")
Back to Archived Journals » Journal of Receptor, Ligand and Channel Research » Volume 8
mTOR signaling in mice with dysfunctional cardiac ryanodine receptor ion channel
Authors Huang T, Zou M, Pasek DA, Meissner G
Received 1 December 2014
Accepted for publication 5 February 2015
Published 20 May 2015 Volume 2015:8 Pages 43—51
DOI https://doi.org/10.2147/JRLCR.S78410
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Trevor W. Stone
Tai-Qin Huang,* Min-Xu Zou,* Daniel A Pasek, Gerhard Meissner
Department of Biochemistry and Biophysics, University of North Carolina, Chapel Hill, NC, USA
*These authors contributed equally to this work
Abstract: Simultaneous substitution of three amino acid residues in the calmodulin binding domain (W3587A/L3591D/F3603A, ADA) of the cardiac ryanodine receptor ion channel (RyR2) impairs calmodulin inhibition of RyR2 and causes cardiac hypertrophy and early death of Ryr2ADA/ADA mice. To determine the physiological significance of growth promoting signaling molecules, the protein and phosphorylation levels of Ser/Thr kinase mTOR and upstream and downstream signaling molecules were determined in hearts of wild-type and Ryr2ADA/ADA mice. Phosphorylation of mTOR at Ser-2448, and mTOR downstream targets p70S6 kinase at Thr-389, S6 ribosomal protein at Ser-240/244, and 4E-BP1 at Ser-65 were increased. However, there was no increased phosphorylation of mTOR upstream kinases PDK1 at Ser-241, AKT at Thr-308, AMPK at Thr-172, and ERK1/2 at Thr-202/Tyr204. To confirm a role for mTOR signaling in the development of cardiac hypertrophy, rapamycin, an inhibitor of mTOR, was injected into wild-type and mutant mice. Rapamycin decreased mouse heart-to-body weight ratio, improved cardiac performance, and decreased phosphorylation of mTOR and downstream targets p70S6K and S6 in 10-day-old Ryr2ADA/ADA mice but did not extend longevity. Taken together, the results link a dysfunctional RyR2 to an altered activity of signaling molecules that regulate cardiac growth and function.
Keywords: calmodulin, cardiac hypertrophy, rapamycin, RyR2
Introduction
Type-2 ryanodine receptor ion channels (RyR2s) release Ca2+ from an intracellular membrane compartment, the sarcoplasmic reticulum, which are required for cardiac muscle contraction. RyR2s are regulated by multiple endogenous effectors that include Ca2+ and associated protein kinases, phosphatases, and calmodulin (CaM).1–3 CaM is a small cytoplasmic Ca2+ binding protein that regulates protein kinases and ion channels. A notable property of the RyR2s is their inhibition by CaM at diastolic and systolic Ca2+ concentrations.4,5
Altered cellular levels of Ca2+ ions induce pathological growth in the heart in response to cellular stress.6–8 We found that simultaneous replacement of three amino acid residues in the CaM binding domain of RyR2 (W3587A/L3591D/F3603A, ADA) causes severe cardiac hypertrophy at postnatal day 10 and death within 2–3 weeks after birth in Ryr2ADA/ADA mice.9 The RyR2 mutations resulted in loss of CaM inhibition of RyR2 at diastolic and systolic Ca2+ concentrations9 and were associated with prolonged Ca2+ transients, lower Cav1.2 current density, calcium-induced Ca2+ release gain, and irregularities in local and global Ca2+ transients,10 which indicated that CaM binding to and CaM inhibition of RyR2 are required for a normally functioning heart.9,10
Mammalian target of rapamycin (mTOR), part of mTORC1 complex, has been implicated in cardiac hypertrophy through regulation of cell size, cell growth, and protein synthesis.11,12 Pathways associated with activation of mTOR through phosphorylation involve upstream tuberin 2 (TSC2) which is regulated by phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K) – phosphoinositide-dependent kinase-1 (PDK1) – AKT, 5′ AMP-activated protein kinase (AMPK), and extracellular signal-regulated kinases 1 and 2 (ERK1/2) signaling. mTOR activation has been linked to cardiac hypertrophy by phosphorylating downstream targets including eukaryotic initiation factor 4E binding protein 1 (4E-BP1) and ribosomal S6 kinase (p70S6K), which leads to increased phosphorylation of ribosomal protein S6 (S6).13 Rapamycin forms a complex with FK506 binding protein that binds to mTOR and inhibits the phosphorylation of mTOR.11,12,14 mTOR is also part of a second complex, mTORC2. The mTORC2 complex has been implicated in cytoskeletal organization, and in contrast to mTORC1, is poorly inhibited by rapamycin.12
The present study asked whether mTOR upstream signaling molecules and downstream targets are upregulated and contribute to cardiac hypertrophy of a mouse model with RyR2 impaired in regulation by CaM. We report that phosphorylation of mTOR at Ser-2448, p70S6 at Thr-389, S6 at Ser-240/244, and 4E-BP1 at Ser-65 increased in Ryr2ADA/ADA mice compared to wild-type mice. Treatment with rapamycin at postnatal days 3, 6, and 10 inhibited phosphorylation of mTOR, p70S6K, and S6; decreased heart size; and improved cardiac function; but did not extend the lifespan of Ryr2ADA/ADA mice.
Materials and methods
Ethics statement
The study was carried out in accordance with recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. Protocols were approved by the Animal Use Committee of the University of North Carolina at Chapel Hill.
Materials
Antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA) with the exception of TSC2-S664 Bioss Products (Atlanta, GA, USA). Chemicals were obtained from Sigma-Aldrich Co (St Louis, MO, USA), unless otherwise specified.
Animals
Ryr2ADA/ADA and wild-type mice were obtained by mating Ryr2+/ADA mice.9 Mice were backcrossed at least ten times to 129/SvEv genetic background.
Rapamycin treatment
Rapamycin at a concentration of 50 μg/mL was dissolved in 0.2% Na carboxymethylcellulose and 0.25% polysorbate.15 Three-day-old pups were separated randomly into four groups. Rapamycin (0.5 μg/g body weight) was injected intraperitoneally in 3-, 6-, and 10-day-old wild-type and mutant pups, unless otherwise indicated. Vehicle injection served as the negative control. Animals were sacrificed 4–5 hours after the last injection.
Immunoblots
Hearts (ventricles and atria) of 1-day- and 10-day-old mice were homogenized in 20 mM imidazole, pH 7.0, 0.3 M sucrose, 0.15 M NaCl, 1 mM ethylene glycol tetraacetic acid, protease and phosphatase inhibitor cocktails (Sigma-Aldrich), 25 mM β-glycerophosphate, 5 mM NaF, and 2.5 mM NaVO4, using a Tekmar Tissumizer for 3×7 seconds at a setting of 13,500 rpm. Homogenates were stored in small aliquots at −80°C. Protein concentrations were determined using bicinchoninic acid assay. Homogenates (20 μg protein/lane) were separated by SDS/PAGE, transferred to nitrocellulose membranes, and probed using rabbit antibodies. Western blots were developed using enhanced chemiluminescence and quantified using ImageQuantTL Analysis Software. Data were obtained by analyzing proteins from at least three hearts for each genotype. GAPDH was the loading control.
Echocardiography
To determine left ventricular cardiac function, transthoracic M-mode echocardiography was performed on restrained, unanesthetized 10-day-old mice, using 770 high-resolution imaging system VisualSonics (Toronto, ON, Canada) with a 40 MHz probe. Mice were restrained by taping down gently to a warmed mouse board made by Indus Industries for VisualSonics.
[3H]Ryanodine binding
Ryanodine specifically binds to ryanodine receptors and is widely used as a probe of channel activity.16 Effects of rapamycin on RyR2 activity were assessed by incubating cardiac muscle homogenates for 20 hours at 24°C in 20 mM imidazole, pH 7.0, 0.25 M KCl, 100 μM Ca2+, 5 mM oxidized glutathione, and 5 nM [3H]ryanodine in absence and presence of 0.5 μg/mL rapamycin.9 Nonspecific binding was determined using 1,000-fold excess of unlabeled ryanodine.
Statistical analysis
Statistical analysis was performed using Sigma-plot 11.0. A t-test was used to compare two groups and analysis of variance to analyze significance among multiple groups. Statistical significance is indicated by P<0.05.
Results
Protein and phosphorylation levels of mTOR and downstream targets
Mice impaired in CaM regulation of the cardiac muscle RyR2 ion channel (Ryr2ADA/ADA mice) develop cardiac hypertrophy and reduced cardiac function by postnatal day 1, and die 2–3 weeks after birth.9 To determine the role of mTOR signaling, hearts were collected from 1-day- and 10-day-old Ryr2ADA/ADA mice. Protein and phosphorylation levels of mTOR and downstream targets were determined by immunoblot analysis (Figure 1A and B). p-mTOR-Ser-2448/mTOR protein ratios increased 1.4- and 1.5-fold in 1-day- and 10-day-old Ryr2ADA/ADA hearts compared to wild-type hearts, respectively (Figure 1C and D), without significant changes in mTOR protein levels (Figure 1E and F).
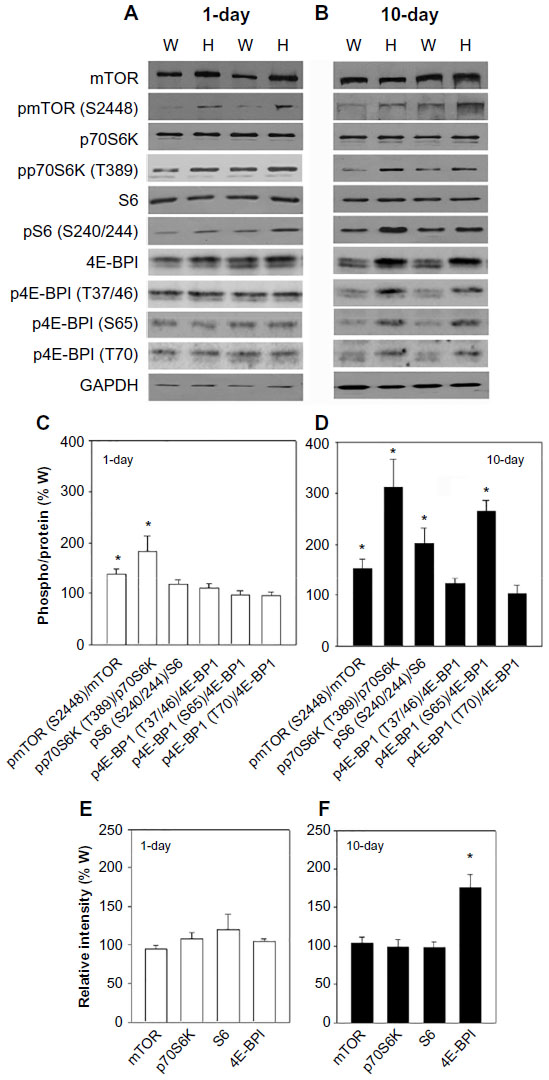 | Figure 1 Protein and phosphorylation levels of mTOR and downstream targets. |
Activation of mTOR leads to the phosphorylation of eukaryotic translation initiation factor eIF4E-binding protein 1 (4E-BP1) and p70S6K which phosphorylates S6.11,12 Figure 1C and D show that pp70S6K-Thr-389/p70SK protein ratios were increased 1.8- and 3.1-fold in 1- and 10-day-old mutant hearts compared to wild-type, respectively. p70S6K protein levels were not altered (Figure 1E and F). pS6-Ser-240/244/S6 protein ratios were comparable in 1-day-old mutant and wild-type hearts but increased twofold in 10-day-old mutant hearts compared to wild-type mice (Figure 1C and D).
4E-BP1 phosphorylation involves an initial phosphorylation of Thr-37 and Thr-46, followed by phosphorylation at Ser-65 and Thr-70.17 Immunoblot analysis showed unaltered protein levels and phosphorylation levels of 4E-BP1 at Thr-37/46, Ser-65, and Thr-70 in 1-day-old mutant hearts compared to wild-type hearts (Figure 1A, C, and E). At day 10, 4E-BP1 protein content increased 1.8-fold, and 4E-BP1 phosphorylation increased 2.2-fold at Thr-37/Thr-46, 4.5-fold at Ser-65, and 1.8-fold at Thr-70, with respective phosphorylation/protein ratios of 1.2, 2.7, and 1.0 (Figure 1B, D, and F). The results of Figure 1 suggest that impaired CaM regulation of RyR2 enhances phosphorylation of mTOR and mTOR downstream targets p70S6K, S6, and 4E-BP1.
Protein and phosphorylation levels of PI3K-PDK1-AKT signaling
The mTORC1 signaling complex is activated by PI3K-PDK1-AKT-TSC2 signaling.11,12 The PI3K family has been implicated in cardiac hypertrophy and heart failure.18 Upon activation by growth factors, PI3Ks phosphorylate the 3-hydroxyl group of phosphatidylinositols, producing phosphatidylinositol (3,4,5)-trisphosphate (PIP3),19 which leads to the phosphorylation and activation of PDK1 and AKT. There were significant increases in the phosphorylation to protein ratio of subunit p85, pPI3Kp85-Tyr-458/PI3Kp85, and the protein content of catalytic subunit p110α in the hearts of 10-day-old Ryr2ADA/ADA mice (Figure 2A–C), but no significant changes in 1-day-old mice (Figure 2D–F), nor were the protein levels of PI3Kp85 significantly increased in 1-day- and 10-day-old mice (Figure 2B and E). Phosphatase and tensin homolog (PTEN) negatively regulates PI3K signaling by dephosphorylating PIP3. It has a carboxy-terminal regulatory domain with several phosphorylation sites including Ser-380 that regulate PTEN stability and activity.20 We observed comparable protein levels and pPTEN-Ser-380/PTEN protein ratios in 1-day- and 10-day-old mutant and wild-type mice (Figure 2).
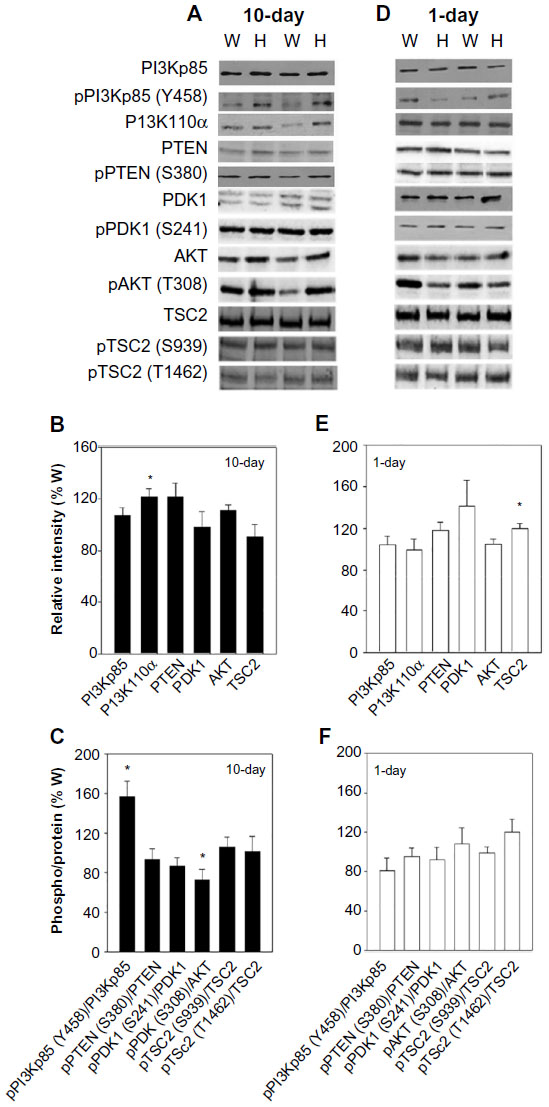 | Figure 2 Protein and phosphorylation levels of PI3K-PDK1-AKT signaling. |
PDK1 and AKT protein levels and pPDK1-Ser-241/PDK1 protein ratios were comparable in 1-day- and 10-day-old wild-type and Ryr2ADA/ADA mice (Figure 2). pAKT-Thr-308/AKT protein ratio was decreased in 10-day-old Ryr2ADA/ADA mice, whereas no changes were detected in the phosphorylation/protein ratio at day 1 (Figure 2C and F). AKT phosphorylation of TSC2 at Ser-939 and Thr-1462 activates mTOR through additional signaling molecules.21 No significant changes in pTSC2-Ser-939/TSC2 and pTSC2-Thr-1462/TSC2 protein ratios were observed in 1-day- and 10-day-old hearts (Figure 2C and F).
Protein and phosphorylation levels of AMPK and ERK1/2 signaling
We considered the possibility that two alternative pathways involving AMPK and ERK1/2 protein kinases regulate mTOR activity by phosphorylating TSC2.11,12 Figure 3A–D show that AMPK protein levels were not altered in 1-day- and 10-day-old hearts. The pAMPK-Thr-172/AMPK protein ratio decreased in 1-day-old mutant hearts, whereas in 10-day-old mutant hearts there were no significant changes in pAMPK- Thr-172/AMPK protein ratio or pTSC2-Ser-1387/TSC2 protein ratio (Figure 3E and F) at a site distinct from those phosphorylated by AKT or ERK.21 Protein and phosphorylation levels of ERK1/2 at Thr-202/Tyr-204 were not altered in 1-day- and 10-day-old hearts (Figure 3). Furthermore, pTSC2-Ser-664/TSC2 protein ratio at a site specific for ERK1/2 was not altered in 1-day-old mutant hearts and decreased in 10-day-old mutant hearts (Figure 3E and F). The results of Figures 2 and 3 suggest that PI3K-PDK1-AKT, AMPK, and ERK1/2 signaling pathways do not have a significant role in the upregulation of mTOR activity in the mutant hearts.
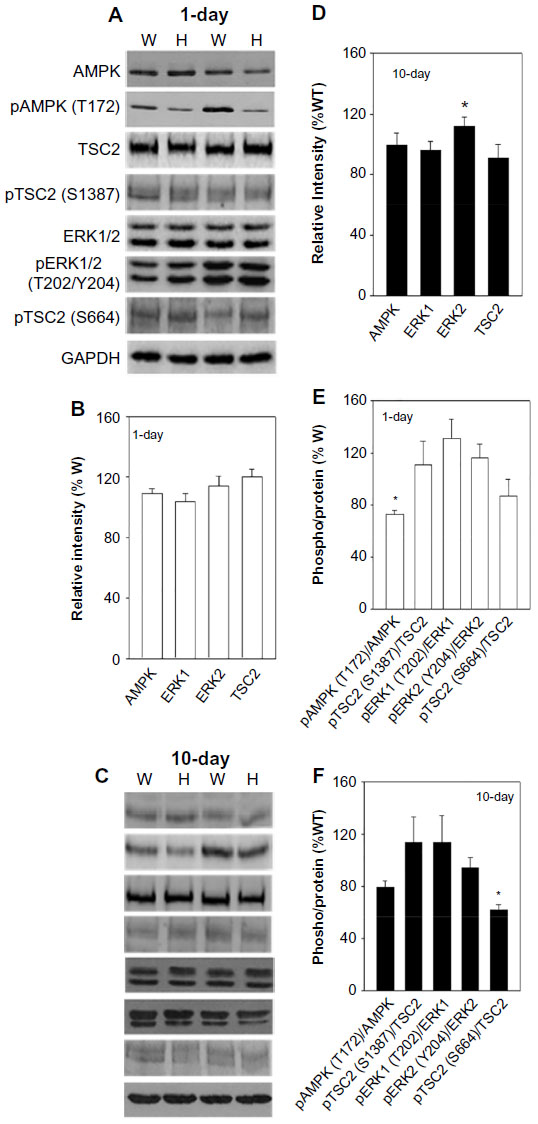 | Figure 3 Protein and phosphorylation levels of AMPK and ERK1/2 signaling. |
Effects of mTOR inhibitor rapamycin
To investigate the in vivo role of mTOR in Ryr2ADA/ADA mice, we used the mTOR inhibitor rapamycin. In initial experiments, rapamycin concentrations ranging from 0.25–10 μg/g body weight were injected in wild-type and mutant mice at postnatal days 3, 6, and 10. Vehicle injection served as a control. An optimal rapamycin concentration was 0.5 μg/g body weight, as it induced no change in the body weight of mutant and wild-type mice (Figure 4). However, heart weight and heart-to-body weight ratio significantly decreased in Ryr2ADA/ADA mice with no significant changes in wild-type mice (Figure 5). Rapamycin did not affect liver- and lung-to-body weight ratios (data not shown).
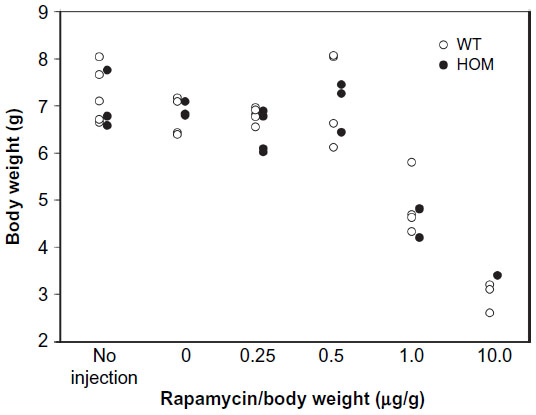 | Figure 4 Body weights of WT and HOM mice as a function of rapamycin concentration. |
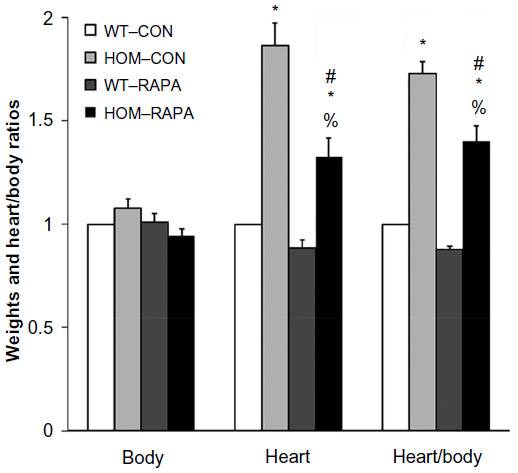 | Figure 5 Body and heart weights of 10-day-old WT and mutant mice without and with rapamycin treatment. |
Rapamycin (0.5 μg/g body weight) did not increase the lifespan of Ryr2ADA/ADA mice, with mean lifetimes of 13.5±0.5 days (n=12) and 13.9±0.5 days (n=10) for mutant mice treated or not treated with rapamycin, respectively. Echocardiography indicated that rapamycin significantly reduced interventricular septum diastolic and systolic thicknesses, left ventricular posterior wall at end systole, and heart rate in wild-type mice, without significantly changing these parameters in Ryr2ADA/ADA mice (Table 1). Rapamycin significantly improved left ventricular dimension at end systole (1.86 vs 2.71 without rapamycin), fractional shortening (20.5% vs 8.9% without rapamycin, as determined by t-test), and ejection fraction (43.0% vs 22.6% without rapamycin) in 10-day-old Ryr2ADA/ADA mice (Table 1). Values obtained for wild-type remained similar with and without rapamycin treatment (0.35 vs 0.36, 72.5% vs 76.3%, and 96.6% vs 97.6%, respectively).
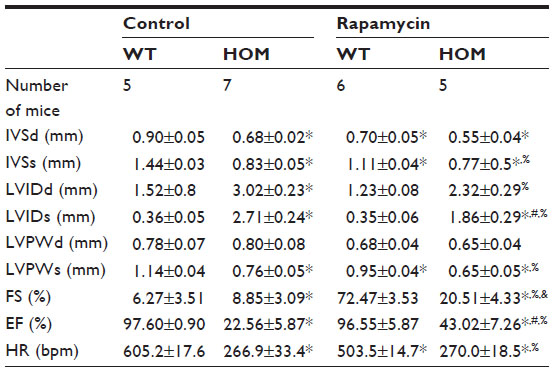 | Table 1 Echocardiography of 10-day-old WT and mutant mice treated without and with rapamycin |
Rapamycin reduced mTOR protein levels in 10-day-old wild-type hearts, whereas p70S6K and S6 protein levels were not significantly altered among the four groups of animals (Figure 6A and C). Phosphorylation/protein ratios were lowered 1.7-fold for mTOR at Ser-2448 and fourfold for p70S6K at Thr-389 in Ryr2ADA/ADA hearts without a significant change in wild-type hearts (Figure 6B). A decrease was detected in the pS6-Ser-240/244/S6 protein ratio in both mutant and wild-type hearts (eightfold in mutant hearts vs fourfold in wild-type). Rapamycin increased 4E-BP1 protein levels in mutant mice (Figure 6A and C). 4E-BP1 phosphorylation levels at Thr-37/Thr-46 (1.4- vs 1.5-fold with rapamycin), Ser-65 (1.8- vs 1.7-fold with rapamycin), and Thr-70 (1.4- vs 2.3-fold with rapamycin) were not significantly altered in mutant hearts compared to wild-type hearts. The respective phosphorylation to protein ratios are shown in Figure 6B. No significant changes were observed between control and rapamycin-treated animals. Taken together, the results suggest that in rapamycin-treated Ryr2ADA/ADA mice, the remaining hypertrophic response may be linked to increased 4E-BP1 protein and phosphorylation levels.
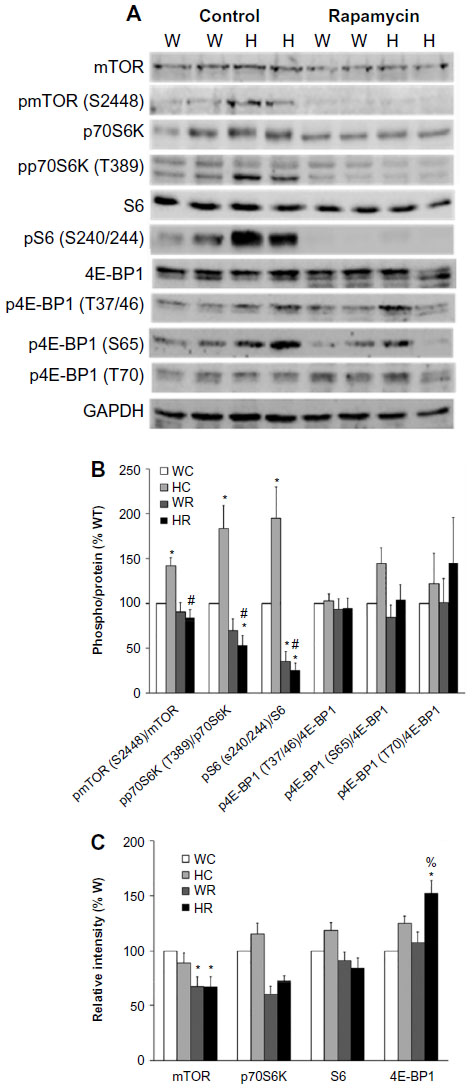 | Figure 6 Protein and phosphorylation levels of mTOR and downstream targets of W and Ryr2ADA/ADA mice treated with and without rapamycin. |
A potential limitation was that rapamycin activated RyR2.22 We found that RyR2 activity, determined by a [3H]ryanodine ligand binding assay using wild-type and mutant heart homogenates (see “Materials and methods”), was not increased (49.4±4.6 and 38.0±3.2 fmol/mg protein of four individual homogenates in absence and presence of rapamycin, respectively, P=0.09) by the low rapamycin concentration (0.5 μg/mL) used in our mouse studies. Furthermore, rapamycin did not increase but rather significantly decreased mTOR protein levels and pS6-Ser-240/244 protein ratio in wild-type mice.
Discussion
The present study investigated the role of signaling molecules associated with the mTORC1 complex in cardiac hypertrophy of a mouse model with well-defined mutations in the cardiac ryanodine receptor. There was a significant increase in mTOR phosphorylation and downstream substrates 4E-BP1 and p70S6K that have been linked to cardiac hypertrophy. On the other hand, upstream kinase activities associated with mTOR activation through phosphorylation were not altered. There was no significant increase in upstream TSC2 phosphorylation which is regulated by PDK1-AKT, AMPK, and ERK signaling pathways (Figure 7). An intriguing finding was that rapamycin reduced heart-to-body weight ratio and improved cardiac function but failed to extend the lifespan of Ryr2ADA/ADA mice. Rapamycin attenuated mTOR, p70S6K, and S6 but not 4E-BP1 phosphorylation (Figure 7). The results suggest that mTOR signaling participates in the development of cardiac hypertrophy in mice with a dysfunctional ryanodine receptor.
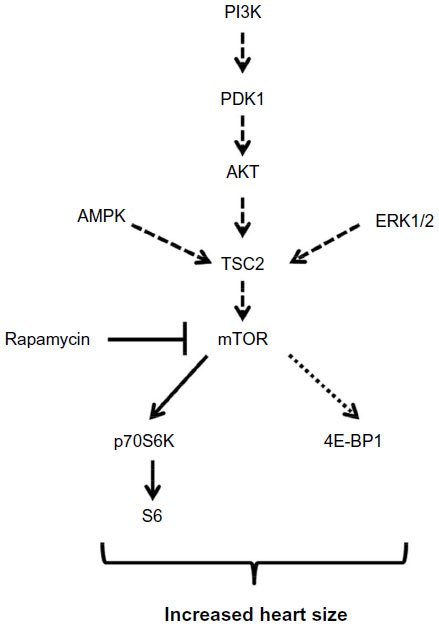 | Figure 7 Schematic representation of mTOR signaling in the hearts of Ryr2ADA/ADA mice. |
An early upregulation of ERK1/2 signaling and reduced GSK3-β activity occurred in hearts of embryonic day-16.5 (E16.5) Ryr2ADA/ADA mice.23 Class II histone deacetylase/myocyte enhancer factor-2 signaling and NFAT transcriptional activity, determined in mice carrying luciferase transgene driven by NFAT promoter, were upregulated in the hearts of 1-day-old hearts but not the hearts of embryonic day-16.5 Ryr2ADA/ADA mice.9,23 Calcineurin A–β ablation modestly decreased heart weight and improved cardiac contractility, and extended twofold the lifespan of Ryr2ADA/ADA mice; however, it did so without suppressing the expression of genes associated with cardiac hypertrophy.23 The results suggest that multiple signaling pathways participate in the development of cardiac hypertrophy and heart failure in mice expressing a cardiac ryanodine receptor impaired in regulation by CaM.
An early role for mTOR signaling in hypertrophy of Ryr2ADA/ADA hearts is suggested by the increase in mTOR-Ser-2448 and p70S6K-Thr-389 phosphorylation at day 1 of age. A nearly twofold reduction in mTOR phosphorylation by rapamycin confirmed a role in the hypertrophic phenotype of Ryr2ADA/ADA mice. In two studies using a pressure-overload mouse model with aortic constriction,15,24 rapamycin reduced left ventricular weight/body weight ratio,24 improved cardiac function,24 and reduced p70S6K15 and S624 phosphorylation levels. In the present study, 4- and 8-fold reduction in pS6-Ser-240/Ser244 phosphorylation to S6 protein ratio in wild-type and mutant hearts, respectively, were associated with a partial reduction in heart weight and improved cardiac performance in 10-day-old Ryr2ADA/ADA mice, with no significant changes in heart weight and performance in wild-type mice. Rapamycin did not reduce 4E-BP1 phosphorylation, in agreement with a study where only transient inhibition of 4E-BP1 phosphorylation by rapamycin was seen.25 The maintained increase in 4E-BP1 phosphorylation may have only partially reduced heart weight in rapamycin-treated Ryr2ADA/ADA mice compared to wild-type mice. Further studies will be required to identify the pathways that upregulate mTOR signaling in Ryr2ADA/ADA mice.
Several signaling pathways upstream of mTOR have been implicated in mTOR activation through phosphorylation of TSC2 and PI3K-PDK1-AKT, AMPK, and ERK signaling pathways.11,12 The pPI3Kp85-Tyr-458 to PI3Kp85 protein ratio and the pI3K110α protein content increased in Ryr2ADA/ADA mice but there was no increase in phosphorylation/protein ratios of TSC2 by AKT, AMPK, and ERK signaling pathways.
In mouse cardiomyocytes, CaM is predominantly bound to RyR2.26 Naturally occurring mutations in the conserved RyR2-CaM binding domain that impair CaM inhibition of sarcoplasmic reticulum Ca2+ release in cardiac pathologies have not been previously reported. However, several RyR2 and CaM mutations are associated with stress-induced ventricular arrhythmias.27–31 PKA-mediated phosphorylation at RyR2-Ser-2808 of the RyR2-Ser-2474 mutant reduced the binding affinity for CaM.32 A modified Gly-Ser-His-CaM that increased CaM binding affinity restored normal function in cAMP-treated mutant cardiomyocytes33 and pacing-induced failing hearts.34 CaM mutants with decreased Ca2+ binding affinity,27,29–31 decreased binding to RyR2,27 and increased single channel activity and binding affinity to RyR235 were associated with ventricular arrhythmias. The results support a regulatory relationship between CaM and RyR2 in hearts.
In conclusion, our studies provide new mechanistic insights into the pathogenesis of a mouse model impaired in CaM regulation of RyR2. Our studies show that upregulation of mTOR signaling contributes to the pathological effects of the RyR2 mutation in Ryr2ADA/ADA mice. Treatment with the mTOR inhibitor rapamycin attenuated heart weight-to-body weight ratio and improved cardiac performance but was not sufficient to extend the lifespan of Ryr2ADA/ADA mice. The results suggest that enlargement of heart size and heart failure are likely mediated through different signaling mechanisms in Ryr2ADA/ADA mice.
Acknowledgment
The work was supported by National Heart, Lung, and Blood Institute Grant HL-073501.
Disclosure
The authors report no conflicts of interest in this work.
References
Wehrens XH, Lehnart SE, Marks AR. Intracellular calcium release and cardiac disease. Annu Rev Physiol. 2005;67:69–98. | |
Meissner G. Molecular regulation of cardiac ryanodine receptor ion channel. Cell Calcium. 2004;35:621–628. | |
Hamilton SL, Serysheva II. Ryanodine receptor structure: progress and challenges. J Biol Chem. 2009;284:4047–4051. | |
Fruen BR, Bardy JM, Byrem TM, Strasburg GM, Louis CF. Differential Ca(2+) sensitivity of skeletal and cardiac muscle ryanodine receptors in the presence of calmodulin. Am J Physiol Cell Physiol. 2000;279:C724–C733. | |
Balshaw DM, Xu L, Yamaguchi N, Pasek DA, Meissner G. Calmodulin binding and inhibition of cardiac muscle calcium release channel (ryanodine receptor). J Biol Chem. 2001;276:20144–20153. | |
Dorn GW 2nd, Force T. Protein kinase cascades in the regulation of cardiac hypertrophy. J Clin Invest. 2005;115:527–537. | |
Eder P, Molkentin JD. TRPC channels as effectors of cardiac hypertrophy. Circ Res. 2011;108:265–272. | |
Kudoh S, Akazawa H, Takano H, et al. Stretch-modulation of second messengers: effects on cardiomyocyte ion transport. Progr Biophys Mol Biol. 2003;82:57–66. | |
Yamaguchi N, Takahashi N, Xu L, Smithies O, Meissner G. Early cardiac hypertrophy in mice with impaired calmodulin regulation of cardiac muscle Ca2+ release channel. J Clin Invest. 2007;117:1344–1353. | |
Arnáiz-Cot JJ, Damon BJ, Zhang XH, et al. Cardiac calcium signaling pathologies associated with defective calmodulin regulation of type 2 ryanodine receptor. J Physiol. 2013;591:4287–4299. | |
Foster KG, Fingar DC. Mammalian target of rapamycin (mTOR): conducting the cellular signaling symphony. J Biol Chem. 2010;285:14071–14077. | |
Ma XM, Blenis J. Molecular mechanisms of mTOR mediated translational control. Nat Rev Mol Cell Biol. 2009;10:307–318. | |
McMullen JR, Shioi T, Zhang L, et al. Deletion of ribosomal S6 kinases does not attenuate pathological, physiological, or insulin-like growth factor 1 receptor-phosphoinositide 3-kinase-induced cardiac hypertrophy. Mol Cell Biol. 2004;24:6231–6240. | |
Banaszynski LA, Liu CW, Wandless TJ. Characterization of the FKBP.rapamycin.FRB ternary complex. J Am Chem Soc. 2005;127:4715–4721. | |
Shioi T, McMullen JR, Tarnavski O, et al. Rapamycin attenuates load-induced cardiac hypertrophy in mice. Circulation. 2003;107:1664–1670. | |
Sutko JL, Airey JA, Welch W, Ruest L. The pharmacology of ryanodine and related compounds. Pharmacol Rev. 1997;49:53–98. | |
Gingras AC, Raught B, Gygi SP, et al. Hierarchical phosphorylation of the translation inhibitor 4E-BP1. Genes Dev. 2001;15:2852–2864. | |
Aoyagi T, Matsui T. Phosphoinositide-3 kinase signaling in cardiac hypertrophy and heart failure. Curr Pharm Des. 2011;17:1818–1824. | |
Vanhaesebroeck B, Stephens L, Hawkins P. PI3K signalling: the path to discovery and understanding. Nat Rev Mol Cell Biol. 2012;13:195–203. | |
Torres J, Pulido R. The tumor suppressor PTEN is phosphorylated by the protein kinase CK2 at its C terminus. Implications for PTEN stability to proteasome-mediated degradation. J Biol Chem. 2001;276:993–998. | |
Huang J, Manning BD. The TSC1-TSC2 complex: a molecular switchboard controlling cell growth. Biochem J. 2008;412:179–190. | |
Kaftan E, Marks AR, Ehrlich BE. Effects of rapamycin on ryanodine receptor/Ca(2+)-release channels from cardiac muscle. Circ Res. 1996;78:990–997. | |
Yamaguchi N, Chakraborty A, Pasek DA, Molkentin JD, Meissner G. Dysfunctional ryanodine receptor and cardiac hypertrophy: role of signaling molecules. Am J Phys Heart Circ Physiol. 2011;300:H2187–H2895. | |
Gao XM, Wong G, Wang B, et al. Inhibition of mTOR reduces chronic pressure-overload cardiac hypertrophy and fibrosis. J Hypertens. 2006;24:1663–1670. | |
Choo AY, Yoon SO, Kim SG, Roux PP, Blenis J. Rapamycin differentially inhibits S6Ks and 4E-BP1 to mediate cell-type-specific repression of mRNA translation. Proc Natl Acad Sci U S A. 2008;105:17414–17419. | |
Yang Y, Guo T, Oda T, et al. Cardiac myocyte Z-line calmodulin is mainly RyR2-bound, and reduction is arrhythmogenic and occurs in heart failure. Circ Res. 2014;114:295–306. | |
Nomikos M, Thanassoulas A, Beck K, et al. Altered RyR2 regulation by the calmodulin F90L mutation associated with idiopathic ventricular fibrillation and early sudden cardiac death. FEBS Lett. 2014;588:2898–2902. | |
Hwang HS, Nitu FR, Yang Y, et al. Divergent regulation of ryanodine receptor 2 calcium release channels by arrhythmogenic human calmodulin missense mutants. Circ Res. 2014;114:1114–1124. | |
Nyegaard M, Overgaard MT, Søndergaard MT, et al. Mutations in calmodulin cause ventricular tachycardia and sudden cardiac death. Am J Hum Genet. 2012;91:703–712. | |
Makita N, Yagihara N, Crotti L, et al. Novel calmodulin mutations associated with congenital arrhythmia susceptibility. Circ Cardiovasc Genet. 2014;7:466–474. | |
Crotti L, Johnson CN, Graf E, et al. Calmodulin mutations associated with recurrent cardiac arrest in infants. Circulation. 2013;127:1009–1017. | |
Xu X, Yano M, Uchinoumi H, et al. Defective calmodulin binding to the cardiac ryanodine receptor plays a key role in CPVT-associated channel dysfunction. Biochem Biophys Res Comm. 2010;394:660–666. | |
Fukuda M, Yamamoto T, Nishimura S, et al. Enhanced binding of calmodulin to RyR2 corrects arrhythmogenic channel disorder in CPVT-associated myocytes. Biochem Biophys Res Comm. 2014;448:1–7. | |
Hino A, Yano M, Kato T, et al. Enhanced binding of calmodulin to the ryanodine receptor corrects contractile dysfunction in failing hearts. Cardiovasc Res. 2012;96:433–443. | |
Hwang SY, Wei J, Westhoff JH, et al. Differential functional interaction of two Vesl/Homer protein isoforms with ryanodine receptor type 1: a novel mechanism for control of intracellular calcium signaling. Cell Calcium. 2003;34:177–184. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.