")
Back to Journals » Drug Design, Development and Therapy » Volume 13
Monosialoganglioside protects against bupivacaine-induced neurotoxicity caused by endoplasmic reticulum stress in rats
Authors Liu B, Ji J, Feng Q , Luo X, Yan X, Ni Y, He Y, Mao Z, Liu J
Received 25 October 2018
Accepted for publication 10 January 2019
Published 19 February 2019 Volume 2019:13 Pages 707—718
DOI https://doi.org/10.2147/DDDT.S192225
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Qiongyu Guo
Benquan Liu,1 Jiemei Ji,1 Qing Feng,1 Xi Luo,1 Xiurong Yan,1 Yuxia Ni,2 Yajun He,1 Zhongxuan Mao,1 Jingchen Liu1
1Department of Anesthesiology, The First Affiliated Hospital of Guangxi Medical University, Nanning 530021, Guangxi, People’s Republic of China; 2Department of Anesthesiology, Langdong Hospital of Guangxi Medical University, Nanning 530021, Guangxi, People’s Republic of China
Background: Local anesthetics in spinal anesthesia have neurotoxic effects, resulting in severe neurological complications. Intrathecal monosialoganglioside (GM1) administration has a therapeutic effect on bupivacaine-induced neurotoxicity. The aim of this study was to determine the underlying mechanisms of bupivacaine-induced neurotoxicity and the potential neuroprotective role of GM1.
Materials and methods: A rat spinal cord neurotoxicity model was established by injecting bupivacaine (5%, 0.12 µL/g) intrathecally. The protective effect of GM1 (30 mg/kg) was evaluated by pretreating the animals with it prior to the bupivacaine regimen. The neurological and locomotor functions were assessed using standard tests. The histomorphological changes, neuron degeneration and apoptosis, and endoplasmic reticulum stress (ERS) relevant markers were analyzed using immunofluorescence, quantitative real-time PCR, and Western blotting.
Results: Bupivacaine resulted in significant neurotoxicity in the form of aberrant neurolocomoter functions and spinal cord histomorphology and neuronal apoptosis. Furthermore, the ERS specific markers were significantly upregulated during bupivacaine-induced neurotoxicity. These neurotoxic effects were ameliorated by GM1.
Conclusion: Pretreatment with GM1 protects against bupivacaine-induced neurotoxicity via the inhibition of the GRP78/PERK/eIF2α/ATF4-mediated ERS.
Keywords: bupivacaine, GM1 ganglioside, ERS, neurotoxicity
Introduction
Spinal anesthesia, a type of regional anesthesia that involves injecting a local anesthetic (LA) into the subarachnoid space, is widely used in surgeries of the lower abdomen, pelvis, and lower extremities.1 LAs provide a good analgesic effect but they have some degree of neurotoxicity and are known to cause neurological complications such as transient neurological symptom, cauda equina syndrome, Guillain–Barre syndrome, and delayed sacral neurosensory disorder.2,3 The patient may be vulnerable to neurotoxicity even with clinically recommended doses of LA and inclusion of adjuvants.4 Although postspinal anesthesia complications are rare with an incidence rate of only ~0.038%,5 they cause irreversible nerve damage and thus significant economic burden on the family and society. Therefore, it is necessary to identify the intrinsic mechanism of LA-induced neurotoxicity.
Bupivacaine, an amide-type LA commonly used in spinal anesthesia, induces neurotoxicity both in vivo and in vitro.6,7 Several mechanisms have been implicated in the pathogenesis of bupivacaine-induced neurotoxicity, such as intracellular calcium release and overload,8 increased p47phox membrane translocation, which results in excessive reactive oxygen species production and neuronal apoptosis,9 activation of the PI3K and MAPK signaling pathways,10,11 and autophagy.12 However, the exact mechanism of bupivacaine-induced neurotoxicity is yet to be elucidated.
Gangliosides are sialic acid-containing membrane glycosphingolipid neurotrophins that are abundant in the central nervous system (CNS). Intrathecal monosialoganglioside (GM1) is a major sialoglycolipid of the neuronal membrane and plays a critical role in its metabolism, plasticity, and regeneration.13 A recent study showed that GM1 inhibited neuronal apoptosis in rats with acute spinal cord injury by downregulating caspase-3 and upregulating the nerve growth factor.14 In addition, GM1 also exerts its neuroprotective effect by activating the PI3K/AKT/Nrf2 pathway and enhancing autophagy.15,16 Our previous study found that treatment with GM1 in intrathecal routes reverses bupivacaine-induced neural injuries and improves the neural dysfunctions;6 however, its potential role in bupivacaine-induced neurotoxicity and the associated mechanisms remains unclear.
The endoplasmic reticulum (ER) is an important organelle in the eukaryotic cells and is involved in protein, lipid, and sterol biosynthesis.17 It stimulates the unfolded protein response (UPR), which results in the ER stress (ERS) response to cellular insults such as ischemia, trauma, hypoxia, glucose deprivation, and oxidative damage.18 The signaling cascade of the ERS is coordinated by three transmembrane protein sensors including PERK, IRE1α and ATF6, which specifically bind to the chaperone glucose-regulated protein 78 (GRP78) under normal conditions.19 ERS is involved in the occurrence and development of several diseases, including osteoporosis, Alzheimer’s disease (AD), Parkinson’s disease (PD), diabetes, cancer, etc.20 Studies also show the involvement of ERS in the toxic effects of bupivacaine,21 but it is unknown whether the GRP78/PERK/eIF2α/ATF4-mediated signaling pathway associated with ERS is also functionally involved.
We established an animal model of bupivacaine-induced neurotoxicity by administering the LA via an intrathecal tube and analyzed the expression pattern of the ERS-related factors in spinal nerves. We also explored the role of GM1 in promoting neurite regrowth, rescuing neuronal apoptosis, and regulating ERS signaling pathways. Our findings will help to determine the neuroprotective mechanisms of GM1 and ERS regulation in LA-induced spinal cord injuries.
Materials and methods
Establishing the animal model and grouping
A total of 180 healthy adult male Sprague–Dawley rats weighing 250–300 g were obtained from the Animal Care Center of Guangxi Medical University (Nanning, People’s Republic of China). All experiments conformed to the Guidelines of the National Institutes of Health (NIH publication, No. 8023) on the ethical use of animals, and studies were approved by the Animal Care and Use Committee of Guangxi Medical University (No. SCXK GUI 2004-0002). The rats were housed in separate cages in environmentally controlled rooms (temperature 25°C ± 5°C and relative humidity 55%–65%) on a 12-hour light/dark cycle, and standard chow and water were provided ad libitum. The animals were acclimatized for 3 days and then randomized into three treatment groups (n=60 each): saline group (group S), bupivacaine group (group B), and pretreated GM1 group (group G).
An intrathecal catheter was inserted using the modified method of Yaksh and Rudy.22 Briefly, the rats were anesthetized with an intraperitoneal injection of sodium pentobarbital (50 mg/kg), and a 1.5 cm longitudinal dorsal midline incision was made in the L5–L6 gap. The subcutaneous tissue was separated and after exposing the spinal dura mater, it was punctured lightly by a 25-gauge needle to access the cerebrospinal fluid (CSF). The subarachnoid space was cannulated with a polyethylene catheter (PE-10) (inside diameter 0.28 mm, outside diameter 0.61 mm, Smiths Medical, Kent, Britain) through the incision and advanced by 1.5 cm in the cephalad direction when the rat’s tail showed a sudden side-swaying motion. The terminal of the catheter was sealed off by heating to prevent CSF leakage and fixed in the neck through the subcutaneous tissue. Penicillin (20,000 U) was subcutaneously administered to prevent infection. The rats were housed in separate cages for 3 days to recover, and then lidocaine (2%, 10 μL) was injected via the catheter. The rats that did not display paralysis of both hind limbs 30 seconds postinjection or that did not recover from paralysis 30 minutes postinjection were excluded from further analyses. Group B rats were injected with 5% bupivacaine (0.12 μL/g, Sigma, MO, USA), group S received equal volumes of 0.9% normal saline, and group G were first pretreated with GM1 (30 mg/kg) and then treated the same way as group B after 24 hours. Following the respective treatments, the ducts were flushed with 10 μL normal saline each time. Each group was analyzed on days 0, 1, 3, 5, 7, and 14 after the last injection.
Neurobehavioral tests
All neurobehavioral measurements were performed in the morning between 9 AM and 11 AM in a double-blind fashion. The tail-flick latency (TFL) test was conducted to evaluate rat tail sensation, using a TF equipment (model YLS-12A; Huaibei ZhengHua Biological Instrument Equipment Co., Anhui, People’s Republic of China). It was fitted with a heat source (32 W infrared ray) that was focused on the tail at a distance of ~4 cm from the tip, and the time (in seconds) taken to withdraw the tail from the heat source was recorded. If the tail was not withdrawn within 16 seconds (cutoff time), the heat source was switched off to prevent tissue damage. Each rat was tested thrice at 5-minute intervals, and the average of the measurements was calculated. The TFL values were converted to percentage maximal possible effect (%MPE) as ([measured value − base value]/[cutoff time − base value]) × 100. The recovery of the locomotor function was assessed using Basso, Beattie, and Bresnahan (BBB) scale before the rats were sacrificed, according to movements of the hip, knee, and ankle joint and were scored between 0 (unable to walk) and 21 (normal movement).23
H&E staining
The rats were anesthetized deeply and their thorax were opened, and then perfused with 0.9% normal saline (4°C) followed by 4% paraformaldehyde (pH 7.4) through the left ventricle. The lumbar enlargement of spinal cord was rapidly removed on ice, and immersed in 4% paraformaldehyde for 24 hours at 4°C before paraffin embedding. The tissue blocks were cut into transverse sections (4 μm thick), dewaxed by a xylene gradient, and then rehydrated by an alcohol gradient. The sections were stained with H&E as per standard protocols and observed for histological and morphological changes under a light microscope (Olympus Microsystems, Tokyo, Japan). The histopathological changes were quantified by evaluating a sum of the distribution and severity scores of histopathological sections based on a previously described method as follows: distribution scores (0 = no lesion, 1 = lesions limited to the posterior root, 2 = lesions in both the posterior root and posterior column) and severity scores (0 = no lesion, 1 = focal disruption of myelin sheath and axons [mild lesion], 2 = moderate lesion, and 3 = diffuse disruption of myelin sheath and axons [severe lesion]).6
Confocal laser microscopy
The paraffin sections were dewaxed and rehydrated, and the tissue surface antigen was retrieved by the high-pressure method. After blocking endogenous peroxidase activity with 3% H2O2 for 15 minutes, the sections were permeabilized with 0.3% Triton X-100 (Solarbio, Beijing, China) for 10 minutes and incubated with 5% normal goat serum for 1 hour to block nonspecific binding. The sections were then incubated overnight with monoclonal rabbit antirat GRP78 (1:200 dilution; Cell Signaling Technology, Danvers, MA, USA) at 4°C, and then with Alexa Fluor 488 goat antirabbit IgG (1:400 dilution; Abcam, Cambridge, MA, USA) for 2 hours at 37°C. Mounting medium containing DAPI (Solarbio) was carefully placed over the sections, which were then observed under the Nikon A1 laser scanning confocal microscope (Tokyo, Japan). The total and fluorescent stained cells were counted in each field from at least three spinal cord sections per group, and the percentage of positive cells was calculated.
Transmission electron microscopy (TEM)
After removing the spinal cord, the posterior horn was cut into 1×1×2 mm pieces and fixed with 2.5% glutaraldehyde phosphate buffer for 24 hours at 4°C. The specimens were then washed thrice with PBS, fixed in 1% osmic acid for 2 hours, rehydrated in an alcohol gradient, and embedded in epoxy resin 618 overnight. Ultrathin sections were cut with an ultramicrotome (Leica UC7, Wetzlar, Germany), double-stained with lead citrate and uranyl acetate, and viewed under a Hitachi H-7650 electron microscope (Hitachi, Japan) at 80 kV.
TUNEL assay
To determine apoptosis in spinal nerve cells, the paraffin sections were dewaxed, rehydrated, and treated with proteinase K (20 μg/mL) for 10 minutes and stained using TUNEL assay kit (Roche, Basel, Switzerland) according to the manufacturer’s protocol. After rinsing thrice with PBS (5 minutes each), the sections were colorized using Converter-POD with 0.03% DAB (Boster, Wuhan, People’s Republic of China). The TUNEL-positive cells were counted in five random regions in each sample under a light microscope at 200× magnification (Olympus Microsystems), and the percentage of the apoptotic cells was calculated.
Quantitative real-time PCR
Total RNA was extracted from spinal cord tissue using Trizol (TaKaRa, Shiga, Japan) according to the manufacturer’s instructions, and the integrity of the RNA was analyzed by agarose gel electrophoresis. The cDNA was synthesized from 1 μg total RNA per sample using PrimeScript™ RT reagent Kit with gDNA Eraser (TaKaRa) in a 20 μL reaction volume according to the manufacturer’s instructions. All cDNAs were diluted 10× with RNase Free dH2O water and used as a template for quantitative real-time PCR. The expression levels of ERS-related genes were measured using a SYBR® Premix Ex Taq™ II kit (TaKaRa) as described previously on an ABI 7500 Real Time PCR System (Applied Biosystems, Carlsbad, CA, USA).7 The 20 μL reaction mix consisted of 10 μL SYBR Premix Ex Taq II, 0.4 μL ROX Reference Dye II, 6 μL H2O, 2 μL cDNAs, and 0.8 μL of each primer (Table 1). The reaction conditions were as follows: 40 cycles of 95°C for 30 seconds, 95°C for 5 seconds, and 60°C for 34 seconds. GAPDH was used as the internal control, and each cDNA sample was tested in triplicates. The relative gene expression was calculated by the 2−ΔΔCT method.
 | Table 1 Sequences of primers used for qRT-PCR |
Western blotting
Spinal cord tissues were ground in liquid nitrogen and homogenized in ice-cold RIPA lysis buffer (Solarbio) containing protease and phosphatase inhibitors. The lysates were centrifuged at 12,000 rpm at 4°C, and protein quantity in the supernatant was determined using BCA protein assay kit (Enhanced BCA Protein Assay Kit; Beyotime). Equal amounts of protein per sample (50 μg) were separated by 10% SDS-PAGE and then transferred to PVDF membranes (Immobilon-P; Millipore, Billerica, MA, USA) via wet transfer method. The membranes were blocked with 5% BSA in 10 mM Tris-buffered saline with 0.05% Tween 20 (TBST) for 1 hour, and incubated overnight with anti-p-PERK (1:1,000, Cell Signaling Technology), anti-p-eIF2α (1:1,000, Abcam), anti-ATF4 (1:1,000; Cell Signaling Technology), and anti-β-tubulin (loading control) at 4°C. The membranes were rinsed thrice with TBST and incubated with infrared-labeled goat antirabbit IgG (H+L) secondary antibody (1:10,000; Cell Signaling Technology) for 1 hour at 4°C. The array image was obtained using an LI-COR Biosciences Odyssey Infrared Imaging System. The pixel intensities of the bands were measured with image analysis software (ImageJ), and the relative expression levels of the ERS proteins were normalized to β-tubulin.
Statistical analyses
All analyses were performed using IBM SPSS Statistics, version 22 (IBM Deutschland GmbH, Ehningen, Germany). The data with normal distribution are presented as mean ± SD. One-way ANOVA was used to compare groups, and Bonferroni test was used for multiple comparisons. Kruskal–Wallis one-way ANOVA and Mann–Whitney U-test were used for data with nonnormal distribution. All tests were two-tailed, and P<0.05 was considered statistically significant.
Results
General characteristics of the rats
Three animals were excluded from the experiment (one due to infection and two due to incorrect catheterization). There were no significant differences between the groups in terms of fundal weight and food and water consumption throughout the study.
GM1 improved neurobehavioral indices
The TFL is a measure of the tail response to heat stimulation. Baseline TFL values were similar across the different groups (4.04±0.206 seconds), and that of the group S did not change significantly during the study. In contrast, the TFL increased remarkably in the groups B and G, although pretreatment with GM1 significantly decreased response latency to infrared heat stimulation compared to the group B. The %MPE values decreased gradually and were nearly restored to the baseline level on the 14th day in the group G (Figure 1A) and were significantly lower than that of the group B after the third day following treatment (P<0.05). Most rats in the group B exhibited behavioral changes, such as paralysis and weakness in both lower extremities, dragging, crawling, and gatism. The behavioral recovery was assessed by the BBB scores, which decreased significantly in the groups B and G compared to the control (Figure 1B; P<0.05). However, the BBB scores in the group G were significantly higher compared to the group G since day 3 (P<0.05).
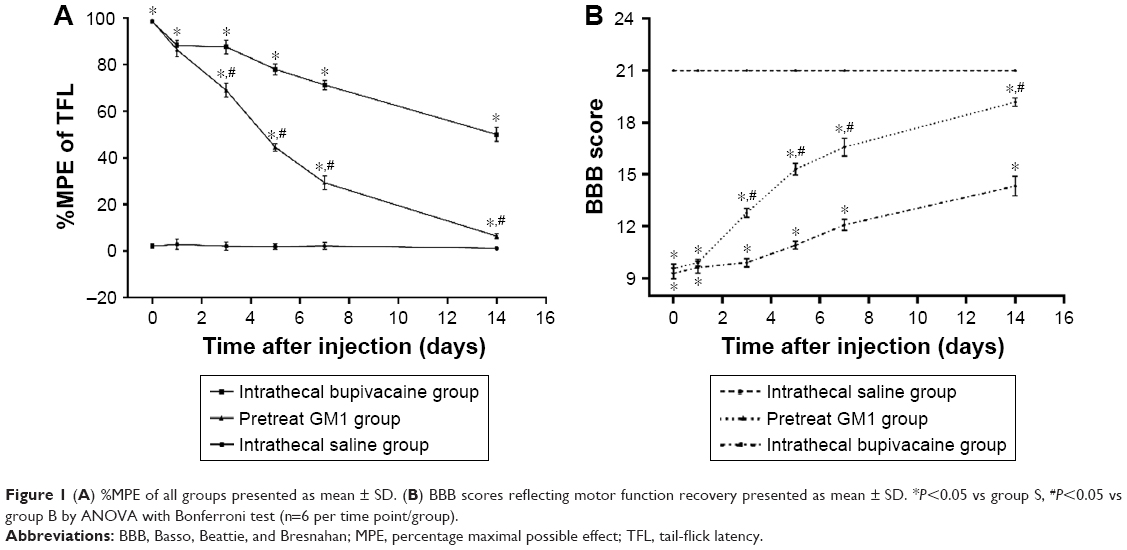 | Figure 1 (A) %MPE of all groups presented as mean ± SD. (B) BBB scores reflecting motor function recovery presented as mean ± SD. *P<0.05 vs group S, #P<0.05 vs group B by ANOVA with Bonferroni test (n=6 per time point/group). |
GM1 attenuated bupivacaine-induced neuronal injuries
Bupivacaine-induced neurotoxicity generally spreads from the posterior root at the entry point into the spinal cord and nearby posterior white matter.24 Therefore, we focused on the histomorphological changes in the posterior root. H&E staining of the spinal cord tissue of the group S showed undamaged structure with clearly visible gray and white matter, no vacuolar and edema changes, and normal neurons and axons (Figure 2A). Several morphological anomalies were observed in the group B, such as central chromatolysis in the motor neurons resulting in ghost cells, degeneration and necrosis of posterior root, vacuoles and edema in glial cells, satellitosis, and demyelination (Figure 2E and F). Although the group G (Figure 2B–D) also showed morphological and pathological changes, they were significantly milder compared to the group B (Figure 2F–H). The histopathological injury score (Figure 2I) was 0 in the group S and increased significantly in the groups B and G. From the fifth day however, injury scores significantly improved in the group G compared to the group B, although GM1 could not completely rescue the damage caused by bupivacaine even 14 days after treatment.
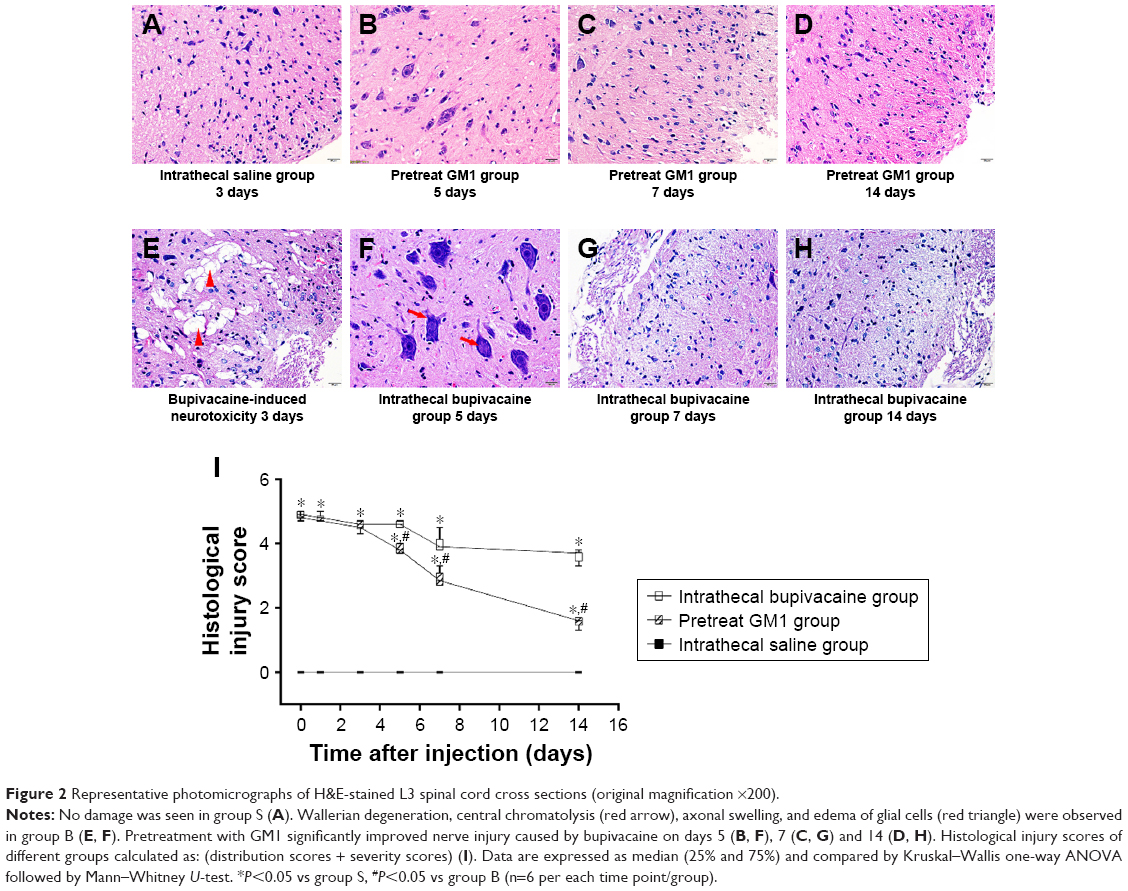 | Figure 2 Representative photomicrographs of H&E-stained L3 spinal cord cross sections (original magnification ×200). |
Transmission electron microscopy also showed ultrastructural differences between the untreated and treated groups (Figure 3). The group S nerve cells showed uniform cytoplasm and chromoplast, abundant organelles and ER, compact myelin sheath, and normal morphology. In contrast, extensive myelin ovoid and axonal degeneration was seen at the proximal end of the posterior root and in the posterior white matter in the group B, in addition, it also appeared nuclear membrane shrinkage, dissolution and peripheral condensation of chromatin, ER dilation and particle detachment, degeneration of the axons of Wallerian and axonal vacuoles. Although similar changes were seen in the group G, they were gradually restored, as seen by the improvement in demyelination and Wallerian degeneration. The protective effect of GM1 was partial, but it effectively regenerated nerve function by the 14th day after treatment.
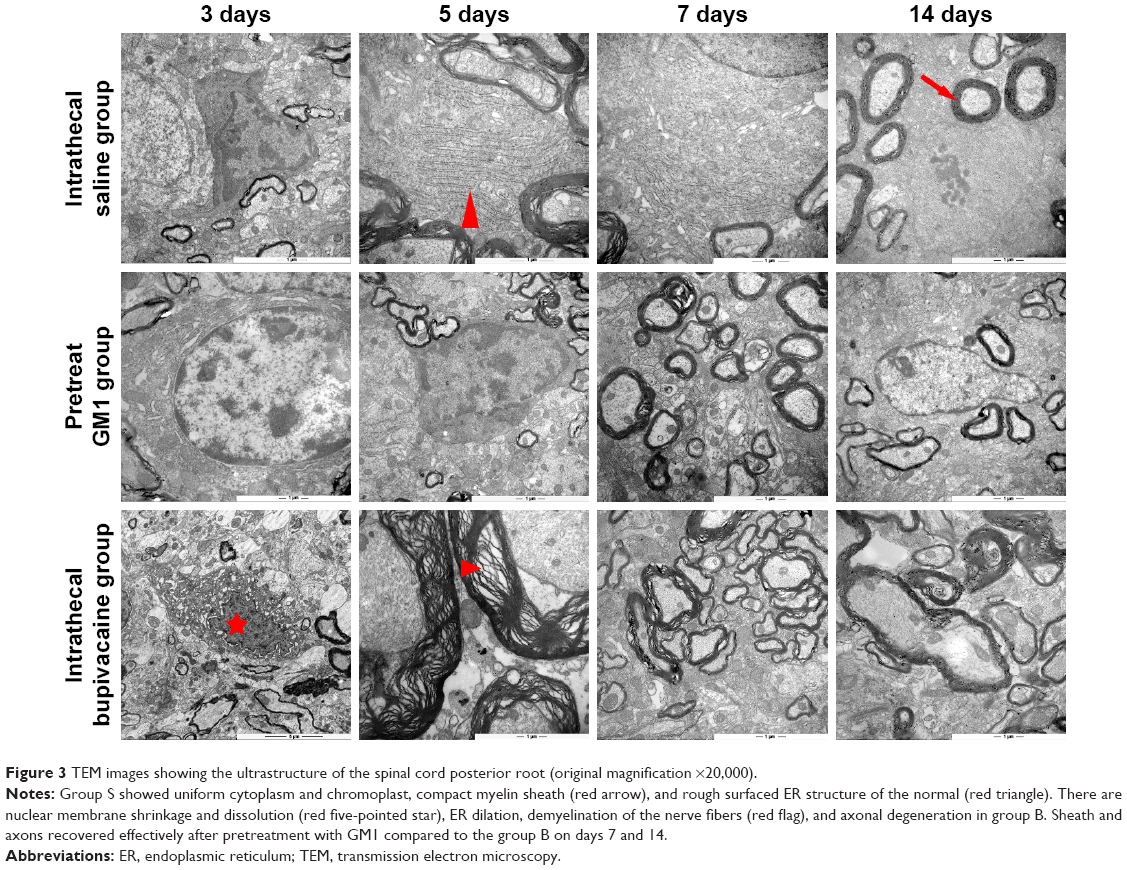 | Figure 3 TEM images showing the ultrastructure of the spinal cord posterior root (original magnification ×20,000). |
GM1 rescued apoptosis induced by bupivacaine
TUNNEL assay was used to evaluate apoptosis on days 0, 1, 3, 5, 7, and 14 after treatment. As shown in Figure 4A, apoptotic cells were appeared brown and the percentage of apoptotic cells increased significantly in the group B, peaking on day 3 after treatment. However, GM1 pretreatment significantly decreased the apoptosis rate on days 7 and 14 after inducing neurotoxicity (Figure 4B).
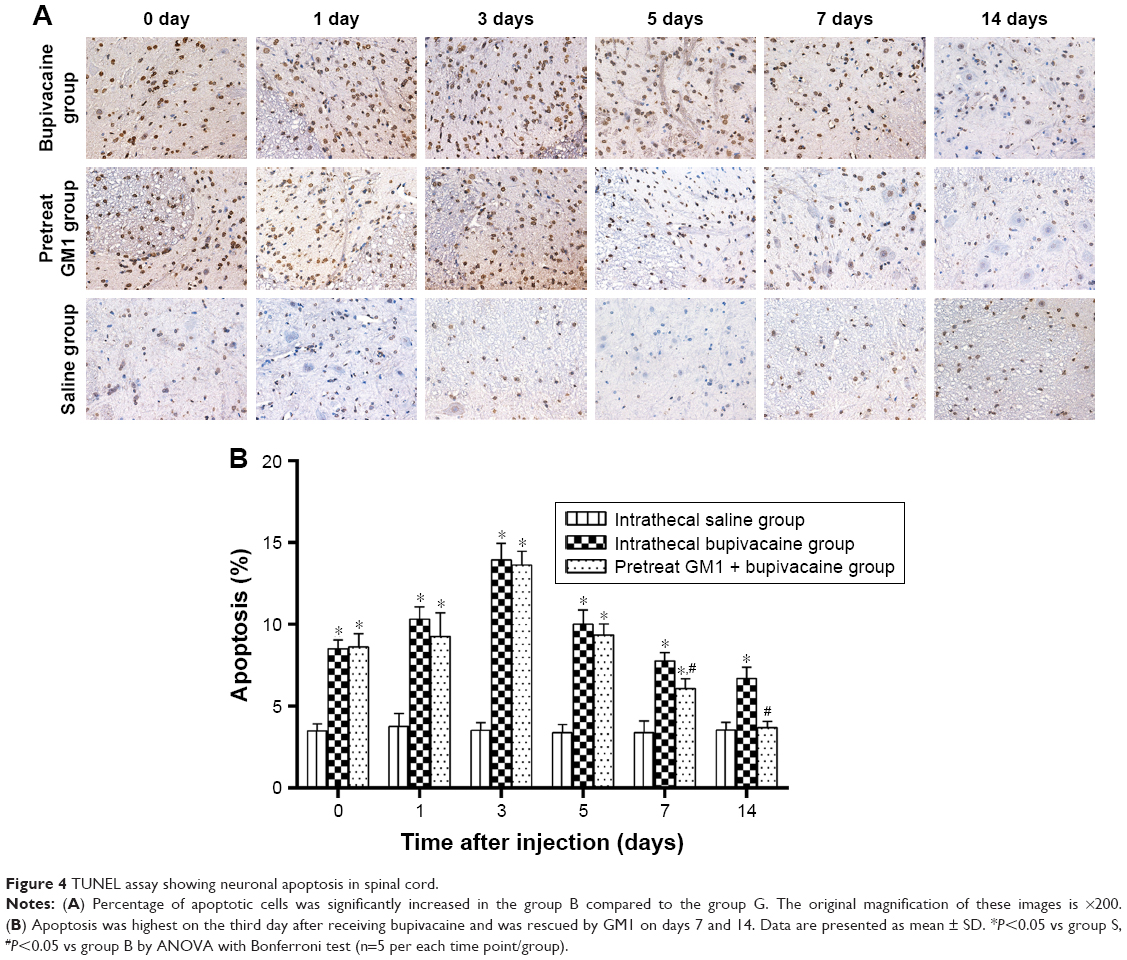 | Figure 4 TUNEL assay showing neuronal apoptosis in spinal cord. |
GM1 downregulated ERS pathway activated by bupivacaine
GRP78 is the common mediator of three pathways that activate the ERS.19 Laser scanning confocal microscopy showed localization of GRP78 protein in the cytoplasm of spinal dorsal horn neurons, which was significantly increased upon bupivacaine exposure (Figure 5A). Pretreatment with GM1 inhibited GRP78 expression on days 7 and 14 after the bupivacaine regimen (Figure 5B and C). To determine the effect of bupivacaine and GM1 on the ERS pathway, we measured the changes in the expression levels of PERK, eIF2α, and ATF4. All three factors were significantly upregulated in the group B compared to the group S at both the mRNA expression (Figure 6A–C) and protein levels (Figure 7A–C). In addition, GM1 pretreatment decreased PERK, eIF2α, and ATF4 mRNA and proteins levels relative to the group B by the 7th day and brought them to the baseline levels on the 14th day (Figure 7D–F). Taken together, bupivacaine induced neurotoxicity by activating the PERK/eIF2α/ATF4 cascade, and GM1 exerted its protective effects by attenuating this cascade.
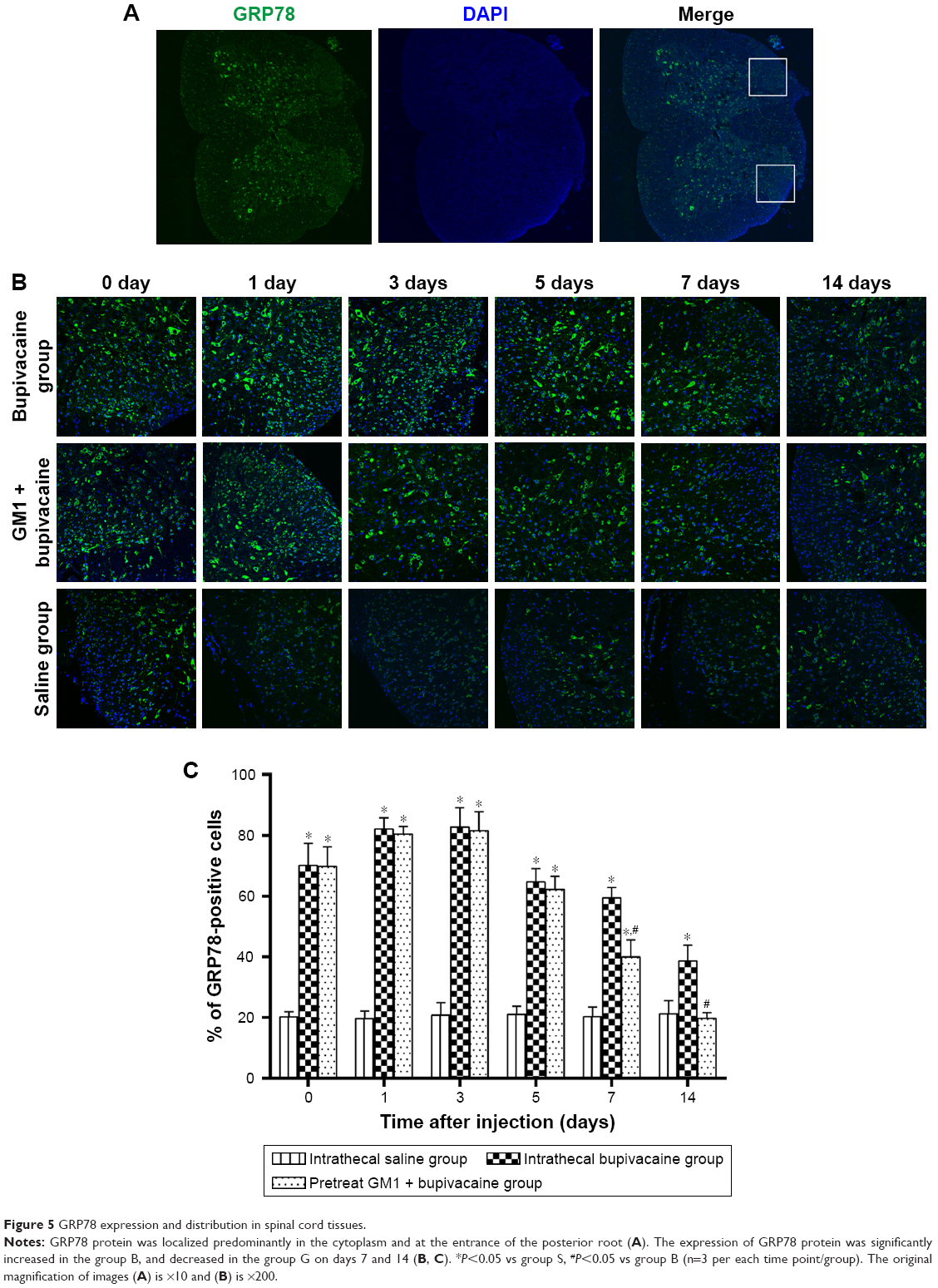 | Figure 5 GRP78 expression and distribution in spinal cord tissues. |
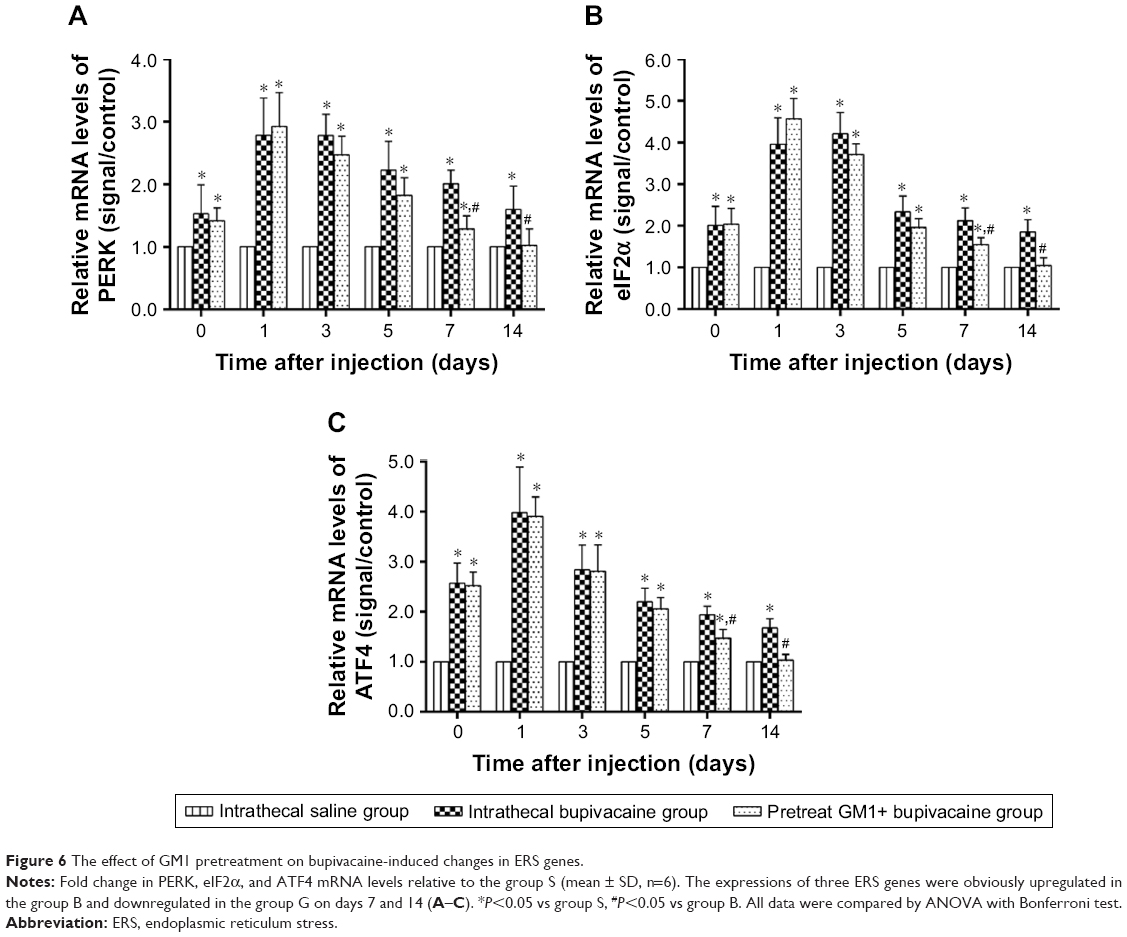 | Figure 6 The effect of GM1 pretreatment on bupivacaine-induced changes in ERS genes. |
 | Figure 7 Western blotting showing levels of p-PERK, p-eIF2α, and ATF4 protein levels at six time points (A–C). Comparing the expression of proteins among three groups on days 5, 7, and 14 (D–F) and quantifying by densitometric analysis. |
Discussion
We established a rat model of spinal cord neurotoxicity by intrathecally injecting rats with bupivacaine, in order to study the pathways involved in bupivacaine-induced neurotoxicity. The ERS pathway, especially the GRP78/PERK/eIF2α/ATF4 axis, was activated upon bupivacaine stimulation, which also resulted in significant histomorphological damage to the spinal cord, neuromotor disturbances, and apoptosis in the dorsal horn neurons. GM1 showed a therapeutic effect against bupivacaine-induced neurotoxicity by inhibiting the GRP78/PERK/eIF2α/ATF4 pathway and consequently ameliorated the sensory and locomotor anomalies.
The success of the intrathecal tube is the vital and prerequisite for the experiment. The precise placement of a catheter can produce a motor block in rats, and this model has been used to study the efficacy of intrathecally administered drugs.24 We chose bupivacaine as the LA to induce neurotoxicity in rat spinal cord since lidocaine and tetracaine show greater neurotoxicity at clinically relevant concentrations.25,26 Takenami et al showed that intrathecally injected 5% bupivacaine severely damaged the posterior root in rats without functional abnormalities.27,28 Another study using 0.5% bupivacaine found no morphological alterations,29 indicating dose-dependent neurotoxicity. The precise mechanisms underlying bupivacaine-induced neurotoxicity have not yet been elucidated so far. The spinal cord nerve root entry zone is extremely vulnerable to large doses of intrathecally administered LA,24 and the limited solubility of the LA in CSF and the restricted vascular supply further slow drug clearance.30,31 Consistent with previous studies, we observed significant histological damage to the posterior roots in rats treated with bupivacaine.
In addition to the inherent vulnerability of the spinal cord posterior root structure to intrathecal drug administration, bupivacaine also has neurotoxic effects. It induces neurotoxicity via the mitochondrial caspase-mediated apoptosis pathway,6,7 which we also observed in the dorsal neurons of rats treated with bupivacaine. In addition to the classical mitochondrial pathway, other signaling pathways may also be involved in bupivacaine-induced apoptosis. We found that bupivacaine significantly upregulated the ERS-related GRP78/PERK/eIF2α/ATF4 signaling pathway in the spinal cord, indicating its potential role in the LA-mediated neurotoxicity.
A recent study showed that neonatal mouse dorsal root ganglion (DRG) treated with lncRNA BDNF-AS rescued bupivacaine-induced neurotoxicity via the activation of the neurotrophin TrkB signaling pathway.32 In addition, the small-molecule GSK-3 inhibitor SB216763 also significantly ameliorated bupivacaine-induced apoptosis and neurite loss in murine DRG.33 Pretreatment of DRG with the antidepressant imipramine also attenuated neurotoxicity by coactivating p-TrkA and p-TrkB.34 Neurotrophins and dexmedetomidine have been shown to partially reverse the morphological injuries to DRG neurons.35,36 GM1, a pleiotropic neurotrophin, promotes CNS regeneration and prevents CNS damage in response to adverse conditions including AD, PD, and hypoxic ischemic encephalopathy.37–39 Furthermore, patients treated with GM1 after posttraumatic spinal cord injury showed considerable improvements in their motor function.40 Consistent with previous findings, we found that pretreatment with GM1 reversed bupivacaine-induced neuronal injury and ameliorated the sensory and locomotor disabilities.6
The ER is a stress sensor in eukaryotic cells, and stimulation of cells with various stresses, such as a high concentration of bupivacaine, results in ERS. Cells have evolved a conserved system known as the UPR to restore intracellular homeostasis and cope with ERS, by decreasing proteins biosynthesis and increasing protein transport and degradation.41 In animal cells, the UPR is predominantly regulated by PERK, which phosphorylates eIF2α, and p-eIF2α in turn upregulates ATF4. All three factors synergistically regulate protein synthesis and restore ER homeostasis.42–44 Therefore, we analyzed the expression of these ERS markers in bupivacaine-treated rats to determine the role of this LA in ERS. The UPR might act as a double-edged sword during ERS; while mild UPR achieves cellular homeostasis by self-regulation, persistent stimulation of UPR triggers a downstream apoptosis cascade.45 We found that UPR was activated in the neurons following stimulation with 5% bupivacaine, with the concomitant upregulation of GRP78, p-PERK, p-eIF2α, and ATF4, which eventually induced neurotoxicity and apoptosis. Interestingly, these factors peaked on the third day after intrathecal bupivacaine, which corresponded to higher apoptosis rates as well, indicating that the ERS reached a fastigium on the third day. Pretreatment with GM1 leads to a significant decrease in the ERS proteins and the rate of apoptosis on days 7 and 14 postbupivacaine and was accompanied by significant improvement in the histopathological and neurobehavioral indices. Therefore, GM1 may ameliorate the bupivacaine-induced neurotoxicity by inhibiting the GRP78/PERK/eIF2α/ATF4 signaling pathway and mitigating ERS and apoptosis.
Our results are consistent with a previous report that showed only a partial improvement of bupivacaine-induced apoptosis and necrosis with GM1 treatment.6 One reason could be that the high concentration of bupivacaine resulted in irreversible damage that could not be overcome by GM1, or that the dose of GM1 was suboptimal. In addition, one study reported a serum half-life of 1.4 hours for GM1,46 but the half-life of GM in cerebrospinal fluid has not been reported. Our pretreatment duration of 24 hours may have affected the pharmacokinetics of GM1 in cavum subarachnoidale and therefore its effective concentration. We only explored the ERS mediated by PERK, while the effects of IRE1α and ATF6, the other two ERS sensing proteins, on bupivacaine-induced neurotoxicity are still unclear. ERS-related protein inhibitors and activators need to be used to bidirectionally verify the specific mechanism of GM1-mediated neuroprotective effect. Future studies should also explore the relationship between ERS and autophagy vis-a-vis bupivacaine-induced neurotoxicity.
Conclusion
Our study demonstrated that pretreatment with GM1 has a certain therapeutic effect on bupivacaine-induced spinal neurotoxicity in rats (after the fifth day), nevertheless, the early treatment effect on LA-induced spinal neurotoxicity is not obvious. The neuroprotective mechanism of GM1 may be related to downregulate the GRP78/PERK/eIF2α/ATF4 of ERS signaling pathway. These findings increase our understanding of the pathogenesis of LA-induced spinal neurotoxicity and lay the foundation for more studies on the pharmacological action of GM1.
Acknowledgment
This study was supported by grants from the National Natural Science Foundation of China (No. 8166130027).
Disclosure
The authors report no conflicts of interest in this work.
References
Fuzier R, Aveline C, Zetlaoui P, Choquet O, Bouaziz H. Spinal anaesthesia in outpatient and conventional surgery: a point of view from experienced French anaesthetists. Anaesth Crit Care Pain Med. 2018;37(3):239–244. | ||
Buowari OY. Permanent neurological damage after spinal anaesthesia. Niger J Med. 2014;23(23):330–334. | ||
Neal JM, Kopp SL, Pasternak JJ, Lanier WL, Rathmell JP. Anatomy and pathophysiology of spinal cord injury associated with regional anesthesia and pain medicine: 2015 update. Reg Anesth Pain Med. 2015;40(5):506–525. | ||
Knight JB, Schott NJ, Kentor ML, Williams BA. Neurotoxicity of common peripheral nerve block adjuvants. Curr Opin Anaesthesiol. 2015;28(5):598–604. | ||
Brull R, McCartney CJ, Chan VW, El-Beheiry H. Neurological complications after regional anesthesia: contemporary estimates of risk. Anesth Analg. 2007;104(4):965–974. | ||
Ji J, Yan X, Li Z, Lai Z, Liu J. Therapeutic effects of intrathecal versus intravenous monosialoganglioside against bupivacaine-induced spinal neurotoxicity in rats. Biomed Pharmacother. 2015;69:311–316. | ||
Liang Y, Ji J, Lin Y, He Y, Liu J. The ganglioside GM-1 inhibits bupivacaine-induced neurotoxicity in mouse neuroblastoma Neuro2a cells. Cell Biochem Funct. 2016;34(6):455–462. | ||
Plank C, Hofmann P, Gruber M, et al. Modification of bupivacaine-induced myotoxicity with dantrolene and caffeine in vitro. Anesth Analg. 2016;122(2):418–423. | ||
Li Y, Zhao W, Yu X, et al. Activation of p47phox as a mechanism of bupivacaine-induced burst production of reactive oxygen species and neural toxicity. Oxid Med Cell Longev. 2017;2017(5):8539026. | ||
Lirk P, Haller I, Colvin HP, et al. In vitro, inhibition of mitogen-activated protein kinase pathways protects against bupivacaine- and ropivacaine-induced neurotoxicity. Anesth Analg. 2008;106(5):1456–1464. | ||
Zhao W, Liu Z, Yu X, et al. iTRAQ proteomics analysis reveals that PI3K is highly associated with bupivacaine-induced neurotoxicity pathways. Proteomics. 2016;16(4):564–575. | ||
Li R, Ma H, Zhang X, et al. Impaired autophagosome clearance contributes to local anesthetic bupivacaine-induced myotoxicity in mouse myoblasts. Anesthesiology. 2015;122(3):595–605. | ||
Bremer EG, Schlessinger J, Hakomori S. Ganglioside-mediated modulation of cell growth. Specific effects of GM3 on tyrosine phosphorylation of the epidermal growth factor receptor. J Biol Chem. 1986;261(5):2434–2440. | ||
Yuan B, Pan S, Zhang WW. Effects of gangliosides on expressions of caspase-3 and NGF in rats with acute spinal cord injury. Eur Rev Med Pharmacol Sci. 2017;21(24):5843–5849. | ||
Dai R, Zhang S, Duan W, et al. Enhanced autophagy contributes to protective effects of GM1 ganglioside against Aβ1-42-Induced neurotoxicity and cognitive deficits. Neurochem Res. 2017;42(8):2417–2426. | ||
Gong G, Yin L, Yuan L, et al. Ganglioside GM1 protects against high altitude cerebral edema in rats by suppressing the oxidative stress and inflammatory response via the PI3K/AKT-Nrf2 pathway. Mol Immunol. 2018;95:91–98. | ||
Srinivasan K, Sharma SS. Edaravone offers neuroprotection in a diabetic stroke model via inhibition of endoplasmic reticulum stress. Basic Clin Pharmacol Toxicol. 2012;110(2):133–140. | ||
Hiramatsu N, Chiang WC, Kurt TD, Sigurdson CJ, Lin JH. Multiple mechanisms of unfolded protein response-induced cell death. Am J Pathol. 2015;185(7):1800–1808. | ||
Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334(6059):1081–1086. | ||
Ghemrawi R, Battagliahsu SF, Arnold C. Endoplasmic reticulum stress in metabolic disorders. Cells. 2018;7(6):63. | ||
Galbes O, Bourret A, Nouette-Gaulain K, et al. N-acetylcysteine protects against bupivacaine-induced myotoxicity caused by oxidative and sarcoplasmic reticulum stress in human skeletal myotubes. Anesthesiology. 2010;113(3):1–569. | ||
Yaksh TL, Rudy TA. Chronic catheterization of the spinal subarachnoid space. Physiol Behav. 1976;17(6):1031–1036. | ||
Basso DM, Beattie MS, Bresnahan JC. A sensitive and reliable locomotor rating scale for open field testing in rats. J Neurotrauma. 1995;12(1):1–21. | ||
Takenami T, Yagishita S, Asato F, Arai M, Hoka S. Intrathecal lidocaine causes posterior root axonal degeneration near entry into the spinal cord in rats. Reg Anesth Pain Med. 2002;27(1):58–67. | ||
Hodgson PS, Neal JM, Pollock JE, Liu SS. The neurotoxicity of drugs given intrathecally (spinal). Anesth Analg. 1999;88(4):797–809. | ||
Sakura S, Kirihara Y, Muguruma T, Kishimoto T, Saito Y. The comparative neurotoxicity of intrathecal lidocaine and bupivacaine in rats. Anesth Analg. 2005;101(2):541–547. | ||
Takenami T, Yagishita S, Murase S, Hiruma H, Kawakami T, Hoka S. Neurotoxicity of intrathecally administered bupivacaine involves the posterior roots/posterior white matter and is milder than lidocaine in rats. Reg Anesth Pain Med. 2005;30(5):464–472. | ||
Takenami T, Wang G, Nara Y. Intrathecally administered ropivacaine is less neurotoxic than procaine, bupivacaine, and levobupivacaine in a rat spinal model. Obstetric Anesthesia Digest. 2012;33(5):456. | ||
Alessi Pissulin CN, Henrique Fernandes AA, Sanchez Orellana AM, Rossi ESRC, Michelin Matheus SM. Low-level laser therapy (LLLT) accelerates the sternomastoid muscle regeneration process after myonecrosis due to bupivacaine. J Photochem Photobiol B. 2017;168:30–39. | ||
Høy K, Hansen ES, He SZ, et al. Regional blood flow, plasma volume, and vascular permeability in the spinal cord, the dural sac, and lumbar nerve roots. Spine. 1994;19(24):2804–2811. | ||
Petterson CA, Olsson Y. Blood supply of spinal nerve roots. An experimental study in the rat. Acta Neuropathol. 1989;78(5):455–461. | ||
Zhang Y, Yan L, Cao Y, Kong G, Lin C. Long noncoding RNA BDNF-AS protects local anesthetic induced neurotoxicity in dorsal root ganglion neurons. Biomed Pharmacother. 2016;80:207–212. | ||
Yu T, Lin W. Small-molecule GSK-3 inhibitor rescued apoptosis and neurodegeneration in anesthetics-injured dorsal root ganglion neurons. Biomed Pharmacother. 2016;84:395–402. | ||
Guo J, Wang H, Tao Q, et al. Antidepressant imipramine protects bupivacaine-induced neurotoxicity in dorsal root ganglion neurons through coactivation of TrkA and TrkB. J Cell Biochem. 2017;118(11):3960–3967. | ||
Memari E, Hosseinian MA, Mirkheshti A, et al. Comparison of histopathological effects of perineural administration of bupivacaine and bupivacaine-dexmedetomidine in rat sciatic nerve. Exp Toxicol Pathol. 2016;68(10):559–564. | ||
Radwan IA, Saito S, Goto F. Neurotrophic factors can partially reverse morphological changes induced by mepivacaine and bupivacaine in developing sensory neurons. Anesth Analg. 2003;97(2):506–511. | ||
Cebecauer M, Hof M, Amaro M. Impact of GM1 on membrane-mediated aggregation/oligomerization of β-amyloid: unifying view. Biophys J. 2017;113(6):1194–1199. | ||
Sheng L, Li Z. Adjuvant treatment with monosialoganglioside may improve neurological outcomes in neonatal hypoxic-ischemic encephalopathy: a meta-analysis of randomized controlled trials. PLoS One. 2017;12(8):e0183490. | ||
Wu G, Lu ZH, Kulkarni N, Ledeen RW. Deficiency of ganglioside GM1 correlates with Parkinson’s disease in mice and humans. J Neurosci Res. 2012;90(10):1997–2008. | ||
Barros TE, Araujo FF, Higino LP, Marcon RM, Cristante AF. The effect of monosialoganglyoside (GM-1) administration in spinal cord injury. Acta Ortop Bras. 2016;24(3):123–126. | ||
Travers KJ, Patil CK, Wodicka L, Lockhart DJ, Weissman JS, Walter P. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell. 2000;101(3):249–258. | ||
Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. 1999;397(6716):271–274. | ||
Marciniak SJ, Garcia-Bonilla L, Hu J, Harding HP, Ron D. Activation-dependent substrate recruitment by the eukaryotic translation initiation factor 2 kinase PERK. J Cell Biol. 2006;172(2):201–209. | ||
Vattem KM, Wek RC. Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc Natl Acad Sci U S A. 2004;101(31):11269–11274. | ||
Hoppins S, Nunnari J. Cell Biology. Mitochondrial dynamics and apoptosis-the ER connection. Science. 2012;337(6098):1052–1054. | ||
Dumontet C, Rebbaa A, Portoukalian J. Kinetics and organ distribution of [14C]-sialic acid-GM3 and [3H]-sphingosine-GM1 after intravenous injection in rats. Biochem Biophys Res Commun. 1992;189(3):1410–1416. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.