myeloid cells by inhibiting CSF-1 receptor: combating tumor evasion of antiangiogenic therapy. Blood. 2010;115(7):1461–1471. | ||
Piao Y, Liang J, Holmes L, et al. Glioblastoma resistance to anti-VEGF therapy is associated with myeloid cell infiltration, stem cell accumulation, and a mesenchymal phenotype. Neuro Oncol. 2012;14(11):1379–1392. | ||
Lu-Emerson C, Snuderl M, Kirkpatrick ND, et al. Increase in tumor-associated macrophages after antiangiogenic therapy is associated with poor survival among patients with recurrent glioblastoma. Neuro Oncol. 2013;15(8):1079–1087. | ||
DeNardo DG, Brennan DJ, Rexhepaj E, et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov. 2011;1(1):54–67. | ||
Nakasone ES, Askautrud HA, Kees T, et al. Imaging tumor-stroma interactions during chemotherapy reveals contributions of the microenvironment to resistance. Cancer Cell. 2012;21(4):488–503. | ||
Shree T, Olson OC, Elie BT, et al. Macrophages and cathepsin proteases blunt chemotherapeutic response in breast cancer. Genes Dev. 2011;25(23):2465–2479. | ||
Ding ZC, Lu X, Yu M, et al. Immunosuppressive myeloid cells induced by chemotherapy attenuate antitumor CD4+ T-cell responses through the PD-1-PD-L1 axis. Cancer Res. 2014;74(13):3441–3453. | ||
Cavnar MJ, Zeng S, Kim TS, et al. KIT oncogene inhibition drives intratumoral macrophage M2 polarization. J Exp Med. 2013;210(13):2873–2886. | ||
Klug F, Prakash H, Huber PE, et al. Low-dose irradiation programs macrophage differentiation to an iNOS(+)/M1 phenotype that orchestrates effective T cell immunotherapy. Cancer Cell. 2013;24(5):589–602. | ||
Bruchard M, Mignot G, Derangere V, et al. Chemotherapy-triggered cathepsin B release in myeloid-derived suppressor cells activates the Nlrp3 inflammasome and promotes tumor growth. Nat Med. 2013;19(1):57–64. | ||
Affara NI, Ruffell B, Medler TR, et al. B cells regulate macrophage phenotype and response to chemotherapy in squamous carcinomas. Cancer Cell. 2014;25(6):809–821. | ||
Sinha P, Clements VK, Bunt SK, Albelda SM, Ostrand-Rosenberg S. Cross-talk between myeloid-derived suppressor cells and macrophages subverts tumor immunity toward a type 2 response. J Immunol. 2007;179(2):977–983. | ||
Vincent J, Mignot G, Chalmin F, et al. 5-Fluorouracil selectively kills tumor-associated myeloid-derived suppressor cells resulting in enhanced T cell-dependent antitumor immunity. Cancer Res. 2010;70(8):3052–3061. | ||
Suzuki E, Kapoor V, Jassar AS, Kaiser LR, Albelda SM. Gemcitabine selectively eliminates splenic Gr-1+/CD11b+ myeloid suppressor cells in tumor-bearing animals and enhances antitumor immune activity. Clin Cancer Res. 2005;11(18):6713–6721. | ||
Rubin P, Johnston CJ, Williams JP, McDonald S, Finkelstein JN. A perpetual cascade of cytokines postirradiation leads to pulmonary fibrosis. Int J Radiat Oncol Biol Phys. 1995;33(1):99–109. | ||
Rubin P, Finkelstein J, Shapiro D. Molecular biology mechanisms in the radiation induction of pulmonary injury syndromes: interrelationship between the alveolar macrophage and the septal fibroblast. Int J Radiat Oncol Biol Phys. 1992;24(1):93–101. | ||
Hallahan DE, Spriggs DR, Beckett MA, Kufe DW, Weichselbaum RR. Increased tumor necrosis factor alpha mRNA after cellular exposure to ionizing radiation. Proc Natl Acad Sci U S A. 1989;86(24):10104–10107. | ||
Weichselbaum RR, Hallahan D, Fuks Z, Kufe D. Radiation induction of immediate early genes: effectors of the radiation-stress response. Int J Radiat Oncol Biol Phys. 1994;30(1):229–234. | ||
Anscher MS, Kong FM, Marks LB, Bentel GC, Jirtle RL. Changes in plasma transforming growth factor beta during radiotherapy and the risk of symptomatic radiation-induced pneumonitis. Int J Radiat Oncol Biol Phys. 1997;37(2):253–258. | ||
Gough MJ, Young K, Crittenden M. The impact of the myeloid response to radiation therapy. Clin Dev Immunol. 2013;2013:281958. | ||
Vatner RE, Formenti SC. Myeloid-derived cells in tumors: effects of radiation. Semin Radiat Oncol. 2015;25(1):18–27. | ||
Napolitano M, D’Alterio C, Cardone E, et al. Peripheral myeloid-derived suppressor and T regulatory PD-1 positive cells predict response to neoadjuvant short-course radiotherapy in rectal cancer patients. Oncotarget. 2015;6(10):8261–8270. | ||
Chen HM, Ma G, Gildener-Leapman N, et al. Myeloid-derived suppressor cells as an immune parameter in patients with concurrent sunitinib and stereotactic body radiotherapy. Clin Cancer Res. 2015;21(18):4073–4085. | ||
Kioi M, Vogel H, Schultz G, Hoffman RM, Harsh GR, Brown JM. Inhibition of vasculogenesis, but not angiogenesis, prevents the recurrence of glioblastoma after irradiation in mice. J Clin Invest. 2010;120(3):694–705. | ||
Kozin SV, Kamoun WS, Huang Y, Dawson MR, Jain RK, Duda DG. Recruitment of myeloid but not endothelial precursor cells facilitates tumor regrowth after local irradiation. Cancer Res. 2010;70(14):5679–5685. | ||
Shiao SL, Ruffell B, DeNardo DG, Faddegon BA, Park CC, Coussens LM. TH2-Polarized CD4(+) T cells and macrophages limit efficacy of radiotherapy. Cancer Immunol Res. 2015;3(5):518–525. | ||
Xu J, Escamilla J, Mok S, et al. CSF1R signaling blockade stanches tumor-infiltrating myeloid cells and improves the efficacy of radiotherapy in prostate cancer. Cancer Res. 2013;73(9):2782–2794. | ||
Formenti SC, Demaria S. Combining radiotherapy and cancer immunotherapy: a paradigm shift. J Natl Cancer Inst. 2013;105(4):256–265. | ||
Crittenden MR, Savage T, Cottam B, et al. The peripheral myeloid expansion driven by murine cancer progression is reversed by radiation therapy of the tumor. PLoS One. 2013;8(7):e69527. | ||
Jain RK, Duda DG, Willett CG, et al. Biomarkers of response and resistance to antiangiogenic therapy. Nat Rev Clin Oncol. 2009;6(6):327–338. | ||
Noy R, Pollard JW. Tumor-associated macrophages: from mechanisms to therapy. Immunity. 2014;41(1):49–61. | ||
Cotechini T, Medler TR, Coussens LM. Myeloid cells as targets for therapy in solid tumors. Cancer J. 2015;21(4):343–350. | ||
Yoshihara K, Shahmoradgoli M, Martinez E, et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun. 2013;4:2612. | ||
Shoemaker JE, Lopes TJ, Ghosh S, Matsuoka Y, Kawaoka Y, Kitano H. CTen: a web-based platform for identifying enriched cell types from heterogeneous microarray data. BMC Genomics. 2012;13:460. | ||
Meng W, Wu Y, He X, et al. A systems biology approach identifies effective tumor-stroma common targets for oral squamous cell carcinoma. Cancer Res. 2014;74(8):2306–2315. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.