")
Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 13
Morphometric analysis of mitochondria in lymphocytes of patients with exacerbations of chronic obstructive pulmonary disease – pilot study
Authors Białas AJ , Liberski PP, Zielińska A, Kumor-Kisielewska A, Szewczyk K, Miłkowska-Dymanowska J, Sitarek P, Piotrowski WJ , Górski P
Received 20 January 2018
Accepted for publication 5 April 2018
Published 30 July 2018 Volume 2018:13 Pages 2313—2318
DOI https://doi.org/10.2147/COPD.S163249
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Richard Russell
Adam J Białas,1 Paweł P Liberski,2 Anna Zielińska,2 Anna Kumor-Kisielewska,1,3 Karolina Szewczyk,1,3 Joanna Miłkowska-Dymanowska,1 Przemysław Sitarek,4 Wojciech J Piotrowski,1 Paweł Górski1
1Department of Pneumology and Allergy, Medical University of Łódź, Łódź, Poland; 2Department of Molecular Pathology and Neuropathology, Medical University of Łódź, Łódź, Poland; 3Laboratory of Respiratory Immunopathology, Department of Pneumology and Allergy, Medical University of Łódź, Łódź, Poland; 4Department of Biology and Pharmaceutical Botany, Medical University of Łódź, Łódź, Poland
Introduction: Exacerbations of chronic obstructive pulmonary disease (ECOPD) are important events in the course of the disease, negatively influencing health status and disease progression. Therefore, there is a strong need for deeper understanding of the pathology of ECOPD to elaborate new therapeutic approaches and ameliorate prognoses. Contributions of mitochondria to pathobiology of COPD are still under investigation, although growing evidence suggests their important role in this disease. The aim of our study was to assess the morphometric parameters of mitochondria in lymphocytes of patients with ECOPD.
Patients and methods: Lymphocytes were isolated from the peripheral blood of patients with COPD. Transmission electron microscopy was used to assess absolute number of mitochondria per cell, mitochondrial content, and morphometric parameters of individual mitochondria. We also counted indexes for elongation and interconnectivity.
Results: Eighteen patients (9 with ECOPD and 9 in the stable period of the disease) were analyzed. We observed significantly lower length of mitochondrion (P=0.03) and significant decrease both in elongation (P=0.03) and interconnectivity indexes (P=0.04) in ECOPD patients.
Conclusions: The morphometric parameters of mitochondria in lymphocytes derived from patients during the early period of ECOPD requiring hospitalization are altered in comparison to patients in the stable period of the disease. This suggests their contribution to pathobiology of ECOPD. These preliminary outcomes should be further validated in larger size samples.
Keywords: mitochondria, exacerbation of chronic obstructive pulmonary disease, ECOPD, T lymphocytes, apoptosis
Introduction
COPD is characterized by persistent respiratory symptoms and airflow limitation that is due to airway and/or alveolar abnormalities, usually caused by significant exposure to noxious particles or gases.1 Summarizing the contemporary knowledge, we should recognize the pathobiology of COPD as involving a complex interaction between several factors, including genetic vulnerability.2–9
Exacerbations of COPD (ECOPD) are important events in the course of the disease. They negatively impact health status, rates of hospitalization and readmission, and disease progression.1,10,11 Exacerbations requiring hospitalization are associated with significant mortality – eg, Connors et al reported that ECOPD with hypercapnia was burdened by in-hospital mortality rate of 11%.12 Long-term prognosis following hospitalization for ECOPD is also poor – according to a meta-analysis by Hoogendoorn et al the average in-hospital mortality rate was 6.7%, the average mortality rates at 3 and 6 months were 18% and 26%, respectively, and 51% at 5 years.13 This is why there is a strong need for deeper understanding of the pathology of ECOPD to elaborate new therapeutic approach and ameliorate prognoses.
There is evidence that adaptive immune response plays an important role in pathogenesis of COPD.14 Curtis et al proposed that the severity and course of ECOPD reflects the success of the adaptive immune response in appropriately modulating the innate response to pathogen-related molecular patterns (“the Goldilocks hypothesis”).15 The fundamental actors in adaptive immunity are T-lymphocytes. T-lymphocytes may contribute to immunopathology of COPD.16 There is also evidence for increased apoptosis of peripheral T-lymphocytes in patients with COPD.17 Apoptosis is a crucial mechanism for the regulation of normal cell turnover; however, increased T-lymphocytes apoptosis may be responsible for defective immune response, which contributes to the high frequency of infections among patients with COPD.18 Moreover, excessive rates of apoptosis may result in unbalanced cellular homeostasis, defective clearance of apoptotic material, secondary necrosis, and perpetuation of the inflammatory response. Lim et al reported significantly higher apoptosis of T-lymphocytes among patients with exacerbation than in the stable period.17
There is also growing evidence for the role of B lymphocytes in COPD – studies have demonstrated increases in B cell counts and in the number and size of B cell-rich lymphoid follicles in COPD lung that correlate directly with COPD severity. There are also increases in lung levels of mediators that promote B cell maturation, activation, and survival in COPD patients.19
Contribution of mitochondria to pathobiology of COPD are still under investigation; however, growing evidence suggests their important role in this disease.8 The aim of our study was to assess the morphometric parameters of mitochondria in lymphocytes of patients with ECOPD.
Methods
Subjects were recruited from the Department of Pneumology and Allergy of the Medical University of Łódź and the Pulmonary Outpatient Service. The study protocol was approved by the Ethical Committee for Human Studies of the Medical University of Łódź (RNN/360/15/KE). All patients provided written informed consent for participation in the study.
The study period was January to September 2016. The study group consisted of patients in early period of ECOPD. We recognized ECOPD as an acute worsening of respiratory symptoms that result in additional therapy.1,10,11 Only patients with exacerbations requiring hospitalization were enrolled to our study. Most of the admitted patients had received initial medical management to which they had not responded; however, patients who took systemic steroids or home oxygen therapy were excluded from our study.
Ex-smoking patients (with smoking history of at least 20 pack-years) above 40 years of age, with confirmed COPD diagnosis were enrolled in our study. The control group consisted of stable COPD patients, defined as patients without exacerbation for the previous 3 months.
The exclusion criteria were: insufficient data confirming COPD diagnosis, other confirmed coexisting chronic pulmonary disease (eg, idiopathic pulmonary fibrosis, sarcoidosis, primary pulmonary hypertension), acute pulmonary embolism, active tuberculosis, home oxygen therapy, pre-admission systemic steroid therapy, obesity (BMI≥30 kg/m2), confirmed α-1-antytrypsin deficiency, any active malignancy, and pharmacotherapy by drugs with any confirmed, significant influence on mitochondrial functioning.
From each included patient, 5.4 mL of peripheral venous blood was collected into tubes containing ethylenediaminetetraacetic acid (S-Monovette, Sarstedt AG & Co. KG, Nümbrecht, Germany). Lymphocytes were isolated at room temperature by means of gradient centrifugation in Gradisol L (Aqua-Med, Łódź, Poland) and immediately transferred to the laboratory of electron microscopy.
The cells were washed in phosphate buffer and in 0.1 M cacodylate buffer (pH 7.4), after which they were fixed for 2 h with 2% glutaraldehyde/0.1 M cacodylate buffer. After rinsing in 0.1 M cacodylate buffer (pH 7.41), postfixation in 1% OsO4/0.1 M cacodylate buffer was performed. Specimens were washed in 0.1 M cacodylate buffer, and dehydrated using a 70%–100% graded ethanol series and propylene oxide. The cells were embedded in Epon 812 (SERVA, Heidelberg, Germany) and ultrathin-sections were made. Specimens were examined with Jeol 1100 transmission electron microscope (JEOL Ltd., Tokyo, Japan).
Electron micrographs were analyzed using ImageJ software (1.51m9; Wayne Rasband, National Institutes of Health, Bethseda, MD, USA). Measurements were generated from images with a magnification of ×20,000. Higher magnifications (up to ×1,000,000) were obtained for detailed morphological assessment. Morphometric parameters were measured by tracing mitochondria. A tracing tablet was used to increase precision of the tracing process. All measurements were performed three times and average of values was counted. We calculated an absolute number of mitochondria per cell and mitochondrial content (percent of cytosol occupied by mitochondria). Using perimeter and areas of individual mitochondria (in μm2), we counted indexes for elongation (square of the length divided by the area), as described by Han et al,20 and for interconnectivity (average area/perimeter ratio), as reported by Dagda et al.21
Additionally, we performed a semi-quantitative morphological assessment, using a classification adopted from a study by Bishop et al22 (Grade 0 – no evidence of cellular pathology or early autolysis, or an occasional mitochondrion with minimal loss of cristae while the remainder of mitochondria appear normal; Grade 1 – discontinuous cristal membranes and/or partial loss of cristae and matrix material in a few mitochondria; Grade 2 – multiple disruptions of the cristal membrane and substantial loss of cristae and matrix in approximately half of the mitochondria; Grade 3 – fragmented cristal membranes and effacement of central architecture in a majority of the mitochondria). The grading data were not subjected to statistical analysis.
Continuous data were presented as the mean with SD or median with interquartile range (IQR), depending on distribution of data. Variables were compared using the bootstrap-boosted inference tests: the unpaired Student’s t-test or the Mann–Whitney’s U-test, depending on data normality and variance homogeneity. Categorical variables were compared using the chi-square test with Yates’s correction for continuity.
Data were analyzed using R software for MacOS.23
Results
Eighteen patients (9 with ECOPD and 9 in the stable period of the disease; 7 females), were analyzed. Study and control groups were not statistically significant different according to age: 63 (60–77) vs 69 (65–72) years (P=0.75), sex (P=1.0) and smoking history: 45 (40–50) vs 40 (40–45) pack-years (P=0.29). All patients in the study presented GOLD “D” category of COPD severity.
We found a lower number of mitochondria per cell and mitochondrial content in ECOPD group; however, results were not statistically significant. On the other hand, we observed significantly lower median of length of mitochondrion in ECOPD patients. The analysis also revealed significant decrease both in elongation and interconnectivity index in ECOPD patients. Additionally, we performed bootstrap-boosted inference, confirming previously observed significances. Detailed results of analyses are presented in Table 1.
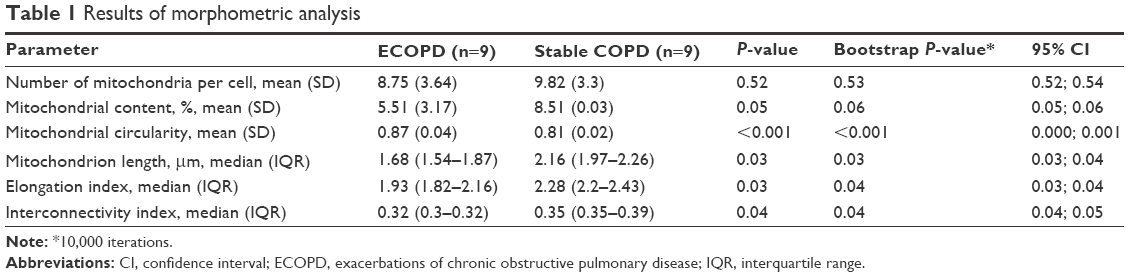 | Table 1 Results of morphometric analysis |
Lymphocytes of patients with ECOPD showed mitochondrial morphological abnormalities, including focal loss of cristae with empty spaces (Figure 1A and B), and swollen mitochondria with severe loss of cristae and matrix (Figure 1C). Elongated mitochondria were more frequently observed in stable COPD patients (Figure 1D). The results of our semi-quantitative evaluation of mitochondrial morphological pathology indicated that mitochondria in ECOPD group showed alterations generally in the 1-to-2-grade range, whereas controls generally exhibited mitochondrial alterations that ranged from grade 0 to 1.
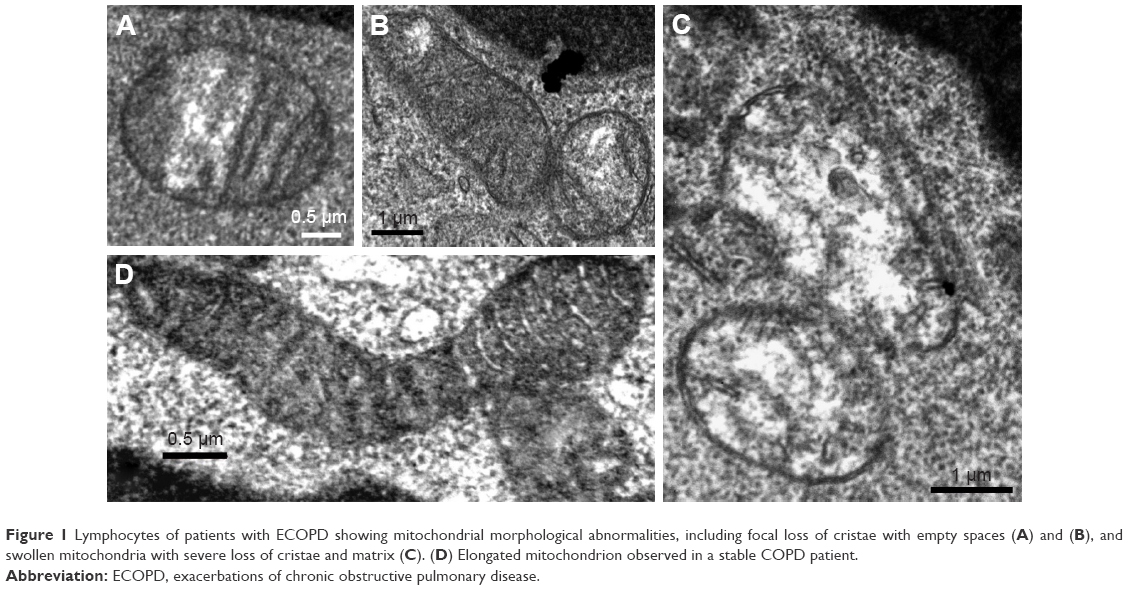 | Figure 1 Lymphocytes of patients with ECOPD showing mitochondrial morphological abnormalities, including focal loss of cristae with empty spaces (A) and (B), and swollen mitochondria with severe loss of cristae and matrix (C). (D) Elongated mitochondrion observed in a stable COPD patient. |
Discussion
We assessed morphological parameters in lymphocytes derived from patients with ECOPD in comparison to patients in the stable period of the disease. We chose the early period of ECOPD requiring hospitalization and applied wide exclusion criteria to avoid or minimize the influence of as many identifiable exogenous factors which would potentially affect mitochondrial morphology as possible.
Evidence for increased apoptosis of peripheral T-lymphocytes in patients with COPD was the rationale for our study.17,18,24 Apoptosis is a crucial mechanism for the regulation of normal cell turnover, but increased T-lymphocyte apoptosis may be responsible for defective immune response, which contributes to the high frequency of infections among patients with COPD.18 Moreover, excessive rates of apoptosis may result in unbalanced cellular homeostasis, defective clearance of apoptotic material, secondary necrosis, and perpetuation of the inflammatory response. Higher apoptosis of T-lymphocytes among patients with exacerbation than in the stable period of COPD was reported as well.17
We found significant differences between the analyzed groups in two parameters of mitochondrial dynamics – elongation and interconnectivity indexes. Both of them were significantly decreased in the ECOPD group, suggesting lower access and communication between mitochondria, and extended fragmentation of mitochondria in ECOPD patients.
Mitochondrial dynamics play an important role in functioning of these organelles, allowing mitochondria to interact with each other; without such dynamics the mitochondrial population consists of autonomous organelles that have impaired function.25 Mitochondrial fusion could be a protective mechanism – it can protect cells from harmful effects of mitochondrial DNA (mtDNA) mutations by allowing functional complementation of mtDNA gene products.25 Moreover, during autophagy, elongated mitochondria are spared from autophagic degradation, possess more cristae, increased levels of dimerization and activity of ATP synthase, and maintain ATP production.26 Hoffmann et al27 reported that prolonged cigarette smoke exposure alters mitochondrial structure and function in airway epithelial cells. The authors observed changes in mitochondrial structure, including fragmentation, branching and quantity of cristae. The majority of these changes were persistent upon cigarette smoke exposure depletion. Also, long-term cigarette smoke exposure significantly increased the expression of specific fission/fusion markers, oxidative phosphorylation proteins, and oxidative stress markers.27 Therefore, our observation of higher elongation index in patients in the stable period of the disease is in line with these findings. To our knowledge this is the first study which aims to analyze mitochondrial morphometries of ECOPD patients.
Mitochondria in lymphocytes of patients with ECOPD in our study showed morphological abnormalities including the presence of swollen mitochondria. This observation is not surprising in the context of pathobiology of ECOPD. Mitochondrial swelling is one of the key players in cytochrome c release associated with apoptotic cell death. Cytochrome c can be released from the mitochondria via both permeability transition pore dependent and independent mechanisms.28 The association between inflammation and apoptosis is well proven. On the other hand, apoptosis further promotes inflammation and oxidative stress,29 forming a kind of vicious cycle.
Several limitations of the current study deserve comment. First, the small sample size determines the character of the report as a pilot study. Such studies are long lasting, mainly due to restrictive exclusion criteria, and expensive. Some authors, to obviate the small sample size limitation, use a cell as a statistical unit. We strongly disagree with this, as it is a violation of assumption of independence of analytical units, and thus is burdened by a high bias, which would potentially influence the results. Trying to improve reliability of our results, in all non-parametric statistical tests we obtained exact P-values, and in the chi-square test we used Yates’s correction for continuity. In normally distributed data, concordantly with all assumptions, we used Student’s t-test – in fact, William Sealy Gosset, known under nickname “Student”, developed the t-test especially for small sample sizes.30 Moreover, there is evidence that the Student’s t-test can be applied even in extremely small size samples.31 Additionally, we performed bootstrap-boosted inference. Despite this, our results should be considered hypothesis generating, raising new issues about the role of mitochondria in the pathobiology of COPD, and deserving further confirmation. Secondly, it would be more valuable to compare mitochondria in lymphocytes in the same patient at two time points, ie, in stable state and during exacerbation, than using a control group. However, taking into account the above-mentioned reasons for the small sample, such an approach would be even harder to accomplish. Finally, our study is a pure morphometric study and provides no information about mitochondrial respiratory parameters, and our results should be further compared with such analyses.
Conclusion
Our study shows that morphometric parameters of mitochondria in lymphocytes derived from patients during the early period of ECOPD requiring hospitalization is altered in comparison to patients in the stable period of the disease. This suggests their contribution to pathobiology of ECOPD and the potential role in abnormalities of lymphocytes observed in ECOPD by authors of previous studies. These preliminary outcomes should be further validated in larger size samples.
Acknowledgments
The authors are grateful to all employees of the Department of Pneumology and Allergy, Laboratory of Respiratory Immunopathology and Molecular Pathology and Neuropathology for their contribution to this study. The costs of this study were defrayed from regular finances of the Department of Pneumology and Allergy and Molecular Pathology and Neuropathology of the Medical University of Łódź.
Author contributions
AJB and PG contributed to the conception and design of the study; AJB, PPL, AZ, PS, AKK, KS, JMD, WJP, and PG took part in the acquisition of data. All authors contributed to analysis and interpretation of data, drafting and critical revision of the manuscript for important intellectual content, gave approval for the version to be published, and agreed to be accountable for all aspects of the work.
Disclosure
The authors report no conflict of interests in this work.
References
Vogelmeier CF, Criner GJ, Martinez FJ, et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive lung disease 2017 report. GOLD Executive Summary. Am J Respir Crit Care Med. 2017;195(5):557–582. | ||
Hancock DB, Eijgelsheim M, Wilk JB, et al. Meta-analyses of genome-wide association studies identify multiple loci associated with pulmonary function. Nat Genet. 2010;42(1):45–52. | ||
Pillai SG, Ge D, Zhu G, et al. A genome-wide association study in chronic obstructive pulmonary disease (COPD): identification of two major susceptibility loci. PLoS Genet. 2009;5(3):e1000421. | ||
Wilk JB, Chen T-H, Gottlieb DJ, et al. A genome-wide association study of pulmonary function measures in the Framingham Heart Study. PLoS Genet. 2009;5(3):e1000429. | ||
Hunninghake GM, Cho MH, Tesfaigzi Y, et al. MMP12, lung function, and COPD in high-risk populations. N Engl J Med. 2009;361(27):2599–2608. | ||
Lambrechts D, Buysschaert I, Zanen P, et al. The 15q24/25 susceptibility variant for lung cancer and chronic obstructive pulmonary disease is associated with emphysema. Am J Respir Crit Care Med. 2010;181(5):486–493. | ||
Zöller B, Li X, Sundquist J, Sundquist K. Familial transmission of chronic obstructive pulmonary disease in adoptees: a Swedish nationwide family study. BMJ Open. 2015;5(4):e007310. | ||
Białas AJ, Sitarek P, Miłkowska-Dymanowska J, Piotrowski WJ, Górski P. The role of mitochondria and oxidative/antioxidative imbalance in pathobiology of chronic obstructive pulmonary disease. Oxid Med Cell Longev. 2016;2016:7808576. | ||
Górka K, Soja J, Jakieła B, et al. Relationship between the thickness of bronchial wall layers, emphysema score, and markers of remodeling in bronchoalveolar lavage fluid in patients with chronic obstructive pulmonary disease. Pol Arch Med Wewn. 2016;126(6):402–410. | ||
Seemungal TA, Donaldson GC, Paul EA, Bestall JC, Jeffries DJ, Wedzicha JA. Effect of exacerbation on quality of life in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1998;157(5 Pt 1):1418–1422. | ||
Wedzicha JA, Seemungal TAR. COPD exacerbations: defining their cause and prevention. Lancet. 2007;370(9589):786–796. | ||
Connors AF, Dawson NV, Thomas C, et al. Outcomes following acute exacerbation of severe chronic obstructive lung disease. The SUPPORT investigators (Study to Understand Prognoses and Preferences for Outcomes and Risks of Treatments). Am J Respir Crit Care Med. 1996;154(4 Pt 1):959–967. | ||
Hoogendoorn M, Hoogenveen RT, Rutten-van Mölken MP, Vestbo J, Feenstra TL. Case fatality of COPD exacerbations: a meta-analysis and statistical modelling approach. Eur Respir J. 2011;37(3):508–515. | ||
Gadgil A, Zhu X, Sciurba FC, Duncan SR. Altered T-cell phenotypes in chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2006;3(6):487–488. | ||
Curtis JL, Freeman CM, Hogg JC. The immunopathogenesis of chronic obstructive pulmonary disease: insights from recent research. Proc Am Thorac Soc. 2007;4(7):512–521. | ||
Gadgil A, Duncan SR. Role of T-lymphocytes and pro-inflammatory mediators in the pathogenesis of chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis. 2008;3(4):531–541. | ||
Lim SC, Ju JY, Chi SY, et al. Apoptosis of T lymphocytes isolated from peripheral blood of patients with acute exacerbation of chronic obstructive pulmonary disease. Yonsei Med J. 2011;52(4):581–587. | ||
Hodge SJ, Hodge GL, Reynolds PN, Scicchitano R, Holmes M. Increased production of TGF-beta and apoptosis of T lymphocytes isolated from peripheral blood in COPD. Am J Physiol Lung Cell Mol Physiol. 2003;285(2):L492–L499. | ||
Polverino F, Seys LJM, Bracke KR, Owen CA. B cells in chronic obstructive pulmonary disease: moving to center stage. Am J Physiol Lung Cell Mol Physiol. 2016;311(4):L687–L695. | ||
Han X-J, Lu Y-F, Li S-A, et al. CaM kinase I alpha-induced phosphorylation of Drp1 regulates mitochondrial morphology. J Cell Biol. 2008;182(3):573–585. | ||
Dagda RK, Cherra SJ, Kulich SM, Tandon A, Park D, Chu CT. Loss of PINK1 function promotes mitophagy through effects on oxidative stress and mitochondrial fission. J Biol Chem. 2009;284(20):13843–13855. | ||
Bishop JB, Tani Y, Witt K, et al. Mitochondrial damage revealed by morphometric and semiquantitative analysis of mouse pup cardiomyocytes following in utero and postnatal exposure to zidovudine and lamivudine. Toxicol Sci. 2004;81(2):512–517. | ||
R Core Team. R: A Language and Environment for Statistical Computing. Vienna, Austria; 2016. | ||
Hodge S, Hodge G, Holmes M, Reynolds PN. Increased peripheral blood T-cell apoptosis and decreased Bcl-2 in chronic obstructive pulmonary disease. Immunol Cell Biol. 2005;83(2):160–166. | ||
Chan DC. Mitochondrial fusion and fission in mammals. Annu Rev Cell Dev Biol. 2006;22:79–99. | ||
Gomes LC, Di Benedetto G, Scorrano L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol. 2011;13(5):589–598. | ||
Hoffmann RF, Zarrintan S, Brandenburg SM, et al. Prolonged cigarette smoke exposure alters mitochondrial structure and function in airway epithelial cells. Respir Res. 2013;14:97. | ||
Kaasik A, Safiulina D, Zharkovsky A, Veksler V. Regulation of mitochondrial matrix volume. Am J Physiol Cell Physiol. 2007;292(1):C157–C163. | ||
Tuder RM, Zhen L, Cho CY, et al. Oxidative stress and apoptosis interact and cause emphysema due to vascular endothelial growth factor receptor blockade. Am J Respir Cell Mol Biol. 2003;29(1):88–97. | ||
Student. The probable error of a mean. Biometrika. 1908;(6):1–25. | ||
de Winter JCF. Using the Student’s t-test with extremely small sample sizes. Pract Assess Res Eval. 2013;18(10):1–12. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.