")
Back to Journals » International Journal of General Medicine » Volume 15
Multidimensional Analysis of CHMP Family Members in Hepatocellular Carcinoma
Authors Guo Y, Shang A, Wang S, Wang M
Received 6 December 2021
Accepted for publication 15 February 2022
Published 10 March 2022 Volume 2022:15 Pages 2877—2894
DOI https://doi.org/10.2147/IJGM.S350228
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Yu Guo,1 An Shang,1 Shuang Wang,2 Min Wang1
1Department of General Surgery, Jilin University Second Hospital, Changchun, Jilin, People’s Republic of China; 2Department of Dermatology, Jilin University Second Hospital, Changchun, Jilin, People’s Republic of China
Correspondence: Shuang Wang, Department of Dermatology, Jilin University Second Hospital, 218 Ziqiang Street, Nanguan District, Changchun, Jilin, People’s Republic of China, Tel +86-181-3543-5372, Email [email protected]
Background: EGFR frequently accumulates and mutates simultaneously in various cancers. Ubiquitinated EGFR proteins can be degraded by the endosomal sorting complex required for transport. Among them, ESCRTIII is mainly composed of CHMP family members.
Methods: A total of 424 samples from the TCGA-LIHC data set were used to explore the relationship between CHMPs and liver hepatocellular carcinoma (LIHC). Oncomine, the Human Protein Altas, cBioPortal, TISIDB, TIMER, Metascape, and R software were used to facilitate analysis of the role played by CHMPs in the pathogenesis of LIHC. The role of CHMPs in the development of LIHC was analyzed in terms of differential expression, survival, mutation, immunoinfiltration, functional enrichment, and drug sensitivity.
Results: Differential expression analysis showed that CHMPs were significantly more expressed in LIHC tumor tissue, and the high expression of some CHMPs was closely correlated with clinicopathological stage. The prognosis was worse in the group with high expression of CHMPs. Among them, CHMP4C was considered to play a major role. Gene-mutation analysis and DNA promoter–methylation analysis further revealed possible mechanisms for the aberrant amplification of CHMPs. Immunoinfiltration analysis indicated that CHMPs were closely associated with multiple immune cells and exhibited resistance to various drugs when highly expressed.
Conclusion: CHMPs were found to be significantly elevated in LIHC and strongly associated with immune-cell infiltration, poor prognosis, multiple star pathways, and drug resistance.
Keywords: hepatocellular carcinoma, ESCRT, EGFR, CHMP4C
Introduction
Liver cancer is one of the most common malignancies worldwide, especially in Asia, Africa, and southern Europe.1–3 Liver hepatocellular carcinoma (LIHC) accounts for a majority of primary liver cancers, is the fourth-leading cause of cancer-related deaths, and ranks sixth in terms of number of new cases globally.4,5 Several current treatments for LIHC, such as surgery and hepatic artery embolization, have rather limited effects. Molecularly targeted drugs have also failed to reach expected targets, due to the lack of drug-producing properties of major LIHC mutations (such as TP53, TNNB1, and TERT promoter).6–9 Recently, the article “EGFR activation limits the response of hepatocellular carcinoma to lenvatinib” was published online as a cover paper in the prestigious international journal Nature.10 It showed that the combination of an EGFR inhibitor and lenvatinib had a significant killing effect on a model of HC with high expression of EGFR. Inspired by this study, we tried to explore the association between genes affecting EGFR degradation and LIHC.
EGFR frequently accumulates and is concomitantly mutated in a variety of cancers.11–13 Besides the classical degradation pathway that relies on EGF induction, EGF-independent proteasomal degradation plays a major regulatory role in maintaining EGFR protein homeostasis.14,15 The EGF–EGFR complex is internalized into the cell and passes through early endosomes, late endosomes, and multivesicular bodies (MVBs) before reaching the lysosome, where it is degraded, thereby attenuating the EGFR-mediated signal.16,17 Endosomal sorting complexes required for transport (ESCRT) are involved in this process, sorting the ubiquitinated receptor at the MVB to facilitate lysosomal degradation.18 The main function of the ESCRT system is to promote the degradation of ubiquitin-labeled membrane proteins,19 and it is also involved in cell division, virus germination, cellular autophagy, and fungal pH sensing.20–22 ESCRT consists of multiple protein complexes, most of which have been structurally resolved. Among them, ESCRT-III is responsible for shearing the neck of the budding body, thereby releasing the vesicles into the lumen of the endosome and completing the budding process.
ESCRT-III is composed of two parts — a core subunit and a regulatory subunit — and its function is essential for ESCRT to sort ubiquitinated proteins.23 That is why the proteins involved in the composition of ESCRT-III were selected as the target of this paper to explore the effects played by their abnormal expression or mutation in LIHC. Unlike yeast, its human-derived proteins mainly include 12 species: CHMP1A, CHMP1B, CHMP2A, CHMP2B, CHMP3 (VPS24), CHMP4A,CHMP4B, CHMP4C, CHMP5, CHMP6, CHMP7, and IST1 (CHMP8). In oncological studies, CHMP1A is implicated as a tumor-suppressor gene with antiproliferative effects in pancreatic cancer.24,25 CHMP4A is suspected to be associated with ovarian cancer and prostate cancer.26,27 CHMP4C plays an important role in the development of cervical cancer and is associated with the radiosensitivity of lung cancer tumor cells.28,29
In this paper, with the help of bioinformatic techniques, patient samples from the Cancer Genome Atlas (TCGA) database were selected for analysis. The relationship between ESCRT-III components and HC was analyzed in terms of differential expression, survival analysis, functional enrichment, and tumor drug resistance.
Methods
Data Source
TCGA was initiated by the National Cancer Institute and the National Human Genome Research Institute in 2006. It is an important source of clinical data, genomic variation, mRNA expression, miRNA expression, methylation, and other data for cancer researchers.30 LIHC samples from TCGA were included in the study.
Differential Expression Analysis
Oncomine (https://www.oncomine.com) takes tumor-related microarray data from various sources and processes them with a standardized analysis process. The results of the data analysis can be queried and visualized through web services, thus facilitating data utilization and tumor research.31 Oncomine and R software (version 3.6.3) were employed for differential expression analysis. The eleven components of ESCRT were analyzed for differential expression at the mRNA level in LIHC tumor tissue and normal tissue. The Human Protein Atlas (HPA) utilizes transcriptomic and proteomic technologies to explore protein expression at the RNA and protein levels in different human tissue types and organs. The HPA database is a convenient way to explore the expression of protein-coding genes in normal and tumor tissue/organs.
Survival Analysis
R software was applied to preprocess the screened data for quality control, background correction, normalization, and gene annotation. The Survival package of R software was implemented to perform survival analysis and plot Kaplan–Meier curves and risk tables. Downloaded sample information was divided into high- and low-expression groups according to the CHMP-expression percentile, and log-rank tests were performed to analyze whether there were any differences in the overall survival (OS) and disease-free survival (DFS) of patients between the high- and low-expression groups.
Mutation Analysis
The cBio cancer genomics portal (cBioPortal) facilitates the exploration of multidimensional cancer-gene data, allowing visual analysis across genes, samples, and data types. Researchers can visualize patterns of genetic alterations across numerous samples in a particular cancer study and compare the frequency of genetic alterations in multiple cancer studies or summarize and summarize all relevant genomic alterations in a single tumor sample.32 cBioPortal was exploited for mutational analysis. The main mutation types and mutation frequencies of the 12 components of ESCRT-III in LIHC were analyzed.
Methylation Analysis
DNA methylation is a form of chemical modification of DNA. Numerous studies have indicated that DNA methylation induces changes in chromatin structure, DNA conformation, DNA stability, and the way DNA interacts with proteins, which in turn controls gene expression.33,34 R software (version 3.6.3) was applied for methylation analysis and visualization to compare whether there were significant differences in promoter-methylation levels of ESCRT-III–related genes between tumor tissue and normal tissue.
Immunoinfiltration Analysis
TISIDB (http://cis.hku.hk/TISIDB) is a powerful website containing a wealth of tumor immunorelated data that facilitates comprehensive study of immune cell–tumor interactions. The site collates 4,176 records from 2,530 publications, in which 988 genes associated with antitumor immunity are documented. For 30 TCGA cancer types, associations between genes and immunounction (such as lymphocytes, immunomodulators, and chemokines) have been precomputed. Tumor immunoinfiltration refers to the movement of immune cells from the blood to tumor tissue and exerting their effects. Immunoinfiltration in tumors is closely related to clinical outcomes, and immune cells infiltrating tumors are most likely to be used as drug targets to improve patient survival. The Tumor Immune Estimation Resource (TIMER; https://cistrome.shinyapps.io/timer) can detect immunoinfiltration in tumor tissue using RNA-Seq expression-profiling data.35
Functional Enrichment Analysis
Functional enrichment analysis is the process of grouping together genes with similar functions in a gene list and associating them with biological phenotypes. Gene Ontology (GO) is a semantic vocabulary standard for qualifying and describing the functions of genes and proteins for all species and can be updated as research progresses. The Kyoto Encyclopedia of Genes and Genomes (KEGG) is a comprehensive database that integrates genomic, chemical, and phylogenetic functional information. Selected genes coexpressed with CHMPs were screened for GO and KEGG enrichment analysis using String and GEPIA. R software was employed to analyze the relationship of genes involved in the formation of the ESCRT-III complex with some star pathways, such as apoptosis, cell cycle, and DNA-damage response.
Drug-Sensitivity Analysis
The Cancer Therapeutics Response Portal (CTRP) bridges genetic, genealogical, and other cellular characteristics of cancer cell lines with small-molecule sensitivity to support future patient-matched drug discovery based on predictive biomarkers.36 The Genomics of Drug Sensitivity in Cancer (GDSC) database characterizes 1,000 human cancer cell lines, screens them with >100 compounds, and tags drug-response data with sensitivity genomes.37 The CTRP and GDSC are employed to collect small molecules.
To analyze the correlation between gene expression and drug sensitivity, we downloaded the area under the dose–response curve values of drugs and gene-expression profiles of all cancer cell lines from the CTRP and GDSC and performed resistance analysis of the gene sets from GDSC/CTRP drug data. Spearman correlation analysis indicates the correlation between gene expression and drug sensitivity. A positive correlation implies that high gene expression confers resistance to the drug and a negative one that it does not.
Data Analysis
The log-rank test was chosen to analyze differences in OS between high and low gene–expression groups. Spearman analysis was employed to analyze correlations of gene-expression levels with drug sensitivity. All the data analysis in the article, such as the differential expression of each gene, correlations of gene-expression levels with pathological staging, and correlations with immunoinfiltration, were considered significant at P<0.05.
Results
Differential Expression Analysis
A total of 424 samples from the TCGA-LIHC data set were selected for this study: 50 of normal tissue and 374 of HC tissue. The Wilcoxon rank-sum test was used to compare differences in expression of CHMPs between normal and tumor tissue, and the results showed that CHMP1A, CHMP2A, CHMP2B, CHMP3 (VPS24), CHMP4A, CHMP4B, CHMP4C, CHMP5, CHMP6, CHMP7,andCHMP8 (IST1) were all highly expressed in tumor tissue (P<0.05, Figure 1A). Paired sample t-tests were used for paired-sample analysis, and 50 normal-tissue and the matched 50 HC-tissue samples were retained. The results showed that the CHMPs remained highly expressed, except for CHMP4A (P<0.05; Figure 1B). Further analysis of the correlation between differences in expression of CHMPs and clinicopathological staging revealed that the higher the CHMP1A, CHMP2B, CHMP3, CHMP4B, CHMP5, and CHMP7 expression, the more advanced the clinical staging of the patients (P<0.05, Figure 1C–H). In contrast, there was no significant correlation between CHMP1B, CHMP2A, CHMP4A, CHMP4C, and CHMP6 expression content and clinicopathological stage (P>0.05). The HPA database contains proteomic, transcriptomic, and system-biology data covering normal and tumor tissue. Differential expression data at the protein level of CHMPs were obtained from the HPA database, and immunohistochemical results demonstrated a significant increase in CHMP expression in tumor tissue, consistent with the results of differential gene-expression analysis (Figure 2).
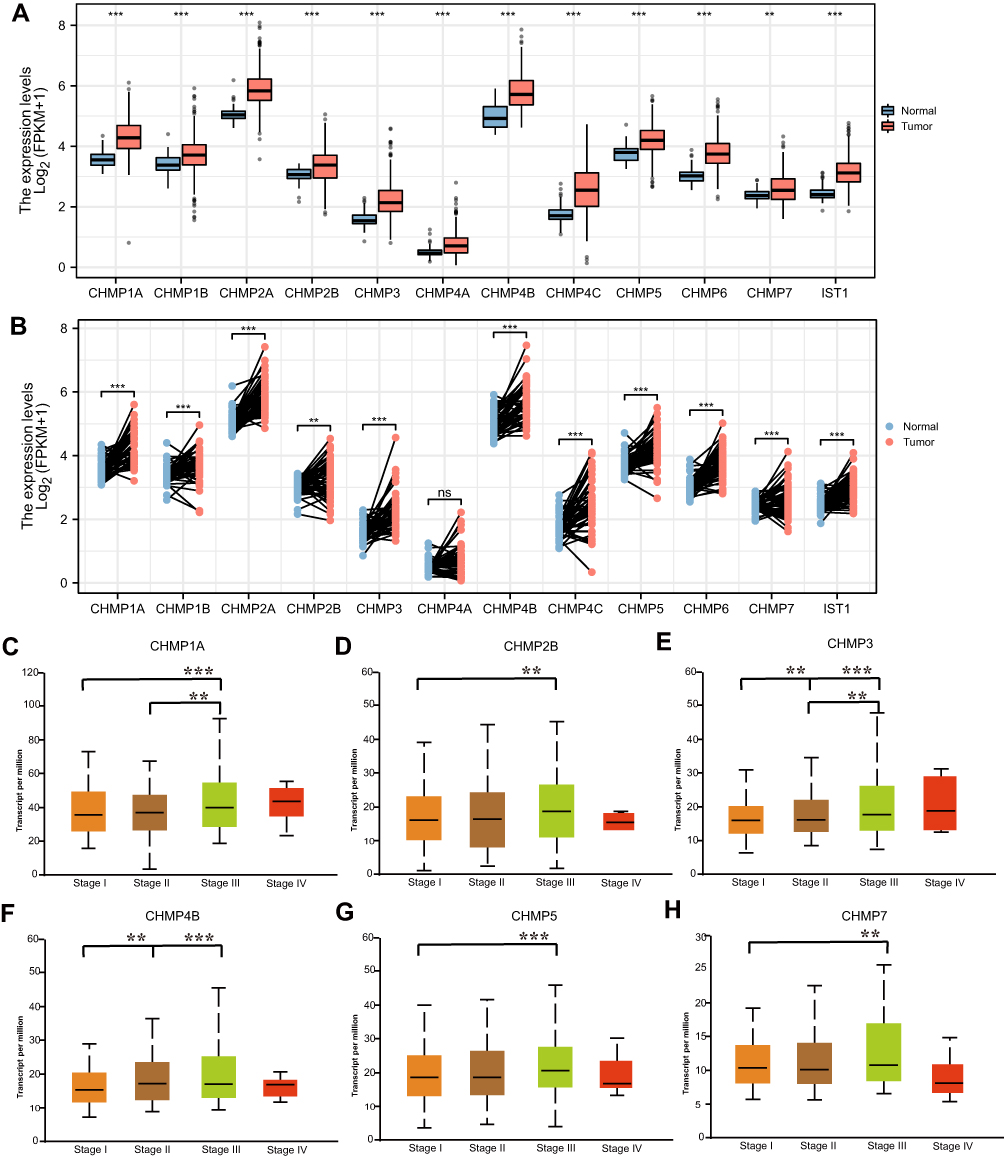 |
Figure 1 Differential expression of CHMPs. (A) Differential expression analysis of CHMPs in TCGA-LIHC data set in 424 samples (normal 50, cancer 374); (B) differential expression analysis of CHMPs in paired samples of the TCGA-LIHC data set; (C–H) Correlations of CHMP expression with clinicopathological staging. *P<0.05; **P<0.01; ***P<0.001. |
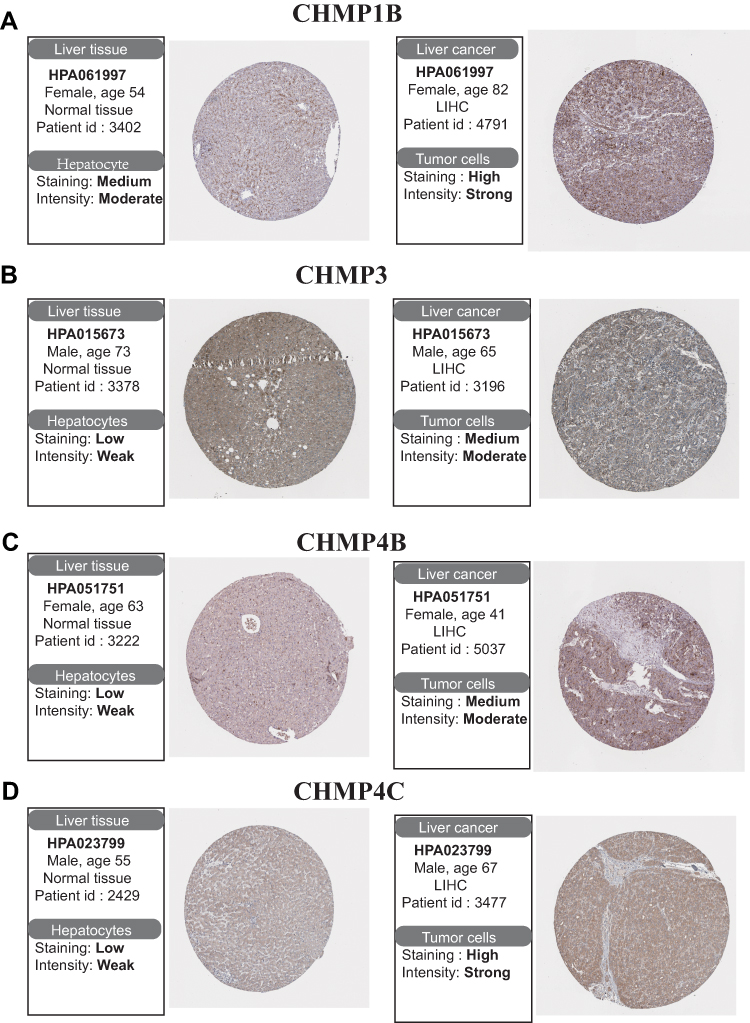 |
Figure 2 Differential expression analysis of CHMPs in the HPA database. (A–D) Immunohistochemistry was employed to analyze differences in expression of CHMP1B, CHMP3, CHMP4B, and CHMP4C in normal and tumor liver tissue. |
Survival Analysis
Data from TCGA-LIHC were divided into high- and low-expression groups based on the percentage of CHMPs expressed. Log-rank tests were employed for survival analysis to compare whether there was a discrepancy in OS and DFS between the groups. As shown in Figure 3A, in terms of OS, CHMP1A (P=0.008), CHMP2B (P=0.013), CHMP3 (P=0.012), CHMP4A (P=0.004), and CHMP4B (P=0.002) expression levels significantly correlated with prognosis, with shorter OS in the high-expression group. Regarding DFS, high expression of CHMP2B (P=0.036), CHMP3 (P=0.031), CHMP4A (P=0.01), and CHMP4B (P=0.002) were associated with poor prognosis (Figure 3B). This result was generally consistent with the differential gene-expression results.
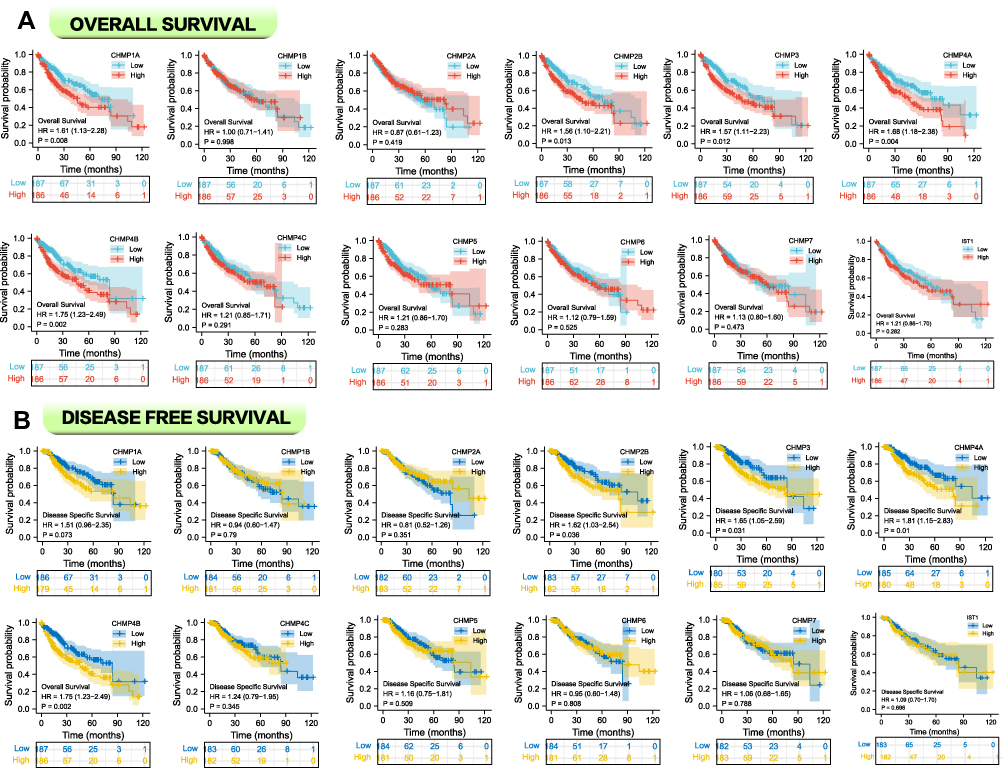 |
Figure 3 Survival analysis with risk tables. (A) Correlation analysis between CHMP expression and OS; (B) correlation analysis between CHMP expression and DSS. |
Mutation Analysis
A total of 814 samples from TCGA PanCancer Altas LIHC (372 samples) and TCGA Firehose Legacy LIHC (442 samples) were included in the study, and the mutation with the highest frequency in CHMPs was found to be amplification (Figure 4A). Further detailed analysis of mutations in each gene in the LIHC samples demonstrated that CHMP4C mutations had the highest frequency of 10% and were primarily amplification mutations. The next-highest frequency of CHMP7 mutations was 7%, which were mainly deep deletion (Figure 4B). Taking the survival analysis further, it was found that the mutation group had a poorer prognosis (Figure 4C, P=0.0465). The heterozygous and homozygous copy-number variation (CNV) statistics for LIHC are displayed as pie charts in Figure 4D.
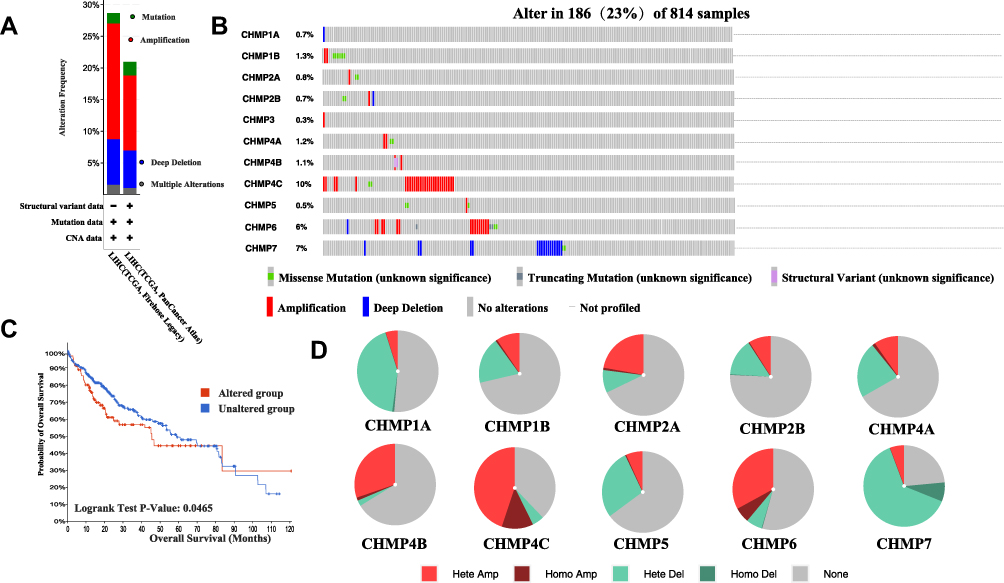 |
Figure 4 Mutation analysis. (A) Mutation frequency and types of CHMP in LIHC tumor tissue; (B) separate presentation of mutation type and frequency of CHMP members in LIHC tumor tissue; (C) OS of CHMP-altered group was significantly lower than that of unaltered group; (D) CNV of CHMPs. |
Methylation Analysis
Methylation analysis was performed with the R software to analyze the differences in promoter-methylation levels of CHMPs between LIHC normal and tumor tissue (Figure 5A–J). The results showed that CHMP1A (P=1.62–12), CHMP1B (P=1.83–12), CHMP2A(P=1.62–12), CHMP3(P=6.7–7), and CHMP4C (P<1–12) tumor tissue had lower promoter-methylation levels, while CHMP2B, CHMP4A, CHMP4B, CHNMP5, and CHMP6 did not differ significantly (P>0.05).
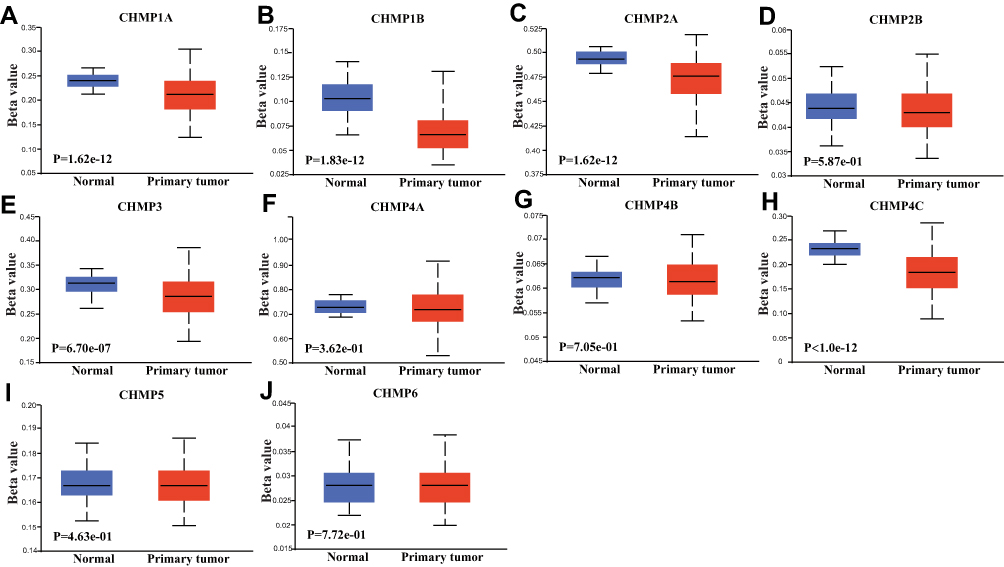 |
Figure 5 Methylation analysis. (A–J) Comparative analysis of differences in DNA promoter–methylation levels of CHMP1A, CHMP1B, CHMP2A, CHMP2B, CHMP3, CHMP4A, CHMP4B, CHMP4C, CHMP5, and CHMP6 in normal and LIHC tumor tissue, respectively. |
Immunorelated Analysis
“The immune landscape of cancer”,38 an article published in the journal Immunity in April 2018, based on differences in macrophage or lymphocyte characteristics, Th1:Th2 cell ratio, degree of tumor heterogeneity, aneuploidy, degree of neoantigen load, cell proliferation, expression of immunoregulatory genes, prognosis, and other indicators among different subtypes, classified all tumors into six immunosubtypes, namely wound healing (C1), IFNγ-dominant (C2), inflammatory (C3), lymphocyte depleted (C4), immunologically quiet (C5), and TGFβ-dominant (C6). The distribution of CHMP expression in immunosubtypes of LIHC was analyzed as depicted in Figure 6. Correlations between the expression of CHMPs in tumor tissue and the infiltration of immune cells were assayed with the help of RNA-Seq expression-profiling data. The results obtained are shown in Figure 7, where CHMP expression was positively correlated with immune-cell infiltration (B cells, CD4+ T cells, CD8+ T cells, neutrophils, macrophages, and dendritic cells; P<0.05) and insignificantly correlated with tumor purity (P>0.05).
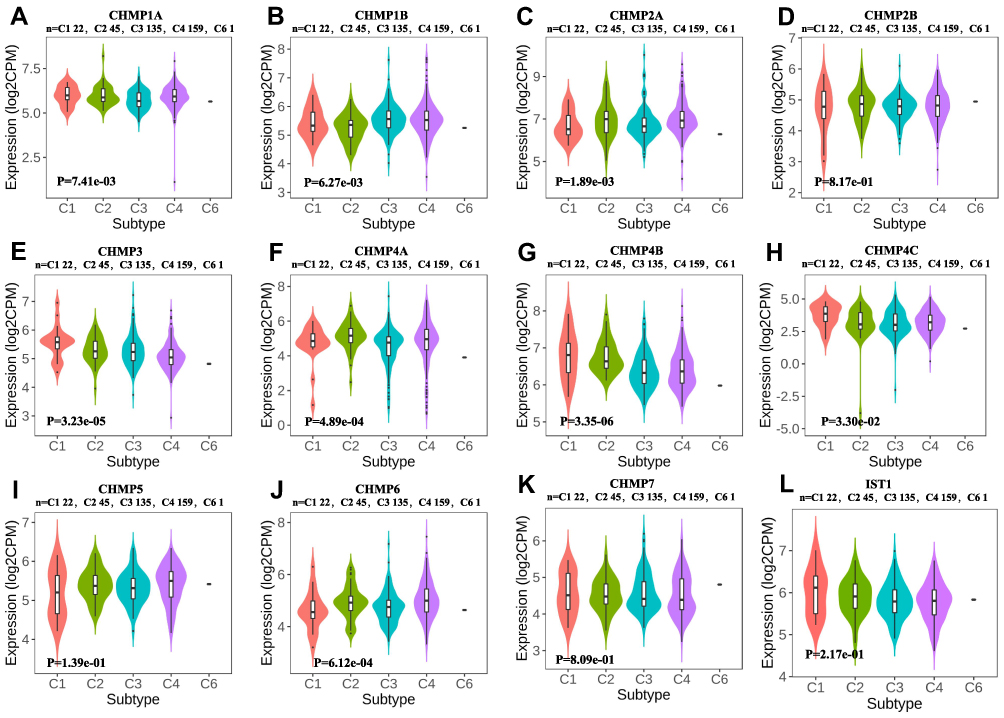 |
Figure 6 Immunological subtypes. (A–L) Differential expression analysis of CHMPs in immunologic subtypes of LIHC. |
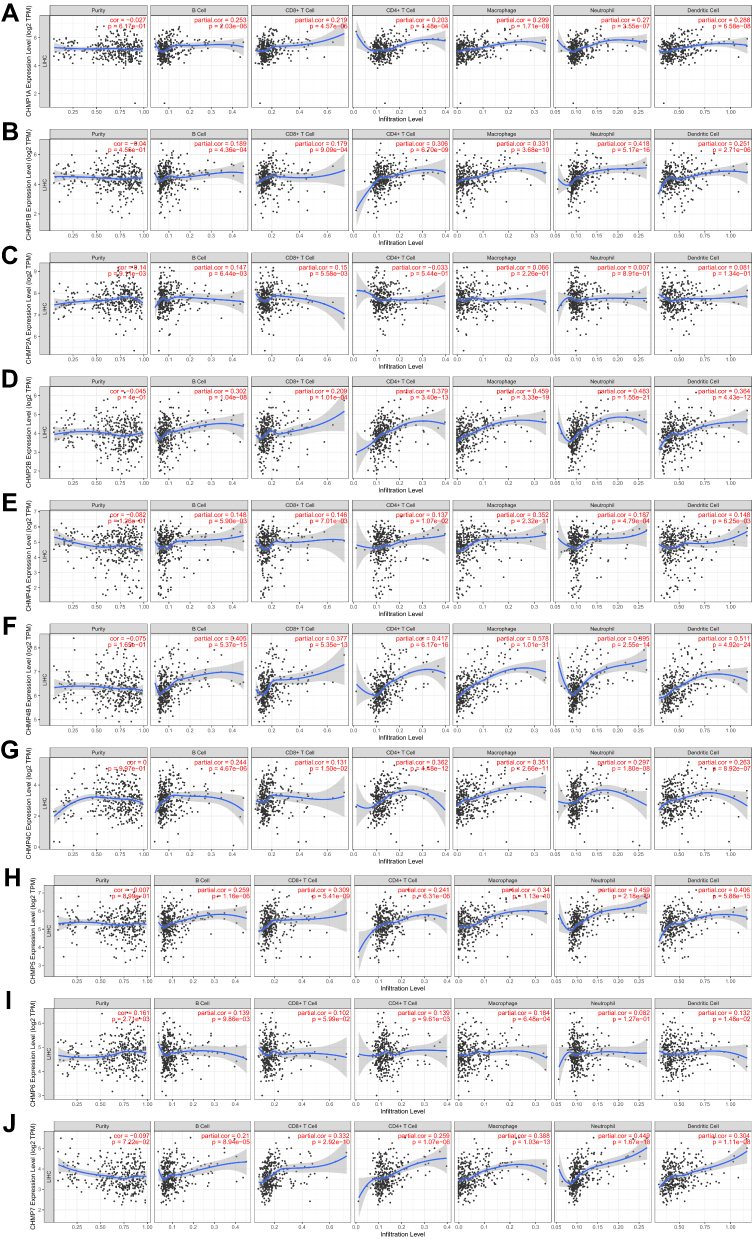 |
Figure 7 Immunoinfiltration analysis. (A–J) Correlations between gene expression of CHMPs in LIHC and infiltration of immune cells (B cells, CD4+ T cells, CD8+ T cells, neutrphils, macrophages, and dendritic cells). |
Functional Enrichment Analysis
The top 100 genes that were coexpressed with CHMPs were screened using GEPIA and the results visualized and analyzed with a looping plot. The purple curves link identical genes (Figure 8A) and the blue curves connected genes that belong to the same enriched ontology term (Figure 8B). To further catch the relationships between terms, we selected an enriched subset of terms and presented it as a network graph, where terms with similarity >0.3 are connected by edges. We selected terms with the best P-value from each of the 20 clusters, with the restriction that there were no more than 15 terms per cluster and no more than 250 terms in total. The network was visualized using Cytoscape 7, with each node representing an enriched term, colored first with its cluster ID (Figure 8C) and then with its P-value (Figure 8D). The network of enriched terms are represented as pie charts and color-coded based on the identities of the gene lists (Figure 8E). Combined with P-value analysis, CHMPs and their coexpressed gene GO were enriched for MVB assembly, proteolysis involved in cellular protein catabolic processes, cell division, and autophagy. KEGG enriched pathways were membrane trafficking, metabolism of RNA, and cell cycle (Table 1).
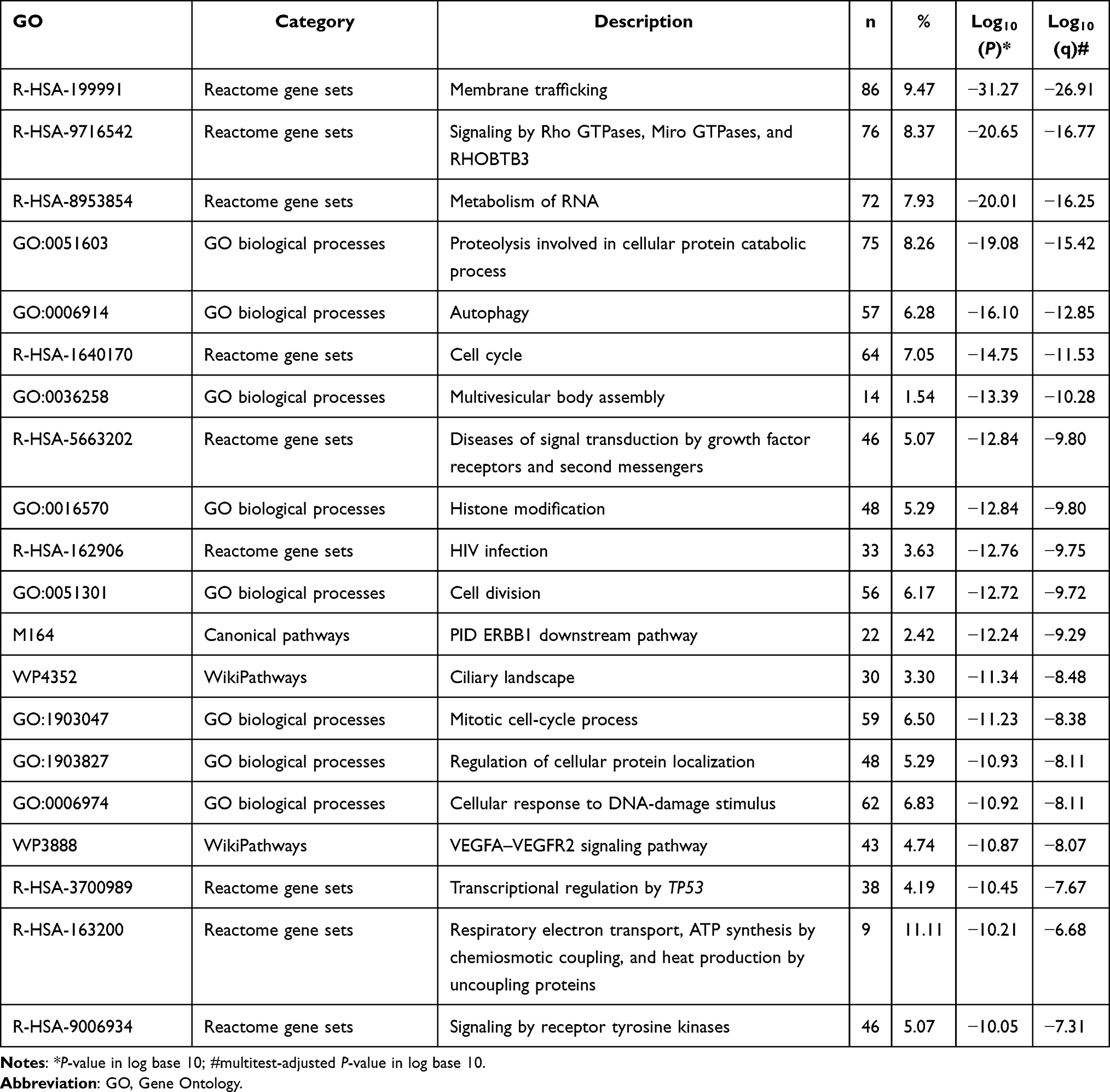 |
Table 1 Top 20 Clusters with Their Representative Enriched Terms of CHMPs |
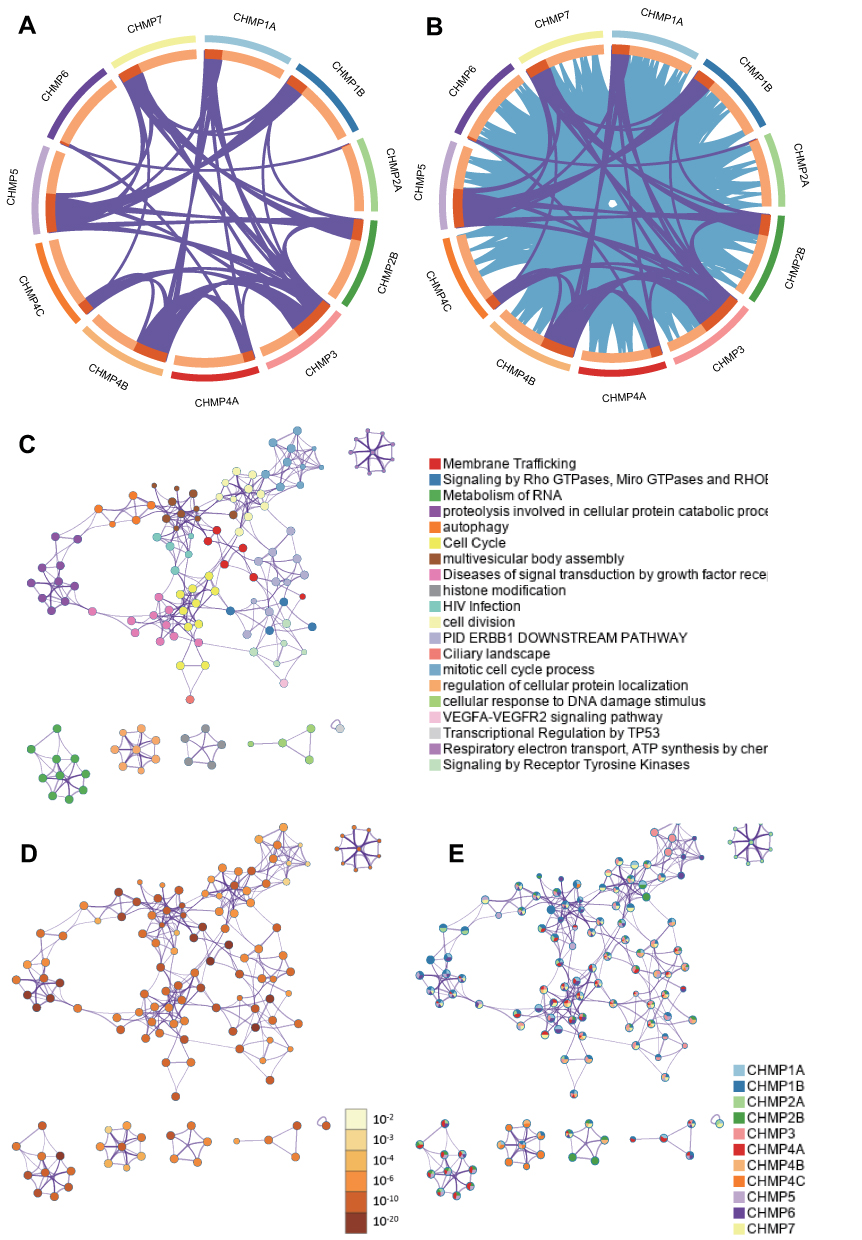 |
Figure 8 Functional enrichment analysis of CHMPs. (A) The top 100 genes coexpressed with CHMPs were screened using GEPIA and the results were visualized and analyzed with a loop plot. At the gene level, where purple curves link identical genes; (B) blue curves link genes that belong to the same enriched ontology term; (C) colored by cluster ID, where nodes with the same cluster ID are usually close to each other; (D) colored by P-value, where terms containing more genes tend to have greater significance; (E) network of enriched terms represented as pie charts, where pies are color-coded based on the identities of the gene lists. |
The role of CHMP members in influencing the star pathway in tumorigenesis development was further specifically analyzed (Figure 9A and B). Included were TSC–mTOR, RTK, RAS–MAPK, PI3K–Akt, hormone ER, hormone AR, epithelial–mesenchymal transition, DNA-damage response, cell cycle, and apoptosis pathways. These are all famous cancer-related pathways. RBN RPPA data were median-centered and normalized by standard deviation across all samples for each component to obtain the relative protein level. The pathway score was the sum of the relative protein level of all positive regulatory components minus that of negative regulatory components in a particular pathway.
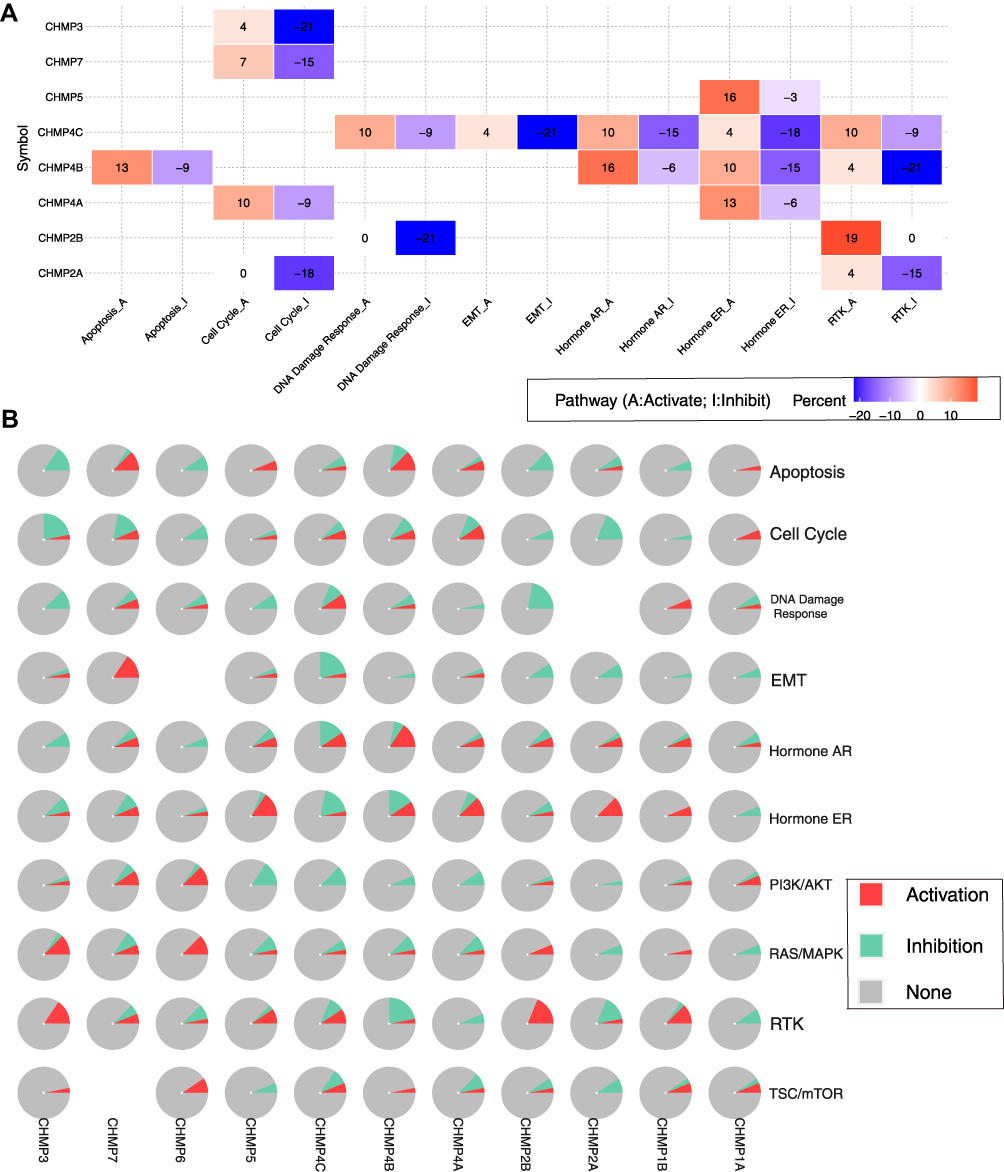 |
Figure 9 CHMP function with activation or inhibition of the star pathway. (A) Heat map showing genes with function (suppression or activation); (B) global percentage of cancers in which a gene has an effect on the pathway. |
Gene expression was divided into two groups (high and low) by median expression, differences in pathway activity score (PASs) between groups were defined using Student’s t-test, P-value was adjusted by FDR, and FDR ≤0.05 was considered significant. When PAS (gene A, high group) > PAS (gene A, low group), we considered that gene A may have an activating effect on a pathway and otherwise an inhibiting effect. The results demonstrated that CHMP4C affected several pathways, the most significant of which was inhibition of epithelial–mesenchymal transition. CHMP4B inhibit RTK, and CHMP2B inhibit DNA-damage response.
Drug-Sensitivity Analysis
We analyzed gene-set drug resistance from GDSC/CTRP IC50 drug data. The Spearman correlation represents gene-expression correlation with the drug. A positive correlation means that high gene expression confers resistance to the drug and a negative one that it does not. Analysis of CTRP and GDSC source data revealed that CHMP4C, CHMP4B, CHMP2B, and CHMP2A showed resistance to multiple drugs when highly expressed (Figure 10A and B). GO and KEGG enrichment analysis was performed on CHMP4C and its coexpressed genes (Table 2). They were primarily enriched in viral budding via the host ESCRT complex, viral budding, midbody abscission, MVB organization, and MVB assembly in biological processes (BPs; Figure 11A), while genes in molecular function (MF; Figure 11B) were mainly enriched in purine-nucleotide binding, GTPase activity, purine-ribonucleoside binding, GTP binding, and structural constituents of the cytoskeleton. Genes in cellular component (CC) were mainly enriched in late endosome membrane, endosome membrane, microtubules, ESCRT complex, and ESCRT-III complex (Figure 11C). KEGG pathway-enrichment analysis showed that the main enrichment pathways that made up this modular gene were endocytosis, Salmonella infection, phagosome, and gap junction (Figure 11D).
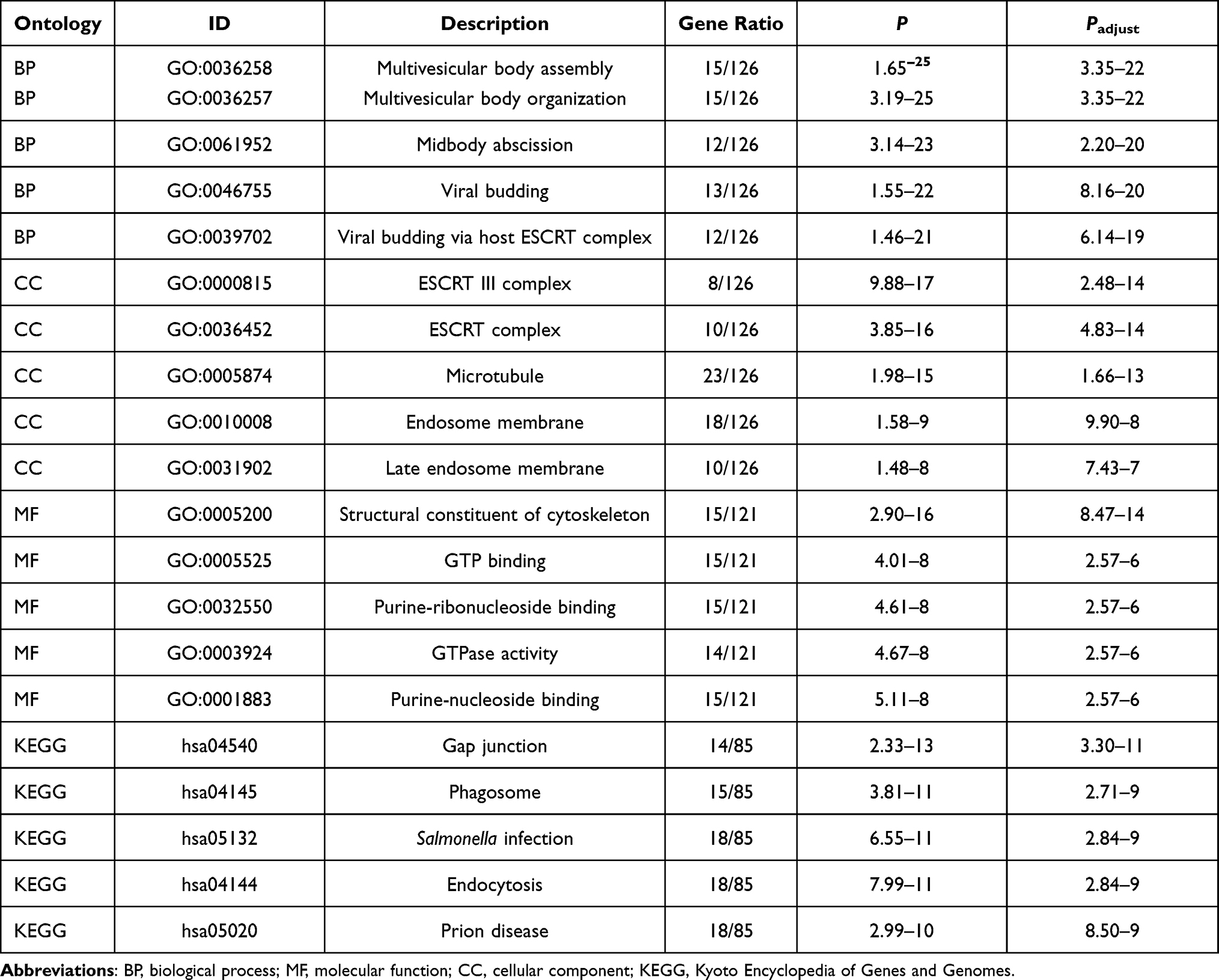 |
Table 2 Functional enrichment analysis of CHMP4C |
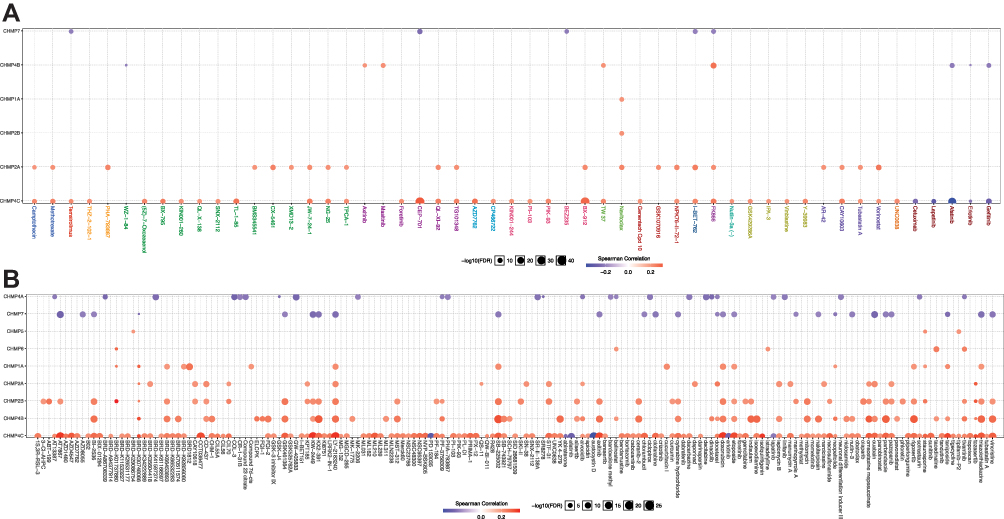 |
Figure 10 Drug-sensitivity analysis. (A) Gene-set drug-resistance analysis from GDSC IC50 drug data; (B) gene-set drug-resistance analysis from CTRP IC50 drug data. Spearman correlations represent correlations gene expression with the drug. A positive correlation means that high gene expression confers resistance to the drug and a negative one that it does not. |
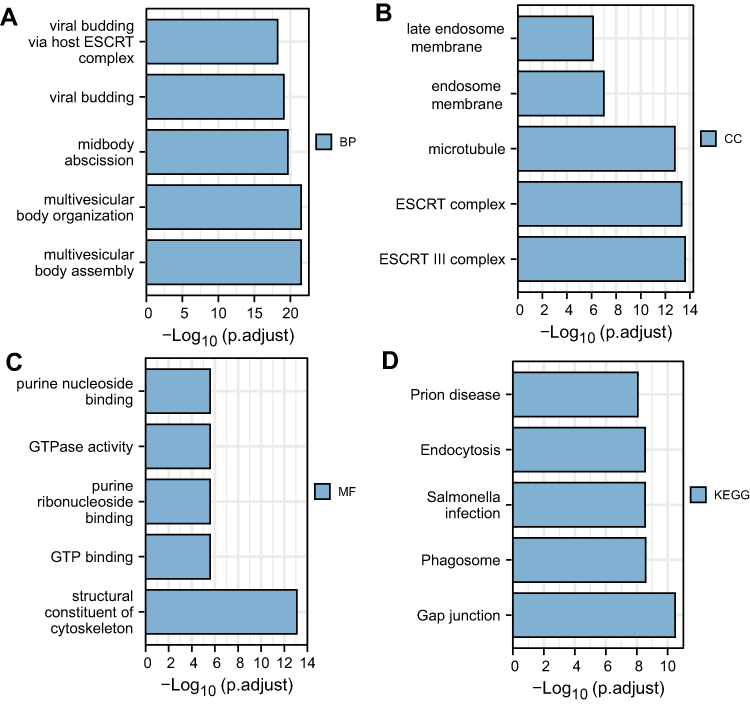 |
Figure 11 Functional enrichment analysis of CHMP4C. (A–D) GO and KEGG enrichment analysis of CHMP4C and its coexpressed genes |
Discussion
EGFR accumulates abnormally in multiple diseases, and its downregulation is mediated through the ESCRT system. Mutations in the cCbl binding region of EGFR can lead to impairment of its ubiquitination and thus prevent its degradation through the ESCRT system, leading to accumulation.39 Several EGFR mutants associated with cancer lack their cCbl binding region and are thus not regulated by the ESCRT system.40 The ESCRT system is an essential molecular mechanism for membrane protein sorting in eukaryotic cells.41 It has been demonstrated that ubiquitin-tagged membrane proteins are first transported to the endosomal membrane by cytokinesis and then invaginated by the ESCRT system, which releases membrane components containing these proteins into the endosomal lumen to form intraluminal vesicles. The endosomes at this point are called MVBs. Afterward, through fusion with lysosomes, the endosomal vesicles and the proteins on their membranes are degraded and the protein-bound ESCRT complex proteins degraded and recycled for reuse. The ESCRT complex mediates cell-membrane remodeling and fission responses. The pathway comprises five core complexes: ESCRT-0, ESCRT-I, ESCRT-II, ESCRT-III, and Vps4.42
ESCRT-III is composed of two parts — the core subunit and the regulatory subunit — whose main function is to complete the shearing of the budding neck to expel MVBs out of the cell. Humans express 12 related ESCRT-III proteins: CHMP1A, CHMP1B, CHMP2A, CHMP2B, CHMP3, CHMP4A, CHMP4B, CHMP4C, CHMP5, CHMP6, CHMP7, and IST1 (CHMP8). Unlike Saccharomyces cerevisiae, some human protein families contain several homologous gene products (such as CHMP2A-B, CHMP4A-C). At least one member of the CHMP4 and CHMP2 families is required for the inside-out ESCRT-dependent response in human cells, while other subunits play secondary or more specific roles, such as maintenance of the shedding checkpoint (CHMP4C)43 and nuclear membrane closure (CHMP7).44 The ESCRT system plays an instrumental role in several ways and is closely related to human health. It has been demonstrated to be associated with several neurodegenerative diseases, eg, mutations in CHAP2B have been found to be associated with frontotemporal dementia and hereditary spastic paraplegia.45
CHMPs have been sparsely studied in oncology. In this paper, the TCGA-LIHC data set was targeted and the role of CHMPs in the development of LIHC analyzed from different aspects with the help of bioinformatic knowledge and technology. We first performed differential expression analysis to compare whether CHMPs had meaningfully altered expression in normal and tumor tissue, and the results revealed that the expression of CHMPs was significantly higher in LIHC tumor tissue. Further analysis of the correlation between the difference in expression of CHMPs and clinicopathological staging revealed that the higher the CHMP1A, CHMP2B, CHMP3, CHMP4B, CHMP5, and CHMP7 expression, the more advanced the clinical staging of the patients (P<0.05). Then, LIHC samples were divided into high- and low-expression groups according to the expression percentage, and the KM curves displayed that the CHMP1A, CHMP2B, CHMP3, CHMP4A, and CHMP4B high-expression group had a worse prognosis, which was consistent with the results of differential gene-expression analysis. It is suggested that high expression of CHMPs may cause impaired degradation of ubiquitinated proteins, which may contribute to the accumulation of suchcytokines as EGFR and induce neoplasms.
Mutation analysis was performed to explore correlations between mutations and changes in the expression of CHMPs. The results revealed that the most frequent mutation of CHMPs in LIHC samples was amplification, with the highest frequency of CHMP4C mutations at 10%. Patients in the mutation group had a worse prognosis. DNA methylation is a major form of epigenetic modification that can alter genetic expression without altering the DNA sequence.33 DNA methylation may silence genes and thus disable their functions. Analysis of promoter methylation in CHMPs revealed that CHMP1A, CHMP1B, and CHMP4C promoters were significantly less methylated in tumor tissue. Combined with the results of previous analyses, it was considered that the lower promoter methylation of CHMPs in tumor tissue might lead to active DNA function, and in addition irregular CNV amplification, both of which led to elevated expression of CHMPs and might be closely related to the occurrence and progression of LIHC. Mutation analysis and DNA promoter–methylation analysis revealed possible mechanisms of abnormal amplification of CHMPs in tumor tissue. The impaired methylation of CHMPs in tumor tissue leads to persistent active DNA function, which affects the implementation of normal ESCRT functions.
Possible reasons for the poor prognosis of LIHC patients when CHMPs are expressed abnormally were further discussed. CHMPs were differentially expressed in different immunosubtypes, and the expression quantity was positively correlated with the level of immune-cell infiltration. CHMPs may have a further impact on tumor progression and patient prognosis by influencing tumor immunoinfiltration. Tumor-infiltrating lymphocytes (TILs) are lymphocytes that leave the bloodstream and enter the tumor, constituting an important component of the tumor microenvironment. They are independent predictors of cancer-outpost lymph-node status and survival.46 TILs can be divided into at least three cell types: effector, regulatory, and inflammatory cell. They can every interact with one another through cytokines and soluble factors. CD8+ T cells, also known as cytotoxic T lymphocytes (CTLs), are recognized as the major antitumor immunoeffector cells. They can bind to tumor cells and produce perforin and other cytotoxins that kill cancer cells, but do not affect normal cells. CD4+ Th cells provide cytokine mediation to help CTLs proliferate with enhancing its virulence. Regulatory T cells make up only ~5% of the CD4+ Th-cell subpopulation, but play an important role in regulating the in situ immunoresponse.
Drug-sensitivity analysis revealed that LIHC tumor tissue showed resistance to multiple drugs when CHMP4C levels were increased. Combined with the previous analysis, CHMP4C was found to be the most frequently mutated in tumor tissue, mainly in the form of amplification. Its promoter methylation was significantly reduced, and patients in the CHMP4C high-expression group had a worse prognosis. Functional enrichment revealed that CHMP4C was able to interact with multiple star pathways. CHMP4C is a key component of the cytokinesis checkpoint, a process required to delay abscission to prevent both premature resolutions of intercellular chromosome bridges and accumulation of DNA damage.43 Amplification mutations and reduced promoter-methylation levels of CHMP4C lead to abnormally high expression, which may affect the normal function of ESCRT-III and lead to the accumulation of ubiquitinated proteins or EGFR, and on the other hand also lead to tumor insensitivity to multiple drugs.
In this article, the TCGA-LIHC data set was targeted to explore the relationship between CHMPs and LIHC. The role of CHMPs in the development of LIHC was analyzed in terms of differential expression, survival, mutation, immunoinfiltration, functional enrichment, and drug-sensitivity. CHMP4C was considered to play a major role. This may suggest a new direction for future studies to discover new targets for the treatment of LIHC from the EGFR-degradation pathway. The biggest limitation of this study is the lack of relevant experimental evidence, which will be the major direction of our next work.
Conclusion
A multifaceted analysis revealed the critical role of CHMPs in the pathogenesis of LIHC and their close association with poor prognosis and drug resistance. The properties exhibited by CHMP4C deserve our attention and may provide new ideas or targets for further treatment.
Abbreviations
ESCRT, endosomal sorting complexes required for transport; MVBs, multivesicular bodies; TCGA, the Cancer Genome Atlas; LIHC, liver hepatocellular carcinoma; OS, overall survival; DFS, disease-free survival; GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; CNV, copy-number variation; RTK, receptor tyrosine kinase; BP, biological process; MF, molecular function; CC, cellular component; CTLs, cytotoxic T lymphocytes.
Data Sharing
The data sets used and analyzed during the current study are available from the corresponding author on reasonable request. If any data related to the study of this paper is desired, you may contact Yu Guo ([email protected]) to obtain it.
Ethics Approval and Consent to Participate
An application was submitted to the Ethics Review Committee of Jilin University Hospital stating that the data used in this paper were obtained from an online public database and thus did not involve ethical or moral issues, and a statement of approval was obtained.
Consent for Publication
Written consent was obtained from patients for publication of this study and accompanying images.
Disclosure
The authors declare that they have no competing interests in this work.
References
1. Anwanwan D, Singh SK, Singh S, et al. Challenges in liver cancer and possible treatment approaches. Biochim Biophys Acta Rev Cancer. 2020;1873(1):188314. doi:10.1016/j.bbcan.2019.188314
2. Islami F, Miller KD, Siegel RL, et al. Disparities in liver cancer occurrence in the United States by race/ethnicity and state. CA Cancer J Clin. 2017;67(4):273–289. doi:10.3322/caac.21402
3. Villanueva A, Longo DL. Hepatocellular carcinoma. N Engl J Med. 2019;380(15):1450–1462. doi:10.1056/NEJMra1713263
4. Jiří T, Igor K. Hepatocellular carcinoma future treatment options. Klin Onkol. 2020;33(Supplementum 3):26–29. doi:10.14735/amko20203S26
5. Kulik L, El-Serag HB. Epidemiology and management of hepatocellular carcinoma. Gastroenterology. 2019;156(2):477–491.e1. doi:10.1053/j.gastro.2018.08.065
6. Villanueva A, Hoshida Y, Battiston C, et al. Combining clinical, pathology, and gene expression data to predict recurrence of hepatocellular carcinoma. Gastroenterology. 2011;140(5):1501–12.e2. doi:10.1053/j.gastro.2011.02.006
7. Ruiz de Galarreta M, Bresnahan E, Molina-Sánchez P, et al. β-catenin activation promotes immune escape and resistance to anti-PD-1 therapy in hepatocellular carcinoma. Cancer Discov. 2019;9(8):1124–1141. doi:10.1158/2159-8290.CD-19-0074
8. Li W, Yan Y, Zheng Z, et al. Targeting the NCOA3-SP1-TERT axis for tumor growth in hepatocellular carcinoma. Cell Death Dis. 2020;11(11):1011. doi:10.1038/s41419-020-03218-x
9. Rizzo A, Dadduzio V, Ricci AD, et al. Lenvatinib plus pembrolizumab: the next frontier for the treatment of hepatocellular carcinoma? Expert Opin Investig Drugs. 2021;1–8. doi:10.1080/13543784.2021.1948532
10. Jin H, Shi Y, Lv Y, et al. EGFR activation limits the response of liver cancer to lenvatinib. Nature. 2021;595(7869):730–734. doi:10.1038/s41586-021-03741-7
11. Harrison PT, Vyse S, Huang PH. Rare epidermal growth factor receptor (EGFR) mutations in non-small cell lung cancer. Semin Cancer Biol. 2020;61:167–179. doi:10.1016/j.semcancer.2019.09.015
12. Nicholson RI, Gee JM, Harper ME. EGFR and cancer prognosis. Eur J Cancer. 2001;37(Suppl 4):S9–15. doi:10.1016/S0959-8049(01)00231-3
13. Tomas A, Futter CE, Eden ER. EGF receptor trafficking: consequences for signaling and cancer. Trends Cell Biol. 2014;24(1):26–34. doi:10.1016/j.tcb.2013.11.002
14. Yao N, Wang C-R, Liu M-Q, et al. Discovery of a novel EGFR ligand DPBA that degrades EGFR and suppresses EGFR-positive NSCLC growth. Signal Transduct Target Ther. 2020;5(1):214. doi:10.1038/s41392-020-00251-2
15. Niu M, Xu J, Liu Y, et al. FBXL2 counteracts Grp94 to destabilize EGFR and inhibit EGFR-driven NSCLC growth. Nat Commun. 2021;12:5919. doi:10.1038/s41467-021-26222-x
16. Qu X, Liu H, Song X, et al. Effective degradation of EGFR(L858R+T790M) mutant proteins by CRBN-based PROTACs through both proteosome and autophagy/lysosome degradation systems. Eur J Med Chem. 2021;218:113328. doi:10.1016/j.ejmech.2021.113328
17. Conte A, Sigismund S. Methods to investigate EGFR ubiquitination. Methods Mol Biol. 2017;1652:81–100.
18. Henne WM, Buchkovich NJ, Emr SD. The ESCRT pathway. Dev Cell. 2011;21(1):77–91. doi:10.1016/j.devcel.2011.05.015
19. McCullough J, Frost A, Sundquist WI. Structures, functions, and dynamics of ESCRT-III/Vps4 membrane remodeling and fission complexes. Annu Rev Cell Dev Biol. 2018;34:85–109. doi:10.1146/annurev-cellbio-100616-060600
20. Juan T, Fürthauer M. Biogenesis and function of ESCRT-dependent extracellular vesicles. Semin Cell Dev Biol. 2018;74:66–77. doi:10.1016/j.semcdb.2017.08.022
21. Lefebvre C, Legouis R, Culetto E. ESCRT and autophagies: endosomal functions and beyond. Semin Cell Dev Biol. 2018;74:21–28. doi:10.1016/j.semcdb.2017.08.014
22. Stoten CL, Carlton JG. ESCRT-dependent control of membrane remodelling during cell division. Semin Cell Dev Biol. 2018;74:50–65. doi:10.1016/j.semcdb.2017.08.035
23. Horváth P, Müller-Reichert T. A structural view on ESCRT-mediated abscission. Front Cell Dev Biol. 2020;8:586880. doi:10.3389/fcell.2020.586880
24. Li J, Orr B, White K, et al. Chmp 1A is a mediator of the anti-proliferative effects of all-trans retinoic acid in human pancreatic cancer cells. Mol Cancer. 2009;8:7. doi:10.1186/1476-4598-8-7
25. Li J, Belogortseva N, Porter D, et al. Chmp1A functions as a novel tumor suppressor gene in human embryonic kidney and ductal pancreatic tumor cells. Cell Cycle. 2008;7(18):2886–2893. doi:10.4161/cc.7.18.6677
26. Fujita K, Kume H, Matsuzaki K, et al. Proteomic analysis of urinary extracellular vesicles from high Gleason score prostate cancer. Sci Rep. 2017;7:42961. doi:10.1038/srep42961
27. Barlin JN, Jelinic P, Olvera N, et al. Validated gene targets associated with curatively treated advanced serous ovarian carcinoma. Gynecol Oncol. 2013;128(3):512–517. doi:10.1016/j.ygyno.2012.11.018
28. Lin SL, Wang M, Cao -Q-Q, et al. Chromatin modified protein 4C (CHMP4C) facilitates the malignant development of cervical cancer cells. FEBS Open Bio. 2020;10(7):1295–1303. doi:10.1002/2211-5463.12880
29. Li K, Liu J, Tian M, et al. CHMP4C disruption sensitizes the human lung cancer cells to irradiation. Int J Mol Sci. 2015;17(1):18. doi:10.3390/ijms17010018
30. Blum A, Wang P, Zenklusen JC. SnapShot: TCGA-analyzed tumors. Cell. 2018;173(2):530. doi:10.1016/j.cell.2018.03.059
31. Rhodes DR, Yu J, Shanker K, et al. ONCOMINE: a cancer microarray database and integrated data-mining platform. Neoplasia. 2004;6(1):1–6. doi:10.1016/S1476-5586(04)80047-2
32. Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–404. doi:10.1158/2159-8290.CD-12-0095
33. Moore LD, Le T, Fan G. DNA methylation and its basic function. Neuropsychopharmacology. 2013;38(1):23–38. doi:10.1038/npp.2012.112
34. Koch A, Joosten SC, Feng Z, et al. Analysis of DNA methylation in cancer: location revisited. Nat Rev Clin Oncol. 2018;15(7):459–466. doi:10.1038/s41571-018-0004-4
35. Li T, Fu J, Zeng Z, et al. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 2020;48(W1):W509–W514. doi:10.1093/nar/gkaa407
36. Rees MG, Seashore-Ludlow B, Cheah JH, et al. Correlating chemical sensitivity and basal gene expression reveals mechanism of action. Nat Chem Biol. 2016;12(2):109–116. doi:10.1038/nchembio.1986
37. Yang W, Soares J, Greninger P, et al. Genomics of Drug Sensitivity in Cancer (GDSC): a resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 2013;41(Database issue):D955–61. doi:10.1093/nar/gks1111
38. Thorsson V, Gibbs DL, Brown SD, et al. The immune landscape of cancer. Immunity. 2018;48(4):812–830.e14. doi:10.1016/j.immuni.2018.03.023
39. Waterman H, Katz M, Rubin C, et al. A mutant EGF-receptor defective in ubiquitylation and endocytosis unveils a role for Grb2 in negative signaling. EMBO J. 2002;21(3):303–313. doi:10.1093/emboj/21.3.303
40. Saksena S, Emr SD. ESCRTs and human disease. Biochem Soc Trans. 2009;37(Pt 1):167–172. doi:10.1042/BST0370167
41. Schöneberg J, Lee I-H, Iwasa JH, et al. Reverse-topology membrane scission by the ESCRT proteins. Nat Rev Mol Cell Biol. 2017;18(1):5–17. doi:10.1038/nrm.2016.121
42. Christ L, Raiborg C, Wenzel EM, et al. Cellular functions and molecular mechanisms of the ESCRT membrane-scission machinery. Trends Biochem Sci. 2017;42(1):42–56. doi:10.1016/j.tibs.2016.08.016
43. Carlton JG, Caballe A, Agromayor M, et al. ESCRT-III governs the Aurora B-mediated abscission checkpoint through CHMP4C. Science. 2012;336(6078):220–225. doi:10.1126/science.1217180
44. Vietri M, Schink KO, Campsteijn C, et al. Spastin and ESCRT-III coordinate mitotic spindle disassembly and nuclear envelope sealing. Nature. 2015;522(7555):231–235. doi:10.1038/nature14408
45. Skibinski G, Parkinson NJ, Brown JM, et al. Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in frontotemporal dementia. Nat Genet. 2005;37(8):806–808. doi:10.1038/ng1609
46. Dieci MV, Radosevic-Robin N, Fineberg S, et al. Update on tumor-infiltrating lymphocytes (TILs) in breast cancer, including recommendations to assess TILs in residual disease after neoadjuvant therapy and in carcinoma in situ: a report of the International Immuno-Oncology Biomarker Working Group on Breast Cancer. Semin Cancer Biol. 2018;52(Pt 2):16–25. doi:10.1016/j.semcancer.2017.10.003
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.