")
Back to Journals » Clinical, Cosmetic and Investigational Dermatology » Volume 15
Multiple Bullous and Ulcers as Cutaneous Manifestations of Wegener’s Granulomatosis: A Rare Case Report
Authors Pangastuti M , Rizqandaru T , Suwarsa O , Dharmadji HP, Sutedja E
Received 8 August 2022
Accepted for publication 1 October 2022
Published 7 October 2022 Volume 2022:15 Pages 2159—2164
DOI https://doi.org/10.2147/CCID.S385464
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Jeffrey Weinberg
Miranti Pangastuti, Trustia Rizqandaru, Oki Suwarsa, Hartati Purbo Dharmadji, Endang Sutedja
Department of Dermatology and Venereology, Faculty of Medicine, Universitas Padjadjaran - Dr. Hasan Sadikin Hospital, Bandung, Indonesia
Correspondence: Miranti Pangastuti, Department of Dermatology and Venereology, Faculty of Medicine, Universitas Padjadjaran - Dr. Hasan Sadikin Hospital, Jl. Pasteur 38, Bandung, West Java, 40161, Indonesia, Tel +6281223114874, Email [email protected]
Abstract: Bullous dermatoses is a heterogeneous group of blistering skin disorders that can either be inherited or acquired. Subepidermal blisters may result in ulceration and scarring following their rupture. Wegener’s granulomatosis (WG) is a granulomatous necrotizing vasculitis affecting small- to medium-sized blood vessels. It is associated with anti-neutrophil cytoplasmic antibodies (ANCA) and can be manifested cutaneously as multiple bullous and ulcers. A case of WG was reported in an 18-year-old man presented with multiple skin bullous and ulcers. The patient was diagnosed with WG based on the findings from nasopharyngoscopy examination that revealed crusts in his nasal cavity; necrotizing granulomatous appearance on chest radiograph; hematuria on urinalysis; and positive ANCA blood test. This patient received a combination of methylprednisolone and methotrexate, resulting in improvement within four weeks of therapy. His multiple skin ulcers were treated with a combination of dialkyl carbamoyl chloride, hydrocolloid, and hydrogel dressings. This patient was in complete remission state after six months of treatment, which later followed by a relapse episode that occurred within one year. WG with multiple skin bullous and ulcers can mimic other diseases. Various examinations such as histopathology, direct immunofluorescence, and ANCA blood test may aid in determining the etiology of skin bullous and ulcers.
Keywords: anti-neutrophil cytoplasmic antibodies, bullous, ulcers, Wegener’s granulomatosis
Introduction
Bullous skin disease comprises a large group of clinically polymorphic and sometimes devastating diseases.1 Subepidermal blister may result in multiple ulcers and scarring following their rupture.2 Bullous disease’s etiology can be differentiated into non-acquired (inherited) or acquired, and immunologically or non-immunologically mediated. Immunologically mediated diseases that could present cutaneously as multiple bullous and ulcers are bullous pemphigoid, linear IgA dermatosis, epidermolysis bullosa, dermatitis herpetiformis, bullous systemic lupus erythematosus, and anti-neutrophil cytoplasmic antibodies (ANCA)-associated vasculitis.3 Wegener’s granulomatosis (WG) is an autoimmune disease affecting small- to medium-sized blood vessels, and it is related to ANCA.4,5 This is a rare condition with an incidence rate of 2–12 cases per one million people annually.5 Skin involvement might occur in 50% of WG cases6 in forms of purpura, pustules, bullae, subcutaneous nodules, ulcer, and gangrene.4,5 This case report aims to describe a rare case of WG with cutaneous manifestations of multiple bullous and ulcers.
Case
An 18-year-old male was admitted to our hospital with a chief complaint of multiple painful bullae and ulcers on both arms, hands, legs, and feet. There was no history of skin trauma, malignancy, established autoimmune disease, and risk factors of sexually transmitted infections. Two weeks prior to admission, he reported erythematous macules which then transformed into painful bullae on both arms and legs. Some of the bullae had ruptured and formed an ulcer. He also reported other symptoms for the past couple of months, such as weight loss, headache, and stuffed nose.
On physical examination, we found skin discharge from the crusts in his nasal cavity and weakness in his extremities. Dermatological assessment revealed erythematous macules, vesicles, bullae, erosions, pustular crusts, and scales on both hands, back, buttocks, legs, and feet (Figure 1A–F). His blood test result showed anemia, neutrophilia, lymphocytosis, hypoalbuminemia, hyponatremia, and hypocalcemia. Microscopic examination using Gram staining from the sample obtained from his right lower leg showed Gram-negative cocci.
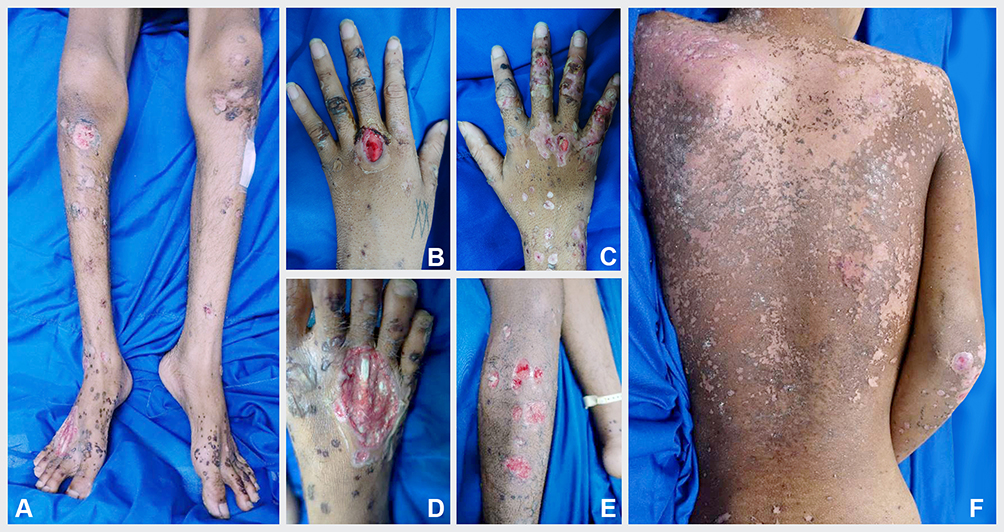 |
Figure 1 Skin disorder in forms of multiple bullous and ulcers on both of the legs (A and E), hands (B and C), and feet (D). On the back, there were erythematous macules and scale lesions (F). |
On the third day of admission, new ulcers appeared on the location of previous skin bullae, with irregular, non-indurated border and granulation tissue as their base. After 6 days of admission, the ulcer on the right hand and right foot were infiltrated further with tendon as its base. Neurological examination showed reduced motor function in both arms and legs. Blood serologic tests resulted in negative Human Immunodeficiency Virus, Venereal Disease Research Laboratory, and Treponema Pallidum Hemagglutination Assay. Urinalysis revealed hematuria with red blood cell (RBC) count of more than 50 per high power field and bacteriuria. Chest radiography showed a thick-walled lucent shadow filled with infiltrates on the upper- to middle-left lung field, which is supporting the appearance of necrotizing granulomatous (Figure 2A). A contrasted head computerized tomography (CT) scan was carried out due to declining motor function and the result was unremarkable.
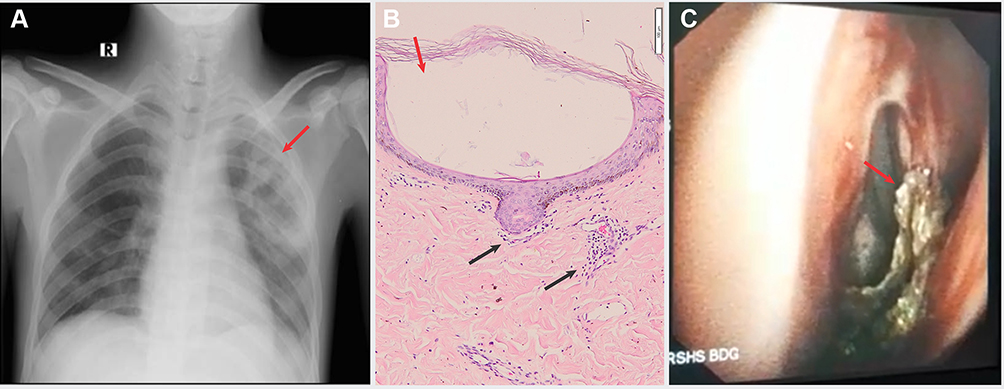 |
Figure 2 (A) Chest radiographic examination showed necrotizing granulomatous appearance (red arrow). (B) Histopathological examination revealed vesiculobullous reaction on subcorneal area (red arrow). Inflammatory cell and extravasation of erythrocytes in papillary dermis (black arrow). (C) Nasopharyngoscopy examination showed crusts (red arrow) that bleeds easily on nasal cavity. |
Skin biopsy of the bullae was taken for histopathological and direct immunofluorescence (DIF) examination. The results showed vesiculobullous reactions in subcorneal areas containing inflammatory cells, lymphocytes, neutrophils, and debris. There was also inflammatory cell and extravasation of the erythrocyte on papillary dermis (Figure 2B). The DIF test resulted in no IgG, IgM, IgA, C3, C1q, and fibrinogen cast. Nasopharyngoscopy revealed crusts in both nasal cavities that bleed easily (Figure 2C). Blood test showed negative antinuclear-antibody (ANA) but positive for p-ANCA with a 1:10 titer. Based on these findings, the patient was then diagnosed with Wegener’s granulomatosis. The diagnosis was established since he fulfilled three of four diagnosis criteria by the American College of Rheumatology (ACR): hematuria of more than 5 RBC per high-power field, abnormal findings on the chest radiography, and nasal discharge.
The patient was hospitalized for two weeks and treated with a systemic antibiotic for one week. Another systemic treatment was prescribed by the Internal Medicine Department, including methotrexate 7.5 mg/week, methylprednisolone 40 mg/day which was then tapered off, folic acid 1 mg/day, and calcium carbonate 500 mg twice a day. We provided modern wound dressing for skin ulcers using dialkyl carbamoyl chloride, hydrocolloid, and hydrogel dressings. His skin lesions showed improvement after four weeks of therapy. The patient was in complete remission state after six months of treatment (Figure 3A–F), followed by a relapse episode that manifested as new skin ulcers and hematuria one year later.
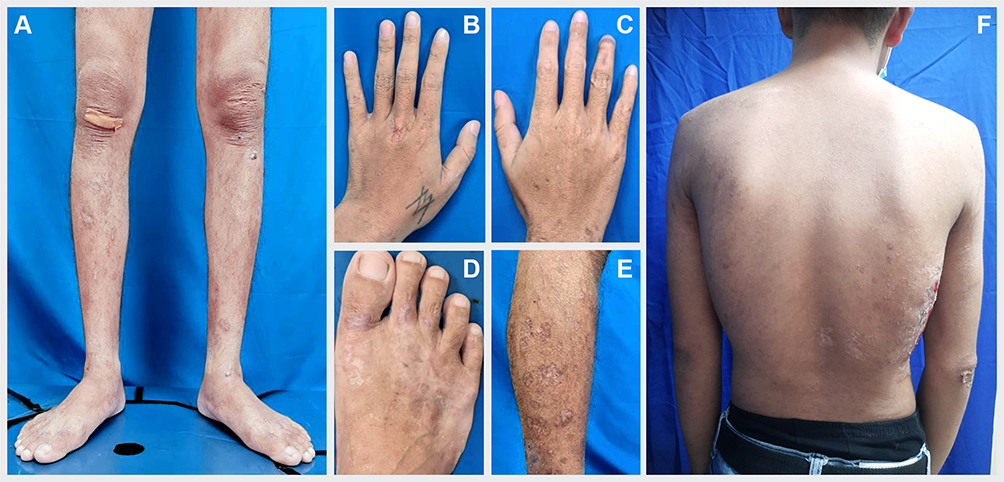 |
Figure 3 After 6-month of treatment, multiple bullous and ulcers in both legs (A and E), hands (B and C), and feet (D) disappeared. Some of the macule and scale lesions at back also disappeared (F). |
Discussion
The etiology of bullous dermatoses can be differentiated into non-acquired (inherited) or acquired; and immunologically and non-immunologically mediated. Based on the depth of blister formation, they can be classified into intraepidermal or subepidermal blisters.3 If systemic vasculitis is suspected as the etiology of skin disorder, a full systemic screening test is crucial to determine and treat the underlying disease in order to prevent internal organ damage.3
Wegener’s granulomatosis is a rare ANCA-associated vasculitis5,7 with prevalence of 23–160 per one million annually. There is no remarkable difference in the incidence of WG between males and females. The highest incidence rate is in 45- to 60-year age group.3,6 WG is rarely found in childhood or young adult age,5,8 with prevalence of 14.57% in people aged less than 19 year-old.9 The patient in our study is an 18-year-old male, a rare finding considering the patient’s age.
The etiology of WG has not been established yet, but the precipitating factors might include bacterial, mycobacterial, fungal, or viral infection in ear, nose, or respiratory tract.5,10 They can induce proinflammatory cytokines and ANCA secretion in people who are genetically predisposed to this disease.5 Proinflammatory cytokines and ANCA will trigger classic symptoms of WG, including necrotic granuloma formation in respiratory tract, vasculitis, and glomerulonephritis.4 WG might also affect mucosa, skin, eyes, cardiovascular, and gastrointestinal system.4,5 The patient in our study suffered from WG with skin, respiratory tract, and renal manifestation.
The most frequent cutaneous manifestations in WG are purpura6,11 and polymorphic lesions in forms of papule, nodule, vesicle, bullae, and ulcer with livedo reticularis.6 In 1994, Frances et al12 reported that amongst 75 WG cases, 35 of them had skin involvement where 14.3% of cases had skin ulcers. In our patient, his dermatological involvements were bullae and multiple ulcers with irregular non-indurated borders, and the depth of these ulcers varied from subcutaneous tissue up to tendon as their base. These findings were consistent with those observed in WG.
The common diagnostic criteria of WG is the 1990 ACR criteria.4,5 ACR criteria yielded 88% sensitivity and 92% specificity if two out of four criteria were established, ie, inflammation on the nose or mouth with a non-tender ulcer or purulent nasal discharge, abnormality of chest radiography showing nodule, infiltrate, or cavity on the lungs, abnormal urine sediment in forms of microscopic hematuria with or without erythrocyte crystal, granulomatous inflammation on biopsy result of the artery and perivascular area.5 Our patient’s clinical findings fulfilled the ACR criteria since we observed easily bleeding crusts in nasal cavity based on nasopharyngoscopy examination and a necrotizing granulomatous appearance on chest x-ray. In patients with WG, ANCA directed against proteinase 3 (PR-3) or c-ANCA was found positive in 80% of the patients, while ANCA directed against myeloperoxidase (MPO) or p-ANCA was merely found positive in 10% of the patients.5 In our subject, his blood test was positive for p-ANCA with 1:10 titer; supporting the diagnosis of WG.
The histopathological appearance in WG usually showed non-specific forms of inflammatory cell infiltration to dermis and the perivascular area of the skin.6,14 The most distinctive histopathological feature of WG is the appearance of granulomatous vasculitis.5 However, this particular finding only appeared in 20% of cases.13 The inflammation of blood vessels in WG is shown by leukocytoclastic vasculitis marked by fibrinoid necrosis in small blood vessel wall.6,13 Our patient’s skin histopathological examination result did not support the diagnosis of WG since it revealed a vesiculobullous reaction in subcorneal area involving inflammatory cells, such as lymphocytes, neutrophils, and debris. However, since the other ACR criteria were fulfilled, our subject was still diagnosed with WG.
The treatment of WG is divided into two phases: induction and maintenance phase. Induction therapy is administered until remission state and should be followed by maintenance therapy to prevent recurrence and to avoid adverse effects.4,7 Induction therapy involves a combination of corticosteroids and immunosuppressant. The standard WG therapy is a combination of cyclophosphamide and systemic corticosteroid.7 The usage of immunosuppressant is adjusted based on the disease severity. The drug of choice for early systemic WG is a combination of corticosteroid and methotrexate.14,15 Our patient received a combination of steroid (prednisone) prescribed at 1 mg/kg BW which was tapered off by 20–30% every 2 weeks and methotrexate 7.5 mg/week. Clinical improvement was achieved following four weeks of treatment. Our patient finally reached a complete remission state within six months.
The cause of mortality in Wegener’s granulomatosis is due to organ failure, particularly renal.4 In this case, internal organ damage was not reported. We monitor his vital organs by performing routine investigation. Multiple skin ulcers in WG can heal and leave scars containing fibrous tissue.16 Unfortunately, systemic corticosteroid and methotrexate might not control the disease completely, as relapse might occur in 12–50% of cases.12 This is reflected in our patient’s case as he reported relapse episode that occurred one year after he reached complete remission state.
Conclusion
Multiple bullous and ulcers may present as skin manifestations of various diseases, including WG. If systemic vasculitis is suspected, a thorough history taking, physical, serological, and histopathological examination should be conducted to establish proper diagnosis and prevent internal organ damage.
Consent for Publication
The patient has signed the consent forms for the use of case details, images for publication, and for scientific purposes. Institutional approval has been obtained to publish the case details. The case report has been approved by the institutional ethics committee of Dr. Hasan Sadikin General Hospital, Bandung, Indonesia.
Acknowledgments
The authors would like to thank the staff at the Dermatology and Venereology Department, Faculty of Medicine, Universitas Padjadjaran, Bandung, West Java, Indonesia.
Funding
The authors declare that this study has received no financial support.
Disclosure
The authors report no conflicts of interest to disclose in this work.
References
1. Tintle SJ, Cruse AR, Brodell RT, Duong B. Classic findings, mimickers, and distinguishing features in primary blistering skin disease. Arch Pathol Lab Med. 2020;144(2):136–147. doi:10.5858/arpa.2019-0175-RA
2. Bickle KM, Roark TK, Hsu S. Autoimmune bullous dermatoses: a review. Am Fam Physician. 2002;65(9):1861.
3. Taghipour K, Perera GK. Autoimmune blistering skin diseases. Medicine. 2013;41(7):387–393. doi:10.1016/j.mpmed.2013.04.021
4. Greco A, Marinelli C, Fusconi M, et al. Clinic manifestations in granulomatosis with polyangiitis. Int J Immunopathol Pharmacol. 2016;29(2):151–159. doi:10.1177/0394632015617063
5. Lutalo PM, D’Cruz DP. Diagnosis and classification of granulomatosis with polyangiitis (aka Wegener’s granulomatosis). J Autoimmun. 2014;48:94–98. doi:10.1016/j.jaut.2014.01.028
6. Marzano AV, Raimondo MG, Berti E, Meroni PL, Ingegnoli F. Cutaneous manifestations of ANCA-associated small vessels vasculitis. Clin Rev Allergy Immunol. 2017;53(3):428–438.
7. Pagnoux C, Guillevin L. Treatment of granulomatosis with polyangiitis (Wegener’s). Expert Rev Clin Immunol. 2015;11(3):339–348. doi:10.1586/1744666X.2015.1008455
8. Flossmann O, Berden A, de Groot K, et al. Long-term patient survival in ANCA-associated vasculitis. Ann Rheum Dis. 2011;70(3):488–494. doi:10.1136/ard.2010.137778
9. Rottem M, Fauci AS, Hallahan CW, et al. Wegener granulomatosis in children and adolescents: clinical presentation and outcome. J Pediatr. 1993;122(1):26–31. doi:10.1016/S0022-3476(05)83482-1
10. Scott DG, Watts RA. Systemic vasculitis: epidemiology, classification and environmental factors. Ann Rheum Dis. 2000;59(3):161–163. doi:10.1136/ard.59.3.161
11. Martínez-Morillo M, Grados D, Naranjo-Hans D, Mateo L, Holgado S, Olivé A. Granulomatosis con poliangeítis (Wegener). Descripción de 15 casos. Reumatol Clin. 2012;8(1):15–19. doi:10.1016/j.reuma.2011.04.009
12. Francès C, Piette J-C, Saada V, et al. Wegener’s granulomatosis: dermatological manifestations in 75 cases with clinicopathologic correlation. Arch Dermatol. 1994;130(7):861–867. doi:10.1001/archderm.1994.01690070055008
13. Weedon D. Vasculitis with Granulomatosis. Weedon’s Skin Pathology.
14. Jayne D. Conventional treatment and outcome of Wegener’s granulomatosis and microscopic polyangiitis. Cleve Clin J Med. 2002;69(2):SII110–SII115. doi:10.3949/ccjm.69.Suppl_2.SII110
15. Tarzi RM, Pusey CD. Current and future prospects in the management of granulomatosis with polyangiitis (Wegener’s granulomatosis). Ther Clin Risk Manag. 2014;10:279. doi:10.2147/TCRM.S41598
16. Mekkes J, Loots M, Van Der Wal A, Bos J. Causes, investigation and treatment of leg ulceration. Br J Dermatol. 2003;148(3):388–401. doi:10.1046/j.1365-2133.2003.05222.x
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.