")
Back to Journals » Neuropsychiatric Disease and Treatment » Volume 10
Multifocal motor neuropathy: a review of pathogenesis, diagnosis, and treatment
Received 17 November 2013
Accepted for publication 13 January 2014
Published 5 April 2014 Volume 2014:10 Pages 567—576
DOI https://doi.org/10.2147/NDT.S39592
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Victoria H Lawson,1 W David Arnold1,2
1Division of Neuromuscular Disorders, Department of Neurology, 2Department of Physical Medicine and Rehabilitation, Wexner Medical Center at The Ohio State University, Columbus, Ohio, USA
Abstract: Multifocal motor neuropathy (MMN) is an uncommon, purely motor neuropathy associated with asymmetric deficits with predilection for upper limb involvement. Even in the early descriptions of MMN, the associations of anti-GM1 antibodies and robust response to immunomodulatory treatment were recognized. These features highlight the likelihood of an underlying autoimmune etiology of MMN. The clinical presentation of MMN can closely mimic several neurological conditions including those with more malignant prognoses such as motor neuron disease. Therefore early and rapid recognition of MMN is critical. Serological evidence of anti GM-1 antibodies and electrodiagnostic findings of conduction block are helpful diagnostic clues for MMN. Importantly, these diagnostic features are not universally present, and patients lacking these characteristic findings can demonstrate similar robust response to immunodulatory treatment. In the current review, recent research in the areas of diagnosis, pathogenesis, and treatment of MMN and needs for the future are discussed. The characteristic findings of MMN and treatment implications are reviewed and contrasted with other mimicking disorders.
Keywords: autoimmune, conduction block, electrodiagnosis, motor neuron, nerve, inflammatory
Background and prevalence
There are few disorders that are confined to nerves and produce a purely motor phenotype. It is important to differentiate the most recognized and fatal form, motor neuron disease, from other treatable or less malignant forms of motor neuropathy. Multifocal motor neuropathy (MMN) is much rarer but can closely mimic motor neuron disease. The accurate differentiation of MMN has very important clinical implications for the patient because of the obvious differences in prognosis and treatment. Prevalence estimates for MMN range from 0.3 to 3 cases in 100,000 depending on the case series examined and how estimates were derived;1–4 but MMN is clearly underrecognized. In a longitudinal study of patients with MMN, the correct diagnosis was made by the referring neurologist in only 6 of 46 patients (13%) referred to a tertiary neuromuscular center, attesting to the importance of a good understanding of this disorder.5
Clinical features
MMN is a purely motor neuropathy characterized by motor deficits in the distribution of single nerves without associated sensory loss. Motor neuron disease is also characterized by motor deficits without sensory loss, but an important distinction from MMN is that it occurs in the distribution of spinal segments rather than single nerves. MMN predominantly affects males, almost three times as often as females (2.7:1).1 Onset is generally before the age of 50 years (80% of cases, with a mean age of onset of 40 years, range 20–70 years) and seldom occurs in children.1,6,7 Consensus criteria delineate slowly progressive or stepwise weakness that is asymmetric and involving at least two separate motor nerve distributions to make a definite diagnosis.8 Compared with the more common autoimmune neuropathy, chronic inflammatory demyelinating neuropathy (CIDP), MMN is more asymmetric and does not evolve into generalized weakness as rapidly as CIDP. Multifocal acquired demyelinating sensory and motor neuropathy (MADSAM) is an uncommon CIDP variant that can look like MMN because it also affects single nerves; however, it is distinguished clinically and on electrodiagnostic studies by sensory involvement. It is characteristic for MMN to involve predominantly the upper limbs, such that this characteristic feature is included as one of the supportive diagnostic criteria by European Federation of Neurological Societies guidelines (Table 1). The ulnar, median, and radial nerves are the most commonly affected, but there may be striking differences in the severity of involvement of different muscles supplied by a common nerve; this is presumably due to fascicular involvement.7 In two thirds of all cases, weakness begins in the distal upper limbs, and patients often present with difficulties in wrist and finger extension or reduced hand grip.1 Symptoms initially occur in the distal lower extremities in approximately one quarter of patients and rarely in the proximal upper extremities.1 Weakness occurs in MMN without associated objective sensory loss; however, mild sensory disturbance may occur, and patients occasionally complain of vague numbness and tingling in the affected limb. Sensory symptoms are not accompanied by alterations in the sensory nerve action potentials on electrodiagnostic study.5,9 In a small study of five patients diagnosed with MMN, reduction in sensory nerve action potentials was observed after a mean lag of 7.2 years, but these patients may have represented an overlap syndrome between MMN and MADSAM.10 Features of MMN and other chronic neuromuscular conditions that should be distinguished from MMN (for prognostic and treatment implications) are reviewed in Table 2.
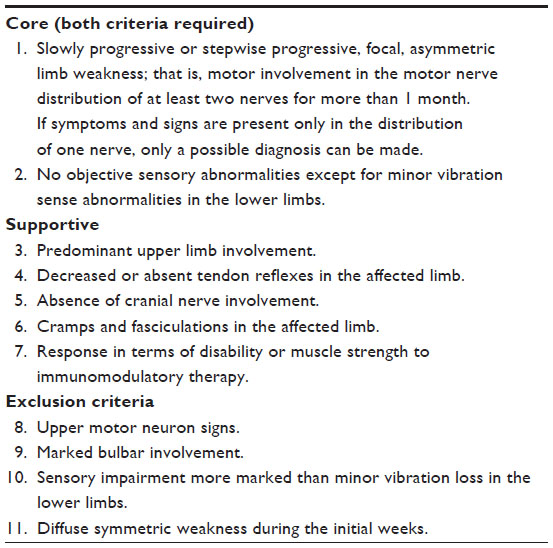 | Table 1 Clinical criteria for the diagnosis of MMN8 |
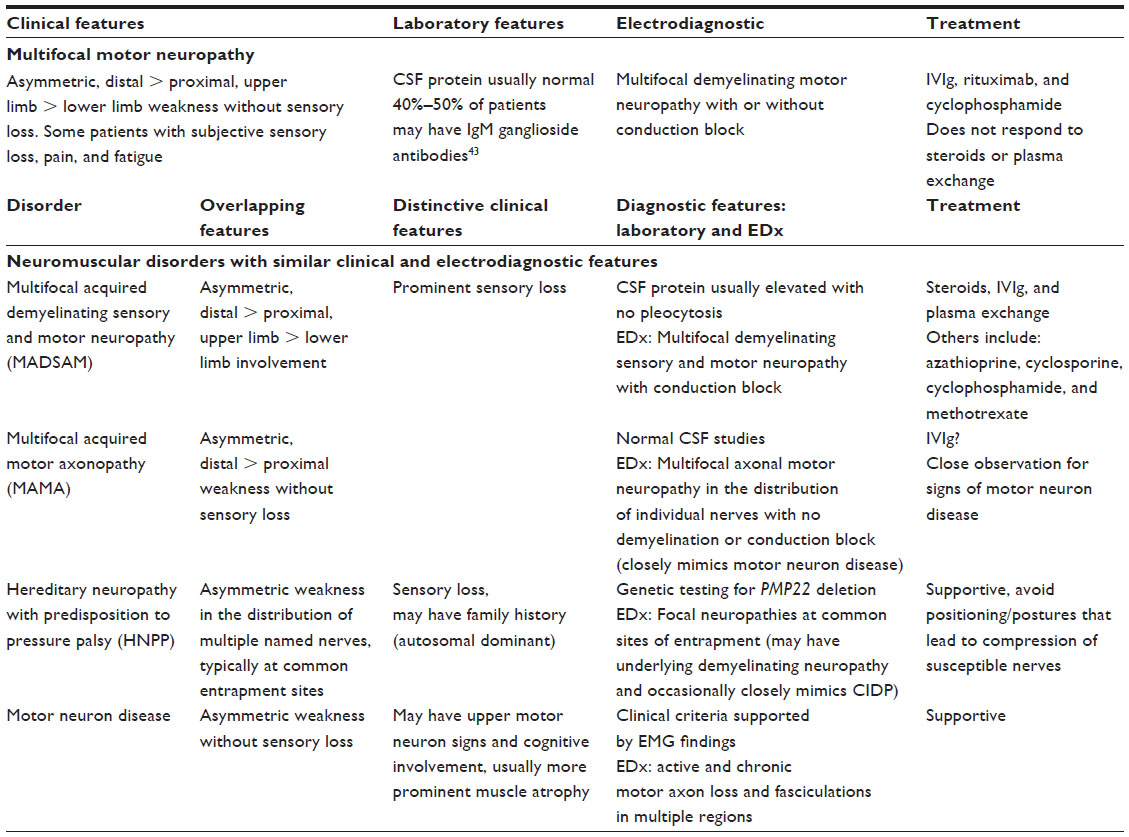 | Table 2 Classic features of multifocal motor neuropathy and other neuromuscular disorders with similar clinical and electrodiagnostic features |
MMN can be difficult to distinguish from motor neuron disease because, as noted above, both present with asymmetric, progressive, distal weakness without numbness. There are, however, features that distinguish MMN from motor neuron disease (amyotrophic lateral sclerosis): 1) MMN affects predominantly (but not exclusively) the upper limbs; 2) MMN usually lacks bulbar or respiratory involvement; 3) muscle weakness in MMN is associated with less atrophy, unless it becomes severe or longstanding; 4) cramps and fasciculations can occur in motor neuron disease but are less prominent than in MMN, occurring in up to 50% of patients with MMN; 5) MMN has no upper motor neuron signs; and 6) MMN has a characteristic electrophysiological pattern of motor conduction block.11 Although typically associated with motor neuron disease, MMN can be associated with normal (20%) or even brisk (8%) deep tendon reflexes and, rarely, respiratory insufficiency due to phrenic nerve involvement.6,12–15 Therefore, care must be taken to consider the clinical picture as a whole. The most important distinguishing feature of MMN from motor neuron disease is conduction block as this, in the correct clinical context, is virtually pathognomonic of MMN.
The diagnosis of MMN rests on predominantly clinical criteria (Table 1) paired with electrophysiologic (Table 3) criteria. Additional supportive criteria help to establish the diagnosis as definite, probable, or possible (Tables 4 and 5). These criteria were established by joint task forces from the European Federation of Neurological Societies and the Peripheral Nerve Society and are shown in Table 3A and B. The minimal criteria for diagnosis requires core clinical criteria, fulfillment of all exclusion criteria (Table 1), and normal sensory nerve conductions in the distribution of affected motor nerves.8
 | Table 3 Electrophysiological criteria for conduction block8 |
 | Table 4 Diagnostic criteria for definite or probable MMN8 |
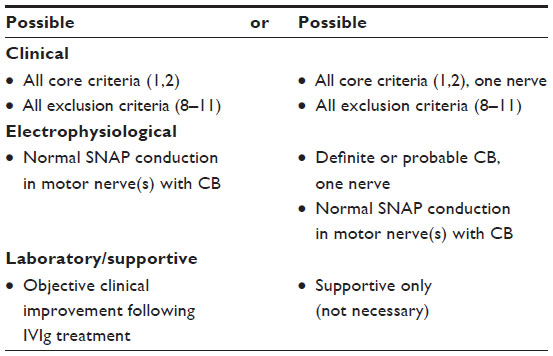 | Table 5 Diagnostic criteria for possible MMN8 |
Electrophysiological features
Motor conduction block is defined as a reduction of compound muscle action potential amplitude recorded from a muscle or group of muscles following proximal nerve stimulation (across the site of conduction failure) as compared with distal nerve stimulation (distal to the site of conduction failure). This drop of amplitude indicates a localized failure of action potential conduction along a structurally intact axon and is consistent with a myelinopathy. This can also occur with focal compression of nerves at compressible sites such as the elbow, wrist, fibular head, etc. In MMN, conduction block (CB) occurs at noncompressible sites along the nerve axons and does not affect sensory fibers; this is the diagnostic hallmark. However, MMN without CB has also been described and CB is not specific for MMN as it can be seen with other types of inflammatory myelinopathies.16,17 Various nonconventional electrophysiological techniques have been used to investigate the pathophysiology of CB by indirectly assessing axonal excitability. Mixed findings have suggested either hyperpolarization or depolarization may occur at the site of CB in MMN.18,19
Importantly, patients with a clinical appearance consistent with MMN but without CB respond to intravenous immunoglobulin (IVIg) treatment similarly to those with CB; they also have a similar long term prognosis. The absence of CB in some patients may represent limitations of electrodiagnostic testing for identification of conduction block along very proximal or very distal segments of nerve; it may also represent other technical considerations. The possibility that MMN exists in a purely axonal form that is responsive to immunomodulatory therapy was raised in a report of a series of patients with axonal electrophysiology in the clinical setting of MMN.20 The authors proposed this as a variant of classic MMN.
Etiology
Early in its description, MMN was associated with elevated serum levels of IgM antibodies against ganglioside GM1. These findings, along with the response of MMN to immunomodulatory therapy, implicate an autoimmune etiology in its pathogenesis.21–23 There appears to be a link between the GM1 antibody and a motor predominant phenotype. The association with GM1 antibodies is seen not only in MMN but also in other immune-mediated neuropathies; in particular, the pure motor variant of Guillain-Barré syndrome (GBS), acute motor axonal neuropathy (AMAN).24,25 The presence of immunoglobulin deposition and inflammatory demyelination in motor roots from autopsy cases further underscores a likely autoimmune etiology.26
The relative abundance of GM1 in the myelin of motor nerves as compared to sensory nerves may provide a potential basis for the sparing of sensory nerve function.27 This is also supported by the finding of perivascular demyelination from motor nerve biopsies that are absent from sensory nerve biopsies.28,29 A recent report describes an increased frequency of a human leukocyte antigen haplotype in MMN patients that is seen in patients with multiple sclerosis.30 First degree relatives of patients with MMN have a significantly increased incidence of other autoimmune disorders including Hashimoto’s thyroiditis, celiac disease, and type 1 diabetes.31 Taken together, these lines of evidence provide supporting evidence for an autoimmune etiology for MMN.
Interestingly, there are cases of toxic motor neuropathy with CB that are indistinguishable from MMN following treatment with infliximab; a human/murine immunoglobulin G1 antitumor necrosis factor-a (TNF) antibody.32–35 The mechanism by which the infliximab triggers the characteristic immune attack that occurs in MMN patients is unknown, but it differs from idiopathic MMN in that it is more rapidly progressive; some of the cases evolve rapidly into axonal loss.36,37 In the cases described, there was inconsistent presence of GM1 antibodies32,33 and most patients were treated with IVIg to resolve the weakness. Although there are also reports of patients improving spontaneously after withdrawal of the TNF-inhibitor,38 most of the patients reported required IVIg, implicating an immune mechanism triggered by toxic exposure.
Natural history
MMN presents with weakness in the distribution of single peripheral nerves accounting for its asymmetry; because weakness derives from CB rather than axonal loss, muscle atrophy does not occur despite fairly pronounced symptoms. There is slow progression with time to a more confluent and segmental pattern that may be more difficult to distinguish from motor neuron disease. Chronicity is associated with axonal loss and, at that point, patients may develop muscle wasting, mimicking motor neuron disease. Longitudinal assessments of treated MMN patients were performed using a validated instrument, the Neuropathy Impairment Score (NIS), in a study of 46 patients with MMN-CB.5 Using the NIS (W) (referring to the portion of the scale measuring weakness), muscle power is assessed as normal (0) or complete paralysis (4) with intermediate grades; 1 representing a muscle that is deemed 25% weak by clinical strength testing, 2 as 50% weak, 3 as 75% weak, 3.25 as movement against gravity, 3.50 as movement with gravity eliminated, and 3.75 as muscle flicker.39 The NIS is a clinically based tool that has been used in epidemiologic and controlled clinical trials in diabetes and other neuropathies; the NIS (W) is comprised of a composite strength score that summates the weakness of head, trunk, and limb muscles. Using this metric, authors of the study of 46 MMN patients identified worsening of the median NIS (W) strength score over a period of 88 months. This was also demonstrated by a composite electrophysiological score in which median summated compound muscle action potential amplitudes unequivocally diminished over time.5 The 46 patients with MMN demonstrated progression of their disease at a rate of 1.3 points on the NIS (W) per year after initiating therapy as compared to 4.2 points per year prior to IVIg. These findings suggest a beneficial effect of therapy on the natural history of MMN. Despite the progressive nature of the weakness, none of the studied patients lost their job or their ability to comply with their professional duties as a result of the disease, attesting to a relatively modest effect of the disease on function as compared to some other inflammatory neuropathies. The number of years without IVIg therapy and the degree of axonal loss are identified as important determinants of disability and permanent weakness in MMN patients.1,5,40 Life expectancy in MMN is normal but conversely spontaneous remission has not been reported.1,5,40 Life-threatening respiratory weakness associated with hypoventilation and respiratory insufficiency due to phrenic nerve involvement with diaphragmatic paresis has been described but is thought to be a rare manifestation of MMN.13,15
As noted above, MMN with CB does not differ clinically from MMN without CB.2,17 In a comparative study of these two groups conducted over a 7-year period, the distribution of peripheral nerves involved, weakness as measured by a composite Medical Research Council (MRC) sum score, and ancillary symptoms of cramps and fasciculations were similar at initial assessment and at follow-up examinations. Subtle differences were described but these were not significant, and both groups responded equally well to IVIg. It is not clear if patients seropositive for anti-GM1 antibodies have a different clinical course than seronegative patients. Independent determinants for more severe weakness included axon loss and chronicity of disease; a greater proportion of patients with anti-GM1 antibodies had axon loss than those without antibodies.1 There have been reports of seropositive patients responding more robustly to IVIg than seronegative patients;41,42 however, other studies do not corroborate this finding.7 It is generally well accepted, however, that early recognition of MMN and initiation of therapy delays progression to axonal loss and more severe disability.
Pathophysiology
One of the compelling immunologic features of MMN is the presence of serum IgM antibodies against the ganglioside GM1 in up to half of MMN patients.43 GM1 localizes to both the axolemma and myelin of peripheral nerves, being found in greatest abundance at the nodes of Ranvier and adjacent paranodes.44 Although ubiquitous, GM1 is found most abundantly in peripheral motor rather than sensory nerves, and it concentrates in cholesterol-enriched domains of the plasma membrane.25,27 These domains have a number of functions, the most relevant of which is paranodal stabilization and ion channel clustering.25,44 GM1 is thought to facilitate the maintenance of tight junctions through this paranodal stabilization as it also provides an anchor for potassium channels and concentrates sodium channels.25,44 These functions are important for action potential propagation and maintenance of conduction velocity in the peripheral nerves. Disruption of these functions would be expected to induce failure of conduction across paranodal regions; thus, the mechanism of CB may be a primary dysfunction of the axon at the paranodes or a dysfunction of the myelin sheath causing failure of action potential propagation.45 As these changes would be expected to have more widespread effects than just at the sites of conduction block, this provides a mechanism for the progressive nature of the disorder.
In animal models of AMAN, IgG GM1 activates the complement cascade leading to the production of a membrane attack complex (MAC).25 MAC compromises the plasma membrane, depositing at the nodal regions first, and then spreading to internodal regions of the nerve. This results in not only disruption of the node and sodium channel dispersion, but also axonal damage.46 An animal model of MMN does not exist, but the mechanism of damage from IgM GM1 is thought to be similar.46 This could provide an explanation for the conduction failure followed by axon loss; that is, early MAC deposition, ion channel dispersion leading to conduction failure, followed by axonal damage. Sera from MMN patients with IgM GM1 antibodies trigger the complement cascade; the extent of complement deposition correlates with anti-GM1 titers and is reduced by IVIg therapy.47,48 Complement activation, then, may be the most important immunologic effect producing pathologic effects.
Although a large proportion of MMN patients possess GM1 IgM antibodies, greater than half in some reports do not; this raises the question of whether other antigenic targets exist that may exert similar effects to anti-GM1 antibodies or a different pathogenic mechanism is responsible for MMN, at least in patients without GM1 antibodies. The clinical similarities between the two groups and their response to IVIg make the latter explanation less desirable. However, the findings of antiganglioside complexes, which may enhance or attenuate GM1 binding, may provide an explanation. GM1 in combination with galactocerebroside has been identified as a sensitive marker for MMN.49 Enhanced IgM GM1 binding was also found by a different set of authors with a complex of GM1, galactocerebroside, and cholesterol.50 Thus, interaction of different types of gangliosides may alter antibody reactivity. Other candidate molecules for which there is little direct evidence include disulfated heparin disaccharide, GM1 chaperone molecules, or as yet unidentified glycolipids with functions similar to GM1.49–51
Treatment
Intravenous immunoglobulin
The treatment of choice continues to be IVIg in MMN with good response seen in as many as 80% of patients.52 The proposed mechanisms of action are myriad but generally thought to be related to the inhibition or consumption of complement depositing at nodes of Ranvier.47 Improvement in weakness after the initial dose is seen rapidly, usually in the first week, and especially early when pathology is predominantly CB. The efficacy of IVIg on muscle strength has been demonstrated by randomized double-blind placebo-controlled trials,23,53–55 the largest of which being comprised of a total of 44 patients.56 Dosing is generally initiated at a total of 2 g/kg administered intravenously over 2–5 days; although response may be rapid, it is seldom sustained and maintenance dosing is often required. Maintenance dosing is subject to institutional variability as there have been no studies establishing the optimal dose or interval for maintenance therapy. However, it is generally initiated at a lower dose with intervals of 1–6 weeks depending on clinical response. Some authors advocate waiting until clinical improvement has reached a plateau or patients begin to experience weakness before initiating maintenance therapy45 while others institute a specific regimen every two weeks with adjustment according to clinical response over time.57 In general, however, maintenance is 0.4 g/kg every week – 1–2 g/kg every 2–6 weeks depending on response and tolerance.58 This requires close monitoring over the period immediately following initial infusion in order to gauge strength and tolerance of IVIg. Renal function should be monitored regularly, and patients should be offered symptomatic therapy for the common flu-like reaction that can occur variably related to the specific infusion-infusion mix of immunoglobulin. Serious side effects to IVIg include an increased incidence of thrombotic events including stroke and myocardial infarction, and aseptic meningitis.
Although many MMN patients respond to IVIg therapy, the response is not universal or complete; clinical improvement has been associated with reductions in conduction block but not in reductions in GM1 antibody titers. Most patients become less responsive to IVIg over time, requiring higher doses or more frequent dosing intervals to maintain their responsiveness. This loss of responsiveness correlates with progressively lower compound muscle action potential amplitudes, supporting the idea that loss of responsiveness to IVIg occurs because of the development of axonal loss with progression of the disease.59–62
Subcutaneous immunoglobulin
The subcutaneous form of immunoglobulin provides the advantage of greater convenience and possibly fewer side effects in patients requiring frequent infusions. Its efficacy has been demonstrated in both open-label studies63,64 and a single randomized single-blinded trial.65 The greatest drawback to this therapy is short-term and potential long-term effects of large subcutaneous volumes required for maintenance therapy.
Steroids and plasma exchange
In acute inflammatory demyelinating neuropathy, or Guillain-Barré syndrome, it is now well accepted that corticosteroids do not have a beneficial effect, unlike CIDP.66,67 Plasma exchange, however, has been proven to be equivalent in its efficacy compared to IVIg in the treatment of GBS.68–70 In MMN there is similarly little evidence for the efficacy of steroids, attesting to its overlap with GBS and AMAN. In a proportion of patients with MMN, steroids worsened weakness, sometimes dramatically.4,71 Surprisingly, plasma exchange is also ineffective in MMN and may result in clinical and electrophysiological worsening.72 This puzzling finding underscores the unique pathophysiology of MMN as an entity distinct from motor predominant GBS, or other variants of inflammatory neuropathy, and remains one of the interesting aspects of MMN yet to be elucidated. In contrast, patients with the clinical phenotype of MMN but with axonal features on electrodiagnostic testing rather than CB, referred to as multifocal acquired motor axonopathy, respond not only to IVIg but also to steroids.17,20 This implies a pathogenesis more similar to CIDP than to MMN and establishes MMN as distinctive.
Other therapies
Other agents such as cyclophosphamide, rituximab, mycophenolate mofetil, β-interferon, cyclosporine, azathioprine, and infliximab have been used for treatment of MMN; however, insufficient controlled trials support their use.73 Of these, there is most support for cyclophosphamide, even in patients unresponsive to IVIg. High dose intravenous cyclophosphamide has been shown to be effective in up to 50% of patients, but, interestingly, low dose cyclophosphamide does not demonstrate efficacy.21,22,74–76 The use of high-dose cyclophosphamide is limited by its toxicity in a non-life-threatening illness and the lack of randomized controlled trials demonstrating its effectiveness. Toxicities include bone marrow suppression, alopecia, hemorrhagic cystitis, delayed bladder cancer, teratogenicity, azoospermia, and infection risk.
Rituximab is a monoclonal antibody directed against CD20 surface antigen for peripheral B cell depletion. Treatment with rituximab has been shown to achieve clinical improvement even in patients with a declining or insufficient response to IVIg in five of seven reports and carries much less risk of toxicity compared with cyclophosphamide.77–80 The reports of its efficacy, however, are based on a small cumulative number of patients (total n=28) and are not consistent. The largest study (n=14) demonstrated a 13% improvement in strength (versus 3% in controls) over 1 year and a 23% strength improvement over 2 years.81 The next largest study (n=6), however, demonstrated no significant change in strength in IVIg responsive patients and did not allow for reduction in the IVIg dose.82 The dose is 375 mg/m2 weekly for 2–4 weeks to achieve B cell depletion. Single booster doses at 375 mg/m2 are administered as maintenance therapy if patients develop increased weakness.
Although single case reports exist regarding efficacy of other immunosuppressant agents, as noted above, these are small, and further randomized controlled trials are needed to gauge both efficacy and risk to patients with MMN.
Patient focused perspectives
MMN has major impacts on patient function, particularly in patients with generalized disease, but, interestingly, in one cohort, as many as 94% of patients remained employed.83 The focus of most studies to date has centered mainly on the motor effects of MMN.83 Not unexpectedly, the majority of impact on quality of life in patients with MMN is related to motor deficits. Impairment is mainly in the upper limb function but, with progression of the disease, there can be a more generalized impact. Cranial nerve impairment is rare.12,13,84 Somewhat surprisingly, sensory symptoms such as pain and dysesthesia are not uncommon. In some series, up to a quarter of patients present with neuropathic pain as a prominent feature, although, not surprisingly, pain was not an indicator of poor prognosis.85 Many patients will describe mild sensory disturbance, in agreement with studies that have shown sensory nerve pathology.29 Despite this, sensory nerve action potentials remain intact on electrodiagnostic assessments, attesting to the unique motor predominance of this disorder. Fatigue is a common but often overlooked feature of MMN. Interestingly, some patients are more disabled by the effects of fatigue than by the effects of the weakness. Electrophysiological assessment has demonstrated activity-dependent axonal failure,86 but whether symptoms of fatigue are directly related to motor deficits deserves further attention.
Conclusion
MMN is an important treatable cause of neuropathy. Early treatment is one of the most important factors in long term functional outcome. This makes recognition vital as MMN must be differentiated from other mimicking conditions for which immunomodulatory therapy is ineffective. Although good diagnostic criteria exist, an accurate diagnosis can be difficult to make due to significant overlap with other conditions and the rarity of MMN. Current treatment options, primarily IVIg, offer significant disease modulation in most patients, but these treatments have significant economic impact and, despite disease modification, patients typically develop gradual worsening with less robust response over time. Ongoing work is needed to enhance the current understanding of the underlying pathophysiological mechanism of MMN in order to develop more targeted and economical therapies that provide sustained treatment effect.
Disclosure
The authors report no conflicts of interest in this work.
References
Cats EA, van der Pol WL, Piepers S, et al. Correlates of outcome and response to IVIg in 88 patients with multifocal motor neuropathy. Neurology. 2010;75(9):818–825. | |
Chaudhry V. Multifocal motor neuropathy. Semin Neurol. 1998;18(1):73–81. | |
Matsui N. [Multifocal motor neuropathy: current review of epidemiology and treatment]. Rinsho Shinkeigaku. 2012;52(11):920–922. Japanese. | |
Nobile-Orazio E, Cappellari A, Priori A. Multifocal motor neuropathy: current concepts and controversies. Muscle Nerve. 2005;31(6):663–680. | |
Taylor BV, Wright RA, Harper CM, Dyck PJ. Natural history of 46 patients with multifocal motor neuropathy with conduction block. Muscle Nerve. 2000;23(6):900–908. | |
Nobile-Orazio E. Multifocal motor neuropathy. J Neuroimmunol. 2001;115(1–2):4–18. | |
Slee M, Selvan A, Donaghy M. Multifocal motor neuropathy: the diagnostic spectrum and response to treatment. Neurology. 2007;69(17):1680–1687. | |
Joint Task Force of the EFNS and the PNS. European Federation of Neurological Societies/Peripheral Nerve Society guideline on management of multifocal motor neuropathy. Report of a joint task force of the European Federation of Neurological Societies and the Peripheral Nerve Society – first revision. J Peripher Nerv Syst. 2010;15(4):295–301. | |
Parry GJ. AAEM case report #30: multifocal motor neuropathy. Muscle Nerve. 1996;19(3):269–276. | |
Lambrecq V, Krim E, Rouanet-Larriviere M, Lagueny A. Sensory loss in multifocal motor neuropathy: a clinical and electrophysiological study. Muscle Nerve. 2009;39(2):131–136. | |
Bentes C, de Carvalho M, Evangelista T, Sales-Luis ML. Multifocal motor neuropathy mimicking motor neuron disease: nine cases. J Neurol Sci. 1999;169(1–2):76–79. | |
Beydoun SR, Copeland D. Bilateral phrenic neuropathy as a presenting feature of multifocal motor neuropathy with conduction block. Muscle Nerve. 2000;23(4):556–559. | |
Boonyapisit K, Katirji B. Multifocal motor neuropathy presenting with respiratory failure. Muscle Nerve. 2000;23(12):1887–1890. | |
Cavaletti G, Zincone A, Marzorati L, Frattola L, Molteni F, Navalesi P. Rapidly progressive multifocal motor neuropathy with phrenic nerve paralysis: effect of nocturnal assisted ventilation. J Neurol. 1998;245(9):613–616. | |
Kyriakides T, Papacostas S, Papanicolaou E, Bagdades E, Papathanasiou ES. Sleep hypoventilation syndrome and respiratory failure due to multifocal motor neuropathy with conduction block. Muscle Nerve. 2011;43(4):610–614. | |
Pakiam AS, Parry GJ. Multifocal motor neuropathy without overt conduction block. Muscle Nerve. 1998;21(2):243–245. | |
Delmont E, Azulay JP, Giorgi R, et al. Multifocal motor neuropathy with and without conduction block: a single entity? Neurology. 2006;67(4):592–596. | |
Kiernan MC, Guglielmi JM, Kaji R, Murray NM, Bostock H. Evidence for axonal membrane hyperpolarization in multifocal motor neuropathy with conduction block. Brain. 2002;125(Pt 3):664–675. | |
Priori A, Bossi B, Ardolino G, et al. Pathophysiological heterogeneity of conduction blocks in multifocal motor neuropathy. Brain. 2005;128(Pt 7):1642–1648. | |
Katz JS, Barohn RJ, Kojan S, et al. Axonal multifocal motor neuropathy without conduction block or other features of demyelination. Neurology. 2002;58(4):615–620. | |
Feldman EL, Bromberg MB, Albers JW, Pestronk A. Immunosuppressive treatment in multifocal motor neuropathy. Ann Neurol. 1991;30(3):397–401. | |
Pestronk A, Cornblath DR, Ilyas AA, et al. A treatable multifocal motor neuropathy with antibodies to GM1 ganglioside. Ann Neurol. 1988;24(1):73–78. | |
Van den Berg LH, Kerkhoff H, Oey PL, et al. Treatment of multifocal motor neuropathy with high dose intravenous immunoglobulins: a double blind, placebo controlled study. J Neurol Neurosurg Psychiatry. 1995;59(3):248–252. | |
van Sorge NM, Yuki N, Jansen MD, et al. Leukocyte and complement activation by GM1-specific antibodies is associated with acute motor axonal neuropathy in rabbits. J Neuroimmunol. 2007;182(1–2):116–123. | |
Susuki K, Rasband MN, Tohyama K, et al. Anti-GM1 antibodies cause complement-mediated disruption of sodium channel clusters in peripheral motor nerve fibers. J Neurosci. 2007;27(15):3956–3967. | |
Oh SJ, LaGanke C, Powers R, Wolfe GI, Quinton RA, Burns DK. Multifocal motor sensory demyelinating neuropathy: inflammatory demyelinating polyradiculoneuropathy. Neurology. 2005;65(10):1639–1642. | |
Ogawa-Goto K, Funamoto N, Ohta Y, Abe T, Nagashima K. Myelin gangliosides of human peripheral nervous system: an enrichment of GM1 in the motor nerve myelin isolated from cauda equina. J Neurochem. 1992;59(5):1844–1849. | |
Kassiotis G, Kollias G. Uncoupling the proinflammatory from the immunosuppressive properties of tumor necrosis factor (TNF) at the p55 TNF receptor level: implications for pathogenesis and therapy of autoimmune demyelination. J Exp Med. 2001;193(4):427–434. | |
Corse AM, Chaudhry V, Crawford TO, Cornblath DR, Kuncl RW, Griffin JW. Sensory nerve pathology in multifocal motor neuropathy. Ann Neurol. 1996;39(3):319–325. | |
Sutedja NA, Otten HG, Cats EA, et al. Increased frequency of HLA-DRB1*15 in patients with multifocal motor neuropathy. Neurology. 2010;74(10):828–832. | |
Cats EA, Bertens AS, Veldink JH, van den Berg LH, van der Pol WL. Associated autoimmune diseases in patients with multifocal motor neuropathy and their family members. J Neurol. 2012;259(6):1137–1141. | |
Cocito D, Bergamasco B, Tavella A, et al. Multifocal motor neuropathy during treatment with infliximab. J Peripher Nerv Syst. 2005;10(4):386–387. | |
Rodriguez-Escalera C, Belzunegui J, Lopez-Dominguez L, Gonzalez C, Figueroa M. Multifocal motor neuropathy with conduction block in a patient with rheumatoid arthritis on infliximab therapy. Rheumatology (Oxford). 2005;44(1):132–133. | |
Singer OC, Otto B, Steinmetz H, Ziemann U. Acute neuropathy with multiple conduction blocks after TNFalpha monoclonal antibody therapy. Neurology. 2004;63(9):1754. | |
Tektonidou MG, Serelis J, Skopouli FN. Peripheral neuropathy in two patients with rheumatoid arthritis receiving infliximab treatment. Clin Rheumatol. 2007;26(2):258–260. | |
McCluskey L, Feinberg D, Cantor C, Bird S. “Pseudo-conduction block” in vasculitic neuropathy. Muscle Nerve. 1999;22(10):1361–1366. | |
Sandbrink F, Klion AD, Floeter MK. “Pseudo-conduction block” in a patient with vasculitic neuropathy. Electromyogr Clin Neurophysiol. 2001;41(4):195–202. | |
Jarand J, Zochodne DW, Martin LO, Voll C. Neurological complications of infliximab. J Rheumatol. 2006;33(5):1018–1020. | |
Bril V. NIS-LL: the primary measurement scale for clinical trial endpoints in diabetic peripheral neuropathy. Eur Neurol. 1999;41 Suppl 1:8–13. | |
Muley SA, Parry GJ. Multifocal motor neuropathy. J Clin Neurosci. 2012;19(9):1201–1209. | |
Bouche P, Moulonguet A, Younes-Chennoufi AB, et al. Multifocal motor neuropathy with conduction block: a study of 24 patients. J Neurol Neurosurg Psychiatry. 1995;59(1):38–44. | |
Van den Berg-Vos RM, Franssen H, Wokke JH, Van Es HW, Van den Berg LH. Multifocal motor neuropathy: diagnostic criteria that predict the response to immunoglobulin treatment. Ann Neurol. 2000;48(6):919–926. | |
Nobile-Orazio E, Giannotta C, Musset L, Messina P, Léger JM. Sensitivity and predictive value of anti-GM1/galactocerebroside IgM antibodies in multifocal motor neuropathy. J Neurol Neurosurg Psychiatry. Epub August 1, 2013. | |
Willison HJ, Yuki N. Peripheral neuropathies and anti-glycolipid antibodies. Brain. 2002;125(Pt 12):2591–2625. | |
Vlam L, van den Berg LH, Cats EA, Piepers S, van der Pol WL. Immune pathogenesis and treatment of multifocal motor neuropathy. J Clin Immunol. 2013;33 Suppl 1:S38–S42. | |
Vlam L, van der Pol WL, Cats EA, et al. Multifocal motor neuropathy: diagnosis, pathogenesis and treatment strategies. Nat Rev Neurol. 2012;8(1):48–58. | |
Yuki N, Watanabe H, Nakajima T, Spath PJ. IVIG blocks complement deposition mediated by anti-GM1 antibodies in multifocal motor neuropathy. J Neurol Neurosurg Psychiatry. 2011;82(1):87–91. | |
Piepers S, Jansen MD, Cats EA, van Sorge NM, van den Berg LH, van der Pol WL. IVIg inhibits classical pathway activity and anti-GM1 IgM-mediated complement deposition in MMN. J Neuroimmunol. 2010;229(1–2):256–262. | |
Galban-Horcajo F, Fitzpatrick AM, Hutton AJ, et al. Antibodies to heteromeric glycolipid complexes in multifocal motor neuropathy. Eur J Neurol. 2013;20(1):62–70. | |
Pestronk A, Choksi R, Blume G, Lopate G. Multifocal motor neuropathy: serum IgM binding to a GM1 ganglioside-containing lipid mixture but not to GM1 alone. Neurology. 1997;48(4):1104–1106. | |
Pestronk A, Chuquilin M, Choksi R. Motor neuropathies and serum IgM binding to NS6S heparin disaccharide or GM1 ganglioside. Journal of Neurology, Neurosurgery and Psychiatry. 2010;81(7):726–730. | |
Nguyen TP, Chaudhry V. Multifocal motor neuropathy. Neurol India. 2011;59(5):700–706. | |
Azulay JP, Rihet P, Pouget J, et al. Long term follow up of multifocal motor neuropathy with conduction block under treatment. J Neurol Neurosurg Psychiatry. 1997;62(4):391–394. | |
Federico P, Zochodne DW, Hahn AF, Brown WF, Feasby TE. Multifocal motor neuropathy improved by IVIg: randomized, double-blind, placebo-controlled study. Neurology. 2000;55(9):1256–1262. | |
Leger JM, Chassande B, Musset L, Meininger V, Bouche P, Baumann N. Intravenous immunoglobulin therapy in multifocal motor neuropathy: a double-blind, placebo-controlled study. Brain. 2001;124(Pt 1):145–153. | |
Hahn AF, Beydoun SR, Lawson V. The IVIg in MMN Study Team, et al. A controlled trial of intravenous immunoglobulin in multifocal motor neuropathy. J Peripher Nerv Sys. 2013;18(4):321–330. | |
Nowacek DG, Teener JW. Multifocal motor neuropathy. Semin Neurol. 2012;32(5):500–505. | |
Nobile-Orazio E, Gallia F. Multifocal motor neuropathy: current therapies and novel strategies. Drugs. 2013;73(5):397–406. | |
Van Asseldonk JT, Van den Berg LH, Kalmijn S, et al. Axon loss is an important determinant of weakness in multifocal motor neuropathy. J Neurol Neurosurg Psychiatry. 2006;77(6):743–747. | |
Van den Berg-Vos RM, Franssen H, Wokke JH, Van den Berg LH. Multifocal motor neuropathy: long-term clinical and electrophysiological assessment of intravenous immunoglobulin maintenance treatment. Brain. 2002;125(Pt 8):1875–1886. | |
Baumann A, Hess CW, Sturzenegger M. IVIg dose increase in multifocal motor neuropathy: a prospective six month follow-up. J Neurol. 2009;256(4):608–614. | |
Terenghi F, Cappellari A, Bersano A, Carpo M, Barbieri S, Nobile-Orazio E. How long is IVIg effective in multifocal motor neuropathy? Neurology. 2004;62(4):666–668. | |
Eftimov F, Vermeulen M, de Haan RJ, et al. Subcutaneous immunoglobulin therapy for multifocal motor neuropathy. J Peripher Nerv Syst. 2009;14:93–100. | |
Gardulf A, Nicolay U, Asensio O, et al. Rapid subcutaneous IgG replacement therapy is effective and safe in children and adults with primary immunodeficiencies–a prospective, multi-national study. J Clin Immunol. 2006;26:177–185. | |
Harbo T, Andersen H, Hess A, Hansen K, Sindrup SH, Jakobsen J. Subcutaneous versus intravenous immunoglobulin in multifocal motor neuropathy: a randomized, single-blinded cross-over trial. Eur J Neurol. 2009;16(5):631–638. | |
No authors listed. Double-blind trial of intravenous methylprednisolone in Guillain-Barre syndrome. Guillain-Barré Syndrome Steroid Trial Group. Lancet. 1993;341(8845):586–590. | |
van Koningsveld R, Schmitz PI, Meche FG, Visser LH, Meulstee J, van Doorn PA. Effect of methylprednisolone when added to standard treatment with intravenous immunoglobulin for Guillain-Barre syndrome: randomised trial. Lancet. 2004;363(9404):192–196. | |
No authors listed. Plasmapheresis and acute Guillain-Barré syndrome. The Guillain-Barre syndrome Study Group. Neurology. 1985;35(8):1096–1104. | |
No authors listed. Efficiency of plasma exchange in Guillain-Barré syndrome: role of replacement fluids. French Cooperative Group on Plasma Exchange in Guillain-Barré syndrome. Ann Neurol. 1987;22(6):753–761. | |
No authors listed. Appropriate number of plasma exchanges in Guillain-Barré syndrome. The French Cooperative Group on Plasma Exchange in Guillain-Barré Syndrome. Ann Neurol. 1997;41(3):298–306. | |
Donaghy M, Mills KR, Boniface SJ, et al. Pure motor demyelinating neuropathy: deterioration after steroid treatment and improvement with intravenous immunoglobulin. J Neurol Neurosurg Psychiatry. 1994;57(7):778–783. | |
Carpo M, Cappellari A, Mora G, et al. Deterioration of multifocal motor neuropathy after plasma exchange. Neurology. 1998;50(5):1480–1482. | |
Umapathi T, Hughes RA, Nobile-Orazio E, Léger JM. Immunosuppressant and immunomodulatory treatments for multifocal motor neuropathy. Cochrane Database Syst Rev. 2009;21(1):CD003217. | |
Parry GJ, Clarke S. Multifocal acquired demyelinating neuropathy masquerading as motor neuron disease. Muscle Nerve. 1988;11(2):103–107. | |
Chaudhry V, Corse AM, Cornblath DR, et al. Multifocal motor neuropathy: response to human immune globulin. Ann Neurol. 1993;33(3):237–242. | |
Krarup C, Stewart JD, Sumner AJ, Pestronk A, Lipton SA. A syndrome of asymmetric limb weakness with motor conduction block. Neurology. 1990;40(1):118–127. | |
Stieglbauer K, Topakian R, Hinterberger G, Aichner FT. Beneficial effect of rituximab monotherapy in multifocal motor neuropathy. Neuromuscul Disord. 2009;19(7):473–475. | |
Rojas-García R, Gallardo E, de Andrés I, et al. Chronic neuropathy with IgM anti-ganglioside antibodies: lack of long term response to rituximab. Neurology. 2003;61(12):1814–1816. | |
Ruegg SJ, Fuhr P, Steck AJ. Rituximab stabilizes multifocal motor neuropathy increasingly less responsive to IVIg. Neurology. 2004;63(11):2178–2179. | |
Michaud A, Delmont E, Jeandel PY, Desnuelle C. [Improvement of severe and intravenous immunoglobulin-dependent multifocal motor neuropathy with conduction block after long-term rituximab]. Rev Neurol (Paris). 2011;167(12):916–920. French. | |
Pestronk A, Florence J, Miller T, Choksi R, Al-Lozi MT, Levine TD. Treatment of IgM antibody associated polyneuropathies using rituximab. J Neurol Neurosurg Psychiatry. 2003;74(4):485–489. | |
Chaudhry V, Cornblath DR. An open-label trial of rituximab (Rituxan®) in multifocal motor neuropathy. J Peripher Nerv Syst. 2010;15(3):196–201. | |
Erdmann PG, Lindeman E, Cats EA, van den Berg LH. Functioning of patients with multifocal motor neuropathy. J Peripher Nerv Syst. 2010;15(2):113–119. | |
Axelsson G, Liedholm LJ. [Multifocal motor neuropathy – unusual cause of hypoglossal palsy]. Lakartidningen. 2002;99(13):1448–1450. Swedish. | |
Omoregie E, Chugh A, Lange D. Pain in Multifocal Motor Neuropathy (P01.146). Neurology. 2013;80(Meeting Abstracts 1):P01.146. | |
Kaji R, Bostock H, Kohara N, Murase N, Kimura J, Shibasaki H. Activity-dependent conduction block in multifocal motor neuropathy. Brain. 2000;123(Pt 8):1602–1611. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.