")
Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Multi-small molecule conjugations as new targeted delivery carriers for tumor therapy
Authors Shan L , Liu M, Wu C, Zhao L, Li S, Xu L, Cao W, Gao G, Gu Y, Zhai K
Received 25 March 2015
Accepted for publication 13 May 2015
Published 1 September 2015 Volume 2015:10(1) Pages 5571—5591
DOI https://doi.org/10.2147/IJN.S85402
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Lei Yang
Lingling Shan,1 Ming Liu,2 Chao Wu,1 Liang Zhao,1 Siwen Li,3 Lisheng Xu,1 Wengen Cao,1 Guizhen Gao,1 Yueqing Gu3
1Institute of Pharmaceutical Biotechnology, School of Biology and Food Engineering, Suzhou University, Suzhou, People’s Republic of China; 2Department of Biology, University of South Dakota, Vermillion, SD, USA; 3Department of Biomedical Engineering, School of Life Science and Technology, China Pharmaceutical University, Nanjing, People’s Republic of China
Abstract: In response to the challenges of cancer chemotherapeutics, including poor physicochemical properties, low tumor targeting ability, and harmful side effects, we developed a new tumor-targeted multi-small molecule drug delivery platform. Using paclitaxel (PTX) as a model therapeutic, we prepared two prodrugs, ie, folic acid-fluorescein-5(6)-isothiocyanate-arginine-paclitaxel (FA-FITC-Arg-PTX) and folic acid-5-aminofluorescein-glutamic-paclitaxel (FA-5AF-Glu-PTX), composed of folic acid (FA, target), amino acids (Arg or Glu, linker), and fluorescent dye (fluorescein in vitro or near-infrared fluorescent dye in vivo) in order to better understand the mechanism of PTX prodrug targeting. In vitro and acute toxicity studies demonstrated the low toxicity of the prodrug formulations compared with the free drug. In vitro and in vivo studies indicated that folate receptor-mediated uptake of PTX-conjugated multi-small molecule carriers induced high antitumor activity. Notably, compared with free PTX and with PTX-loaded macromolecular carriers from our previous study, this multi-small molecule-conjugated strategy improved the water solubility, loading rate, targeting ability, antitumor activity, and toxicity profile of PTX. These results support the use of multi-small molecules as tumor-targeting drug delivery systems.
Keywords: multi-small molecules, paclitaxel, prodrugs, targeting, tumor therapy
Introduction
Water solubility is one of the most important physicochemical properties of a medicine or prodrug. Adequate aqueous solubility is important for toxicity studies, drug testing, and clinical application.1 Looking at the physicochemical properties of marketed drugs and those in clinical development, most hydrophobic drugs with poor water solubility have been discontinued.2 Developing a prodrug is one of the best strategies for achieving enhanced chemical or metabolic stability and higher water solubility.3 A prodrug can also have increased targeting ability, improved oral or local absorption, and reduced toxicity, and cause less local irritation.4
Paclitaxel (PTX) is a potential antitumor compound and a front-line chemotherapeutic used in the treatment of a variety of cancers.5 However, the current formulations have severe side effects related to low water solubility (0.4 μg/mL).6 To improve its water solubility, decrease its toxicity, increase its bioavailability, and achieve a better therapeutic effect, nanobased delivery systems have been developed for PTX, using liposomes,7 micelles,8 polymers,9 inorganic nanoparticles,10 human serum albumin,11 and polyethylene glycol–polylactic acid.12 Further, an albumin-bound PTX nanoparticle formulation (Abraxane®) has been approved by the US Food and Drug Administration to treat metastatic breast cancer and non-small-cell lung cancer.13 However, the free drug-loading rates for these prodrug formulations are relatively low. For example, the PTX loading rate for the human serum albumin and polyethylene glycol–polylactic acid formulations is about 10.0 wt% and 16.7 wt%, respectively.14 In clinical practice, some patients also experience significant undesirable side effects because of the emulsifier. Whether Abraxane® could improve survival and overcome P-glycoprotein-mediated drug resistance is still unclear.15 While we have previously developed protein-carrier prodrug formulations to enhance the solubility and targeting ability of PTX, some problems have remained unresolved, such as the low PTX content and high immunotoxicity.16 Therefore, there is still a pressing need to develop alternative PTX formulations. Esterification with amino acids has been shown to be a useful approach for increasing aqueous solubility and bioavailability.17 In this work, PTX was derivatized at its 2′-hydroxy function by esterification with amino acids.
Folic acid (FA) has been identified as a ligand for selective delivery of therapeutic agents to cancer cells.18 Uptake of FA into cells is mediated by the FA receptor.19 The human folate receptor alpha (FR-α) has high affinity for the FA ligand, and is currently considered an essential component in the cellular accumulation of FA used in chemotherapy.20 FR-α expression is very low in most normal cells and tissues or not detectable, but is upregulated and detectable in ovarian, breast, brain, lung, and colorectal cancers.21 In our previous work, we investigated an FA-modified drug delivery system and found that covalently attached FA can increase the ability to target FR-positive tumors.22
Biomedical optics is a rapidly expanding imaging field with direct applications in cellular biology, pharmacology, and diagnosis of disease.23 The small molecule fluorescein is widely used as a fluorescent dye in studies designed to elucidate the structures and mechanisms of drug delivery systems.24 In this work, the water-soluble fluorescent dye, fluorescein-5(6)-isothiocyanate (FITC) and 5-aminofluorescein (5AF) were used to label PTX and detect cellular uptake of the prodrug in vitro, along with its pharmacokinetics and biodistribution. Near-infrared fluorescence imaging has many advantages as a non-invasive technique for real-time in vivo monitoring of biological information in living subjects without use of ionizing radiation.25 Our research group has successfully developed a hydrophilic ICG derivative (ICG-Der-02) to trace the dynamic behavior of prodrugs in nude mice, and its tumor targeting ability has been demonstrated.16,22
In this study, PTX was derivatized at its 2′-hydroxy function using Fmoc-Arg(Pbf)-OH and Fmoc-Glu(OtBu)-OH as linkers. The carboxyl groups of the linkers were covalently attached to FA by formation of amide bonds to produce FA-Arg-PTX and FA-Glu-PTX. Small molecule fluorescent dye, FITC/5AF, was then used to label FA-FITC-Arg-PTX and FA-5AF-Glu-PXT prodrugs by amide bond. The release properties of the parent drug PTX from the ester bond of the PTX linker were investigated. The toxicity of the prodrug was studied by in vitro and acute toxicity (LD50) testing. Dynamic behavior and tumor targeting ability were investigated by in vitro and in vivo optical imaging. Finally, the therapeutic efficacy of the synthesized prodrug was evaluated by in vitro and in vivo studies.
Materials and methods
Materials
PTX (molecular weight 853.9) was purchased from JiangSu Research Institute (Wuxi, People’s Republic of China). FA (molecular weight 441.4), dicyclohexylcarbodiimide, N-hydroxysuccinimide (NHS), Fmoc-L-arginine acid(Pbf)-OH (Fmoc-Arg[Pbf]-OH, molecular weight 648.77), Fmoc-L-glutamic acid 5-tert-butyl ester (Fmoc-Glu[OtBu]-OH, molecular weight 425.49), FITC (molecular weight 389.38), and 5AF (molecular weight 347.32) were purchased from commercial sources. ICG-Der-02 (molecular weight 995) was prepared in our laboratory. All other solvents and reagents used in this study were certified analytical reagent grade and from Sigma Chemicals (St Louis, MO, USA).
Synthesis of multi-small molecule-conjugated PTX targeted complex
Synthesis of FA-Arg(Pbf)-PTX and FA-Glu(OtBu)-PTX
FA-Arg(Pbf)-PTX and FA-Glu(OtBu)-PTX were synthesized in four steps, as shown in Figure 1Aa–Ad and Ba–Bd, respectively. Briefly, PTX (100 mg, 0.117 mmol, 1 equivalent) and Fmoc-Arg(Pbf)-OH (91.08 mg, 0.14 mmol, 1.2 equivalents) were dissolved in 10 mL of CH2Cl2, and 4-dimethylaminopyridine (14.27 mg, 0.117 mmol, 1 equivalent) was subsequently added (Figure 1Aa). Cold EDC (44.85 mg, 0.234 mmol, 2 equivalent, 5mL of CH2Cl2) was added dropwise to the mixture over 20 minutes and stirred at room temperature for 22 hours. The reaction mixture was diluted further with 15 mL of CH2Cl2. The organic layer was washed with water, saturated aqueous NaHCO3, and dried over MgSO4. The residue obtained after evaporation (vacuum) of the organic solvent was purified by recrystallization from diethyl ether. The purified Fmoc-Arg(Pbf)-PTX was obtained as a white solid in 85% yield. This product was used in the next step without further characterization. Next, 44.14 mg of FA (0.1 mmol) in 5 mL of dimethyl sulfoxide was activated with dicyclohexylcarbodiimide and NHS (molar ratio of FA to dicyclohexylcarbodiimide to NHS, 1:1.5:1.5) at room temperature in the dark for 5 hours (Figure 1Ab). The residue was removed by filtration under reduced pressure, and the activated FA was extracted with anhydrous acetone. Fmoc-Arg(Pbf)-PTX (150.3 mg, 0.1 mmol) was dissolved in 10 mL of CH2Cl2, 2 mL of 2-pipecoline was added to the solvent for deprotection, and the mixture was stirred at room temperature for 10 hours (Figure 1Ac). The residue obtained after evaporation (vacuum) of the organic solvent was purified by recrystallization from diethyl ether. The purified NH2-Arg(Pbf)-PTX was obtained as a white solid in 95% yield. The molecular weight of the purified NH2-Arg(Pbf)-PTX was determined by liquid chromatography mass spectrometry (LC-MS), NH2-Arg[Pbf]-PTX: MS (electrospray ionization [ESI], m/z): 1,279.43 ([M + H]+), NH2-Glu(tBut)-PTX: MS (ESI, m/z): 1,056.15 ([M + H]+). NH2-Arg(Pbf)-PTX (127.94 mg, 0.1 mmol) was dissolved in dimethyl sulfoxide, and the activated FA-NHS was added to the mixture, which was then stirred at room temperature in the dark for 24 hours (Figure 1Ad). The reaction mixture was concentrated in a vacuum and purified on a silica gel column. The purified FA-Arg(Pbf)-PTX was obtained as a yellow solid in 85% yield. A procedure similar to that described above was also applied to synthesize the control FA-Glu(OtBu)-PTX (Figure 1Ba–Bd). These two products were used in the next step without further characterization.
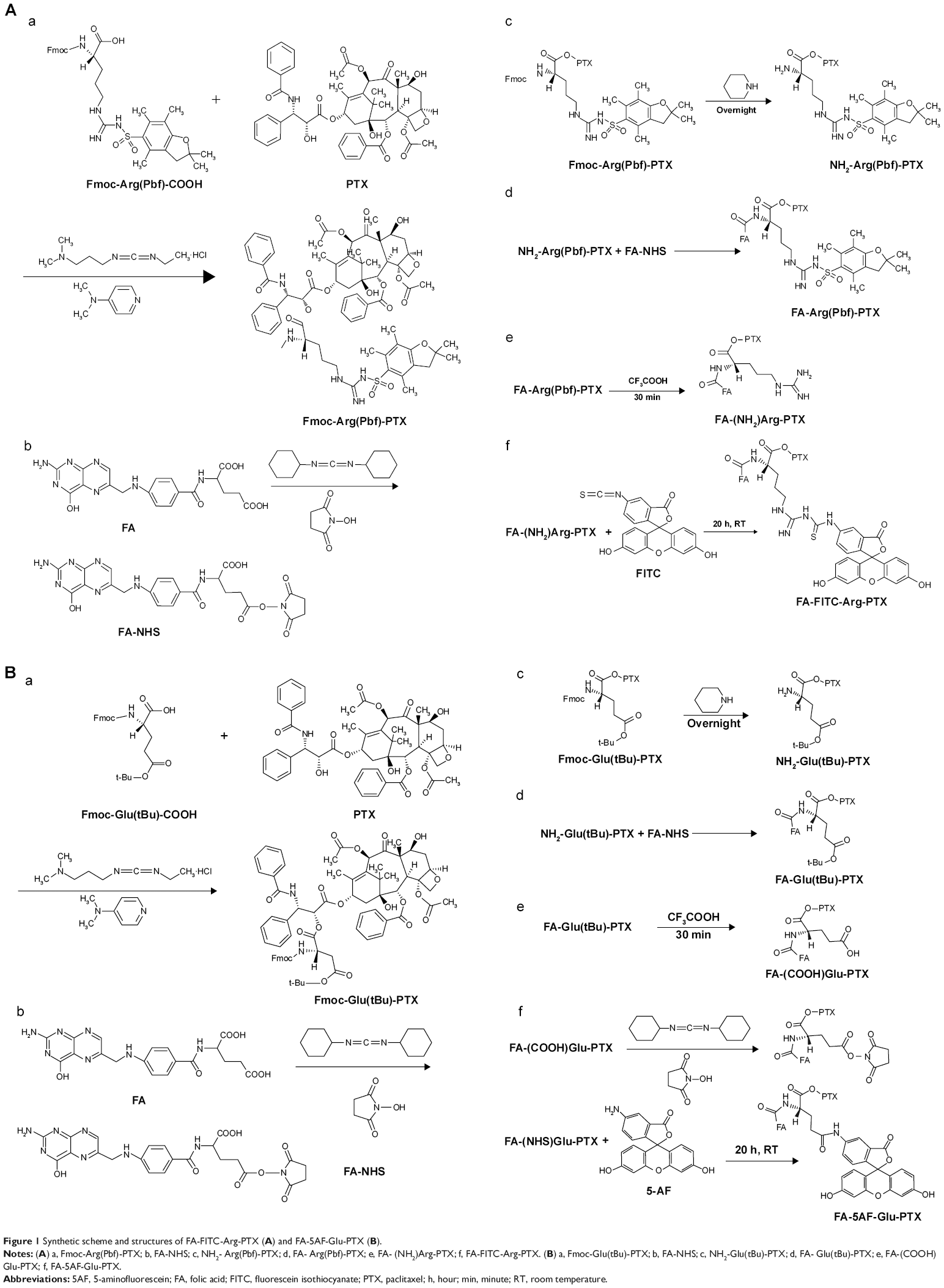 | Figure 1 Synthetic scheme and structures of FA-FITC-Arg-PTX (A) and FA-5AF-Glu-PTX (B). |
Synthesis of FA-FITC-Arg-PTX and FA-5AF-Glu-PTX targeted prodrugs
FA-FITC-Arg-PTX and FA-5AF-Glu-PTX were synthesized in two steps (Figure 1Ae–Af and Be–Bf, respectively). As shown in Figure 1Ae, FA-Arg(Pbf)-PTX (172.1 mg, 0.1 mmol, 1 equivalent) was dissolved in 4 mL of dimethyl sulfoxide, and 3 mL of trifluoroacetic acid was subsequently added. The mixture was stirred at room temperature for 30 minutes. The FA-(NH2)Arg-PTX obtained after evaporation (vacuum) of the organic solvent was purified by recrystallization from diethyl ether. The purified FA-(NH2)Arg-PTX was obtained as a yellow solid in 80% yield. The molecular weight of the purified FA-(NH2)Arg-PTX was determined by LC-MS. FA-(NH2)Arg-PTX: MS (ESI, m/z): 1,468.5 ([M + H]+. Finally, the activated 38.93 mg FITC was added in 5 mL of dimethyl sulfoxide buffer containing FA-(NH2)Arg-PTX (146.85 mg, 0.1 mmol, molar ratio 1:1), as shown in Figure 1Af. After 12 hours, the reaction mixture was purified on a silica gel column. The purified FA-FITC-Arg-PTX was obtained as a brown solid in 65% yield. The molecular weight of the purified FA-FITC-Arg-PTX was determined by LC-MS. FA-FITC-Arg-PTX: MS (ESI, m/z): 1,857.88 [M + H]+. In order to investigate the dynamics of this small molecule-conjugated PTX targeted delivery system, the near-infrared fluorescent dye ICG-Der-02(COOH) was attached to FA-(NH2)Arg-PTX and NH2 -Arg(Pbf)-PTX by a method similar to that described previously. The purity of the FA-ICG02-Arg-PTX and ICG02-(NH2)Arg-PTX was determined by thin layer chromatography.
Next, 3 mL of triethylamine was added to 5 mL of FA-Glu(OtBu)-PTX (in Figure 1Be), and the mixture was stirred at room temperature for 30 minutes. FA-Glu(OtBu)-PTX was removed from the protective-OtBu group to form FA-(COOH)Glu-PTX. The reaction mixture was purified by recrystallization from diethyl ether. The purified FA-(COOH)Glu-PTX was obtained as a yellow solid in 78% yield. The molecular weight of the purified FA-(COOH)Glu-PTX was determined by LC-MS. FA-(COOH)Glu-PTX: MS (ESI, m/z): 1,447.4 ([M + H]+). The activated FA-(NHS)Glu-PTX was dissolved in 5 mL of dimethyl sulfoxide containing 34.73 mg 5AF (molar ratio is 1:1, Figure 1B–f). After 16 hours, the reaction mixture was purified using a silica gel column to remove the unconjugated FA-(NHS)Glu-PTX and 5AF fragments. The purified FA-5AF-Glu-PTX was obtained as a light brown solid in 60% yield. The molecular weight of the purified FA-5AF-Glu-PTX was also determined by LC-MS.
Characterization of FA-FITC-Arg-PTX and FA-5AF-Glu-PTX
To confirm the components of the above synthesized compounds, FA-FITC-Arg-PTX, FA-5AF-Glu-PTX, ICG02-(NH2)Arg-PTX, and FA-ICG02-Arg-PTX were evaluated by spectroscopic measurement at 365 nm, 220 nm, 227 nm, 347 nm, 492 nm, and 780 nm, which correspond to the absorption peaks of FA, Arg, PTX, FITC, 5AF, and ICG-Der-02, respectively. Successful conjugation of PTX to the small molecules was further confirmed by LC-MS and high-performance liquid chromatography (HPLC, Shimadzu).
Physical and chemical properties of FA-FITC-Arg-PTX and FA-5AF-Glu-PTX
The physical and chemical properties of the PTX prodrug formulations, including their drug loading (ie, amount of PTX), aqueous solubility, and stability, were then determined. The amount of conjugated PTX in FA-FITC-Arg-PTX or FA-5AF-Glu-PTX was evaluated using theoretical methods and a standard curve of HPLC peak area versus PTX content (anhydrous ethyl alcohol). The aqueous solubility of the conjugated PTX was determined using a method reported elsewhere.26 At room temperature (25°C), 100 mg of conjugated PTX was placed in a 10 mL volumetric flask to which 500 μL of water was added by injector. After saturation was reached, the aqueous solubility of the two prodrugs was measured. To investigate the stability of the ester bond in the FA-FITC-Arg-PTX and FA-5AF-Glu-PTX compounds, the free PTX released from the derivatives was measured in different types of medium, including phosphate-buffered saline (PBS, pH 7.4) and human plasma at 37°C for up to 12 hours. The PTX-conjugated products (5 mg each of FA-FITC-Arg-PTX and FA-5AF-Glu-PTX) were dissolved in 2.5 mL of each type of medium. Next, 100 μL of each sample was taken at a designated time point. The free PTX was then extracted using 1.5 mL of ethyl acetate and measured by reverse-phase HPLC on a C18 column (300 mm ×3.9 mm; Nova-Pak, Waters) with an acetonitrile/water gradient (mobile phase consisting of methanol to water (45/45), 1 mL per minute at 30°C, PTX: 227 nm).
In vitro studies
Cell culture
Human breast cancer (MDA-MB-231, MCF-7), human lung cancer (A549), and human renal epithelial (HEK293) cell lines were purchased from the American Type Culture Collection and cultured at 37°C in a humidified atmosphere containing 5% CO2 in Dulbecco’s Modified Eagle’s Medium and Roswell Park Memorial Institute 1640 medium supplemented with 10% fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin. The use of these cell lines were approved by the ethics committee of Suzhou University, Suzhou, People’s Republic of China.
Cellular uptake of multi-small molecule-conjugated PTX targeted prodrugs
The above four cell lines (MDA-MB-231, MCF-7, A549, and HEK293) were used to evaluate FA-α receptor protein expression by Western blot analysis, and were prepared as described previously.20 Tumor cells and normal mammary cells were washed twice at room temperature in PBS and harvested by scraping in cold lysis buffer containing PBS, 1% NP40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, 0.1 mg/mL phenylmethyl sulfonyl fluoride, 2 μg/mL aprotinin, and 1 mmol/L sodium orthovanadate. After 20 minutes in an ice bath, the lysates were passed through a 21-gauge needle, incubated on ice for another 30 minutes, and then centrifuged at 15,000× g for 20 minutes at 4°C. Protein concentrations were determined using the Pierce Micro bicinchoninic acid protein assay reagent, and the proteins were stored at −80°C.
The multi-small molecule-conjugated PTX-targeted prodrugs were labeled with visible fluorescent dye (FITC and 5AF) for fluorescence microscopy. The three types of cells were seeded in 6-well plates and incubated in culture medium overnight, followed by incubation in a 200 μL solution of FA-FITC-Arg-PTX or FA-5AF-Glu-PTX (1 mg/mL) for 8 hours. After washing with PBS, the cells were directly visualized by fluorescence microscopy (40× objective magnification).
Cell cytometry was used to further quantify the uptake of FA-FITC-Arg-PTX and FA-5AF-Glu-PTX in the cell lines. Briefly, 5×105 cells were seeded in 6-well plates and incubated in culture medium overnight. Next, 200 μL of 1 mg/mL FA-FITC-Arg-PTX and FA-5AF-Glu-PTX were added into each well. After 8 hours of incubation at 37°C, the cells were resuspended in 500 μL of PBS. The cell suspension was immediately analyzed by flow cytometry.
Cell viability assay
To evaluate the antitumor activity and cytotoxicity of FA-FITC-Arg-PTX and FA-5AF-Glu-PTX, MTT assays were conducted on the MDA-MB-231, MCF-7, A549, and HEK293 cell lines following a standard protocol.18 The cells were plated at a density of 5×103 cells/well in 96-well plates and then incubated for 48 hours with PTX, FA-FITC-Arg-PTX (theoretically conjugated PTX 45.93%), and FA-5AF-Glu-PTX (theoretically conjugated PTX 47.71%) at the same concentrations in the range of 0.3125–1 μg/mL. The concentration of conjugated PTX was 0.01435–0.4771 μg/mL. Each drug was tested in six wells. MTT solution (20 mL, 5 mg/mL) was then added into each well. The absorbance of the solution in each well was measured at 490 nm using a multi-well plate reader.
Apoptosis study
To further investigate the efficacy of the prodrugs in the three tumor cell lines, cell apoptosis was qualitatively and quantitatively studied. Briefly, fluorescence microscopy was used to identify the various morphological changes during apoptosis. Propidium iodide (PI), which binds to DNA once the cell membrane had become permeable, was used to identify cell apoptosis. We also used flow cytometry to quantify apoptosis.20 After 24 hours of incubation with free PTX, FA-FITC-Arg-PTX, or FA-5AF-Glu-PTX (200 μL, 1 mg/mL), the cells were washed three times with PBS (pH 7.0) before addition of 500 μL of binding buffer and 5 μL of PI to the mixture. The cells were directly visualized by fluorescence microscopy. To quantify apoptosis after 24 hours of incubation, the medium was discarded and the cells were resuspended in 500 μL of binding buffer, and 5 μL of PI were added to the mixture. The cell suspension was immediately analyzed by flow cytometry.
In vivo studies
Animal subjects and tumor model
The study protocol was approved by the Animal Ethical and Welfare Committee of Suzhou University, Suzhou, People’s Republic of China. All animal experiments were carried out in compliance with the animal management regulations of the Ministry of Health, People’s Republic of China (document 55, 2001) and the guidelines for the care and use of laboratory animals at Suzhou University.
MDA-MB-231, MCF-7, and A549 tumor cells with relatively high FR-α expression were implanted in the upper right axillary fossa in nude mice (each group, n=5). When the tumors had grown to a diameter of 0.2–0.5 cm, the mice were investigated for drug targeting ability and therapeutic effect.
Acute toxicity (LD50) test in mice
The acute toxicity of PTX, FA-FITC-Arg-PTX, and FA-5AF-Glu-PTX was investigated in normal (Kunming) mice purchased from Charles River Laboratories.27 Briefly, 150 ICR mice (aged 3–4 weeks, weight 18–22 g) were divided into five groups of ten mice each to receive PTX (15, 20, 25, 30, 45 mg/kg), FA-FITC-Arg-PTX, or FA-5AF-Glu-PTX (80, 160, 200, 280, 320 mg/kg). Each group of mice contained equal numbers of males and females. The above prodrugs (in 0.2 mL) were injected via the tail vein in each mouse. The mice were injected with 50% of the lethal dose determined in the LD50 studies. After 14 days, all mice were euthanized by CO2 asphyxiation followed by cervical dislocation to monitor for tissue changes.
Dynamics and targeting ability of PTX based on multi-small molecule carriers in tumor-bearing mice
Nude mice bearing MDA-MB-231 tumors were used to investigate the dynamics and targeting ability of the prodrug formulations (n=5 in each group). ICG02-(NH2)Arg-PTX (0.2 mL, 5 mg/kg) and FA-ICG02-Arg-PTX (0.2 mL, 5 mg/kg) were administered to mice via tail vein injection. Fluorescence imaging was acquired using a near-infrared imaging method as reported previously.16 Tumor-to-normal (T/N) tissue contrast ratios were calculated by using the region of interest function of Living Image® software.22
In addition, the successfully conjugated PTX prodrug including the fluorescent dye (FITC, 5AF), FA-FITC-Arg-PTX (0.2 mL, 5 mg/kg), and FA-5AF-Glu-PTX (0.2 mL, 5 mg/kg) were injected into the mice bearing MDA-MB-231 tumors (n=5) via the tail vein. The mice were euthanized post-injection at a designated time point, and tissue was excised for sectioning and direct visualization by fluorescence microscopy.
Antitumor activity
Nude mice bearing MCF-7 and A549 tumors were randomly assigned to four groups (n=5 per group) and treated every 3 days by tail vein injection of PBS (pH 7.4, control), pure PTX solution (5 mg/kg), FA-FITC-Arg-PTX (5 mg/kg), or FA-5AF-Glu-PTX (5 mg/kg). Five tail vein injections were given every 3 days, with each dose in 0.2 mL of solution. The therapeutic efficacy and systemic toxicity of the two prodrug formulations was assessed by daily recording of tumor volume and body weight.
Statistical analysis
All data are reported as the mean ± standard deviation of n independent measurements. The statistical analysis was performed using the Student’s t-test, with statistical significance assigned at a P-value of <0.05.
Results
Characterization of targeted multi-small molecule-conjugated PTX prodrugs
FA-FITC-Arg-PTX and FA-5AF-Glu-PTX were synthesized according to the procedures described in Figure 1. The absorption and fluorescence spectra for FA-FITC-Arg-PTX and FA-5AF-Glu-PTX showed all absorption peaks of the constituent components (FA, Arg/Glu, PTX, FITC/5AF, and ICG02 at 365 nm, 220 nm, 227 nm, 375/465 nm, and 780 nm, respectively) in the complex, indicating successful conjugation of the two prodrug formulations, which was further confirmed by LC-MS and HPLC, as shown in Figures S1 and S2. FA-FITC-Arg-PTX: MS (ESI, m/z): 1858.88 [M + H]+, FA-5AF-Glu-PTX: MS (ESI, m/z): 1789.75 [M + H]+ (thin layer chromatography, Figure S2). In addition, the fluorescence of ICG-Der-02 was observed from the band of purified FA-ICG02-Arg-PTX and ICG02-(NH2)Arg-PTX using a near-infrared imaging system, which further demonstrated that ICG-Der-02 was attached to FA-(NH2)Arg-PTX or -(NH2)-Arg(Pbf)-PTX (Figure S2D).
Physicochemical properties of FA-FITC-Arg-PTX and FA-5AF-Glu-PTX
PTX loading in the prodrug formulations was evaluated by the standard curve for HPLC peak area versus PTX content. The regression equation for the standard PTX curve was Y (OD) =65535.65035+17388.99822× (R2=0.99922). The linear range of the standard curve was 0.85–95.18 μg/mL. PTX loading in the two prodrug formulations was 38.70%±0.49% and 32.19%±0.35% (n=5), respectively. To quantify the aqueous solubility of the multi-small molecule-conjugated PTX prodrug formulations, the direct observations were followed in the solubility calculations of the PTX prodrug formulations, FA-FITC-Arg-PTX (6.45±0.15 mg/mL, n=4) and FA-5AF-Glu-PTX (6.41±0.18 mg/mL, n=4) as shown in Figure S3. To investigate the cleavage of the linkers (Arg/Glu) in the products we measured the amount of free PTX released from the compounds after incubation of the products in PBS (pH 7.4) and human plasma, respectively, at 37°C. The free PTX was measured by HPLC, as shown in Figure S4A and B. The release profiles are given in Figure 2E. The FA-FITC-Arg-PTX/FA-5AF-Glu-PTX formulation released PTX more rapidly in human plasma (56.72%/54.84%, 4 hours) and released less PTX in PBS (25.8%/23.3%, 4 hours).
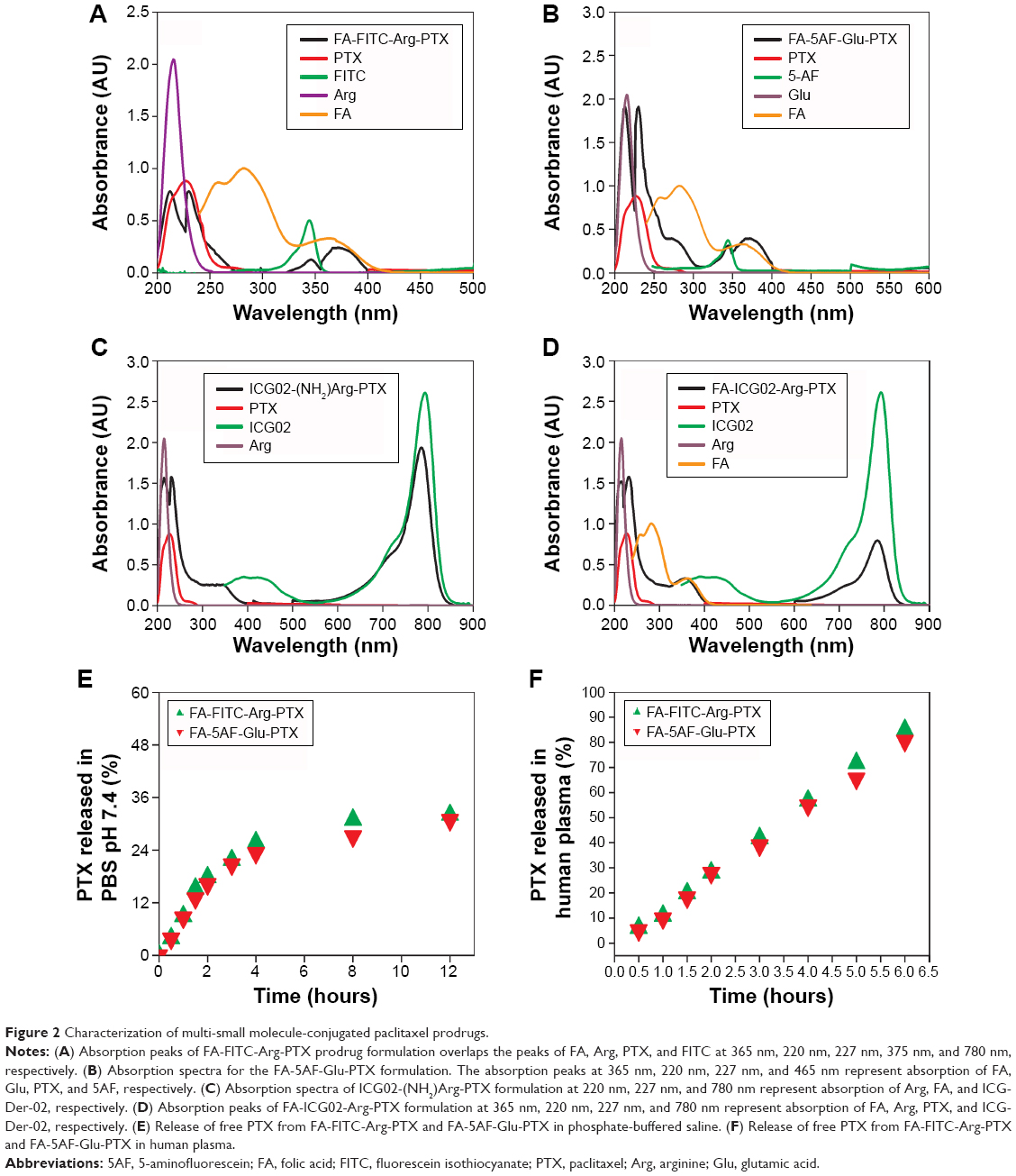 | Figure 2 Characterization of multi-small molecule-conjugated paclitaxel prodrugs. |
In vitro cell targeting
To explain the receptor-mediated targeting of multi-small molecule-conjugated PTX vectors to HEK293, MDA-MB-231, MCF-7, and A549 cells, expression levels of FR-α protein were investigated by Western blot, and were found to be in the following order: MDA-MB-231 > MCF-7 > A549 > HEK293, as shown in Figure 3A.
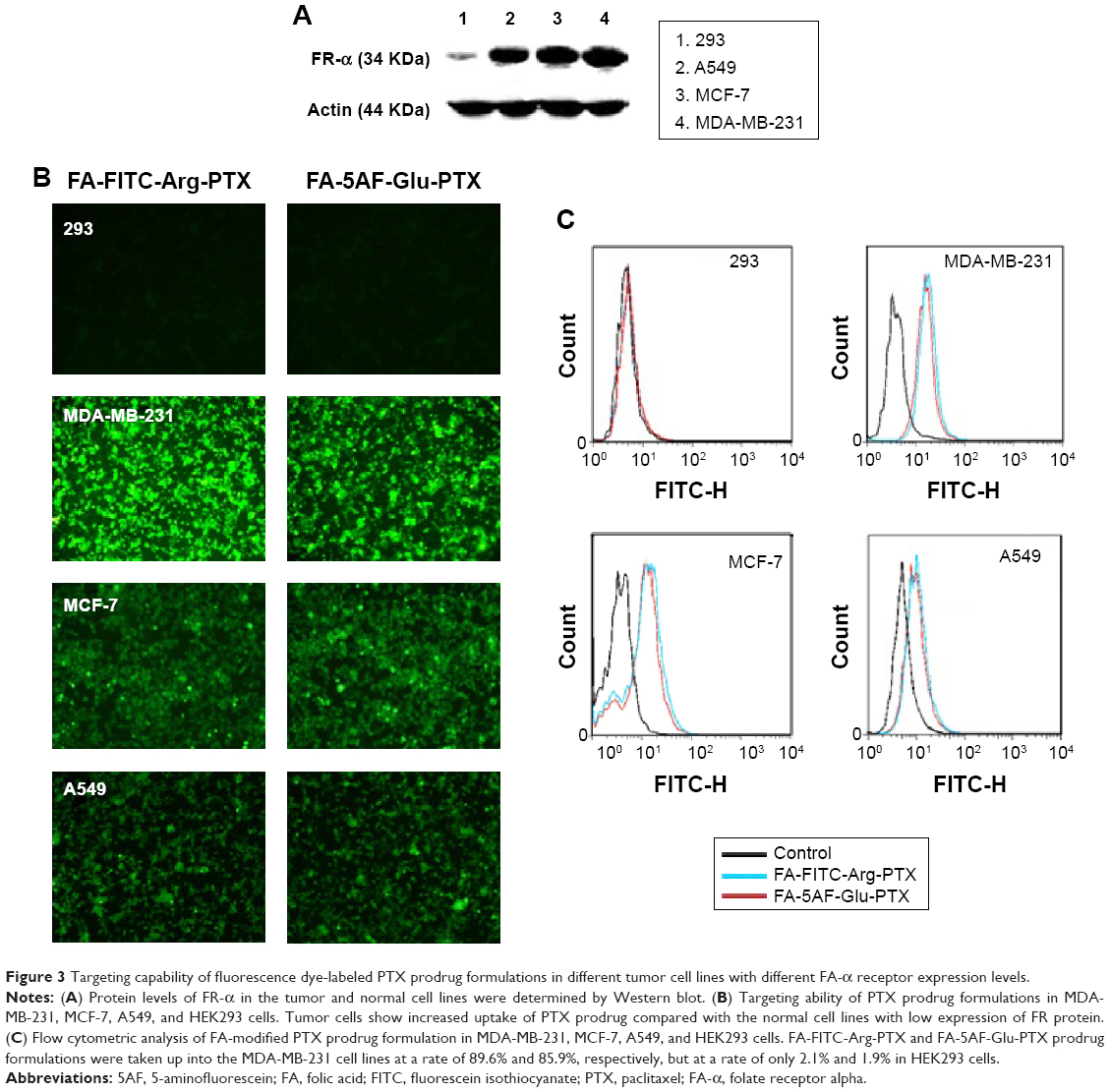 | Figure 3 Targeting capability of fluorescence dye-labeled PTX prodrug formulations in different tumor cell lines with different FA-α receptor expression levels. |
Uptake of these compounds in the cell lines was imaged by fluorescence microscopy and the results are shown in Figure 3B. The two tested PTX prodrug formulations showed increased uptake by tumor cells overexpressing FR-α (ie, MDA-MB-231, MCF-7, and A549) when compared with normal HEK293 cells. Flow cytometry indicated that the FA-FITC-Arg-PTX and FA-5AF-Glu-PTX formulations were taken up into MDA-MB-231 cells at a rate of 89.6% and 85.9%, respectively, and were retained at rate of 78.1% and 75.1% in MCF-7 cells and 63.2% and 59.0% in A549 cells; however, they were taken up by HEK293 cells at a rate of 2.1% and 1.9% (Figure 3C). These findings suggest that the FA modification enhanced the targeting ability of the two PTX prodrug formulations to tumors overexpressing FR-α, and their uptake in the three tumor cell lines was consistent with FR-α protein levels.
In vitro antitumor activity of FA-FITC-Arg-PTX and FA-5AF-Glu-PTX
The cell viability ratio assay was used to qualitatively determine in vitro the antitumor activity and cytotoxicity of FA-FITC-Arg-PTX and FA-5AF-Glu-PTX in MDA-MB-231, MCF-7, and A549 cancer cells. and in normal HEK293 cells. The results are shown in Figure 4A. All the drug formulations showed dose-dependent antitumor activity at the different PTX concentrations. In the three tumor cell lines, the inhibition ratios of FA-FITC-Arg-PTX and FA-5AF-Glu-PTX were over 78.1% of total cells, whereas PTX inhibited a maximum of 62.4% of total cells. Importantly, the concentration of free PTX at each point was 2-fold when compared with the corresponding conjugated PTX formulation. A high concentration of free PTX still showed a lower tumor inhibition rate when compared with the conjugated PTX. In addition, the inhibition rates achieved by FA-FITC-Arg-PTX and FA-5AF-Glu-PTX were not significantly different in any of the cell lines tested. All drugs displayed an increased inhibition ratio for MDA-MB-231 tumor cells with relatively high-level expression of FR-α when compared with MCF-7 and A549. Moreover, the two prodrug formulations showed a very high viability rate in normal HEK293 cells, even at the highest concentration (0.459 μg/mL). These findings suggest that multi-small molecule-conjugated PTX delivery systems have low cytotoxicity to normal cells and enhance the antitumor ability of free PTX in tumors overexpressing FR-α.
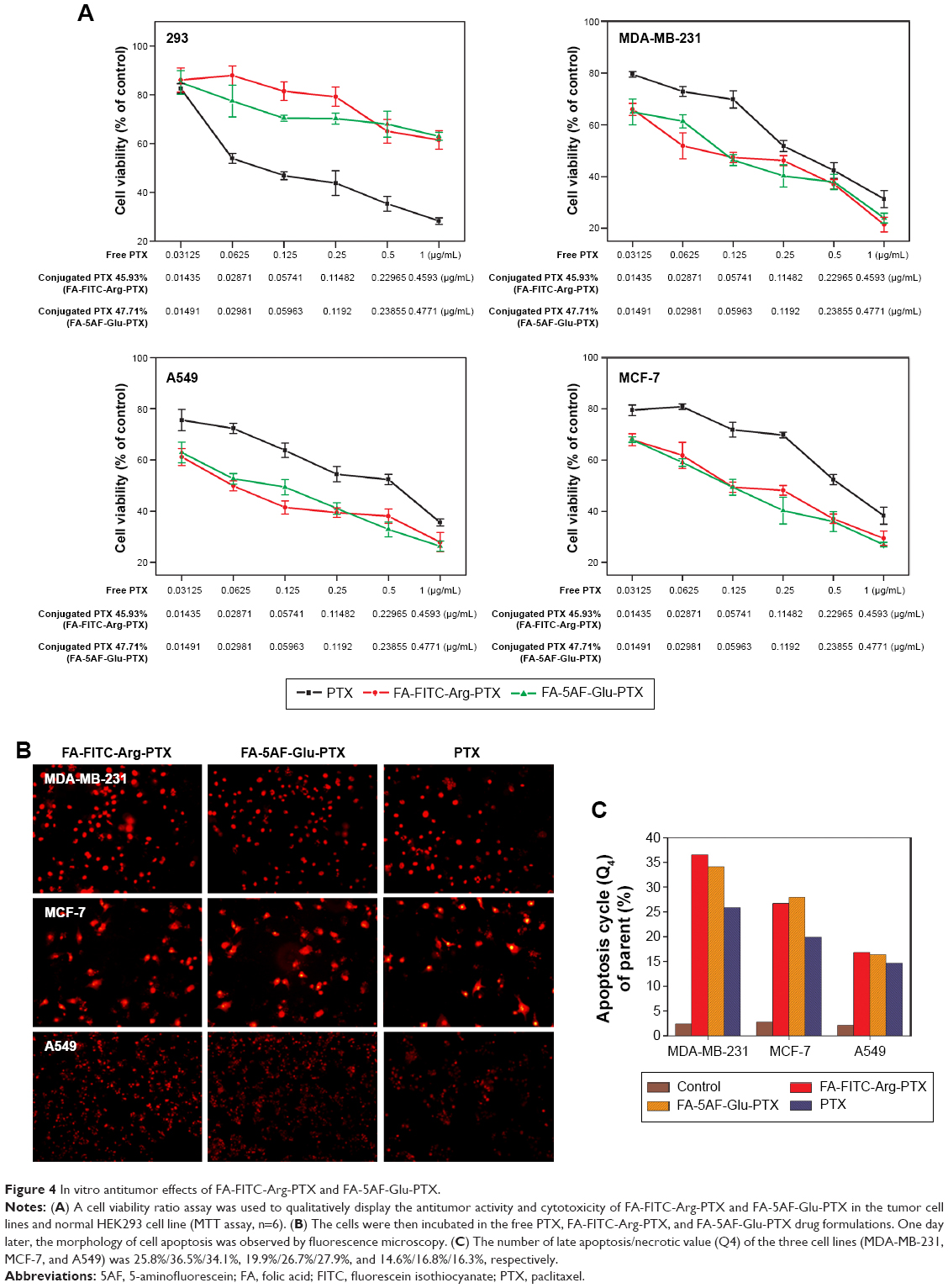 | Figure 4 In vitro antitumor effects of FA-FITC-Arg-PTX and FA-5AF-Glu-PTX. |
By staining cells with PI, it is possible to distinguish and analyze nonapoptotic cells, late apoptotic cells (PI-positive, red), and necrotic cells (PI-positive, red).22 The cells were also examined under fluorescence microscopy. The results of a representative experiment are shown in Figure 4B. The necrotic cells (PI-positive) were clearly displayed in three tumor cell lines with free PTX, FA-FITC-Arg-PTX, and FA-5AF-Glu-PTX. Flow cytometry quantitative analyses indicated that the number of late apoptosis/necrotic value (Q4) of the three cell lines (MDA-MB-231, MCF-7, and A549) was 25.8/36.5%/34.1%, 19.9%/26.7%/27.9%, and 14.6%/16.8%/16.3%, respectively (Figure 4C). The above qualitative and quantitative analyses demonstrate that the PTX-conjugated multi-small molecule greatly enhanced the antitumor activity of low-dose PTX.
Acute toxicity
Surviving animals in the PTX group are indicated with a vertical line on the survival graph (Figure 5). The survival rate of animal in the different dose PTX group (from 15 mg/kg to 45 mg/kg) has decreased from 90% to 0%. Regardless of the dose of FA-FITC-Arg-PTX or FA-5AF-Glu-PTX (from 80 to 320 mg/kg), all groups of animals maintained their original survival rates. No histological difference was observed between the FA-FITC-Arg-PTX and FA-5AF-Glu-PTX group post-injection (data not shown). All results demonstrated that the cytotoxicity of the doses of prodrugs was low to mice.
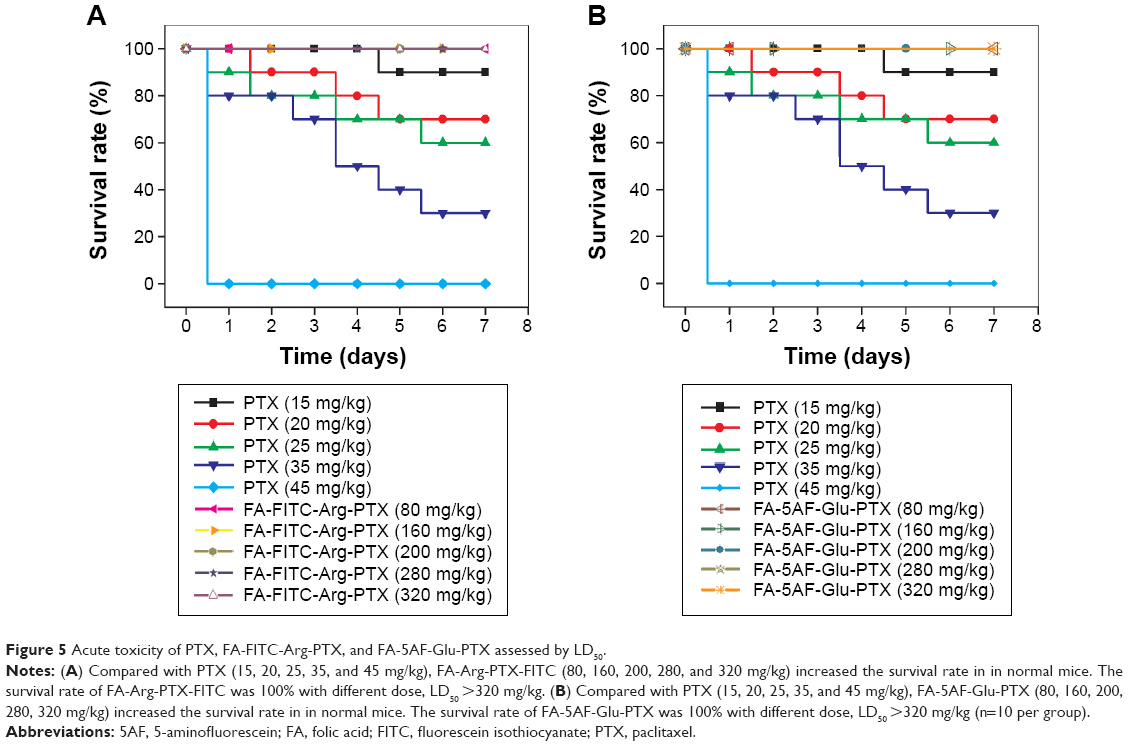 | Figure 5 Acute toxicity of PTX, FA-FITC-Arg-PTX, and FA-5AF-Glu-PTX assessed by LD50. |
Dynamics and targeting ability of multi-small molecule-loaded PTX in tumor-bearing mice
MDA-MB-231 cells with relative overexpression of FR-α were used to investigate the dynamics and tumor targeting ability of multi-small molecule-loaded PTX. Representative near-infrared images after administration of ICG02-(NH2)Arg-PTX and FA-ICG02-Arg-PTX are shown in Figure 6A and B. The fluorescent ICG02-(NH2)Arg-PTX initially spread throughout the whole body at about 30 minutes post-injection and gradually appeared in the tumor, as well as the organs of excretion, including the liver, gastrointestinal tract, and bladder. The fluorescent drug in the tumor gradually washed out, with a decrease in signals from the liver, gastrointestinal tract, and bladder. At 24 hours post-injection, the fluorescent probe has mostly cleared from the body (Figure 6A). The FA-ICG02-Arg-PTX initially distributed all over the body and was subsequently cleared via the hepatobiliary and renal pathways (Figure 6B). However, the tumor sites were identifiable 1 hour after injection of the probes. Over time, the drug increasingly accumulated in the tumors and the fluorescence intensity peaked at about 4 hours. The bright fluorescence signal in the tumor tissue gradually disappeared after 4 days (data not shown).
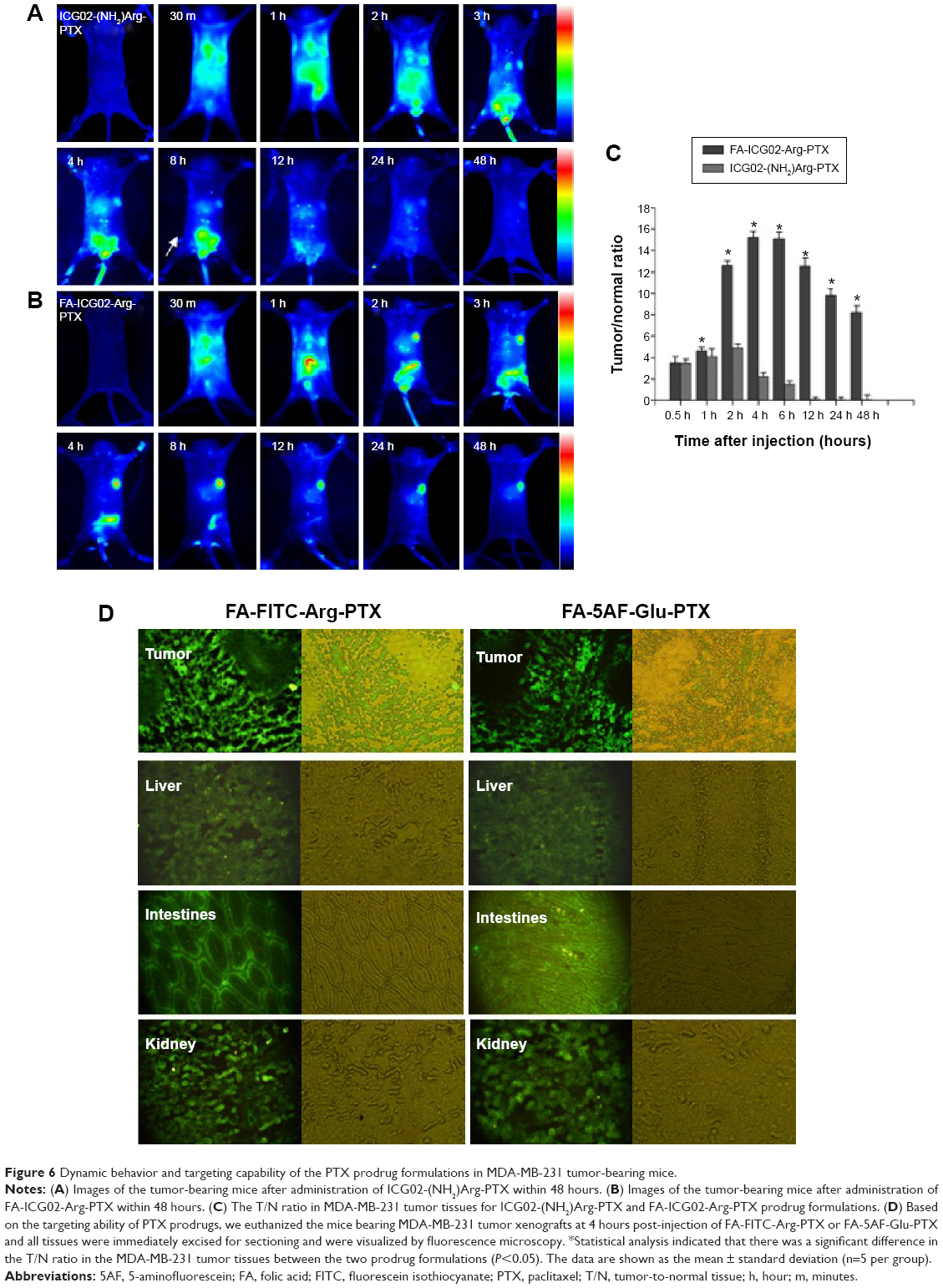 | Figure 6 Dynamic behavior and targeting capability of the PTX prodrug formulations in MDA-MB-231 tumor-bearing mice. |
Tumor contrast as quantified by region of interest analysis of fluorescence intensity shown in Figure 6C indicates that the T/N tissue ratio was 4.42±0.16 at 1 hour post-injection of FA-ICG02-Arg-PTX, reaching a peak at 4 hours post-injection (15.1±0.46) and slowly declined to 8.10±0.71 by 48 hours. In contrast, MDA-MB-231 tumor-bearing mice intravenously injected with the ICG02-(NH2)Arg-PTX formulation showed a T/N ratio of 3.24±0.33 after 0.5 hours; uptake reached a peak of 4.87±0.42 at 2 hours, and slowly declined to 2.18±0.40 by 4 hours. Statistical analysis indicated a significant difference in the T/N ratio in MDA-MB-231 tumors for the two PTX drug formulations (P<0.05). FA-ICG02-Arg-PTX could extend the drug residence time or drug concentration in tumor tissue.
On the basis of the above results, we euthanized the mice bearing MDA-MB-231 tumor xenografts at 4 hours after injection of FA-FITC-Arg-PTX or FA-5AF-Glu-PTX. All tissues were immediately excised for sectioning and were visualized by fluorescence microscopy. Representative images are shown in Figure 6D. Fluorescence images of tissue sections displayed the highest fluorescence signal in tumor tissues, and appeared in the organs of excretion, ie, the liver, gastrointestinal system, and bladder. The drug probably entered the liver and was subsequently transported to the gastrointestinal system. Another fraction of the drug was excreted by the kidney–bladder pathway. Similar results were seen in the in vivo and in vitro studies.
In vivo therapeutic experiment
The in vivo antitumor efficacy of PTX, FA-FITC-Arg-PTX, and FA-5AF-Glu-PTX was evaluated in MCF-7-tumor-bearing mice and A549 tumor-bearing mice by measuring tumor growth rate and body weight. Figure 7A indicates that treatment with PBS produced the fastest tumor growth, while multi-small molecule-conjugated PTX inhibited tumor growth most effectively. MCF-7 tumor growth was inhibited at a rate of 57.63% and 54.02% after administration of FA-FITC-Arg-PTX and FA-5AF-Glu-PTX, respectively, which is higher than that of free PTX (about 38.16%). The inhibition rate of the two prodrug (48.65%/44.52%)-treated A549 tumor was about 1.4/1.3 times higher than that of the free PTX-treated tumor (35.23%, Figure 7).
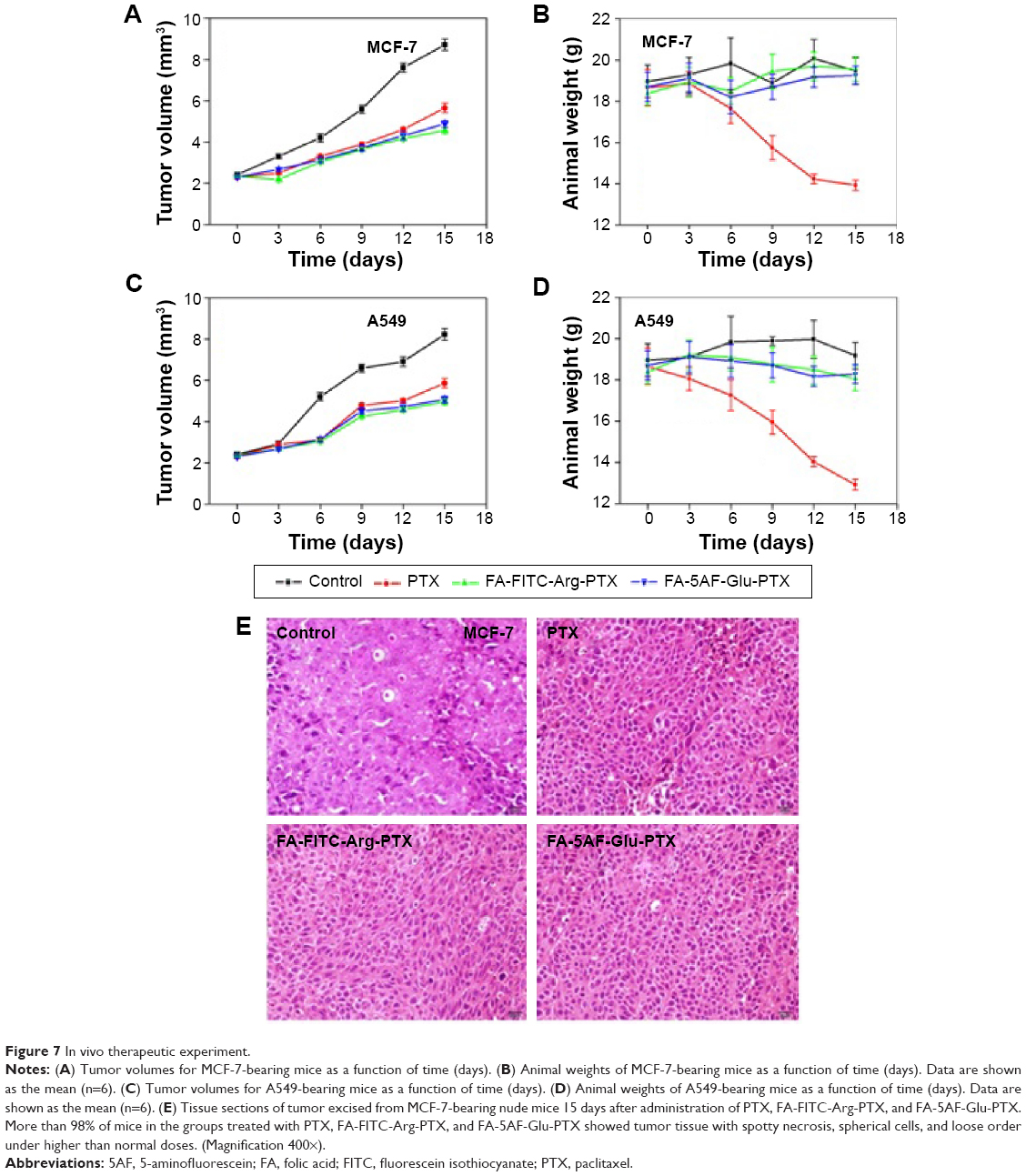 | Figure 7 In vivo therapeutic experiment. |
Body weight loss in the PTX-treated mice bearing MCF-7 tumors was 3.88 g, which was approximately 20% of their original body weight at the end of the treatment period (Figure 7B). A gradual increase in body weight in the PBS, FA-FITC-Arg-PTX and FA-5AF-Glu-PTX groups was observed during the treatment period. The decrease of body weight of the A549 group treated with PTX was 3.92 g, about 25.34% of the original weight. Slightly reduction in body weight also occurred in the two groups of mice treated with FA-FITC-Arg-PTX/FA-5AF-Glu-PTX (about 2%, Figure 7D). These results suggest that conjugation of PTX could significantly decrease the toxicity of the free drug.
To further evaluate the antitumor effect of PTX, FA-FITC-Arg-PTX, and FA-5AF-Glu-PTX in animals, the tumor tissues were excised for pathology. Figure 7E shows representative tissue sections from different mice groups. Tumor tissue in the PTX, FA-FITC-Arg-PTX, and FA-5AF-Glu-PTX groups exhibited spotty necrosis, spherical cells, and loose order. Finally, FA-FITC-Arg-PTX and FA-5AF-Glu-PTX groups possessed loose order in tumor tissue. However, no histological difference was observed in the different mice-bearing A549 tumor groups. All results confirmed that multi-small molecule-conjugation is the most effective tumor-inhibiting strategy when compared with the other groups.
Discussion
PTX is an antitumor compound, with high clinical efficacy. However, its low aqueous solubility remains a major problem, because it can lead to high toxicity and low bioavailability. The currently developed formulations of PTX indicate that these problems are still unresolved.28,29 In this study, we developed a new active vector mediating a multi-small molecule-conjugated PTX prodrug delivery system for targeted tumor therapy. Water-soluble small molecules, amino acids (Glu or Arg), FA, and fluorescent dye (FITC or 5AF), were successfully conjugated to PTX, and greatly enhanced the solubility of synthesized FA-Arg-PTX-FITC (6.45±0.15 mg/mL) and FA-Glu-PTX-5AF (6.61±0.18 mg/mL). The PTX loading rate of these two prodrugs reached 38.70%±0.49% and 32.19%±0.35%, respectively. The release rate for free PTX from the conjugation was an important factor influencing therapeutic efficacy. As shown in Figure 2E, more of the two prodrugs FA-FITC-Arg-PTX and FA-5AF-Glu-PTX was released in human plasma than in PBS, indicating that the faster release of the PTX might be related to an enzyme in plasma.17 Interestingly, high PTX release rates occurred when the product accumulated in tumor cells/tissues (by 4 hours). The slight release of free PTX from the conjugation in PBS supports storage of the prodrug at −20°C.
The toxicity of PTX greatly affects its clinical utility.30 In this study, we assessed the in vitro toxicity of the two prepared prodrugs in both tumor and normal cell lines (Figures 4A and 5). We found decreased cytotoxicity of PTX in both cell lines after multi-small molecule conjugation. In tumor cells, both FA-FITC-Arg-PTX and FA-5AF-Glu-PTX showed comparably strong toxicity (approximately 20% cell viability) at high doses, whereas multi-small molecule-conjugated PTX was much safer in normal cells. It can be easily understood that the cytotoxicity of PTX is higher in normal cells than in tumor cells given that proliferation rates are higher in tumor cells than in normal cells, as are FR-α protein expression levels, resulting in the different response to the lower dose treatment.31 Cell viability rates and FR-α receptor expression in MDA-MB-231, MCF-7, and A549 cells showed a statistically significant correlation between receptor protein expression and tumor cell targeting (Figure 4). The cytotoxicity of PTX and its conjugates was dose-dependent. In the in vivo acute toxicity study, the LD50 of free PTX was 25.48±0.17 mg/kg, whereas the intravenous LD50 of each prodrug was greater than 320 mg/kg. No obvious histological changes were found in either of the PTX prodrug formulations. The above results suggest that multi-small molecule-loaded PTX has lower toxicity than free PTX.
The targeting ability of drugs is also one of the important factors contributing to drug development and clinical application because higher targeting ability can increase therapeutic efficacy and reduce side effects. The cellular uptake ability of multi-small molecule-conjugated PTX was analyzed by qualitative and quantitative analysis (Figure 3). The percentage cell uptake in the tumor groups treated with FA-FITC-Arg-PTX (63.22%–89.63%) and FA-5AF-Glu-PTX (59.01%–85.93%) was much higher than that of normal cells (2.1% and 1.9%, respectively). Our in vitro data clearly showed that the FA-FITC-Arg-PTX/FA-5AF-Glu-PTX prodrugs mediated by FA enhanced cellular uptake and antitumor activity. For the in vivo study, the time-dependent clearance of ICG02-(NH2)Arg-PTX displayed greater uptake of ICG02-(NH2)Arg-PTX by the liver and intestines than by the tumor (Figure 6A). This result is consistent with the finding that PTX can pass through the enterohepatic circulation and undergo first pass metabolism in those organs.22 However, FA-ICG02-Arg-PTX rapidly accumulates in MDA-MB-231 tumors by receptor-mediated uptake, reaching a peak after 4 hours after administration and maintaining a bright signal for up to 2 days. A more significant amount of FA-ICG02-Arg-PTX than ICG02-(NH2)Arg-PTX was observed to accumulate in tumor tissues with positive FR-α receptor expression (Figure 6B). Similar results were also shown in tissue sections treated with FA-FITC-Arg-PTX/FA-Glu-PTX-5AF at 4 hours post-injection (Figure 6D). The tumor targeting ability of the synthesized FA-FITC-Arg-PTX and FA-5AF-Glu-PTX was significantly improved by receptor-mediated uptake on imaging with a fluorescence microscope. These results indicate that PTX-loaded multi-small molecules improved the targeting ability of PTX for FR-positive tumors. Above all, tumor-targeted PTX-conjugated multi-small molecule delivery systems could enhance drug retention time and drug concentrations in tumor tissue.
The in vitro antitumor efficiency of multi-small molecule-conjugated PTX was confirmed by analysis of both cell viability and induction of apoptosis (Figure 4). The cell viability study showed that FA-FITC-Arg-PTX/FA-5AF-Glu-PTX achieved a higher tumor cell inhibition rate when compared with free PTX. The percentage of late apoptotic (PI-positive) cells in the tumor groups was higher. The number of necrotic/dead cells (Q4) was 36.5% for FA-FITC-Arg-PTX and 34.1% for FA-5AF-Glu-PTX, compared with only 25.8% in MDA-MB-231 cells treated with treated free PTX. We also tested the therapeutic activity of these FR-α sensitive prodrugs by injecting the prodrugs and free PTX into tumor-bearing mice five times within 15 days at a dose of 5 mg/kg for the conjugated PTX and 5 mg/kg for the free PTX. Our data indicate that the released multi-small molecule-conjugated PTX was still active after the FR-mediated, which is consistent with the indication that esterification at C-2′ did not significantly influence the activity of PTXs. Tumor growth in the FR-α-mediated prodrug group was significantly inhibited when compared with free PTX. It is also notable that inhibition of tumor growth was closely correlated with the significant apoptosis seen in the tumor tissues (Figure 7). No significant changes were observed after prodrugs treatment with the FR-mediated small-preparation in terms of mouse body weight. The mice treated with free PTX showed a significantly smaller weight decrease (approximately 25.34%) and higher tumor growth volume (approximately 30%). In contrast, the multi-small molecule carriers improved the water solubility, pharmacokinetic profile, and biodistribution of PTX, resulting in less toxicity. Hematoxylin and eosin staining did not show histological signs of toxicity in the major organs from mice treated with the FR-α mediated multi-small molecule preparation. However, necrotic areas were clearly present in tumors treated with the FR-α-sensitive multi-small molecule preparations (Figure 7), in agreement with the antitumor effects observed in Figure 4. The high therapeutic index of the two prodrugs is most likely a result of collaborative functions, including the effect of the water solubility of the multi-small molecules (Arg/Glu, FA, FITC/5AF), the “stealth” characteristics of the amino acid ester, FA/FR-α-mediated intracellular drug delivery, and enhanced penetration/diffusion.
Conclusion
Taken together, the multi-small molecule carrier for PTX used in the current study can be potentially used to increase the therapeutic efficacy of PTX concomitantly with high targeting ability, a high therapeutic index, and low side effects. Furthermore, compared with previous macromolecular PTX formulations, multi-small molecular-conjugated PTX prodrugs also demonstrated a higher water solubility, loading rate, targeting ability, and antitumor activity, as well as lower toxicity. These results suggest that the use of this platform would accelerate the clinical development of this PTX formulation and could potentially provide the basis for a prospective targeting drug delivery system for tumor therapy.
Acknowledgments
The authors thank Wei Zhu and Xiuqin Yang for their expert assistance with the animal experiments, and are grateful for the grants received from the Natural Science Foundation Committee of China (NSFC81220108012, 661335007, 81171395, 81328012), the Natural Science Foundation Committee of Anhui Province (1308085MH136, KJ2014A249, KJ2013A242, KJ2013A243, 2013SQRL086ZD), and the Science Foundation of Suzhou University (2012jb04, 2014jb05, 2014jb06).
Author contributions
LS, YG conceived and designed the study. LS, GG, and WC developed the study methodology. LS, CW, SL, and LX were responsible for acquisition of the data, including acquisition of the study animals and providing facilities. LS and LZ analyzed and interpreted the data, and performed the statistical analysis. Writing, review, and revision of the paper was undertaken by LS and ML. The study was supervised by LS. All authors contributed toward data analysis, drafting and critically revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Llinas A, Glen RC, Goodman JM. Solubility challenge: can you predict solubilities of thirty-two molecules using a database of one hundred reliable measurements. J Chem Inf Model. 2008;48:1289–12303. | ||
Wenlock MC, Austin RP, Barton P, et al. Prodrug approaches for enhancing the bioavailability of drugs with low solubility. J Med Chem. 2003;46:1250–1256. | ||
Williams HD, Trevaskis NL, Charman SA, et al. Strategies to address low drug solubility in discovery and development. Pharmacol Rev. 2013;65:315–499. | ||
Kim EJ, Bhuniya S, Lee H, et al. An activatable prodrug for the treatment of metastatic tumors. J Am Chem Soc. 2014;136:1388–1394. | ||
Singla AK, Garg A, Aggarwal D. Paclitaxel and its formulations. Int J Pharm. 2002;235:179–192. | ||
Dorr RT. Pharmacology and toxicology of Cremophor EL diluent. Ann Pharmacother. 1994;28:S11–S14. | ||
Holvoet C, Vander Heyden Y, Lories G, Plaizier-Vercammen J. Preparation and evaluation of paclitaxel-containing liposomes. Pharmazie. 2007;2:126–132. | ||
Duong HH, Yung LY. Synergistic co-delivery of doxorubicin and paclitaxel using multi-functional micelles for cancer treatment. Int J Pharm. 2013;54:486–495. | ||
Shahin M, Ahmed S, Kaur K, Lavasanifar A. Decoration of polymeric micelles with cancer-specific peptide ligands for active targeting of paclitaxel. Biomaterials. 2011;32:5123–5133. | ||
Yezhelyev M, Yacoub R, O’Regan R. Inorganic nanoparticles for predictive oncology of breast cancer. Nanomedicine (Lond). 2009;4: 83–103. | ||
Paál K, Müller J, Hegedûs L. High affinity binding of paclitaxel to human serum albumin. Eur J Biochem. 2001;268:2187–2191. | ||
Wang H, Zhao Y, Wu Y, et al. Enhanced anti-tumor efficacy by co-delivery of doxorubicin and paclitaxel with amphiphilic methoxy PEG-PLGA copolymer nanoparticles. Biomaterials. 2011;32:8281–8290. | ||
Gupta N, Hatoum H, Dy GK. First line treatment of advanced non-small-cell lung cancer-specific focus on albumin bound paclitaxel. Int J Nanomedicine. 2014;9:209–221. | ||
Wohl AR, Michel AR, Kalscheuer S, Macosko CW, Panyam J, Hoye TR. Silicate esters of paclitaxel and docetaxel: synthesis, hydrophobicity, hydrolytic stability, cytotoxicity, and prodrug potential. J Med Chem. 2014;57:2368–2379. | ||
Ma P, Mumper RJ. Paclitaxel nano-delivery systems: a comprehensive review. J Nanomed Nanotechnol. 2013;18:1000164. | ||
Shan L, Cui S, Du C, et al. A paclitaxel-conjugated adenovirus vector for targeted drug delivery for tumor therapy. Biomaterials. 2012;33:146–162. | ||
Hasabelnaby S, Goudah A, Agarwal HK, abd Alla MS, Tjarks W. Synthesis, chemical and enzymatic hydrolysis, and aqueous solubility of amino acid ester prodrugs of 3-carboranyl thymidine analogs for boron neutron capture therapy of brain tumors. Eur J Med Chem. 2012;55:325–334. | ||
Arote RB, Hwang SK, Lim HT, et al. The therapeutic efficiency of FP-PEA/TAM67 gene complexese via folate receptor-mediated endocytosis in a xenograft mice model. Biomaterials. 2010;31:2435–2445. | ||
Leanmon CP, Low PS. Delivery of macromolecules into living cells: a method that exploits folate receptor endocytosis. Proc Natl Acad Sci U S A. 1991;88:5572–5576. | ||
Jhaveri MS, Rait AS, Chung KN, Trepel JB, Chang EH. Antisense oligonucleotides targeted to the human folate receptor inhibit breast cancer cell growth and sensitize the cells to doxorubicin treatment. Mol Cancer Ther. 2004;3:1505–1512. | ||
Wu M, Gunning W, Ratnam M. Expression of folate receptor type alpha in relation to cell type, malignancy, and differentiation in ovary, uterus, and cervix. Cancer Epidemiol Biomarkers Prev. 1999;8:775–782. | ||
Shan L, Xue J, Guo J, Qian Z, Achilefu S, Gu Y. Improved targeting of ligand-modified adenovirus as a new near infrared fluorescence tumor imaging probe. Bioconjug Chem. 2011;22:567–581. | ||
Zhang J, Chen H, Xu L, Gu Y. The targeted behavior of thermally responsive nanohydrogel evaluated by NIR system in mouse model. J Control Release. 2008;131:34–40. | ||
Amaechi BT, Ramalingam K. Evaluation of fluorescence imaging with reflectance enhancement technology for early caries detection. Am J Dent. 2014;27:111–116. | ||
Mondal SB, Gao S, Zhu N, Liang R, Gruev V, Achilefu S. Real-time fluorescence image-guided oncologic surgery. Adv Cancer Res. 2014; 124:171–211. | ||
Greenwald RB, Pendri A, Bolikal D. Highly water soluble Taxol derivatives: 7-polyethylene glycol carbamates and carbonates. J Org Chem. 1995;60:331–336. | ||
Yoshida M, Maehara Y, Sugimachi K. MST-16, a novel bis-dioxopiperazine anticancer agent, ameliorates doxorubicin-induced acute toxicity while maintaining antitumor efficacy. Clin Cancer Res. 1999;5:4295–4300. | ||
Guillemard V, Saragovi HU. Taxane-antibody conjugates afford potent cytotoxicity, enhanced solubility, and tumor target selectivity. Cancer Res. 2001;61:694–699. | ||
Boztas AO, Karakuzu O, Galante G, et al. Synergistic interaction of paclitaxel and curcumin with cyclodextrin polymer complexation in human cancer cells. Mol Pharm. 2013;10:76–83. | ||
Tsukiyama I, Takeuchi M, Abe F, et al. De-alcoholization of paclitaxel injection for clinical application. Anticancer Res. 2011;31:4339–4346. | ||
Bandara NA, Hansen MJ, Low PS. Effect of receptor occupancy on folate receptor internalization. Mol Pharm. 2014;11:56–66. |
Supplementary materials
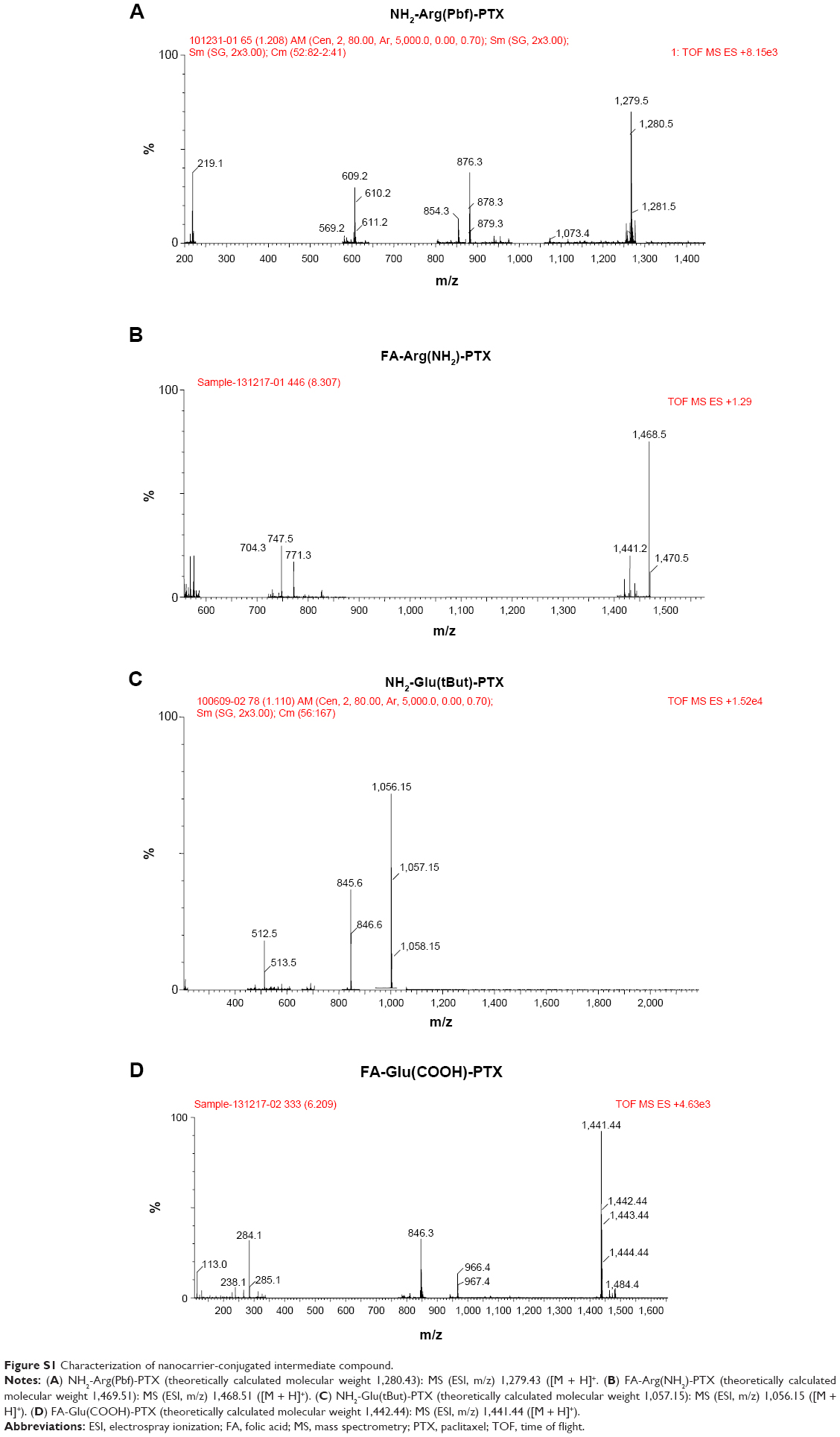 | Figure S1 Characterization of nanocarrier-conjugated intermediate compound. |
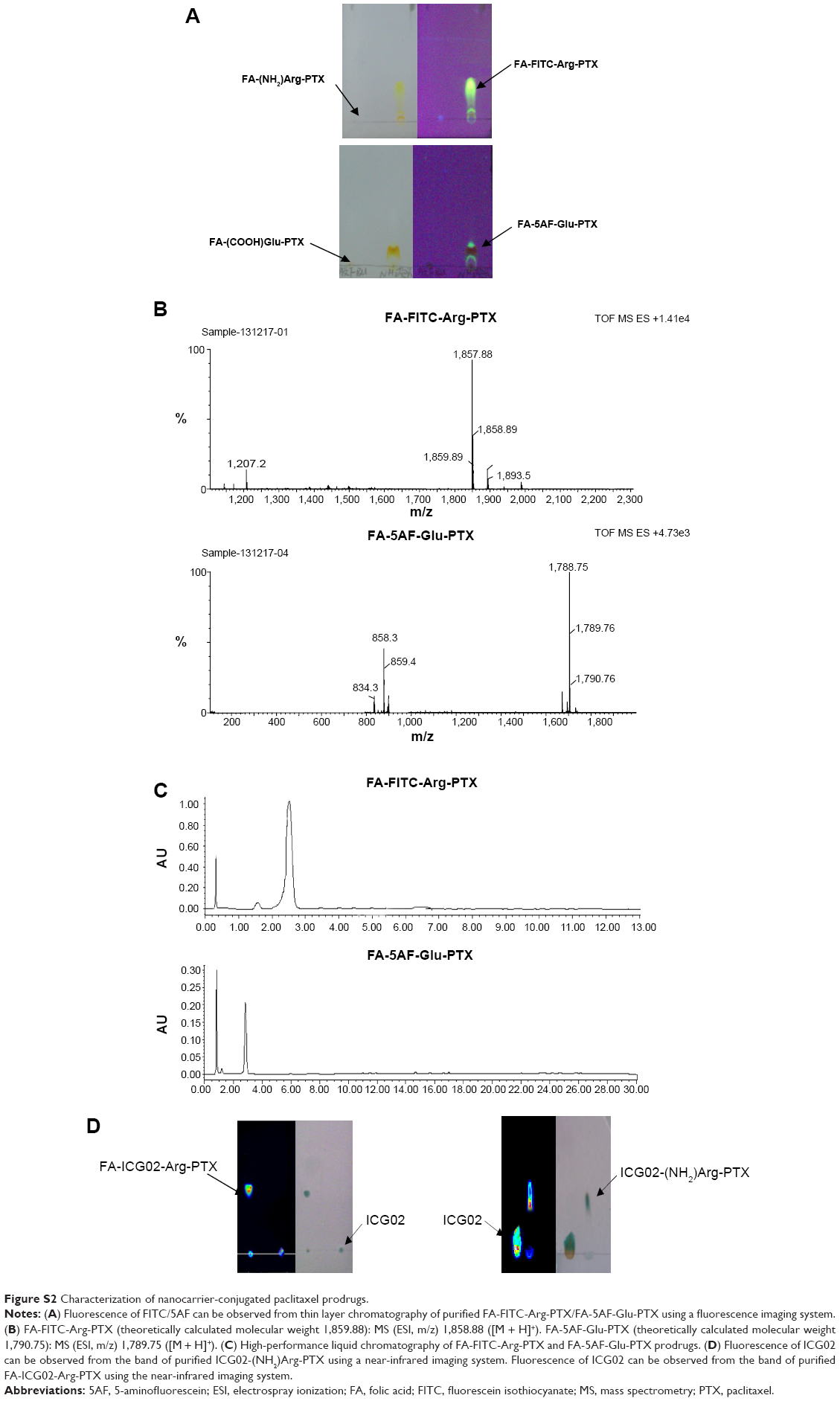 | Figure S2 Characterization of nanocarrier-conjugated paclitaxel prodrugs. |
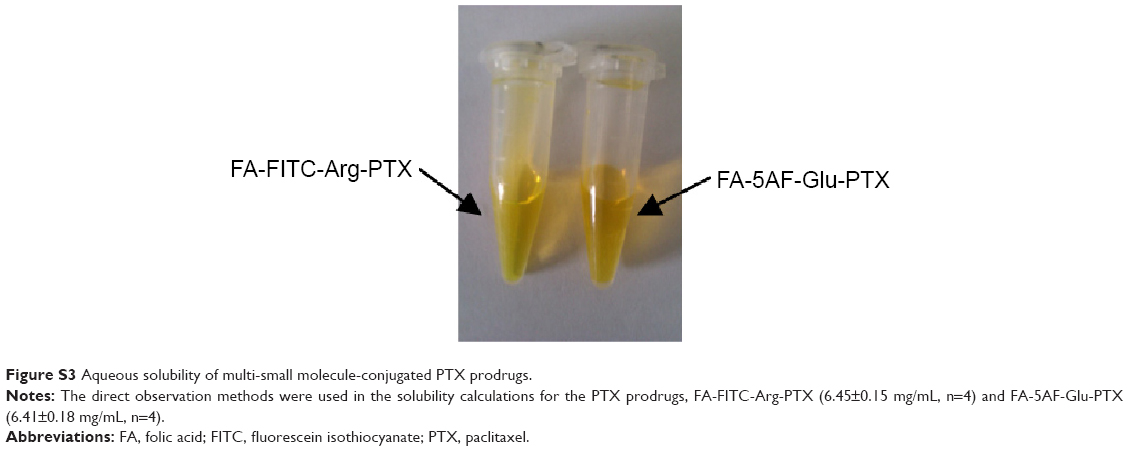 | Figure S3 Aqueous solubility of multi-small molecule-conjugated PTX prodrugs. |
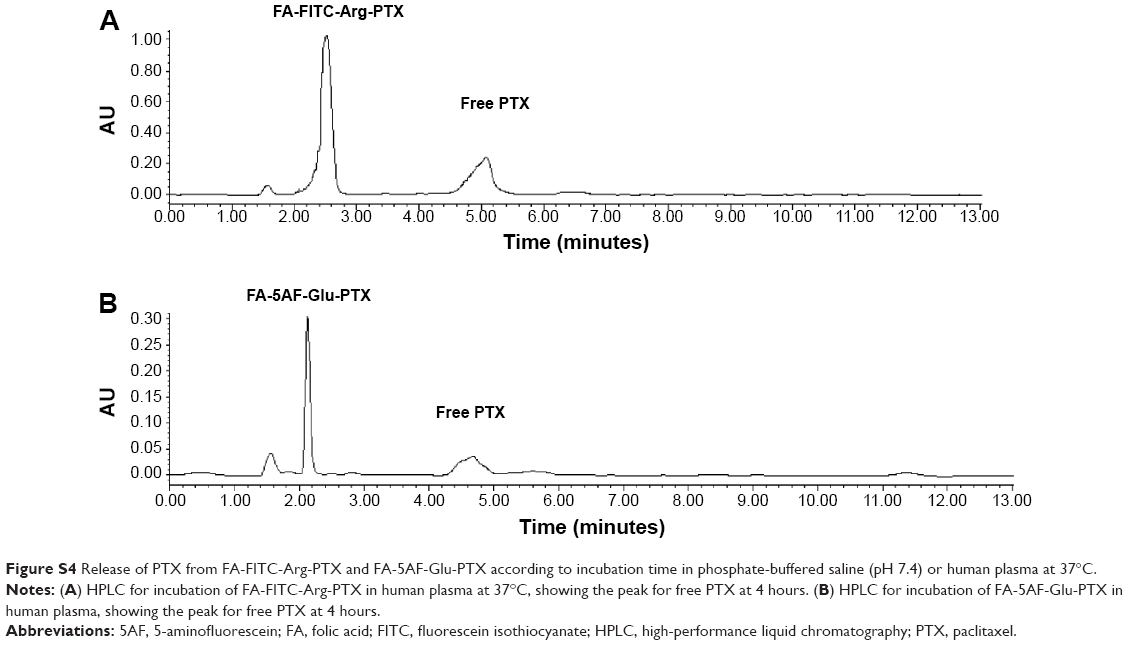 | Figure S4 Release of PTX from FA-FITC-Arg-PTX and FA-5AF-Glu-PTX according to incubation time in phosphate-buffered saline (pH 7.4) or human plasma at 37°C. |
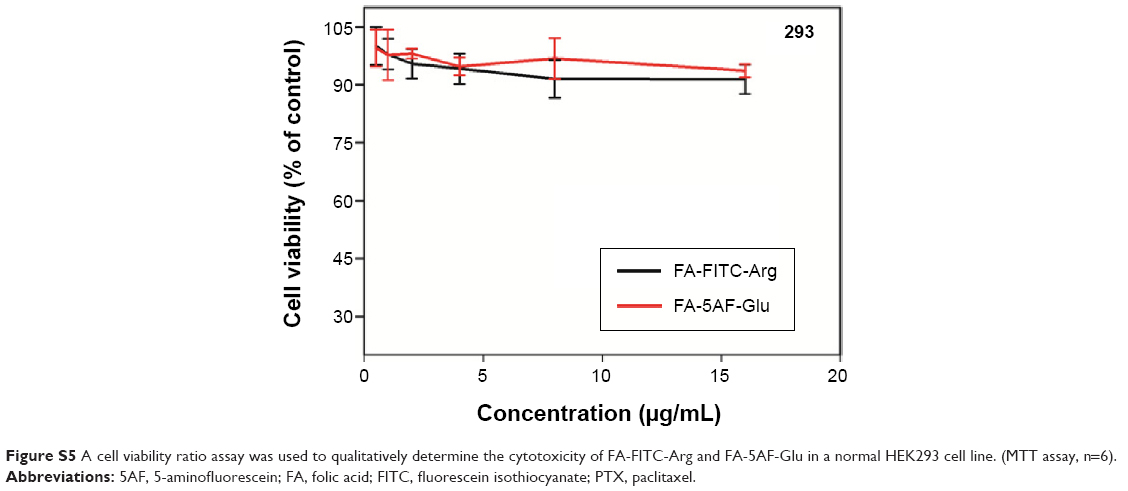 | Figure S5 A cell viability ratio assay was used to qualitatively determine the cytotoxicity of FA-FITC-Arg and FA-5AF-Glu in a normal HEK293 cell line. (MTT assay, n=6). |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.