")
Back to Journals » OncoTargets and Therapy » Volume 14
Multi Cytogenetic Changes in a Patient as Co-Existing MDS and CLL Progresses
Authors Li X , Ma J, Wang L, Yan S, Li F, Wang L, Wang L, Li G, Ma D, Li H
Received 13 September 2020
Accepted for publication 22 December 2020
Published 8 January 2021 Volume 2021:14 Pages 177—186
DOI https://doi.org/10.2147/OTT.S281800
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Federico Perche
Xiangxin Li, Jiale Ma, Luqun Wang, Shuxin Yan, Fanglin Li, Lingling Wang, Lin Wang, Guosheng Li, Daoxin Ma, Hao Li
Department of Hematology, Qilu Hospital, Cheeloo College of Medicine, Shandong University, Jinan, Shandong, People’s Republic of China
Correspondence: Hao Li
Department of Hematology, Qilu Hospital, Cheeloo College of Medicine, Shandong University, 107# Wenhuaxi Road, Jinan, Shandong 250012, People’s Republic of China
Tel +86 13589136826
Email [email protected]
Background: Chronic lymphocytic leukemia (CLL) and myelodysplastic syndrome (MDS) existing simultaneously in untreated patients is extremely rare. There have only been nine cases of untreated CLL concurrent with or followed by the development of MDS. Of all nine cases, four patients exhibited results of cytogenetic phonotypes all showing more than one abnormal chromosome karyotype. It is unknown whether or not these abnormal chromosome karyotypes change during the development of the disease. Meanwhile, the optimal treatment for the concurrence of CLL with MDS has yet to be identified.
Case Presentation: A 69-year-old Chinese man diagnosed with co-existing CLL with MDS was observed from diagnosis, treatment, relapse to death during an admission period of a total of 158 days. Since being diagnosed with CLL and MDS, he was treated by decitabine and his condition went into remission for three months. Four laboratory tests showed an abnormal chromosome cytogenetic karyotype successively changed during the progression of the disease.
Conclusion: It is the first time the abnormal chromosome karyotype variation significantly associated with the change of the illness was discovered. In the relapse and deterioration stages of the disease, there was t(9;22)(q24; q11.2); add(11)(p15) and other chromosome translocation. Repeated occurrence of TET2 mutation is special at this stage of the disease. Furthermore, decitabine could be beneficial for the treatment of the disease.
Keywords: chronic lymphocytic leukemia, myelodysplastic syndrome, chromosome karyotype, treatment, variation
Background
Chronic lymphocytic leukemia (CLL) is the proliferation of small B-lymphocytes in bone marrow, blood and lymph nodes.1,2 In almost all cases where CLL exist simultaneously with myelodysplastic syndrome (MDS), the condition occurred in patients with CLL after treatment with chemotherapy or radiotherapy. The concurrence of CLL with MDS is very rare3–8(Table 1). Given the paucity of molecular information involving mutation genes in the process of two simultaneous malignant phenomena, the mechanism as to how it occurs is unclear. The optimal treatment for the concurrence of CLL with MDS has yet to be identified, and the survival time is very poor.4–9 Here we report a case of a patient diagnosed with CLL and MDS. He was successfully treated by decitabine and his condition went into remission for 3 months. We found changes involving chromosomal abnormalities during the course of his disease progression. The characteristics of this patient during the process is shown in Table 2.
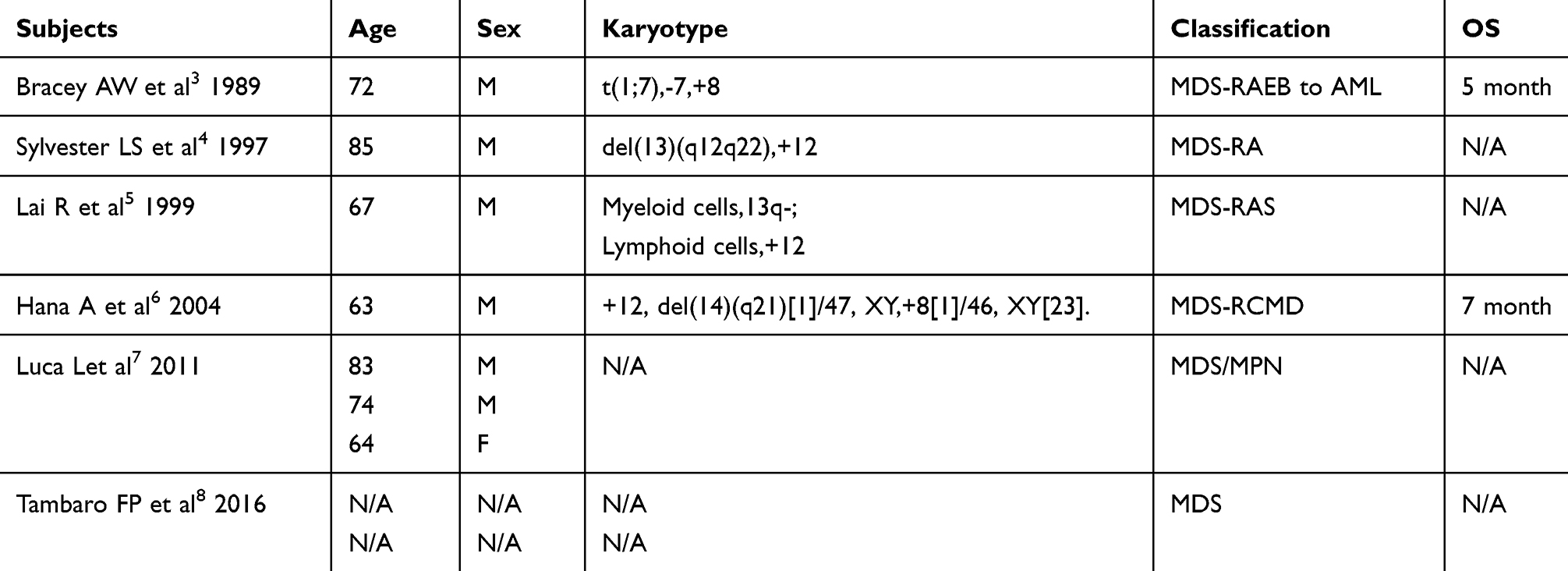 |
Table 1 Documented Clinicopathological Characteristics of Patients Co-Existing CLL and MDS |
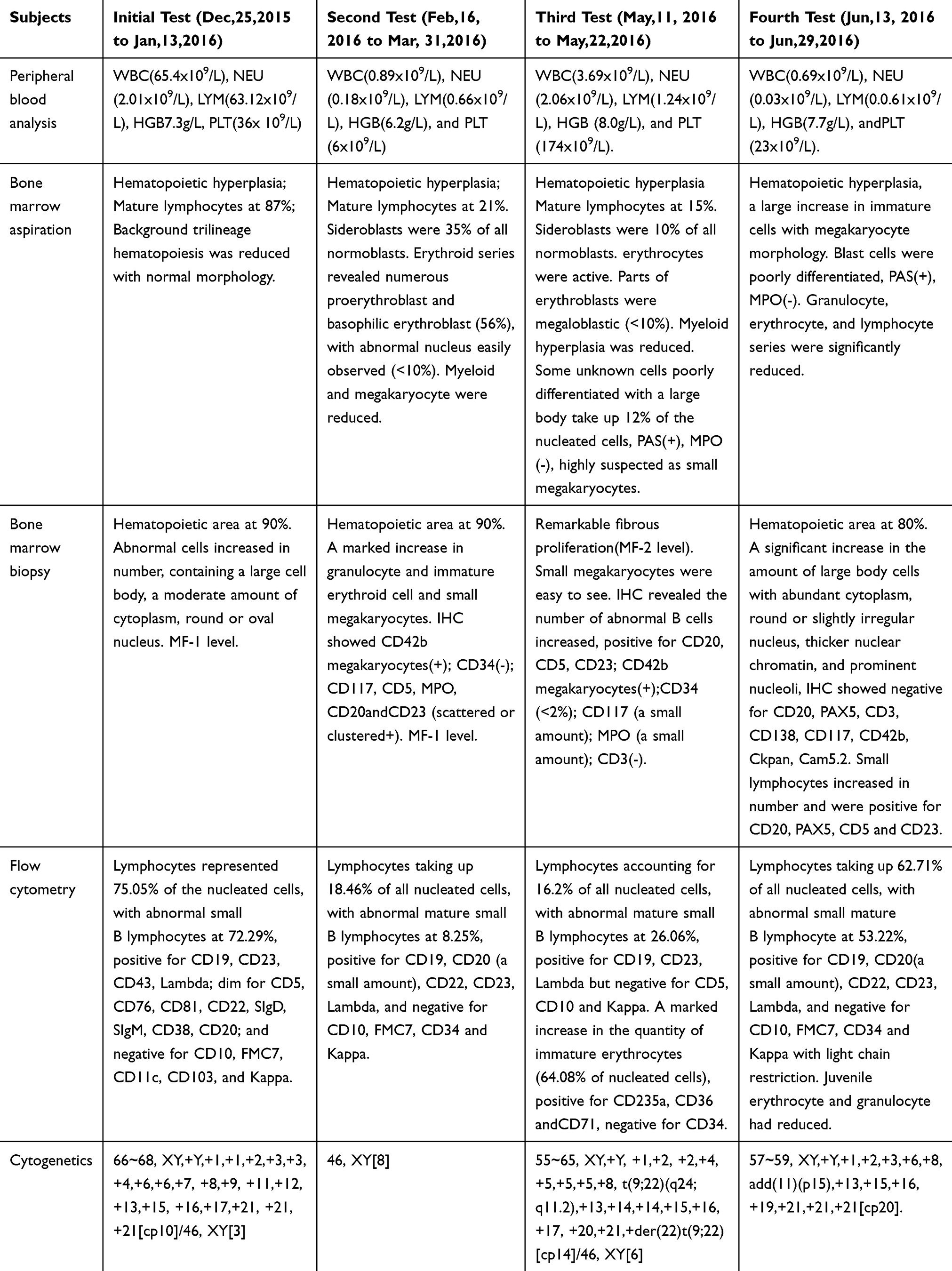 |
Table 2 Patient Characteristics During the Process |
Case Presentation
On 24 December 2015, a 69-year-old Chinese man with three months’ history of fatigue, dyspnea, heart palpitations after activities presented himself at the hospital. He had no fever, but reported weight loss. His past medical history was significant for hypertension for 20 years and diabetes for 7 years, with blood pressure stabilized at 150/90 mmHg and blood glucose controlled in the normal range. He has no history of a previously known hematologic disease or long-term history of exposure to chemical or radioactive substances. During a physical examination, the following conditions were discovered: lymph node enlargement with a size of 1–3cm, mild hepatomegaly (3cm below the costal margin) and moderate splenomegaly (5cm below the costal margin) with sternal tenderness. Other observations included: normal B-type natriuretic peptide, coagulation, hepatic and renal function, with nothing particularly remarkable about the physical examination to report.
Initial peripheral complete blood cell count (CBC) examination demonstrated hemoglobin (HGB) (7.3g/L), white blood cell count (WBC) (65.4x109/L), neutrophil (NEU) (2.01x109/L), lymphocyte (LYM) (63.12x109/L), and platelet (PLT) (36x109/L) (Figure 1).
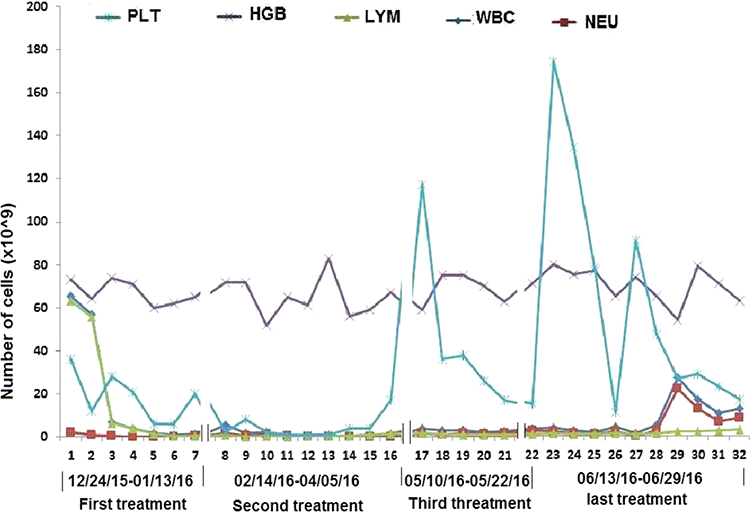 |
Figure 1 Tendency of peripheral blood cells during hospitalization and treatment. |
The initial laboratory tests (taken for the first time from 25 December 2015 to 13 January 2016) found the following information:
- The bone marrow (BM) aspirate smears showed a significant increase of hypercellular marrow,87% of which are mature lymphocytes. Background trilineage hematopoiesis was reduced with normal morphology (Figure 2A).
- Pathological examination of BM biopsy showed extreme hypercellularity with hematopoietic area at 90%. Abnormal cells containing a large cell body, a moderate amount of cytoplasm, round or oval nucleus increased in number. Nuclear staining region was crude, partially visible nucleoli were observed. Granulocytes, erythrocytes, and megakaryocytes were normal in number (Figure 2E).
- Flow cytometry analysis of the BM aspirate cells showed (antibodies come from BECKMAN COULTER; BD; Test instrument came from BC. NAVIOS) that the population of mature lymphocytes reviewed represented approximately 75.05% of the nucleated cells, with abnormal cells at 72.29%. The abnormal cells tested positive for CD19, CD23, CD43, Lambda;dim for CD5, CD76, CD81, CD22, SIgD, SIgM, CD38, CD20; and were negative for CD10, FMC7, CD11c, CD103, and Kappa (See attachment file 1–1 for flow cytometry). Matutes/Catovsky score=4 (SmIg dim+, CD5+, CD23+, FMC7-).
- Cytogenetic test found in chromosome and multi-color fluorescent in situ hybridization (mFISH) analyses showed no changes in 11q22.3,13q14,13q14.3,17p13.1 and+12, while long time 72–96 hours stimulated cultures with CpG-OPN+IL-2(LTSC) of bone marrow showed:66~68, XY,+Y,+1,+1,+2,+3,+3,+4,+6,+6,+7,+8,+9,+11,+12,+13,+15,+16,+17,+21,+21,+21[cp10]/46, XY[3] (Figure 3A).
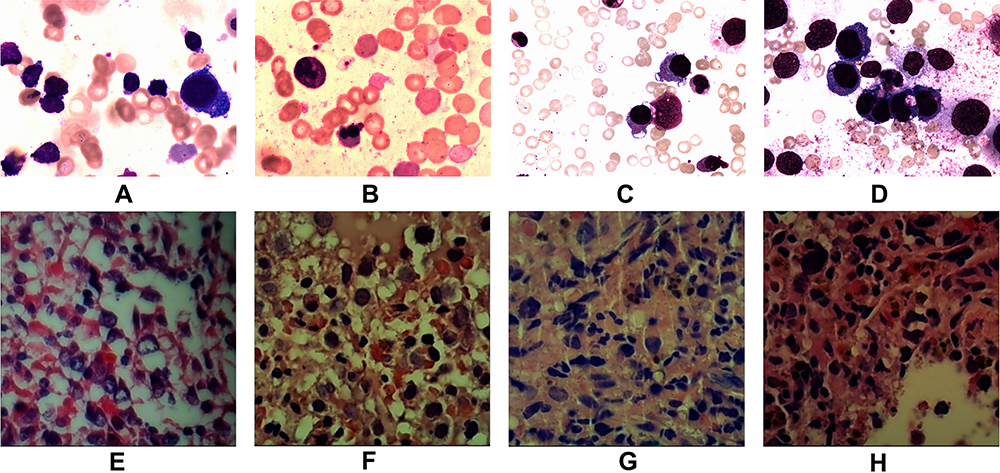 |
Figure 2 Images A & E, B & F, C & G, and D & H show the initial, second, third and fourth bone marrow (BM) aspirate cells smear.(H & E X400) and biopsy pathological tests (H & E X200) respectively. Cells characters: (A) (12/25/2015) Hematopoietic hyperplasia; Mature lymphocytes at 87%; Background trilineage hematopoiesis was reduced with normal morphology (E) (12/25/2015) Hematopoietic area at 90%. Abnormal cells increased in number, containing a large cell body, a moderate amount of cytoplasm, round or oval nucleus. MF-1 level. (B) (03/31/2016) Hematopoietic hyperplasia;Mature lymphocytes at 21%. Iron stain positive Sideroblasts were 35% of all normoblasts. Erythroid series revealed numerous proerythroblasts and basophilic erythroblast (56%), with abnormal nucleus easily observed (<10%). Myeloid and megakaryocyte were reduced. (F) BM biopsy (03/31/2016) demonstrated Hematopoietic area at 90%. A marked increase in granulocyte and immature erythroid cell and small megakaryocytes. Immunohistochemistry showed CD42b megakaryocytes(+); CD34(-);CD117, CD5, MPO, CD20andCD23 (scattered or clustered+). MF-1 level. (C) (05/11/2016) Hematopoietic hyperplasia Mature lymphocytes at 15%. Sideroblasts were 10% of all normoblasts. Erythrocytes were active. Parts of erythroblasts were megaloblastic (<10%). Myeloid hyperplasia was reduced. (G) (05/11/2016) BM biopsy showed increased fibrosis, and granulocyte, erythrocyte, and megakaryocyte cells were easily observed. Lymphocytes were focally or dispersedly distributed. Immunohistochemistry revealed abnormal B cells had increased in quantity, reacting positively for CD20, CD5, CD23, and 42b megakaryocytes, CD34 (<2%), CD117 (a small amount), MPO (a small amount), but negative for CD3 Remarkable fibrous proliferation (MF-2 level). Small megakaryocytes were easy to see. Immunohistochemistry revealed the number of abnormal B cells increased, positive for CD20, CD5, CD23; CD42b megakaryocytes(+); CD34 (<2%); CD117 (a small amount); MPO (a small amount); CD3(-). (D) (06/13/2016) Hematopoietic area at 80%. A significant increase in the amount of large body cells with abundant cytoplasm, round or slightly irregular nucleus, thicker nuclear chromatin, and prominent nucleoli, IHC showed negative for CD20, PAX5, CD3, CD138, CD117, CD42b, Ckpan, Cam5.2. Small lymphocytes increased in number and were positive for CD20, PAX5, CD5 and CD23. (H) BM biopsy showed Lymphocytes taking up 62.71% of all nucleated cells, with abnormal small mature B lymphocyte at 53.22%, positive for CD19, CD20 (a small amount), CD22, CD23, Lambda, and negative for CD10, FMC7, CD34 and Kappa with light chain restriction. Juvenile erythrocyte and granulocyte had reduced. |
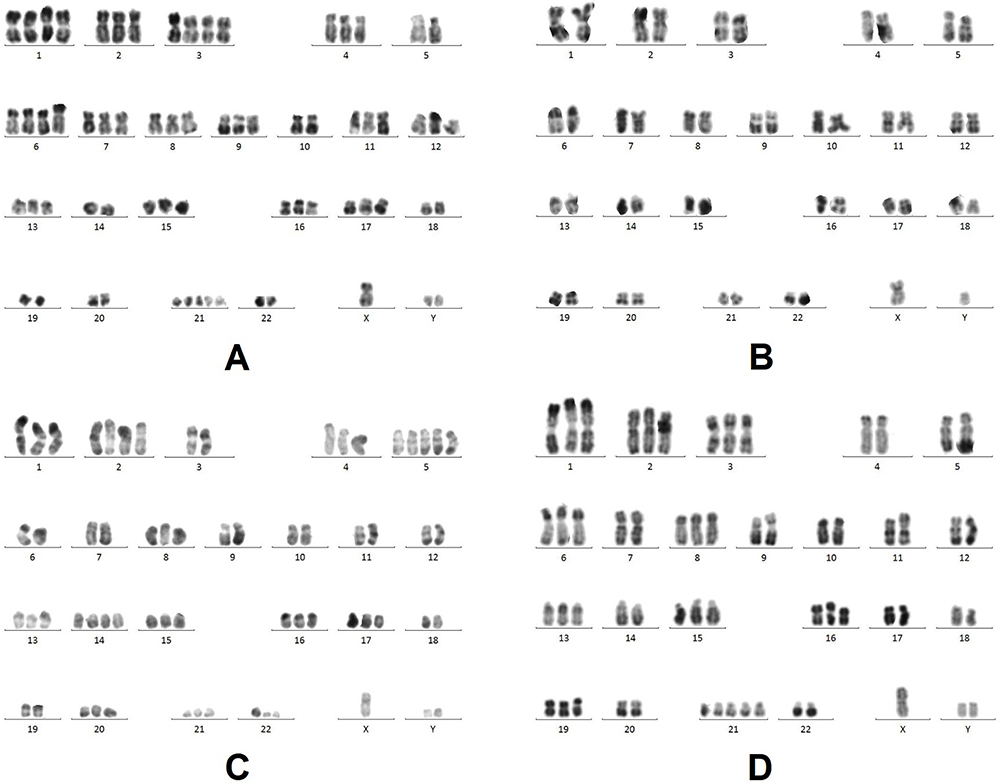 |
Figure 3 A, B, C, and D represent the initial, second, third and fourth results taken during conventional cytogenetic analysis of bone marrow. RHG-banded metaphases were analysed and karyotyped according to ISCN (2013). (A) Karyotype on 25 December 2015, LTSC showing:66~68, XY,+Y,+1,+1,+2,+3,+3,+4,+6,+6,+7, +8,+9, +11,+12, +13,+15, +16,+17,+21, +21,+21[cp10]/46, XY[3]. (B) Karyotype on 1 April 2016, USSTC showing less cells with poor proliferation: 46, XY[8]. (C) Karyotype on 11 May 2016, LTSC showing 55~65, XY,+Y, +1,+2, +2,+4, +5,+5,+5,+8, t(9;22)(q24; q11.2), +13,+14, +14,+15, +16, +17,+20,+21,+der(22)t(9;22)[cp14]/46, XY[6]. (D) Karyotype on 14 June 2016, LTSC showing 57~59, XY,+Y,+1,+2,+3,+6,+8, add(11)(p15), +13,+15,+16,+19,+21,+21, +21[cp20]. |
These results were considered consistent with CLL, and MDS was just suspected. Treatment of the patient was initiated with FCR (fludarabine: 25 mg/m2 on days 2–4, cyclophosphamide: 250 mg/m2 on days 2–4 and rituximab: 375 mg/m2 on day 1) on 28 December 2015 for one cycle. However, the patient’s symptoms did not improve and his complete peripheral blood counts decreased day by day. On 9 January,2016, two weeks after diagnosis of CLL, the CBC showed HGB (6.2 g/L), WBC (0.89x 109/L), NEU (0.18x 109/L), LYM (0.66x 109/L), and PLT (6x 109/L) (Figure 1).
A subsequent laboratory test (taken on 16 February 2016 through to 31 March 2016) was performed and the following information was obtained:
- The subsequent BM aspirate smears demonstrated hematopoietic hyperplasia with mature lymphocytes at 21%. The extracellular staining was positive, and sideroblasts were 35% of all normoblasts. The presence of trilineage hematopoiesis showed that myeloid and megakaryocyte hyperplasia has reduced, but erythrocytes were active. Of note, the erythroid series revealed numerous proerythroblast and basophilic erythroblast (56%), with abnormal nucleus easily observed (<10%) (Figure 2B).
- Pathological examination of BM biopsy showed a hypercellular (about 90% cellularity) bone marrow with a marked increase in granulocyte and immature erythroid cells, while megakaryocytes remained the same in number. Immunohistochemistry tests showed positive expression for CD42b megakaryocytes, CD34, scattered or clustered positive expression for CD117, CD5, MPO, CD20 and CD23 (Figure 2F).
- Flow cytometric immunophenotypic analysis showed a hypercellular (about 87.29% cellularity) bone marrow with lymphocytes taking up 18.46% of all nucleated cells phenotype. It also showed the population of mature small B lymphocyte grew, representing approximately 8.25% of the nucleated cells, positive for CD19, CD20 (a small amount), CD22, CD23, Lambda, and negative for CD10, FMC7, CD34 and Kappa, with light chain restriction (See attachment file 1–2 for flow cytometry).
- A gene rearrangement examination demonstrated that IgH and IgK gene rearrangement were positive, while TCRβ, TCRγ were negative. Other common leukemia fusion genes were negative. A second-generation gene sequencing was conducted at the same time with TET2 mutation (62.68%, exon3: c.G652A: p.V218M rs 6,843,141), which commonly appears in Chronic myelomonocytic leukemia (CMML).
- Conventional cytogenetic analysis of chromosome karyotypes of bone marrow with unstimulated short time culture (USSTC) showed 46, XY[8] (Figure 3B).
Considering all above laboratory results, as well as the inefficiency of the FCR regimen in the recovery of marrow dyshaematopoiesis especially of erythroid and megakaryocytic lines, the patient suffering from CLL with MDS-multilineage dysplasia (MLD) was newly diagnosed.10 We thereby deactivated the above drugs, and turned to decitabine for the patient’s treatment. During admission from 17 February 2016 on which cycle 1 of FCR was administered, through to Day 60–9 April 2016, he received decitabine treatment (20 mg/m2/day for 5 days every 28 days) for two cycles. His clinical symptoms, as well as peripheral CBC test improved and a very good clinical outlook was achieved. His condition went into remission for three months. On 10 May 2016, 83 days after the diagnosis of CLL concurrent with MDS, laboratory testing showed HGB (8.0 g/L), WBC (3.69x 109/L), NEU (2.06x 109/L), LYM (1.24x 109/L) and PLT (174x 109/L) (Figure 1).
A subsequent laboratory test was performed for the third time over the period of 11 May 2016 to 22 May 2016, the following was observed:
- The BM aspirate smears demonstrated hematopoietic hyperplasia with mature lymphocytes accounted for 15% of cells present. Sideroblasts were 10% of all normoblasts. The presence of trilineage hematopoiesis showed that myeloid hyperplasia had reduced, but erythrocytes were active. Parts of erythroblasts were megaloblastic. Megakaryocyte was normal. Of note, some unknown cells with a large body that are poorly differentiated take up 12% of the nucleated cells, reacting positive for PAS, but negative for MPO, the morphology of which was highly suspected as that of small megakaryocytes (Figure 2C).
- The pathological test of BM biopsy showed granulocyte, erythrocyte, and megakaryocyte cells were easily observed. Lymphocytes were focally or dispersedly distributed. Immunohistochemistry testing revealed the number of abnormal B cells increased, which were positive for CD20, CD5, CD23, and 42b megakaryocytes, CD34 (<2%), CD117 (a small amount), MPO (a small amount), but negative for CD3 (Figure 2G).
- Flow cytometric immunophenotypic analysis showed a hypercellular (about 80.06% cellularity) bone marrow with lymphocytes accounting for 16.2% of all nucleated cell phenotypes. It also showed an increase in the population of abnormal mature small B lymphocyte phenotype, representing approximately 26.06% of the nucleated cells, positive for CD19, CD23, Lambda but lack CD5, CD10 and Kappa, and light chain was expressed restrictively. Additionally, there was a marked increase in the quantity of immature erythrocytes (64.08% of nucleated cells), which were positive for CD235a, CD36 and CD71 but lack CD34 positive progenitor cells. (See attachment file 1–3 for flow cytometry).
- The subsequent cytogenetic karyotypes examination with LTSC of bone marrow showed two clones: 55~65, XY,+Y,+1,+2,+2,+4,+5,+5,+5,+8, t(9;22)(q24;q11.2),+13,+14,+14,+15,+16,+17,+20,+21,+der(22)t(9;22) [cp14]/46, XY[6] (Figure 3C).
- A second-generation gene sequencing was conducted for the second time with TET2 mutation (52.86%, exon3: c.G652A: p.V218M rs 6,843,141).
These results indicated the patient was still suffering from CLL with MDS-MLD. After three months of remission, on 13 May 2016, during the third cycle of chemotherapy, decitabine (20 mg/m2/day for 5 days) combined with cytarabine (0.02g/d d3-9) was given to the patient. Repetitive laboratory observations showed that peripheral blood CBC appeared to have the tendency of looking like a “drop-return-drop-return” model with some symptomatic and supportive measures. Three weeks following the treatment, the patient’s leukocyte gradually increased, while the number of platelets decreased. Based on the performance score system of the Eastern Cooperative Oncology Group (ECOG), the patient’s condition was deteriorating. The next course of treatment for the patient was decitabine combined with chlorambucil. However, the patient’s condition failed to significantly improved. He was better for a while and then had a relapse.
The fourth laboratory tests taken on 13 June 2016 through to 29 June 2016 documented the following results:
- BM aspirate smears showed a hypercellular bone marrow with a large increase in immature cells with megakaryocyte morphology. These blast cells were poorly differentiated, reacting strongly positive for PAS, but negative for MPO. Granulocyte, erythrocyte, and lymphocyte series were significantly reduced (Figure 2D).
- Pathological examination of BM biopsy showed a hypercellular (about 80% cellularity) bone marrow with a significant increase in the amount of large body cells with abundant cytoplasm, round or slightly irregular nucleus, thicker nuclear chromatin, and prominent nucleoli, none of which contains antibodies including CD20, PAX5, CD3, CD138, CD117, CD42b, Ckpan, Cam5.2 for B cells, T cells, plasma cells, myeloid precursor cells, and megakaryocytes cells. Small lymphocyte cells increased in number, and were positive for CD20, PAX5, CD5 and CD23. Granulocyte, erythrocyte, and megakaryocyte cells were easily observed (Figure 2H).
- Flow cytometric immunophenotypic analysis showed a hypercellular (about 52.49% cellularity) bone marrow with lymphocytes taking up to 62.71% of all nucleated cells phenotype. It also showed an increase in the population of abnormal small mature B lymphocyte phenotype representing approximately 53.22% of lymphocyte, which showed up positive for CD19, CD20 (a small amount), CD22, CD23, Lambda, and negative for CD10, FMC7, CD34 and Kappa with light chain restriction. Juvenile erythrocyte and granulocyte had reduced. (See attachment file 1–4 for flow cytometry).
- The fourth cytogenetic karyotype examination with LTSC of bone marrow showed one clone: 57~59, XY,+Y,+1,+2,+3,+6,+8, add(11)(p15),+13,+15,+16,+19,+21,+21,+21[cp20] (Figure 3D).
These results showed the patient was still affected with CLL. However, the increased abnormally large cells belonged to an unknown series.
Unfortunately, the patient developed pneumonia. On 2 July 2016 he died of uncontrolled pulmonary infection. He survived a total of 138 days after his diagnosis.
As the patient’s condition progresses, abnormal chromosome karyotype variations significantly associated with the change of the illness were discovered. In the relapse and deterioration stages of the disease, gene translocation as t(9;22)(q24; q11.2), add(11)(p15) and repeated occurrence of TET2 mutation (exon3: c.G652A: p.V218M rs 6,843,141) were successively found.
Discussion
Complex karyotype (CK), defined by the presence of 3 or more chromosome aberrations, represents a prognostic negative biomarker associated with an inferior outcome independent of the International Prognostic Index for patients with CLL (CLL‐IPI),11 and a worse response to treatment including novel agents such as BCL2 inhibitor venetoclax.12 Although CK is detectable in 14–34% of untreated CLL patients,11–13 it seldom brings about abnormal bone marrow morphologic changes like MDS or AML, especially simultaneously in untreated CLL.
As reported, the most frequent chromosome breakpoints in CLL are 13q, followed by 14q, 18q, 17q, and 17p; notably there exists a wide spectrum of translocation partners with 13q. In CK cases, besides co‐existing trisomies of chromosomes 12/18/19 (2.4% in multiple trisomies), which indicates a more favorable clinicobiological feature.13 Other CK especially those with five or more cytogenetic aberrations follow a particularly aggressive clinical course. We reviewed the clinical, immunophenotypic and cytogenetic features of the nine cases of untreated CLL concurrent with MDS reported.3–8 Of all nine MDS patients, four cases recorded the presence of cytogenetic abnormalities: t(1;7),-7,+8;3 del(13)(q12q22),+12;4 Myeloid cells,13q- and lymphoid cells, +12;5 and+12, del(14) (q21)[1]/47, XY,+8[1]/46, XY[23].6
In the study of Tambaro FP,8 the outcomes of CLL diagnosed with AL (n=38) or MDS (n=57), either concurrently (n=5) or subsequently (n=90) were documented, only two of which were concurrently diagnosed with CLL and MDS. The chromosome on metaphase karyotype involved in AML or MDS with CLL are 5,7,8,11 and 17.
Karyotype for MDS and AL was poor, 63% and 68% respectively according to the International Prognostic Scoring System (IPSS) of WHO 2016. 8% of AL patients and 28% of MDS patients received treatments using hypomethylating agents such as azacitidine or decitabine. In this case, the subject was found to have abnormal chromosomes that are involved with almost all chromosomes, and for the first time we found the abnormal chromosome karyotypes variation significantly associated with the change of the illness. In the severe deterioration stage of the disease, a gene translocation t(9;22)(q24; q11.2) was shown. The significance of t(9;22) had not been reported in CLL, and is also a very rare event in MDS. Manish Kumar Singh reported two cases of De novo Philadelphia chromosome positive myelodysplastic syndrome. In literature, 40 cases of Ph+MDS were recorded, of which less than half showed positive Ph at initial diagnosis. The presence of the Philadelphia chromosome probably indicates a poor prognostic group of MDS14. This point is consistent with the temporary effect of decitabine in the process.
More than two genes were identified to have undergone alterations including gene deletion and point mutation in clinical samples of CLL/MDS patients, suggesting that multiple gene mutations are required for leukemogenesis,15,16 such as ASXL1 and SETBP1 or N-Ras in MDS etiology15 In this case, no other gene mutation occurred while TET2 mutation was repeatedly found as the second-generation gene sequencing was conducted twice. TET2 is a major factor in hematopoietic function. Some hematologic malignancies such as myelodysplastic syndrome, acute myelogenous leukemia or chronic myelogenous leukemia are associated with TET2 mutations.17 María HS observed that TET2 was overexpressed in CLL cells compared with healthy donor cells.18 In terms of prognosis, TET2 and IDH1 were observed to be associated with longer TFS than those with low expression19. We therefore hypothesised that TET2 mutation in this patient mainly acted on the proliferation of MDS clones, but had little effect on the maintenance of CLL malignant clones. The temporary efficacy of demethylation drug may also support the diagnosis of MDS rather than CLL co-existing with CK.
Furthermore, it is unknown whether these abnormal genes were involved in the whole process of the disease simultaneously or successively. Or did they drift from initially altered genes to other newly changed genes as the disease develops. From observing the patient’sprogress, we first noticed that multiple chromosomal karyotype mutations one after another are involved in the deterioration process of the disease as the condition of the patient with co-existing MDS and CLL progresses. This observation indicates cell genetic karyotype monitoring is very important in the treatment of leukemia. The findings will be beneficial in proving the pathogenesis of leukemia in the future.
Conclusion
Complex karyotype (CK) as a negative biomarker is detectable in CLL, while it seldom brings about abnormal bone marrow morphologic changes like MDS or AML, especially simultaneously in untreated CLL. In this study, a patient who was simultaneously suffering from CLL and MDS was observed. His condition failed to go into remission after treatment with the FCR regimen, while it went into remission for three months after using decitabine, which supports the diagnosis of CLL co-existing with MDS. An abnormal chromosome karyotype successively changed during the progression of the disease. This novel case is the first time abnormal chromosome karyotype variation significantly associated with the change of the illness was discovered. The relapse and deterioration stages of the disease involved gene translocation as t(9;22)(q24; q11.2) and add(11)(p15), and repetition of TET2 mutation (exon3: c.G652A: p.V218M rs 6,843,141). Monitoring the bone marrow morphological changes and abnormal chromosome karyotype variation had some guiding effect on our choice of medication. The collection of this information will benefit etiological research in the future.
Abbreviations
CLL, chronic lymphocytic leukemia; MDS, myelodysplastic syndrome; CBC, complete blood cell count; HGB, hemoglobin; WBC, white blood cell; NEU, neutrophil; LYM, lymphocyte; PLT, platelet; BM, bone marrow; RA, refractory anemia; RAS, refractory anemia with sideroblasts; RAEB, refractory anemia with excess blasts; CMML, chronic myelomonocytic leukemia; RCMD, refractory cytopenia with multiple dysplasia; MDS-MLD, MDS with multilineage dysplasia; ECOG, Eastern Cooperative Oncology Group.
Data Sharing Statement
All data generated or analyzed during this study are included in this published article.
Ethics Approval and Consent
This study was approved by the Ethics Committee of the Qilu Hospital, Cheeloo College of Medicine, Shandong University. An informed consent form was signed by the son of the patient to publish the case details.
Consent to Publication
Written informed consent was obtained from the son of the patient for publication of this case report and accompanying images.
Acknowledgments
We thank the Laboratory of the Blood Institute of China Academy of Medical Sciences (Tianjin, China) who assisted us in performing the FISH and cytogenetic analysis.
Funding
This work was supported by The Key Research and Development Program of Shandong Province (No. 2017GSF18133, No. 2016GSF201038). Those providing funds had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Fabbri G, Dalla-Favera R. The molecular pathogenesis of chronic lymphocytic leukaemia. Nat Rev Cancer. 2016;16:145–162. doi:10.1038/nrc.2016.8
2. Cramer P, Isfort S, Bahlo J, et al. Outcome of advanced chronic lymphocytic leukemia following different first-line and relapse therapies: a meta-analysis of five prospective trials by the German CLL Study Group (GCLLSG). Haematologica. 2015;100:1451–1459. doi:10.3324/haematol.2015.124693
3. Bracey AW, Maddox AM, Immken L, Hsu SM, Marks ME. Coexistence of myelodysplastic syndrome and untreated chronic lymphocytic leukemia with development of acute myeloid leukemia immediately after treatment of chronic lymphocytic leukemia. Am J Hematol. 1989;30(3):174–180. doi:10.1002/ajh.2830300310
4. Sylvester LS, Nowell PC, Bonner H, Moreau L, Moore JS. Concurrent diagnosis of chronic lymphocytic leukemia and myelodysplastic syndrome. Leuk Res. 1997;21(7):619–621. doi:10.1016/s0145-2126(97)00017-9
5. Lai R, Arber DA, Brynes RK, Chan O, Chang KL. Untreated chronic lymphocytic leukemia concurrent with or followed by acute myelogenous leukemia or myelodysplastic syndrome - a report of five cases and review of the literature. Am J Clin Pathol. 1999;111(3):373–378. doi:10.1093/ajcp/111.3.373
6. Hana A, Denga T, Kasturi D, et al. Simultaneous appearance of trisomy 8 and trisomy12 in different cell populations in a patient with untreated B-cell chronic lymphocytic leukemia and myelodysplasia. Leuk Lymphoma. 2004;45(6):1279–1283. doi:10.1080/10428190310001638869
7. Luca L, Michela T, Ilaria N, et al. The coexistence of chronic lymphocytic leukemia and myeloproliperative neoplasms: a retrospective multicentric GIMEMA experience. Am J Hematol. 2011;86(12):1007–1012. doi:10.1002/ajh.22171
8. Tambaro FP, Garcia-Manero G, O’Brien SM, et al. Outcomes for patients with chronic lymphocytic leukemia and acute leukemia or myelodysplastic syndrome. Leukemia. 2016;30(2):325–330. doi:10.1038/leu.2015.227
9. Cameron Y, Guilin T, Gary L, et al. Del(20q) in patients with chronic lymphocytic leukemia: a therapy-related abnormality involving lymphoid or myeloid cells. Mod Pathol. 2015;28(8):1130–1137. doi:10.1038/modpathol.2015.58
10. Daniel AA, Attilio O, Robert H, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391–2405. doi:10.1182/blood-2016-03-643544
11. International CLL-IPI working group. An international prognostic index for patients with chronic lymphocytic leukaemia (CLL-IPI): a meta-analysis of individual patient data. Lancet Oncol. 2016;17:779–790. doi:10.1016/S1470-2045(16)30029-8
12. Cavallari M, Cavazzini F, Bardi A, et al. Biological significance and prognostic/predictive impact of complex karyotype in chronic lymphocytic leukemia. Oncotarget. 2018;9(76):34398–34412. doi:10.18632/oncotarget.26146
13. Kruzova L, Schneiderova P, Holzerova M, et al. Complex karyotype as a predictor of high-risk chronic lymphocytic leukemia: a single center experience over 12 years. Leuk Res. 2019;85:106218. doi:10.1016/j.leukres.2019.106218
14. Khaliqur R, Manish KS, Ruchi G, Sarjana D, Soniya N. De novo Philadelphia chromosome positive myelodysplastic syndrome: report of two cases with brief literature review. J Can Res Ther. 2020;16:173–176. doi:10.4103/0973-1482.188428
15. Yun S, Vincelette ND, Abraham I, et al. Targeting epigenetic pathways in acute myeloid leukemia and myelodysplastic syndrome: a systematic review of hypomethylating agents trials. Clin Epigenetics. 2016;8(1). doi:10.1186/s13148-016-0233-2
16. Makishima H, Yoshida K, Nguyen N, et al. Somatic SETBP1 mutations in myeloid malignancies. Nat Genet. 2013;45:942–946. doi:10.1007/s12185-017-2241-1
17. Kyoko I, Joun L, Stephanie C, et al. Non-catalytic roles of Tet2 are essential to regulate hematopoietic stem and progenitor cell homeostasis. Cell Rep. 2019;28:2480–2490. doi:10.1016/j.celrep.2019.07.094
18. María HS, Ana ER, Alexander K, et al. TET2 overexpression in chronic lymphocytic leukemia is unrelated to the presence of TET2 variations. Biomed Res Int. 2014;2014:814294. doi:10.1155/2014/814294
19. Michaël VD, Emerence CN, Meuleman MM, et al. Characterization of TET and IDH gene expression in chronic lymphocytic leukemia: comparison with normal B cells and prognostic significance. Clin Epigenetics. 2016;8:132. doi:10.1186/s13148-016-0298-y
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.