")
Back to Journals » OncoTargets and Therapy » Volume 10
Monosomal karyotype in myeloid neoplasias: a literature review
Authors Anelli L , Pasciolla C, Zagaria A, Specchia G, Albano F
Received 4 February 2017
Accepted for publication 23 March 2017
Published 20 April 2017 Volume 2017:10 Pages 2163—2171
DOI https://doi.org/10.2147/OTT.S133937
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Samir Farghaly
Luisa Anelli, Crescenza Pasciolla, Antonella Zagaria, Giorgina Specchia, Francesco Albano
Department of Emergency and Organ Transplantation (D.E.T.O.), Hematology Section, University of Bari, Bari, Italy
Abstract: In 2008, the concept of the monosomal karyotype (MK) in adult acute myeloid leukemia (AML) patients was introduced, defined by the presence of a chromosomal aberration pattern characterized by the presence of at least two autosomal monosomies or of one monosomy plus one or more structural aberrations (not including loss of a chromosome). We present a systematic review of the literature about the influence of the MK on the outcome of patients affected by myeloid malignancies (AML, myelodysplastic syndromes, and primary myelofibrosis). For this review, a comprehensive literature search using the term “monosomal karyotype” was performed, considering articles listed in MEDLINE. This analysis of the literature confirms the negative prognostic impact on survival of the MK in myeloid neoplasias. The detrimental effect of MK on AML patients’ outcome is independent of other variables, including adverse cytogenetic features, supporting the identification of this entity as a challenging subgroup of patients with distinct biologic and clinical features.
Keywords: monosomal karyotype, acute myeloid leukemia, myelodysplastic syndromes, primary myelofibrosis, prognosis
Introduction
In the last few years, the application of cytogenetic and molecular disease markers has redefined the approach to the diagnosis, risk stratification, and treatment of myeloid malignancies, including acute myeloid leukemia (AML), myelodysplastic syndromes (MDS), and primary myelofibrosis (PMF). In 2008, in a cooperative study, the Dutch–Belgian Haemato-Oncology Cooperative Group/Swiss Group for Clinical Cancer Research (HOVON-SAKK)1 proposed the concept of the monosomal karyotype (MK), defined by the presence of a chromosomal aberration pattern characterized by the presence of at least two autosomal monosomies or of one monosomy plus one or more structural aberrations (not including loss of a chromosome). We present a systematic review of the literature concerning the influence of the MK on the outcome of patients affected by myeloid malignancies.
MK in AML
AML is a clinically and biologically heterogeneous hematologic malignancy derived from a clonal transformation of hematopoietic precursors due to the acquisition of chromosomal rearrangements and gene mutations.
The World Health Organization 2008 classification firstly, and the 2016 revision later, incorporated genetic information into diagnostic algorithms for myeloid neoplasms with the aim of redefining clinically distinct disease subtypes that require specific therapeutic interventions.2,3 In this context, subtypes of AML are classified and prognostically determined on the basis of their cytogenetic profiles.4,5 Thus, in clinical practice, the metaphase cytogenetics obtained at the diagnosis of AML has allowed clinicians to classify patients into different risk groups.6,7
In adult AML, cytogenetic risk is the most important independent prognostic factor, predictive of the probability of overall survival (OS) and progression-free survival.4,5 Cytogenetic abnormalities are found in ~60% of patients with AML, and most cytogenetic risk stratification systems subdivide patients, by recurrent cytogenetic abnormalities, into favorable, intermediate, and unfavorable risk groups.6–8
In the HOVON-SAKK study, MK+ AML was prevalent in about 9% of AML patients between 15 and 60 years of age.1 In subsequent studies, MK+ AML has been reported in about 6%–10% patients with newly diagnosed AML and the prevalence rises with increasing age.9–11 For example, the Southwest Oncology Group (SWOG)9 reported a frequency of MK+ AML of about 20% in newly diagnosed patients with AML of age >60 years, 4% in patients of age <31 years, 7% in patients aged between 31 and 40 years, 11% for patients with age ranging from 41 to 50 years, and 13% for patients aged 51–60 years. These data on the frequency of the MK were later confirmed in two other studies that showed a frequency of 10% in patients with AML of age <60 years and 13%–15% in patients with AML of age >60 years.10,11
Types of cytogenetic abnormalities
All studies are in agreement about the frequency of the different types of autosomal monosomies (Figure 1).
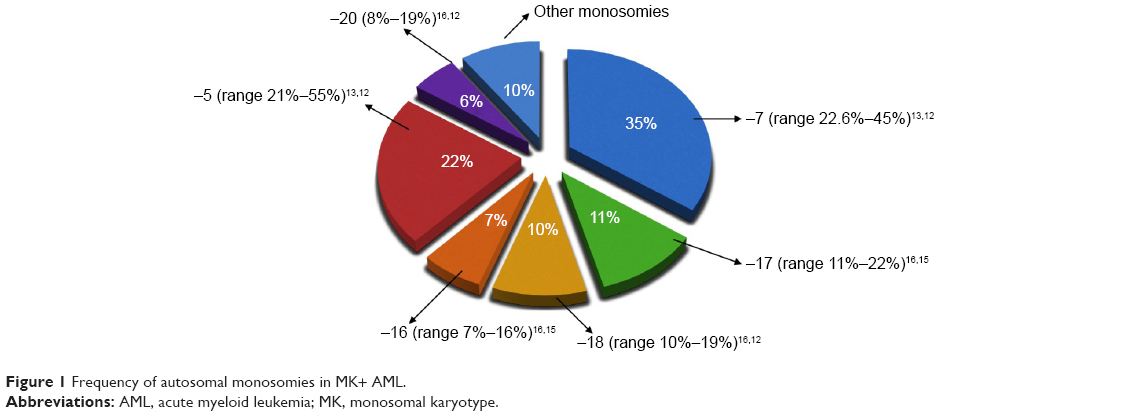 | Figure 1 Frequency of autosomal monosomies in MK+ AML. |
The most common monosomies involved in MK+ AML are −5 and −7. In the German–Austrian AML Study Group analysis of MK+ AML cases, the most frequent chromosome abnormalities were −5 or 5q (55%) and −7 (45%).12 Another paper showed that the most frequent monosomy was −7, followed by monosomies 17, 18, 16, 5, and 3.4 Voutiadou et al also reported monosomy 7 as predominant in terms of frequency, being present in 22.6% of MK cases, followed by monosomy 5 in 21%.13 Furthermore, the HOVON-SAKK study showed, in multiple comparisons, that any type of monosomy in AML was associated with a poor outcome.1 In fact, direct comparison between patients with a single monosomy 7 and other variable single autosomal monosomies revealed an identically poor OS. Therefore, no difference in the prognosis seems to result from the presence of any specific monosomy, in the context of the MK.
MK and TP53 alterations
The mechanisms responsible for MK+ AML are still unclear, but it may be associated with deletions or mutations in TP53 gene and multiple drug resistance. In fact, recent evidence indicated that TP53 alterations occur in 70% of MK+ AML patients and that these abnormalities are more frequent in patients with a complex karyotype (CK+)/MK+ AML than in those with CK+/MK− AML.14 They potentially lead to a chromosome instability pattern that is usually a result of a single catastrophic event known as chromothripsis.15 Moreover, it has been recently reported that chromothripsis-positive AML cases were characterized by a particularly high degree of karyotype complexity (CK, MK, abnl[5q], abnl[7q], and abnl[17p]), TP53 mutations, and dismal prognosis.16 Thus, TP53 alterations appear to be one molecular basis for this MK+ AML subset and, in particular, biallelic alterations suggest an important role for p53 in leukemogenesis. From a prognostic standpoint, TP53 abnormalities have a negative impact on the outcome of these patients due to chemoresistance (lower complete remission (CR) rates and higher rates of refractory disease).14
Prognostic impact of MK in AML
Several recent studies have revealed that AML with the MK are at the extreme end of the unfavorable risk category, and its presence is predictive of the worst possible outcome.
Two major cooperative trial groups, namely, the HOVON-SAKK and the SWOG groups, collected cytogenetic diagnostics at baseline in patients with AML enrolled in their treatment protocols. These works generated datasets in large series of homogeneously treated patients, in whom the prognostic contribution of various cytogenetic abnormalities such as CK could be evaluated. Statistical analysis revealed that the loss of a complete autosomal chromosome conferred a negative prognostic impact, while structural abnormalities negatively influenced the prognosis in association with an autosomal monosomy.1
In particular, among 733 non-core-binding factor AML patients ranging in age from 15 to 60 years, the first HOVON-SAKK group1 paper showed that the presence of a single autosomal monosomy conferred a poor prognosis, regardless of the chromosome involved, as compared with patients without monosomy. Similar results were later reported by the SWOG group;9 in their study, 1344 AML patients ranging in age from 16 to 88 years were included and treated with standard chemotherapy. One hundred seventy-six (13%) patients were classified as having the MK. The median age of patients bearing the MK was 61 years. The median OS of the MK group was 4 months. The CR rate in patients with unfavorable cytogenetics without the MK was 34% vs 18% in patients with the MK, and the 4-year OS of patients with unfavorable cytogenetics but without the MK was 13% vs 3% in the MK group. Thus, these data confirmed that the MK defines a subset of AML patients with unfavorable cytogenetics and poor prognosis.
In two HOVON-SAKK studies10,11 and one by SWOG,9 the CR rates for MK+ AML were no more than 52% in patients aged <60 years and only 34% in those aged >60 years. In particular, a CR rate of 50% was reported in patients aged <31 years (a low percentage for young AML compared to historical controls), 27% in patients aged 31–40 years, 14% in patients from 41 to 50 years old, 24% for patients aged 51–60 years, and 13% in MK+ AML patients aged over 60 years.9 Apart from the CR rates, the survival estimates were also very poor in MK+ AML. In the original study, the HOVON-SAKK group1 showed an OS of 4% at 4 years in patients with MK+ AML of age >60 years; subsequently, the SWOG study9 also reported an OS of 3% at 4 years in patients with MK+ AML aged between 16 and 88 years, compared to a 4-year OS of 13% in patients with an unfavorable karyotype but without the MK. The latest HOVON-SAKK group studies reported 4% OS at 2 years in MK+ patients aged >60 years11 and 7% OS at 5 years in those aged <60 years.10 Of note, in the SWOG9 study, an estimated OS of <1% at 4 years was reported for patients with MK+ AML aged between 41 and 88 years, while in the HOVON-SAKK study, at 5 years, there were no long-term survivors among patients >60 years of age.11 The very poor prognosis of MK+ AML in terms of OS was also shown in a large-scale study of AML patients aged between 16 and 59 years conducted by the UK Medical Research Council:7 the estimated OS at 10 years was 5%. Similarly, the Groupe Ouest Est des Leucemies Aigues et Autres Maladies du Sang17 made a retrospective study of patients aged >60 years with unfavorable cytogenetics. There was a significantly lower CR rate for MK+ AML than for the MK− group (37% vs 64%, respectively), and the OS rate at 2 years was also decreased (7% and 22%, respectively). Moreover, in the subgroup of CK patients, OS was dramatically decreased for MK+ patients (8% vs 28%). These results demonstrate that the MK is a major independent very poor prognosis factor in elderly AML. During the past few years, several large studies have been conducted to analyze the prognostic impact of the MK on the outcome of AML (summarized in Table 1), confirming the previously reported findings.1,5,9–13,17–24 Moreover, a recent report demonstrated that the combination of CK (defined in this study as unrelated chromosomal abnormalities ≥4) and the MK missed the least number of patients with unfavorable prognosis.18
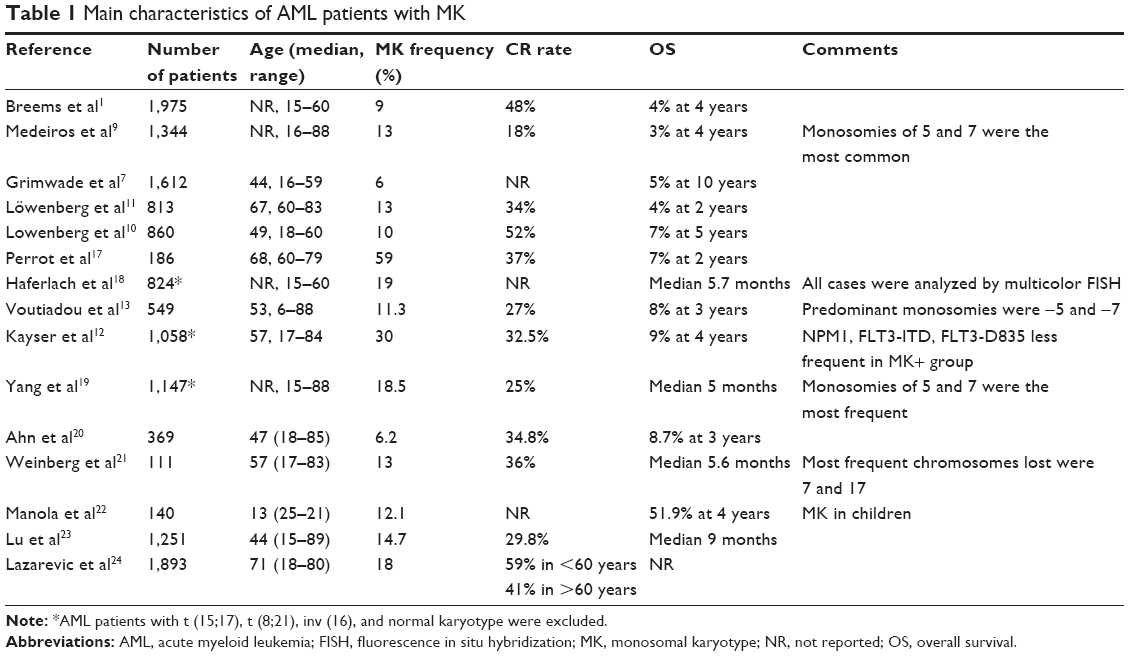 | Table 1 Main characteristics of AML patients with MK |
With regard to the MK in AML, it should be remembered that many monosomies described in chromosome banding analysis may be not real monosomies, but part of chromosomal material hidden in unbalanced translocations or marker chromosomes.14
Therapeutic implications of MK in AML
MK+ AML patients respond poorly to conventional chemotherapy due to resistance to current treatments, resulting in a low CR rate or in an early relapse rate after CR. In the past, daunorubicin dose escalation was shown to yield a higher CR rate and improved survival in patients with AML under 65 years of age.20 In the same way, two recent studies reported that high-dose cytarabine-based chemotherapeutic protocols could improve long-term survival in patients with MK+ AML and showed a potential survival benefit.25–27 In particular, a study conducted by the HOVON-SAKK group26 reported that no significant differences were noted between the intermediate- and the high-dose groups in terms of CR rates (80% and 82%, respectively), probability of relapse-free survival at 5 years (34% and 35%, respectively), or OS (40% and 42%, respectively). However, only in the MK+ subgroup (89 patients, 9% of the total population) were the 5-year event-free survival (13% vs 0%) and OS rates (16% vs 0%) better in the high-dose cytarabine group, compared with the standard-dose group. The impact of different postremission strategies on the outcome of these subsets of patients is not clear. A study by the Fred Hutchinson Cancer Research Center reviewed the experience of allogenic hematopoietic stem cell transplantation (alloHSCT) in 432 patients with AML.28 They showed that alloHSCT could increase the 4-year disease-free survival rate of MK+ AML by up to 25%, and similar results were obtained with matched related and matched unrelated donors. Although this result is still lower than the 56% OS seen in patients without the MK, it is better than the 3%–9% OS seen in patients receiving chemotherapy alone. This study also showed that the OS rate was higher for patients who achieved CR before transplantation, and that the 4-year OS rate was 30% for patients in CR1, 25% for patients in CR2, and 16% for patients in other disease statuses before transplantation.28
Subsequent studies also confirmed that transplantation could improve long-term survival in MK+ AML patients, but it was associated with disadvantages such as a high recurrence rate and a short median time to recurrence.29,30 In 2012, a comparative analysis performed by the HOVON/SAKK group was reported, which included >300 MK+ patients.31 Among the 140 patients who achieved CR after two induction cycles, 107 (76%) proceeded to consolidation therapy and 45 (32%) subsequently received alloHSCT. Finally, MK+ patients in CR1 who received alloHSCT had a long-term OS of 19% at 5 years, compared to 8% among those receiving alternative consolidation chemotherapies.31
Moreover, a study reported by Moon et al confirmed these findings, showing that alloHSCT in MK+ patients with active disease at the time of transplantation had a negative impact on the outcomes.32 So, the remission status at the time of transplantation is critical for all patients, and particularly for MK+ AML patients.
However, allogeneic transplant remains the only potentially curative strategy for MK+ patients who were refractory to the initial therapy. In fact, while the median OS for MK+ patients refractory to induction therapy and who received an allogeneic transplant was 3–9 months, and 10% of patients achieved long-term survival, a shorter median OS and no long-term survivors were reported for refractory MK+ AML patients who did not proceed to transplantation.31
In conclusion, the data available suggest that alloHSCT in first CR is a reasonable treatment to improve the outcomes in this subset of patients. However, the MK+ AML prognosis remains poor even after alloHSCT, and this category of AML patients should be seen as candidates for clinical trials testing novel treatment strategies in order to improve their overall outcome.
MK in MDS
MDS are heterogeneous diseases characterized by bone marrow dysplasia, cytopenias, and a variable risk of evolution to AML.33 The most widely used risk scoring system in MDS is the International Prognostic Scoring System (IPSS), which utilizes clinical and molecular features, including cytogenetics, percentage of blasts, and the number of cytopenias, to risk-stratify patients.34 The IPSS was subsequently revised (IPSS-R) and this version maintained bone marrow cytogenetics, marrow blast percentage, and cytopenias as the basis of the scoring system, but introduced increased stratification within these categories.35 While the IPSS includes only three cytogenetic patterns, the IPSS-R classifies cytogenetic information in MDS in five risk groups with correspondingly different median OS.36
Prognostic impact of MK in MDS
An abnormal karyotype is seen in 50% of de novo MDS and 80%–92% of therapy-related MDS, and it is clear that the poor and the very poor IPSS-R cytogenetic groups include patients with the MK.35,37 Several conflicting reports (summarized in Table 2) on the impact of the MK as an independent predictor of survival in MDS have drawn attention to the importance of considering the prognostic weight of complex aberrations in association with autosomal monosomies.38–43 In a recent study including only CK MDS patients from the Mayo Clinic database, the authors suggested that MK+ is associated with a lower OS:38 the OS was significantly inferior in patients with MK+ compared with those with a CK without monosomies, and the median survival of patients with a CK without monosomies was 13 months vs 7 months in patients with MK+. By contrast, the Spanish group of MDS (GESMD) analyzed 1054 MDS patients with an abnormal karyotype. In their cohort, MK+ was identified with a frequency of 16%, and the majority of these patients (88%) also had a CK.39 To clarify the significance of the presence of MK+, the GESMD study analyzed its impact on both CK patients and those with only two chromosomal abnormalities (ie, at least two autosomal monosomies or of one monosomy plus one structural aberrations), being the minimum necessary to fulfill the criteria for MK+. In CK patients, the MK was not statistically associated with a lower OS in univariate or multivariate analysis, but the risk factors associated with a lower OS in patients with CK were the classic variables (refractory anemia with excess blasts, high IPSS, low hemoglobin level, and low platelet count) together with higher numbers of chromosomal abnormalities.39 In patients with only two abnormalities, although MK+ patients showed a lower OS in univariate analysis, this effect did not persist in multivariate analysis, in which the only variable associated with a lower OS was a higher IPSS risk group. By contrast, the CK adverse prognostic value was retained in MK+ patients also. These results support the hypothesis regarding the predominant role of karyotype complexity in determining the prognosis in patients with MDS. Similarly, in 2013, a study analyzed 431 untreated MDS patients with two or more chromosomal abnormalities from an international MDS database and found that MK+ was associated with a worse OS only in those patients with four or fewer abnormalities, and that in the multivariate analysis, it was not independently associated with OS.40 The authors concluded that the number of chromosomal abnormalities, rather than the presence of MK+, defined the MDS subgroups with a worse prognosis and that the number of chromosomal aberrations in MK+ subgroups of MDS was directly related to OS, and so MK+ was not reflected as an independent prognostic factor.
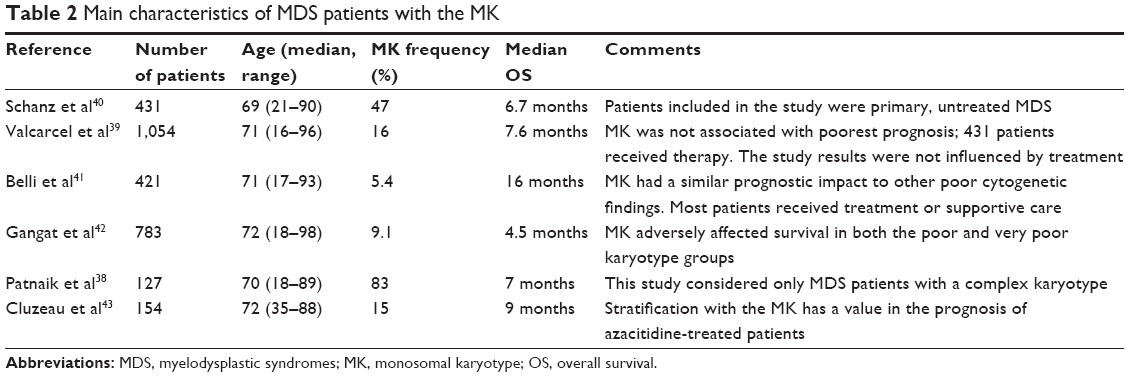 | Table 2 Main characteristics of MDS patients with the MK |
In conclusion, although the cytogenetic prognosis is very important in the IPSS-R, MK+ was not considered as a prognostic subgroup. The Mayo Clinic group suggested that the MK provides additional prognostic information in poor/very poor karyotypes: in their cohort of 783 patients with MDS, there was no significant difference in survival among IPSS-R subgroups (very good, intermediate, poor), but reclassification using MK+ allowed them to obtain a significant difference in OS.42
Therapeutic implication of MK in MDS
Azacitidine (AZA) is a first-line treatment for higher IPSS risk patients with MDS who are ineligible for alloHSCT.44 The prognostic impact of MK+ in AZA-treated patients is unclear. The French group has shown that the IPSS-R is a powerful tool to evaluate the outcome of previously untreated MDS patients treated with AZA.45 A report in 2011 suggested that AZA might reduce the negative impact of MK+ in higher IPSS risk MDS patients:46 in a cohort of 75 patients with high-risk MDS treated with AZA, the median OS was 7.1 months in MK+ and 8.7 months in non-MK patients (no statistically significant difference). Several studies have pointed out that high-risk patients are often referred for hematopoietic cell transplantation, the only potentially curative treatment for MDS.47–49 Many recent studies evaluating the importance of MK+ in alloHCT have reported similar results. In a cohort of 261 MDS patients with chromosome 7 abnormalities, it was shown that MK+ was more predictive of progression-free survival and OS after alloHSCT than complex cytogenetics in 261 MDS patients.49
It has also been reported that MK+ was more predictive of alloHSCT outcomes in MDS patients, compared to other established scoring systems.50 Moreover, the presence of MK+, both at diagnosis and at alloHSCT, was predictive of a worse OS after alloHSCT; in fact, the presence of MK+ at diagnosis was associated with a poor 3-year disease-free survival (27% vs 39%) and OS (29% vs 47%). Therefore, MK+, particularly at diagnosis, seems to be the cytogenetic risk factor most predictive of post-alloHSCT outcomes. A large study by the International Blood and Marrow Transplant Research group analyzed the influence of MK+ on survival in a large cohort of patients with AML and MDS undergoing alloHSCT.51 Among patients with MDS, MK+ MDS was associated with higher disease relapse rates, higher transplant-related mortality, and a worse OS. Subset analyses comparing chromosome 7 abnormalities with or without MK+ demonstrated a higher mortality for MK+ MDS. Koichiro et al reported the outcome of 53 MDS patients; among them, 9 (17%) had MK+ and 4-year OS was 0%, significantly lower than that of MDS patients with a normal karyotype.52 However, the European Bone Marrow Transplantation group reported that the CK is a better predictor of a poor outcome than MK+ after alloHSCT in MDS patients;53 in this study, the OS of patients with CK, with or without MK, was significantly worse than that of patients with isolated MK+.
A work by Gruppo Italiano Trapianto Midollo Osseo analyzed 519 patients with primary MDS undergoing alloHSCT and showed a 5-year OS of MK+ patients of only 10%, significantly worse than that of patients without the MK.54 Moreover, the 5-year incidence of relapse was 49%, significantly greater than that of patients without the MK.
In summary, cytogenetic abnormalities remain the most important predictor of treatment outcome in MDS patients. MK+, which was not considered as a separate category in the scoring system, may represent another subset of high-risk MDS patients with a dismal outcome after alloHSCT. Further evaluation with prospective studies will help to define the importance of MK+ in MDS and improve treatment strategies for this subgroup of patients.
MK in PMF
In clinical practice, the IPSS is used to assess prognosis in PMF.55 The IPSS score was later modified to the Dynamic IPSS (DIPSS) for use at any time during the disease course.56 Most recently, DIPSS was further modified to the DIPSS-plus, with the incorporation of three additional DIPSS-independent risk factors: red cell transfusion need, platelet count <100×109/L, and unfavorable karyotype.57 This category includes CK or one or two abnormalities among +8, −7/7q−, i (17q), inv (3), −5/5q−, 12p-, or 11q23 rearrangement.
Two studies have analyzed the impact of MK+ in PMF patients. In the first, 793 patients were included; among them, 341 (43%) showed an abnormal karyotype, including 41 (12%) with CK and among these, 17 (41%) were classified as having an MK+.58 To determine whether the presence of MK+ conferred additional prognostic significance, the authors compared groups with MK+, CK without monosomies, and with only trisomy 8: median survival was 6, 24, and 20 months, respectively, and the corresponding 2-year leukemic transformation rates were 29.4%, 8.3%, and 0%. Therefore, the study shows that MK+ is equally as bad as PMF in terms of both OS and leukemia-free survival. In fact, the presence of MK+ in PMF signified a worse survival than the rate associated with either the CK without monosomies or a sole trisomy 8, both of which had previously been identified as unfavorable cytogenetic findings in PMF.59 More recently, a large cohort of Chinese patients with PMF60 was analyzed: the MK was found in 12% of cases. The patients were divided into two cytogenetics-based cohorts: a favorable (subjects with a normal karyotype, a CK that was not an MK, +8 only or a balanced translocation only) and an unfavorable karyotype (all the others). The median OS was 52 vs 72 months in patients with a favorable and unfavorable karyotype, respectively.
These findings underscore the importance of paying attention to cytogenetic findings also in PMF and the prudence of early intervention with investigational drug therapy or allogeneic stem cell transplantation in MK+ PMF, although the value of such a treatment strategy in this particular patient population remains unproven.
Conclusion
This analysis of the literature confirms the negative prognostic impact on survival of the MK in myeloid neoplasias. The detrimental effect of the MK on AML patients’ outcome is independent of other variables, including adverse cytogenetic features such as monosomy 7 and CK, supporting the identification of this entity as a challenging subgroup of patients with distinct biologic and clinical features.
All studies on AML and MDS confirm that the MK classification scheme identifies a group with a very poor prognosis for all study endpoints, even after alloHSCT.
The last several years have seen marked improvements, thanks to standardized chemotherapy (including induction therapy and consolidation therapy), in the survival times of AML patients, while alloHSCT is currently used as a salvage therapy for patients with high-risk AML, including MK+ AML. However, MK+ has been shown to be significantly correlated with a worse OS among patients who have undergone alloHSCT.
In conclusion, the presence of MK+ is associated with a worse outcome (in terms of OS and CR rate) compared to other cytogenetic abnormalities. Therefore, research to identify more effective induction regimens, conditioning regimens, and posttransplant treatments for the prevention of relapse is warranted in this high-risk group of patients, in order to improve the number of patients who can benefit from alloHSCT and achieve a better outcome. Moreover, MK+ patients, mostly those with AML, should be enrolled to take part in clinical trials of novel treatment strategies in order to improve the OS in this very poor prognostic group.
Acknowledgment
This work was supported by “Associazione Italiana contro le Leucemie (AIL)-BARI”.
Disclosure
The authors report no conflicts of interest in this work.
References
Breems DA, Van Putten WL, De Greef GE, et al. Monosomal karyotype in acute myeloid leukemia: a better indicator of poor prognosis than a complex karyotype. J Clin Oncol. 2008;26(29):4791–4797. | ||
Campo E, Swerdlow SHCC, Harris NL, et al. WHO classification of tumours of the haematopoietic and lymphoid tissues. Lyon: International Agency for Research on Cancer (IARC); 2008. | ||
Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391–2405. | ||
Smith ML, Hills RK, Grimwade D. Independent prognostic variables in acute myeloid leukaemia. Blood Rev. 2011;25(1):39–51. | ||
Grimwade D, Walker H, Oliver F, et al. The importance of diagnostic cytogenetics on outcome in AML: analysis of 1,612 patients entered into the MRC AML 10 trial. The Medical Research Council Adult and Children’s Leukaemia Working Parties. Blood. 1998;92(7):2322–2333. | ||
Dohner H, Estey EH, Amadori S, et al. Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European Leukemia Net. Blood. 2010;115(3):453–474. | ||
Grimwade D, Hills RK, Moorman AV, et al. Refinement of cytogenetic classification in acute myeloid leukemia: determination of prognostic significance of rare recurring chromosomal abnormalities among 5876 younger adult patients treated in the United Kingdom Medical Research Council trials. Blood. 2010;116(3):354–365. | ||
Slovak ML, Kopecky KJ, Cassileth PA, et al. Karyotypic analysis predicts outcome of preremission and postremission therapy in adult acute myeloid leukemia: A Southwest Oncology Group/Eastern Cooperative Oncology Group Study. Blood. 2000;96(13):4075–4083. | ||
Medeiros BC, Othus M, Fang M, Roulston D, Appelbaum FR. Prognostic impact of monosomal karyotype in young adult and elderly acute myeloid leukemia: the Southwest Oncology Group (SWOG) experience. Blood. 2010;116(13):2224–2248. | ||
Lowenberg B, Pabst T, Vellenga E, et al. Cytarabine dose for acute myeloid leukemia. N Engl J Med. 2011;364(11):1027–1036. | ||
Löwenberg B, Ossenkoppele GJ, Van Putten W, et al. High-dose daunorubicin in older patients with acute myeloid leukemia. N Engl J Med. 2009;361(13):1235–1248. | ||
Kayser S, Zucknick M, Dohner K, et al. Monosomal karyotype in adult acute myeloid leukemia: prognostic impact and outcome after different treatment strategies. Blood. 2012;119(2):551–558. | ||
Voutiadou G, Papaioannou G, Gaitatzi M, et al. Monosomal karyotype in acute myeloid leukemia defines a distinct subgroup within the adverse cytogenetic risk category. Cancer Genet. 2013;206(1–2):32–36. | ||
Rucker FG, Schlenk RF, Bullinger L, et al. TP53 alterations in acute myeloid leukemia with complex karyotype correlate with specific copy number alterations, monosomal karyotype, and dismal outcome. Blood. 2012;119(9):2114–2121. | ||
Rausch T, Jones DT, Zapatka M, et al. Genome sequencing of pediatric medulloblastoma links catastrophic DNA rearrangements with TP53 mutations. Cell. 2012;148(1–2):59–71. | ||
Bochtler T, Granzow M, Stölzel F, et al. Marker chromosomes can arise from chromothripsis and predict adverse prognosis in acute myeloid leukemia. Blood. 2017;129(10):1333–1342. | ||
Perrot A, Luquet I, Pigneux A, et al. Dismal prognostic value of monosomal karyotype in elderly patients with acute myeloid leukemia: a GOELAMS study of 186 patients with unfavorable cytogenetic abnormalities. Blood. 2011;118(3):679–685. | ||
Haferlach C, Alperman T, Schnittger S, et al. Prognostic value of “monosomal karyotype” in comparison to “complex aberrant karyotype” in acute myeloid leukemia: study on 824 cases with aberrant karyotype. Blood. 2012;119(9):2122–2125. | ||
Yang XF, Sun AN, Yin J, et al. Monosomal karyotype among 1147 chinese patients with acute myeloid leukemia: prevalence, features and prognostic impact. Asian Pac J Cancer Prev. 2012;13(11):5421–5426. | ||
Ahn HK, Jang JH, Kim K, et al. Monosomal karyotype in acute myeloid leukemia predicts adverse treatment outcome and associates with high functional multidrug resistance activity. Am J Hematol. 2012;87(1):37–41. | ||
Weinberg OK, Ohgami RS, et al. Acute myeloid leukemia with monosomal karyotype. Am J Clin Pathol. 2014;142(2):190–195. | ||
Manola KN, Panitsas F, Polychronopoulou S, et al. Cytogenetic abnormalities and monosomal karyotypes in children and adolescents with acute myeloid leukemia: correlations with clinical characteristics and outcome. Cancer Genet. 2013;206(3):63–72. | ||
Lu QS, Xu N, Zhou X, et al. Prognostic significance of monosomal karyotype in adult patients with acute myeloid leukemia treated with risk-adapted protocols. Clin Lymphoma Myeloma Leuk. 2015;15(12):790–796. | ||
Lazarevic V, Horstedt AS, Johansson B, et al. Incidence and prognostic significance of karyotypic subgroups in older patients with acute myeloid leukemia: the Swedish population-based experience. Blood Cancer J. 2014;4:e188. | ||
Fernandez HF, Sun Z, Yao X, et al. Anthracycline dose intensification in acute myeloid leukemia. N Engl J Med. 2009;361(13):1249–1259. | ||
Lowenberg B, Pabst T, Vellenga E, et al. Cytarabine dose for acute myeloid leukemia. N Engl J Med. 2011;364(11):1027–1036. | ||
Medeiros BC, Othus M, Appelbaum FR. Cytarabine dose for acute myeloid leukemia. N Engl J Med. 2011;364(22):2167–2168. | ||
Fang M, Storer B, Estey E, et al. Outcome of patients with acute myeloid leukemia with monosomal karyotype who undergo hematopoietic cell transplantation. Blood. 2011;118(6):1490–1494. | ||
Oran B, Dolan M, Can Q, et al. Monosomal karyotype provides better prognostic prediction after allogenic stem cell transplantation in patients with acute myelogenous. Biol Blood Marrow Transplant. 2011;17(3):356–364. | ||
Yanada M, Kurosawa S, Yamaguchi T, et al. Prognosis of acute myeloid leukemia harboring monosomal karyotype in patients treated with or without allogenic hematopoietic cell transplantation after achieving complete remission. Haematologica. 2012;97(6):915–918. | ||
Cornelissen JJ, Breems D, Lowenberg B, et al. Comparative analysis of the value of allogeneic hematopoietic stem-cell transplantation in acute myeloid leukemia with monosomal karyotype versus other cytogenetic risk categories. J Clin Oncol. 2012;30(17):2140–2146. | ||
Moon JH, Lee YJ, Seo SK, et al. Outcomes of allogeneic hematopoietic cell transplantation in acute myeloid leukemia patients with monosomal karyotypes. Acta Haematol. 2015;133(4):327–335. | ||
Tefferi A, Vardiman JW. Myelodysplastic syndromes. N Engl J Med. 2009;361(19):1872–1885. | ||
Greenberg P, Cox C, LeBeau MM, et al. International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood. 1997;89(6):2079–2088. | ||
Greenberg PL, Tuechler H, Schanz J, et al. Revised International Prognostic Scoring System (IPSS-R) for myelodysplastic syndromes. Blood. 2012;120(12):2454–2465. | ||
Schanz J, Tuchler H, Sole F, et al. New comprehensive cytogenetic scoring system for primary myelodysplastic syndromes (MDS) and oligoblastic acute myeloid leukemia after MDS derived from an international database merge. J Clin Oncol. 2012;30(8):820–829. | ||
Haase D, Germing U, Schanz J, et al. New insights into the prognostic impact of the karyotype in MDS and correlation with subtypes: evidence from a core dataset of 2124 patients. Blood. 2007;110(13):4385–4395. | ||
Patnaik MM, Hanson CA, Hodnefield JM, et al. Monosomal karyotype in myelodysplastic syndromes, with or without monosomy 7 or 5, is prognostically worse than an otherwise complex karyotype. Leukemia. 2011;25(2):266–270. | ||
Valcarcel D, Adema V, Sole F, et al. Complex, not monosomal, karyotype is the cytogenetic marker of poorest prognosis in patients with primary myelodysplastic syndrome. J Clin Oncol. 2013;31(7):916–922. | ||
Schanz J, Tuchler H, Sole F, et al. Monosomal karyotype in MDS: explaining the poor prognosis? Leukemia. 2013;27(10):1988–1995. | ||
Belli CB, Bengio R, Aranguren PN, et al. Partial and total monosomal karyotypes in myelodysplastic syndromes: comparative prognostic relevance among 421 patients. Am J Hematol. 2011;86(7):540–545. | ||
Gangat N, Patnaik MM, Begna K, et al. Evaluation of revised IPSS cytogenetic risk stratification and prognostic impact of monosomal karyotype in 783 patients with primary myelodysplastic syndromes. Am J Hematol. 2013;88(8):690–693. | ||
Cluzeau T, Mounier N, Karsenti JM, et al. Monosomal karyotype improves IPSS-R stratification in MDS and AML patients treated with Azacitidine. Am J Hematol. 2013;88(9):780–783. | ||
Fenaux P, Mufti GJ, Hellstrom-Lindberg E, et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: a randomised, open-label, phase III study. Lancet Oncol. 2009;10(3):223–232. | ||
Lamarque M, Raynaud S, Itzykson R, et al. The revised IPSS is a powerful tool to evaluate the outcome of MDS patients treated with azacitidine: the GFM experience. Blood. 2012;120(25):5084–5085. | ||
Itzykson R, Thepot S, Eclache V, et al. Prognostic significance of monosomal karyotype in higher risk myelodysplastic syndrome treated with azacitidine. Leukemia. 2011;25(7):1207–1209. | ||
Chang C, Storer BE, Scott BL, et al. Hematopoietic cell transplantation in patients with myelodysplastic syndrome or acute myeloid leukemia arising from myelodysplastic syndrome: similar outcomes in patients with de novo disease and disease following prior therapy or antecedent hematologic disorders. Blood. 2007;110(4):1379–1387. | ||
Appelbaum FR, Anderson J. Allogeneic bone marrow transplantation for myelodysplastic syndrome: outcomes analysis according to IPSS score. Leukemia. 1998;12(Suppl 1):S25–S29. | ||
van Gelder M, de Wreede LC, Schetelig J, et al. Monosomal karyotype predicts poor survival after allogeneic stem cell transplantation in chromosome 7 abnormal myelodysplastic syndrome and secondary acute myeloid leukemia. Leukemia. 2013;27(4):879–888. | ||
Ustun C, Trottier BJ, Sachs Z, et al. Monosomal karyotype at the time of diagnosis or transplantation predicts outcomes of allogeneic hematopoietic cell transplantation in myelodysplastic syndrome. Biol Blood Marrow Transplant. 2015;21(5):866–872. | ||
Pasquini MC, Zhang MJ, Medeiros BC, et al. Hematopoietic cell transplantation outcomes in monosomal karyotype myeloid malignancies. Biol Blood Marrow Transplant. 2016;22(2):248–257. | ||
Koichiro M, Shigematsu A, Nakata M, et al. Monosomal karyotype in myeloid malignancies is prognostically worse even after allogeneic hematopoietic stem cell transplantation; results from two transplant centers. Blood. 2011;118(21):Abstract 4154. | ||
Brands-Nijenhuis A, Van Gelder M, De Witte T, et al. Complex karyotype predicts outcome better than monosomal karyotype in patients with MDS/secondary acute leukemia treated with allogeneic hematopoietic stem cell transplantation (HSCT): a retrospective survey on behalf of the chronic leukemia working party of the european group for blood and marrow transplantation (EBMT). Blood. 2010;116(21):Abstract 578. | ||
Della Porta MG, Alessandrino EP, Bacigalupo A, et al. Predicative factors for the outcome of allogenic transplantation in patients with myelodysplastic syndrome stratified according to the revised IPSS-R. Blood. 2014;123(15):2333–2342. | ||
Cervantes F, Dupriez B, Pereira A, et al. New prognostic scoring system for primary myelofibrosis based on a study of the International Working Group for Myelofibrosis Research and Treatment. Blood. 2009;113(13):2895–2901. | ||
Passamonti F, Cervantes F, Vannucchi AM, et al. A dynamic prognostic model to predict survival in primary myelofibrosis: a study by the IWG-MRT (International Working Group for Myeloproliferative Neoplasms Research and Treatment). Blood. 2010;115(9):1703–1708. | ||
Gangat N, Caramazza D, Vaidya R, et al. DIPSS-Plus: a refined dynamic international prognostic scoring system (DIPSS) for primary myelofibrosis that incorporates prognostic information from karyotype, platelet count and transfusion status. J Clin Oncol. 2011;29(4):392–397. | ||
Vaidya R, Caramazza D, Begna KH, et al. Monosomal karyotype in primary myelofibrosis is detrimental to both overall and leukemia-free survival. Blood. 2011;117(21):5612–5615. | ||
Caramazza D, Begna KH, Gangat N, et al. Refined cytogenetic risk categorization for overall and leukemia-free survival in primary myelofibrosis: a single center study of 433 patients. Leukemia. 2011;25(1):82–88. | ||
Li B, Xu J, Li C, et al. Cytogenetic studies and their prognostic contribution in 565 Chinese patients with primary myelofibrosis. Am J Hematol. 2014;89(11):1043–1046. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.