")
Back to Journals » OncoTargets and Therapy » Volume 9
Monitoring cancer stem cells: insights into clinical oncology
Authors Lin S, Xu Y, Gan Z, Han K, Hu H, Yao Y, Huang M, Min D
Received 18 September 2015
Accepted for publication 1 January 2016
Published 11 February 2016 Volume 2016:9 Pages 731—740
DOI https://doi.org/10.2147/OTT.S96645
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr William C. Cho
ShuChen Lin,1,* YingChun Xu,2,* ZhiHua Gan,1 Kun Han,1 HaiYan Hu,3 Yang Yao,3 MingZhu Huang,4 DaLiu Min1
1Department of Oncology, Shanghai Sixth People’s Hospital East Campus, Shanghai Jiao Tong University, 2Department of Oncology, Renji Hospital, Shanghai Jiao Tong University, 3Department of Oncology, The Sixth People’s Hospital, Shanghai Jiao Tong University, 4Department of Medical Oncology, Cancer Hospital of Fudan University, Shanghai, People’s Republic of China
*These authors contributed equally to this work
Abstract: Cancer stem cells (CSCs) are a small, characteristically distinctive subset of tumor cells responsible for tumor initiation and progression. Several treatment modalities, such as surgery, glycolytic inhibition, driving CSC proliferation, immunotherapy, and hypofractionated radiotherapy, may have the potential to eradicate CSCs. We propose that monitoring CSCs is important in clinical oncology as CSC populations may reflect true treatment response and assist with managing treatment strategies, such as defining optimal chemotherapy cycles, permitting pretreatment cancer surveillance, conducting a comprehensive treatment plan, modifying radiation treatment, and deploying rechallenge chemotherapy. Then, we describe methods for monitoring CSCs.
Keywords: cancer stem cells, glycolytic inhibition, watchful waiting, rechallenge, immunotherapy
Introduction
In 1889, Paget proposed the “seed-and-soil” theory of cancer based on autopsy records of 735 women with fatal breast cancer.1 He found that metastasis was common in some organs because the “seed” (tumor cells) and “soil” (specific organs) were compatible. Recently, this theory was refreshed with the idea that cancer arises from cancer stem cells (CSCs), which are a small fraction of tumor cells.2 This cell subset can repopulate the entire tumor tissue and is the root of tumor progression, disease recurrence, and metastases. In addition, CSCs are predictive of poor prognosis.3–6 For example, colorectal cancer patients with stem-like subtypes (high expression of Wnt signaling and myoepithelial and mesenchymal genes, and low expression of differentiation markers) had the shortest disease-free survival.3 Liu et al6 identified a 186-gene “invasiveness” gene signature by comparing differential gene expression of breast CSCs and normal breast epithelia. Interestingly, “invasiveness” gene signature was associated with poor prognosis for several cancers such as medulloblastoma and breast, lung, and prostate cancers.
CSCs (“seeds”) reside in and are regulated by niches (“soil”) composed of an extracellular matrix, and differentiated and stromal cells, such as fibroblasts, vascular endothelial and inflammatory cells, and mesenchymal and hematopoietic stem cells.7 These cells produced factors that trigger multiple signaling pathways in CSCs to promote self-renewal, invasion, metastasis, and tumorigenicity.8,9 In addition, niches induced differentiated cancer cells subsequently to dedifferentiate into CSCs,8 and niches sheltered CSCs from xenobiotics, X-rays, and ions, which explains CSC resistance to chemotherapy and conventional radiotherapy.7
Recent findings in pancreatic ductal adenocarcinomas indicate that niche function is complicated. First, stroma was a physical barrier to chemotherapy against CSCs,10,11 but stroma restrained tumor growth, as evidenced in studies of transgenic mice with pancreatic cancer and deleted myofibroblasts. The animals had less overall immune infiltration, and increased CD4+/CD25+ regulatory T cell infiltration in the tumor stroma, as well as a greater CSCs phenotype and decreased survival.12 Also, Sonic hedgehog-deficient tumors had less stroma, which accelerated tumor growth.13 These data will hopefully inform novel strategies to treat cancer.
Methods for eradicating CSCs
As previously described, CSCs are essential for tumor initiation and progression. Therefore, selectively targeting and reducing chemotherapy-resistant and radio-resistant CSCs may reduce cancer. CSCs may be eradicated by the following methods.
Surgery
Theoretically, surgically resecting a tumor can directly remove CSCs. However, there are some issues that need to be addressed. Circulating tumor cells (CTCs) are present in peripheral blood and have CSC features associated with poor progression-free and overall survival in metastatic disease.14 CTCs are reported to appear in preoperative peripheral blood of patients with operable diseases, including breast15 and colorectal cancers16 and hepatocellular carcinoma.17 Therefore, existing CTCs may proliferate and cause tumor recurrence. This idea was confirmed through observations that a high proportion of these cells were associated with shorter disease-free survival.16,18 Work by Kim et al19 indicated that CTCs not only caused distant metastases but also led to primary site relapse via “tumor self-seeding”. Recently, a subset of CSCs in CTCs was identified in operable hepatocellular carcinomas,17,20 and colorectal cancer,21 and they were correlated with disease recurrence.20 Recent reports indicate that surgical manipulation immediately increased circulating hepatocellular CSCs,17 as well as created a favorable microenvironment that triggered the activation of signal transduction and activation of the transcription 3 (STAT3) signal pathway and this promoted CSC self-renewal.22
Glycolytic inhibition
Tumors undergo metabolic shifts involving the pentose phosphate pathway, glutamine-transporter genes, the tricarboxylic acid cycle, and acetyl-CoA carboxylase.23 Cancer treatments that target metabolic enzymes are under study and include targeting nucleic-acid and lipid synthesis, amino-acid metabolism/protein synthesis, glycolysis, the tricarboxylic acid cycle and mitochondrial metabolism, and fatty-acid and NAD metabolism.24 Of these, the Warburg effect (shift from oxidative phosphorylation to glycolysis) is an important metabolic change essential for cancer development.25 Data show that CSCs, unlike differentiated cells, rely on the Warburg effect or function and have increased enzymatic activity. CSCs also consumed more glucose and produced less lactate.26–29 Inhibiting glucose uptake by interfering with glucose transporter 1 expression30 or glucose transporter 3 expression31 significantly decreased self-renewal and tumorigenicity. Similar results were observed after direct inhibition of glycolysis.26,29
Driving CSC proliferation
Cancer cell dormancy contributes to treatment failure,32 and may be regulated by several mechanisms including the serine/threonine protein kinase AKT pathway, the P38 pathway, and noncoding RNAs.32 Specifically, CSCs are quiescent, so encouraging proliferation will increase chemotherapeutic efficacy.32 Recent data indicate that various mechanisms underlie the switch between CSC dormancy and proliferation.33–35 For example, the Fos family transcription factor Fos-related antigen 1 decreased the proportion of CSCs in vitro, causing dormancy loss and increasing chemosensitivity in vitro and in vivo.33 The bone morphogenetic protein (BMP) 7/BMP receptor 2/p38/N-myc downstream-regulated gene 1 axis was also implicated in dormancy and recurrence of prostate CSCs in bone, and withdrawal of BMP7 induced CSC growth.34 Farnesyl transferase inhibitors are necessary for autophagy of breast CSCs to maintain dormancy and avoid apoptosis. Therefore, withdrawal of farnesyl transferase inhibitors offers hope for driving CSC proliferation.35
Immunotherapy
In niches, CSCs were surrounded by many functional immune cells. CSC marker Oct4-reactive CD4+ and CD8+ T cells appeared in peripheral blood from both healthy individuals and ovarian cancer patients, and ascites contained Oct4-specific T cells,36 which can kill ovarian CSCs. In studies of prostate cancer, dendritic cells (DCs) pulsed with CSCs had greater tumor-specific immune responses both in vitro and in vivo, compared with DCs pulsed with differentiated tumor cells.37 Similar results were observed by Pellegatta et al,38 who reported that in glioma, DCs loaded with CSCs could cure both neurosphere-derived tumors and adherent cell-derived tumors, whereas DCs loaded with adherent cells did not cure neurosphere-derived tumors. Aldehyde dehydrogenase isoform 1 (ALDH1A1)+-specific cytotoxic T lymphocytes (CTLs) could target ALDH+ CSCs from head and neck squamous cell carcinoma, breast carcinoma, and pancreatic carcinoma, decreased tumor growth and metastases, and prolonged survival.39 Autologous tumor-infiltrating lymphocytes and a peripheral CTL clone recognized and killed CSCs of malignant fibrous histiocytoma.40
However, CSCs use several mechanisms to escape tumor immune responses; specifically, they expressed small amounts of major histocompatibility complex class I and II molecules and greater amounts of anti-apoptotic proteins such as bcl-2, bcl-xL, or survivin. Also, CSCs release immunosuppressive factors such as transforming growth factor beta, indoleamine deoxygenase, galectin-3, B7-H1, interleukin-4, and interleukin-10.41,42 Moreover, in pancreatic cancer, regulatory T cell function has been confirmed to be promotion of CSC self-renewal.12 In addition, differentiated tumor cells around CSCs comprise a protective barrier between them and CTLs, and compete with the latter for metabolic resources such as glucose.43 Therefore, many immune cells such as CTLs, CD4+ T cells, DCs, and cytokines may be involved in immunoregulation of CSCs. Targeting CSCs or destroying the immunosuppressive microenvironment may be a potential therapy for cancer.
Hypofractionated radiotherapy
Several studies indicate that CSCs are radio-resistant compared with non-stem cells due to greater fractions of CSCs observed after irradiation.44–47 However, irradiating glioma stem cells (2 Gy) does not alter tumorigenicity, but 5 Gy irradiation does. Similar results were noted for CSC survival after irradiation.44
Monitoring CSCs in clinical oncology
CSCs are key to tumorigenesis and prognosis,2 so monitoring them can yield data for treatment decisions. According to the Response Evaluation Criteria in Solid Tumors (RECIST), response effects include complete response, partial response, stable response, and progressive response. Tumor volumes can be measured, but few studies focus on CSC population changes after treatment. In a pilot study by Sprenger et al,48 biopsy and tumor specimens were compared, and 99 patients with rectal cancer with tumor regression after preoperative radio-chemotherapy had unique changes in fractions of CD133-expressing cells (44 patients had more; 55 had less). Increased CD133+ cancer cells were correlated with increased distant cancer recurrence, poorer disease-free survival, and worse cancer-specific overall survival. Targeted treatment such as surgery, glycolytic inhibition, driving CSC proliferation, immunotherapy, or hypofractionated radiotherapy is used, and CSCs may be reduced or stabilized, which helps with tumor control, or may be increased, which causes treatment failure (Figure 1).
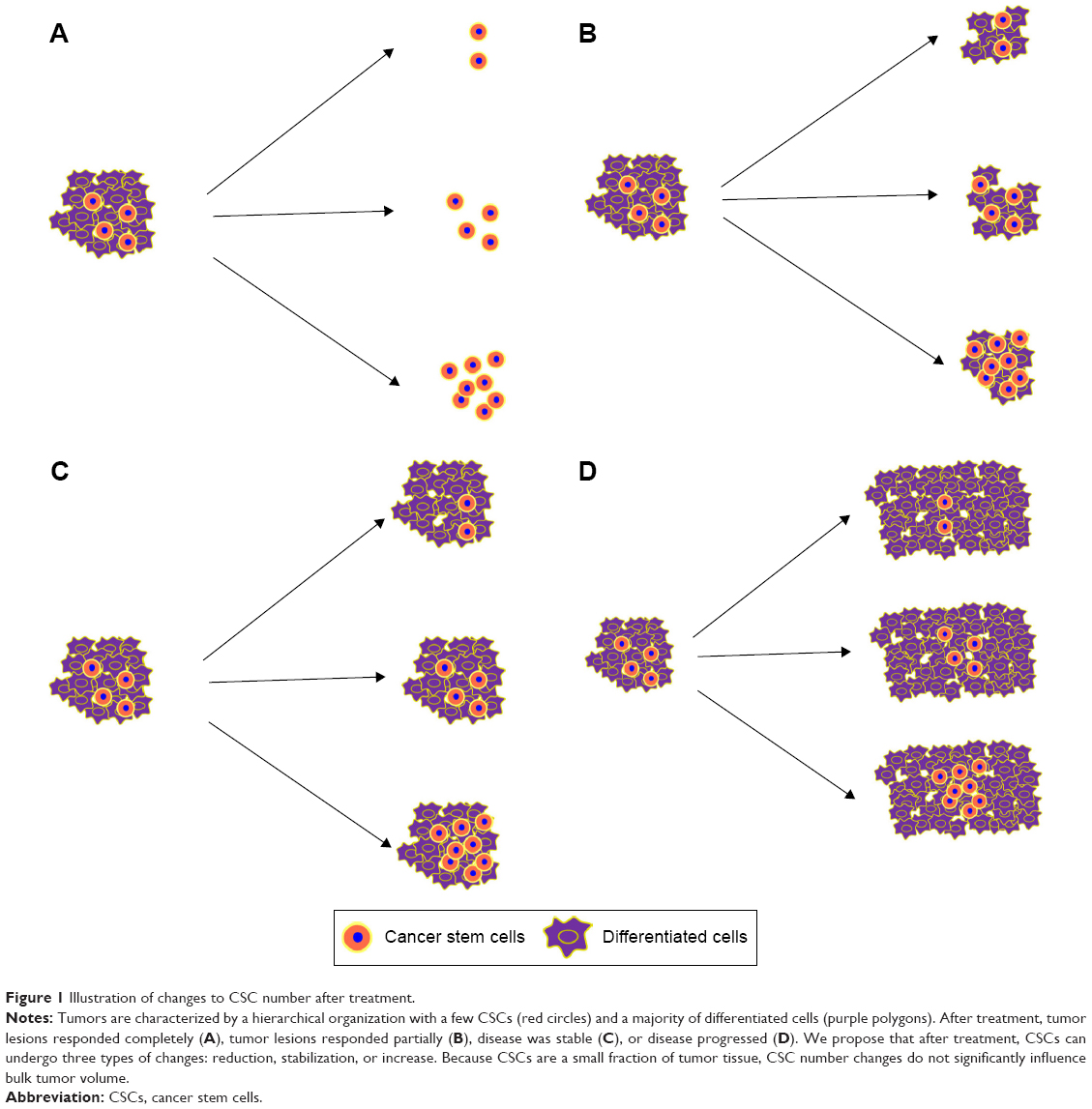 | Figure 1 Illustration of changes to CSC number after treatment. |
CSCs undergo either asymmetrical or symmetrical division. Asymmetrical division generates two different daughter cells, a CSC and a progenitor cell, and the CSC number is the same. When CSCs proliferate through symmetrical division, CSCs increase, and ultimately, the tumor progresses.49 Therefore, when CSCs divide asymmetrically, chemotherapy and conventional radiotherapy are appropriate treatments; when CSCs divide symmetrically, targeted treatments depicted earlier are recommended. Therefore, symmetrical division of CSCs in cancer patients is undesirable, but how division pathways are selected is unclear.
Selecting optimal chemotherapy cycles
Most advanced tumors, such as stage IV lung cancer and locally advanced breast cancer, should be treated with a chemotherapy-based treatment with cycle number determined by clinical trial results or recommendations of an expert panel. Instead, cycle numbers can be guided by CSC monitoring. Specifically, when proliferative cells are killed by chemotherapy, CSC homeostasis may be disturbed, and CSCs may be triggered to divide. When CSCs are quiescent or are dividing asymmetrically, chemotherapy could be continued until CSCs begin symmetrically dividing or when side effects are unacceptable.
Genomic abnormalities are associated with tumor initiation, tumor progression, and therapeutic resistance. Deep sequencing can be used to identify transcriptional events such as gene fusions or deletions, novel or cancer-associated isoforms, and putative novel noncoding RNAs, the functions of which could be further studied.50 Due to technical progress with isolation of single tumor cells, cell lysis, nucleic acid extraction, and amplification, single-cell sequencing can be used to study intratumoral heterogeneity.51 Using genomic profiles of CSCs and differentiated cells, a specific tumor type can be subdivided into several genomic subtypes that may have different prognoses and different treatment responses to drugs. Combined with real-time CSC and differentiated cell monitoring, drug resistance can be identified, drug treatment can be timed to coincide with CSC division patterns, and cycle length and number can be tailored to the patient.
Watchful waiting
For some tumors (prostate and ovarian cancers), a treatment period that can prevent recurrence is unknown, so watchful waiting is best. For example, in ovarian cancer, ~60% of patients with advanced disease suffered recurrence after undergoing first-line chemotherapy.52 The MRC OV05/EORTC 55955 trial assessed the most effective time to administer chemotherapy for relapsed ovarian cancer, comparing early treatment prompted by increased tumor marker CA125 with delayed treatment in response to clinical symptoms or symptomatic relapse. Early initiation of chemotherapy did not improve overall survival or quality of life compared with delayed chemotherapy.53 However, treatment effects targeting CSCs were not assessed. In many studies, the prevalence of ovarian CSCs was positively correlated with recurrence and a shortened disease-free interval after complete remission.54–56 Therefore, early treatment to target CSCs may offer better clinical outcomes compared to delayed treatment at symptomatic relapse.
CSCs may be reliable surrogates for treatment response
Poor treatment results occur when tumors shrink but CSCs increase; better responses involve tumor growth and fewer CSCs. When complete response is achieved, the CSC population should be measured to confirm treatment success. When a recovered immune system can eliminate CSCs after chemotherapy, observation may be rational, or further treatments (such as glycolytic inhibition and immunotherapy) may be required.
Modifying radiation
Tumor biology-guided adaptive radiotherapy is promising for treating cancer. With the recognition of different biological characteristics of CSCs and differentiated-like cells, therapeutic strategy has been proposed theoretically. Alfonso et al57 developed an individual cell-based model in which they assumed that CSCs and differentiated-like cells are randomly distributed before treatment, and the length of CSCs cycle is significantly longer than that of differentiated-like cells. Then, they become concentrated as the tumor grows. Therefore, different radiation doses can be used to enhance tumor control, especially by boosting radiation in areas of numerous CSCs. However, this model was not based on the real distribution of CSCs. Helical intensity-modulated radiotherapy (H-IMRT) integrates spiral computed tomography and a linear accelerator, which delivered radiation from 360° angles with a moving couch and intensity modulation through a binary multileaf collimator. This offered better intensity-modulated radiation dose distributions than other advanced photon radiotherapy methods. Also, image guidance can monitor patient/organ displacement and inform strategies for daily treatment modifications.58,59 A successful example is that H-IMRT could overwhelmingly minimize hippocampus dose to decrease the nervous system impairment when delivering whole-brain radiotherapy.60 We believe that the integration of monitoring CSCs and H-IMRT may guide radiation treatment modifications as CSCs could be irradiated with hypofractionated radiotherapy, and other cancer cells could be treated with conventional radiotherapy. This may offer better local control and fewer adverse events.
Rechallenge chemotherapy
In general, tumors that relapse >6 months after treatment may still be sensitive to previously applied chemotherapeutic agents and tumors that relapse <6 months are considered to be resistant. Rechallenge chemotherapy (RC) is a chemotherapeutic regimen identical to the first-line protocol. Nagano et al61 evaluated RC efficacy as second-line chemotherapy for patients with relapsed non-small-cell lung cancer (NSCLC). They reviewed 28 cases of consecutive NSCLC patients who received RC as second-line chemotherapy and compared outcomes with those of 38 consecutive NSCLC patients who were treated with docetaxel, a standard second-line chemotherapy agent. Median survival and 1-year survival in the RC group were significantly better than that of the docetaxel group, and for many in the RC group, the interval from the end of first-line chemotherapy to relapse was <6 months (median 5.0 months; range 1.6–36.1 months). This implied that there are still many NSCLC patients who relapsed <6 months after first-line chemotherapy, and may benefit from RC. Why this occurred is unclear. In our opinion, the CSC theory may suggest a potential mechanism. We proposed that RC was efficacious for those who relapsed <6 months because CSCs can differentiate into different daughter cells. Some daughter cells are sensitive to chemotherapeutic agent X and were killed after this first-line therapy was applied. In contrast, daughter cells sensitive to chemotherapeutic agent Y would be killed by that drug. Due to the plasticity of CSCs, upon relapse, three events may have transpired – A: CSCs differentiated into daughter “X” cells would still be sensitive to drug X; B: If CSCs divided symmetrically, the tumor may be multidrug resistant; and C: if CSCs differentiated into daughter “Y” cells, drug X would not be useful, but drug Y would (Figure 2). Therefore, upon relapse, RC may be effective for event A, but methods for differentiating among these events have not been established.
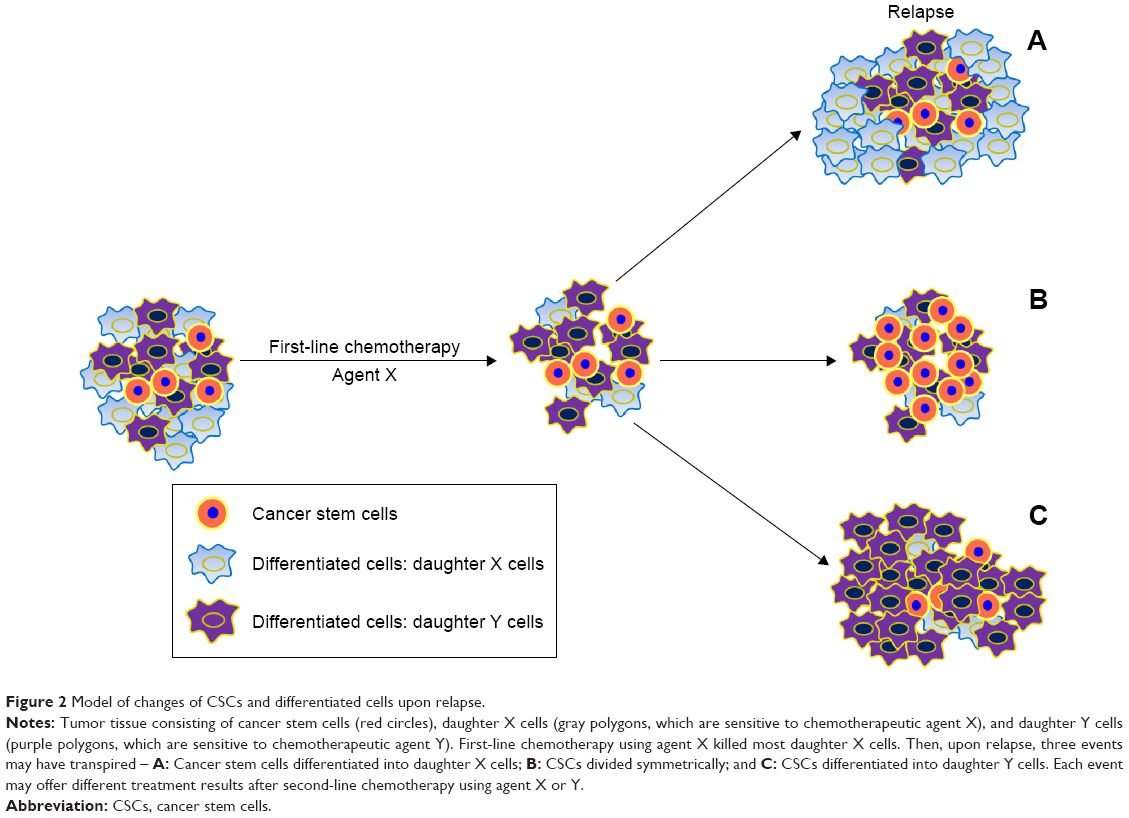 | Figure 2 Model of changes of CSCs and differentiated cells upon relapse. |
Targeted therapy
Several signaling pathways, including the Wnt, Notch, Hedgehog, and phosphatase and tensin homolog (PTEN)/mammalian target of rapamycin (mTOR)/STAT3 pathways, regulate CSC self-renewal.62 Agents inhibiting these pathways induce apoptosis and decrease tumorigenicity of CSCs.63,64 Because CSCs are a small fraction of tumor tissue, RECIST is not a sensitive method for evaluating the effect of these agents. In particular, there is growing recognition that improvements in our ability to measure the stem cell pool might offer additional insights into a tumor’s response to treatment, beyond what can be currently measured by RECIST. For example, the PTEN/mTOR/STAT3 pathway is very important for survival, proliferation, and tumorigenicity of breast CSCs.65 Everolimus, an oral inhibitor of mTOR, alone or in combination with other agents has antitumor effects on breast CSCs.66–68 Treatment with everolimus plus docetaxel significantly decreased tumor volume, whereas everolimus alone was less effective.66 Nodal and activin, members of the transforming growth factor beta superfamily, were recently recognized to be important regulators of self-renewal and tumorigenicity of pancreatic CSCs.69,70 As expected, adding a nodal/activin receptor inhibitor to gemcitabine treatment did not affect pancreatic cell growth initially; however, at long-term follow-up, it did reduce tumor cell growth.70 Thus, RECIST-based judgment of treatment effects should be interpreted with caution, and new methods of imaging CSCs should be applied.
In lung cancers with epidermal growth factor receptor (EGFR)-activating mutations, ALDH1A1-negative cancer cells are more sensitive to tyrosine kinase inhibitors (TKIs) than ALDH1A1-positive CSCs. In vitro, an EGFR TKI-resistant cell line overexpressed ALDH1A1 and induced CSC proliferation. Clinical studies indicate that lung cancers resistant to EGFR TKIs and chemotherapeutic drugs had more ALDH1A1-positive cells compared with other lung cancers.71–73 These data indicate that monitoring CSCs in tumors may reveal acquired resistance to TKIs. More studies are needed to confirm whether CSCs are reliable predictors of targeted therapy agent.
Rational methods of CSC monitoring
Many are working on better methods to detect CSCs and to determine whether they can function as reliable surrogates for response, and whether they might represent important targets for novel drug therapies. However, there are extremely few studies that help us better understand the ultimate role of CSCs in cancer diagnosis, response evaluation, drug development, and prognosis. Positron-emission tomography/computed tomography (CT) is superior to other diagnostic imaging systems, such as CT and magnetic resonance imaging (MRI) because it depicts glucose uptake of tumor cells which can distinguish between viable tumor cells and fibrotic lesions. Yoshii et al74 observed that CSCs could be overlooked by positron-emission tomography/CT due to low uptake of radiolabeled [18F]-2-fluoro-2-deoxy-d-glucose by CSCs. Then investigators made efforts to apply the reliable markers of CSCs to image them in vivo, and had gained some initial achievement.
Labeling CSCs with fluorescent protein, luciferase, superparamagnetic iron oxide (SPIO) nanoparticles, ferritin, or micron-sized iron oxide particles, and observing CSC behavior with optical imaging or MRI is also under study.75–80 Wang et al75 reported that SPIO has low toxicity to CSCs and can be used to tracked glioblastoma CSCs. However, iron not only distributes to CSC daughter cells but also can be absorbed by nearby macrophages, so this will diminish the sensitivity and specificity of the signal.81 Using the MRI reporter gene ferritin, instead of SPIO, is a solution.76 Liu et al77 suggested that a labeled dual-function bioluminescence system (modified codon-optimized version of luciferase fused with fluorescent proteins) was more sensitive. As few as ten breast CSCs were identified in vivo. Intravital microscopy offers a resolution of up to 1 μm, so this was used to observe single-labeled CSCs and determine a relationship between CSCs and the microenvironment. CSCs and their descendants were noted to be close to the vasculature, offering the first in vivo evidence that CSCs are responsible for tumor propagation.78 Choi et al79 induced colon tumors in Lgr5–eGFP (Lgr5, intestinal CSC marker) mice with azoxymethane and dextran sodium sulfate. Using confocal laser endomicroscopy, they observed that over time, Lgr5+ cells migrated to the colonic luminal surface, expanded, and formed adenomas. This work provided in vivo images of early colon tumorigenesis and may be useful for early tumor prevention and treatment. With micron-sized iron oxide particles, high-resolution MRI could be used to measure metastasizing breast carcinoma cells in the brain at the single-cell level.80 However, no reports are available to describe high-resolution MRI for visualizing single CSCs. Therefore, the progressing techniques make monitoring single cell of CSCs in vivo possible. However, the requirement of in vitro cell labeling suggests that human use is a few years away.
The second strategy includes constructing a fluorescent ligand or magnetic nanocrystal complex, and ligand–receptor interactions permit visualization of CSCs by MRI or optical imaging. Tsurumi et al82 used a fluorescently labeled CD133-specific monoclonal antibody AC133.1 to quantify CD133-positive glioblastoma stem cells and colon CSCs. There was initial evidence that noninvasive antibody-based in vivo imaging of tumor-associated CD133 is feasible. A similar method was used to image glioma tumors as small as 2–3 mm.83 Thereafter, other molecular tracers were used to track different CSCs. For example, a GSC-targeting peptide-binding nestin protein was used to count glioma stem cells,84 and hyaluronic acid-binding CD44 was used to measure gastric CSCs.85 For a single-luciferase model, at least 2,500 luciferase-positive cells are required.86
The third imaging method was established using biological characteristics of CSCs. For example, CSC phenotypes in glioma and breast cancer had reduced 26S proteosome activity, so CSCs could be easily tracked in vitro and in vivo when cells were transfected with ZsGreen (fluorescent protein and substrate of 26S proteosome).87 64Cu-diacetyl-bis (N4-methylthiosemicarbazone, 64Cu-ATSM) is an imaging agent that targets hypoxic tumors as well as a potential agent for internal radiotherapy of tumors due to its beta-particle and auger electron emission.88 64Cu-ATSM localized preferentially to regions with a high density of CD133+ colon CSCs and decreased CD133+ cells and metastatic ability.
The studies presented here were animal studies, so applications in humans are pending more research. Also, sensitivity, specify, safety, and noninvasiveness must be established. With the development and integration of innovative techniques, tracking single CSCs in humans may be practical.
Recently, Miranda-Lorenzo et al89 reported that CSCs of pancreatic ductal adenocarcinoma, colorectal carcinoma, hepatocellular carcinoma, and NSCLC could emit autofluorescence and that autofluorescent cells have CSCs phenotypes (ie, self-renewal capacity, expression of pluripotency-associated genes, high tumorigenicity, invasiveness, and chemoresistance). Autofluorescence was due to riboflavin accumulation in membrane-bound cytoplasmic structures of CSCs which had high ABCG2 transporter activity. Therefore, autofluorescence is an intrinsic characteristic of CSCs, and a novel method for isolating or monitoring CSCs in vivo.
CTCs, which have stem cell-like characteristics, may be important to gastric cancer metastasis, and identifying stem cell-like CTC subsets may offer more useful information than CTCs.90 A promising approach is to identify stem cell-like CTCs in the peripheral blood in an adjuvant setting and before and after salvage therapy, but this method requires large blood samples. Also, removal of the cells from cancer patients may alter their intrinsic properties. To address these potential problems, Galanzha et al91 used in vivo flow cytometry and intrinsic photothermal and photoacoustic characteristics of stem cell-like CTCs, labeling them with golden carbon nanotubes bioconjugated with folate and antibodies specific to the CD44 receptor. This enabled high sensitivity quantification of circulating stem cell-like CTCs in live breast cancer-bearing mice. More work is required to confirm whether these low toxic molecular contrast agents can be used in humans.
Conclusion
CSCs have many malignant features such as chemotherapuetic and radiotherapeutic resistance and tumorigenicity and are responsible for tumor progression, disease recurrence, and metastasis. We suggest that CSCs may reflect the true treatment response and monitoring them may be helpful to resolve a lot of clinical problems.
Acknowledgments
The authors thank LetPub (www.letpub.com) for its linguistic assistance during the preparation of this manuscript. This work was supported by the National Natural Science Foundation of China (Grant No 81301858) and funded by the Shanghai Sixth People’s Hospital East Campus (Grant No 2013021).
Disclosure
The authors report no conflicts of interest in this work.
References
Paget S. The distribution of secondary growths in cancer of the breast. 1889. Cancer Metastasis Reviews. 1989;8(2):98–101. | ||
Ajani JA, Song S, Hochster HS, Steinberg IB. Cancer stem cells: the promise and the potential. Seminars in Oncology. 2015;42 Suppl 1:S3–S17. | ||
Sadanandam A, Lyssiotis CA, Homicsko K, et al. A colorectal cancer classification system that associates cellular phenotype and responses to therapy. Nature Medicine. 2013;19(5):619–625. | ||
Sadanandam A, Gray J, Hanahan D. Reply to Colorectal cancer classification based on gene expression is not associated with FOLFIRI response. Nature Medicine. 2014;20(11):1231–1232. | ||
Calon A, Lonardo E, Berenguer-Llergo A, et al. Stromal gene expression defines poor-prognosis subtypes in colorectal cancer. Nature Genetics. 2015;47(4):320–329. | ||
Liu R, Wang X, Chen GY, et al. The prognostic role of a gene signature from tumorigenic breast-cancer cells. The New England Journal of Medicine. 2007;356(3):217–226. | ||
Lin S, Fu S. The correlation of hematopoietic stem cells with cancer stem cells through the regulation of stromal cells in tumor microenvironment. Medical Hypotheses. 2013;80(4):494–497. | ||
Ping YF, Zhang X, Bian XW. Cancer stem cells and their vascular niche: do they benefit from each other? Cancer Letters. 2015. Epub 2015 May 10. | ||
Plaks V, Kong N, Werb Z. The cancer stem cell niche: how essential is the niche in regulating stemness of tumor cells? Cell Stem Cell. 2015;16(3):225–238. | ||
Feig C, Gopinathan A, Neesse A, Chan DS, Cook N, Tuveson DA. The pancreas cancer microenvironment. Clinical Cancer Research: An Official Journal of the American Association for Cancer Research. 2012;18(16):4266–4276. | ||
Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, Hingorani SR. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;21(3):418–429. | ||
Ozdemir BC, Pentcheva-Hoang T, Carstens JL, et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell. 2014;25(6):719–734. | ||
Rhim AD, Oberstein PE, Thomas DH, et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell. 2014;25(6):735–747. | ||
Tinhofer I, Saki M, Niehr F, Keilholz U, Budach V. Cancer stem cell characteristics of circulating tumor cells. International Journal of Radiation Biology. 2014;90(8):622–627. | ||
Lang JE, Mosalpuria K, Cristofanilli M, et al. HER2 status predicts the presence of circulating tumor cells in patients with operable breast cancer. Breast Cancer Research and Treatment. 2009;113(3):501–507. | ||
Torino F, Bonmassar E, Bonmassar L, et al. Circulating tumor cells in colorectal cancer patients. Cancer Treatment Reviews. 2013;39(7):759–772. | ||
Li W, Zhou X, Huang Z, et al. Laparoscopic surgery minimizes the release of circulating tumor cells compared to open surgery for hepatocellular carcinoma. Surgical Endoscopy. 2015;29(11):3146–3153. | ||
Lang JE, Hall CS, Singh B, Lucci A. Significance of micrometastasis in bone marrow and blood of operable breast cancer patients: research tool or clinical application? Expert Review of Anticancer Therapy. 2007;7(10):1463–1472. | ||
Kim MY, Oskarsson T, Acharyya S, et al. Tumor self-seeding by circulating cancer cells. Cell. 2009;139(7):1315–1326. | ||
Fan ST, Yang ZF, Ho DW, Ng MN, Yu WC, Wong J. Prediction of posthepatectomy recurrence of hepatocellular carcinoma by circulating cancer stem cells: a prospective study. Annals of Surgery. 2011;254(4):569–576. | ||
Kantara C, O’Connell MR, Luthra G, et al. Methods for detecting circulating cancer stem cells (CCSCs) as a novel approach for diagnosis of colon cancer relapse/metastasis. Laboratory Investigation; a Journal of Technical Methods and Pathology. 2015;95(1):100–112. | ||
Segatto I, Berton S, Sonego M, et al. Surgery-induced wound response promotes stem-like and tumor-initiating features of breast cancer cells, via STAT3 signaling. Oncotarget. 2014;5(15):6267–6279. | ||
Creighton CJ, Morgan M, Gunaratne PH, et al. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. 2013;499(7456):43–49. | ||
Vander Heiden MG. Targeting cancer metabolism: a therapeutic window opens. Nature Reviews. Drug Discovery. 2011;10(9):671–684. | ||
Jang M, Kim SS, Lee J. Cancer cell metabolism: implications for therapeutic targets. Experimental & Molecular Medicine. 2013;45:e45. | ||
Ciavardelli D, Rossi C, Barcaroli D, et al. Breast cancer stem cells rely on fermentative glycolysis and are sensitive to 2-deoxyglucose treatment. Cell Death & Disease. 2014;5:e1336. | ||
Vlashi E, Lagadec C, Vergnes L, et al. Metabolic differences in breast cancer stem cells and differentiated progeny. Breast Cancer Research and Treatment. 2014;146(3):525–534. | ||
Liao J, Qian F, Tchabo N, et al. Ovarian cancer spheroid cells with stem cell-like properties contribute to tumor generation, metastasis and chemotherapy resistance through hypoxia-resistant metabolism. PLoS One. 2014;9(1):e84941. | ||
Liu PP, Liao J, Tang ZJ, et al. Metabolic regulation of cancer cell side population by glucose through activation of the Akt pathway. Cell Death and Differentiation. 2014;21(1):124–135. | ||
Shibuya K, Okada M, Suzuki S, et al. Targeting the facilitative glucose transporter GLUT1 inhibits the self-renewal and tumor-initiating capacity of cancer stem cells. Oncotarget. 2015;6(2):651–661. | ||
Flavahan WA, Wu Q, Hitomi M, et al. Brain tumor initiating cells adapt to restricted nutrition through preferential glucose uptake. Nature Neuroscience. 2013;16(10):1373–1382. | ||
Crea F, Nur Saidy NR, Collins CC, Wang Y. The epigenetic/noncoding origin of tumor dormancy. Trends in Molecular Medicine. 2015;21(4):206–211. | ||
Lu D, Chen S, Tan X, et al. Fra-1 promotes breast cancer chemosensitivity by driving cancer stem cells from dormancy. Cancer Research. 2012;72(14):3451–3456. | ||
Kobayashi A, Okuda H, Xing F, et al. Bone morphogenetic protein 7 in dormancy and metastasis of prostate cancer stem-like cells in bone. The Journal of Experimental Medicine. 2011;208(13):2641–2655. | ||
Chaterjee M, van Golen KL. Breast cancer stem cells survive periods of farnesyl-transferase inhibitor-induced dormancy by undergoing autophagy. Bone Marrow Research. 2011;2011:362938. | ||
Di J, Massuger LF, Duiveman-de Boer T, Zusterzeel PL, Figdor CG, Torensma R. Functional OCT4-specific CD4 and CD8 T cells in healthy controls and ovarian cancer patients. Oncoimmunology. 2013;2(5):e24271. | ||
Jachetti E, Mazzoleni S, Grioni M, et al. Prostate cancer stem cells are targets of both innate and adaptive immunity and elicit tumor-specific immune responses. Oncoimmunology. 2013;2(5):e24520. | ||
Pellegatta S, Poliani PL, Corno D, et al. Neurospheres enriched in cancer stem-like cells are highly effective in eliciting a dendritic cell-mediated immune response against malignant gliomas. Cancer Research. 2006;66(21):10247–10252. | ||
Qian X, Ma C, Nie X, et al. Biology and immunology of cancer stem(-like) cells in head and neck cancer. Critical Reviews in Oncology/Hematology. 2015;95(3):337–345. | ||
Kano M, Tsukahara T, Emori M, et al. Autologous CTL response against cancer stem-like cells/cancer-initiating cells of bone malignant fibrous histiocytoma. Cancer Science. 2011;102(8):1443–1447. | ||
Bruttel VS, Wischhusen J. Cancer stem cell immunology: key to understanding tumorigenesis and tumor immune escape? Frontiers in Immunology. 2014;5:360. | ||
Maccalli C, Volonte A, Cimminiello C, Parmiani G. Immunology of cancer stem cells in solid tumours. A review. European Journal of Cancer (Oxford, England: 1990). 2014;50(3):649–655. | ||
Kareva I. Immune evasion through competitive inhibition: the shielding effect of cancer non-stem cells. Journal of Theoretical Biology. 2015;364:40–48. | ||
Bao S, Wu Q, McLendon RE, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444(7120):756–760. | ||
Piao LS, Hur W, Kim TK, et al. CD133+ liver cancer stem cells modulate radioresistance in human hepatocellular carcinoma. Cancer Letters. 2012;315(2):129–137. | ||
Al-Assar O, Muschel RJ, Mantoni TS, McKenna WG, Brunner TB. Radiation response of cancer stem-like cells from established human cell lines after sorting for surface markers. International Journal of Radiation Oncology, Biology, Physics. 2009;75(4):1216–1225. | ||
Phillips TM, McBride WH, Pajonk F. The response of CD24(-/low)/CD44+ breast cancer-initiating cells to radiation. Journal of the National Cancer Institute. 2006;98(24):1777–1785. | ||
Sprenger T, Conradi LC, Beissbarth T, et al. Enrichment of CD133-expressing cells in rectal cancers treated with preoperative radiochemotherapy is an independent marker for metastasis and survival. Cancer. 2013;119(1):26–35. | ||
Berger F, Gay E, Pelletier L, Tropel P, Wion D. Development of gliomas: potential role of asymmetrical cell division of neural stem cells. The Lancet. Oncology. 2004;5(8):511–514. | ||
Guffanti A, Iacono M, Pelucchi P, et al. A transcriptional sketch of a primary human breast cancer by 454 deep sequencing. BMC Genomics. 2009;10:163. | ||
Sun HJ, Chen J, Ni B, Yang X, Wu YZ. Recent advances and current issues in single-cell sequencing of tumors. Cancer Letters. 2015;365(1):1–10. | ||
Marchetti C, Palaia I, De Felice F, et al. Tyrosine-kinases inhibitors in recurrent platinum-resistant ovarian cancer patients. Cancer Treatment Reviews. 2016;42:41–46. | ||
Rustin GJ, van der Burg ME, Griffin CL, et al. Early versus delayed treatment of relapsed ovarian cancer (MRC OV05/EORTC 55955): a randomised trial. Lancet. 2010;376(9747):1155–1163. | ||
Liu M, Mor G, Cheng H, et al. High frequency of putative ovarian cancer stem cells with CD44/CK19 coexpression is associated with decreased progression-free intervals in patients with recurrent epithelial ovarian cancer. Reproductive Sciences (Thousand Oaks, Calif.). 2013;20(5):605–615. | ||
Meng E, Long B, Sullivan P, et al. CD44+/CD24- ovarian cancer cells demonstrate cancer stem cell properties and correlate to survival. Clinical & Experimental Metastasis. 2012;29(8):939–948. | ||
Steffensen KD, Alvero AB, Yang Y, et al. Prevalence of epithelial ovarian cancer stem cells correlates with recurrence in early-stage ovarian cancer. Journal of Oncology. 2011;2011:620523. | ||
Alfonso JC, Jagiella N, Nunez L, Herrero MA, Drasdo D. Estimating dose painting effects in radiotherapy: a mathematical model. PLoS One. 2014;9(2):e89380. | ||
Welsh JS, Lock M, Harari PM, et al. Clinical implementation of adaptive helical tomotherapy: a unique approach to image-guided intensity modulated radiotherapy. Technology in Cancer Research & Treatment. 2006;5(5):465–479. | ||
Rong Y, Welsh JS. Dosimetric and clinical review of helical tomotherapy. Expert Review of Anticancer Therapy. 2011;11(2):309–320. | ||
Rong Y, Evans J, Xu-Welliver M, et al. Dosimetric evaluation of intensity-modulated radiotherapy, volumetric modulated arc therapy, and helical tomotherapy for hippocampal-avoidance whole brain radiotherapy. PLoS One. 2015;10(4):e0126222. | ||
Nagano T, Kim YH, Goto K, et al. Re-challenge chemotherapy for relapsed non-small-cell lung cancer. Lung Cancer (Amsterdam, Netherlands). 2010;69(3):315–318. | ||
Korkaya H, Wicha MS. Selective targeting of cancer stem cells: a new concept in cancer therapeutics. BioDrugs: Clinical Immunotherapeutics, Biopharmaceuticals and Gene Therapy. 2007;21(5):299–310. | ||
Takebe N, Miele L, Harris PJ, et al. Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells: clinical update. Nature Reviews. Clinical Oncology. 2015;12(8):445–464. | ||
Islam F, Gopalan V, Smith RA, Lam AK. Translational potential of cancer stem cells: a review of the detection of cancer stem cells and their roles in cancer recurrence and cancer treatment. Experimental Cell Research. 2015;335(1):135–147. | ||
Zhou J, Wulfkuhle J, Zhang H, et al. Activation of the PTEN/mTOR/STAT3 pathway in breast cancer stem-like cells is required for viability and maintenance. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(41):16158–16163. | ||
Zhang X, Zhang S, Liu Y, et al. Effects of the combination of RAD001 and docetaxel on breast cancer stem cells. European Journal of Cancer (Oxford, England: 1990). 2012;48(10):1581–1592. | ||
Zhu Y, Zhang X, Liu Y, et al. Antitumor effect of the mTOR inhibitor everolimus in combination with trastuzumab on human breast cancer stem cells in vitro and in vivo. Tumour Biology: The Journal of the International Society for Oncodevelopmental Biology and Medicine. 2012;33(5):1349–1362. | ||
Liu Y, Zhang X, Liu J, Hou G, Zhang S, Zhang J. Everolimus in combination with letrozole inhibit human breast cancer MCF-7/Aro stem cells via PI3K/mTOR pathway: an experimental study. Tumour Biology: The Journal of the International Society for Oncodevelopmental Biology and Medicine. 2014;35(2):1275–1286. | ||
Lonardo E, Frias-Aldeguer J, Hermann PC, Heeschen C. Pancreatic stellate cells form a niche for cancer stem cells and promote their self-renewal and invasiveness. Cell Cycle (Georgetown, Tex.). 2012;11(7):1282–1290. | ||
Lonardo E, Hermann PC, Mueller MT, et al. Nodal/Activin signaling drives self-renewal and tumorigenicity of pancreatic cancer stem cells and provides a target for combined drug therapy. Cell Stem Cell. 2011;9(5):433–446. | ||
Arasada RR, Amann JM, Rahman MA, Huppert SS, Carbone DP. EGFR blockade enriches for lung cancer stem-like cells through Notch3-dependent signaling. Cancer Research. 2014;74(19):5572–5584. | ||
Huang CP, Tsai MF, Chang TH, et al. ALDH-positive lung cancer stem cells confer resistance to epidermal growth factor receptor tyrosine kinase inhibitors. Cancer Letters. 2013;328(1):144–151. | ||
Shien K, Toyooka S, Yamamoto H, et al. Acquired resistance to EGFR inhibitors is associated with a manifestation of stem cell-like properties in cancer cells. Cancer Research. 2013;73(10):3051–3061. | ||
Yoshii Y, Kiyono Y, Waki A, et al. Hypoxia imaging agent 64Cu-ATSM visualizes cancer stem cell-rich regions within tumors. Journal of Nuclear Medicine. 2009;50(Suppl 2):1554. | ||
Wang X, Wei F, Liu A, et al. Cancer stem cell labeling using poly(L-lysine)-modified iron oxide nanoparticles. Biomaterials. 2012;33(14):3719–3732. | ||
Choi Y, Kim HS, Cho KW, et al. Noninvasive identification of viable cell populations in docetaxel-treated breast tumors using ferritin-based magnetic resonance imaging. PLoS One. 2013;8(1):e52931. | ||
Liu H, Patel MR, Prescher JA, et al. Cancer stem cells from human breast tumors are involved in spontaneous metastases in orthotopic mouse models. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(42):18115–18120. | ||
Lathia JD, Gallagher J, Myers JT, et al. Direct in vivo evidence for tumor propagation by glioblastoma cancer stem cells. PLoS One. 2011;6(9):e24807. | ||
Choi JW, Kim JK, Choi M, Kim YR, Yun SH. In vivo imaging of Lgr5-positive cell populations using confocal laser endomicroscopy during early colon tumorigenesis. Endoscopy. 2014;46(12):1110–1116. | ||
Heyn C, Ronald JA, Ramadan SS, et al. In vivo MRI of cancer cell fate at the single-cell level in a mouse model of breast cancer metastasis to the brain. Magnetic Resonance in Medicine. 2006;56(5):1001–1010. | ||
Heryanto YD, Achmad A, Taketomi-Takahashi A, Tsushima Y. In vivo molecular imaging of cancer stem cells. American Journal of Nuclear Medicine and Molecular Imaging. 2015;5(1):14–26. | ||
Tsurumi C, Esser N, Firat E, et al. Non-invasive in vivo imaging of tumor-associated CD133/prominin. PLoS One. 2010;5(12):e15605. | ||
Gaedicke S, Braun F, Prasad S, et al. Noninvasive positron emission tomography and fluorescence imaging of CD133+ tumor stem cells. Proceedings of the National Academy of Sciences of the United States of America. 2014;111(6):E692–E701. | ||
Beck S, Jin X, Yin J, et al. Identification of a peptide that interacts with Nestin protein expressed in brain cancer stem cells. Biomaterials. 2011;32(33):8518–8528. | ||
Park J, Ku M, Kim E, et al. CD44-specific supramolecular hydrogels for fluorescence molecular imaging of stem-like gastric cancer cells. Integrative Biology: Quantitative Biosciences from Nano to Macro. 2013;5(4):669–672. | ||
Sweeney TJ, Mailander V, Tucker AA, et al. Visualizing the kinetics of tumor-cell clearance in living animals. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(21):12044–12049. | ||
Vlashi E, Kim K, Lagadec C, et al. In vivo imaging, tracking, and targeting of cancer stem cells. Journal of the National Cancer Institute. 2009;101(5):350–359. | ||
Yoshii Y, Furukawa T, Kiyono Y, et al. Internal radiotherapy with copper-64-diacetyl-bis (N4-methylthiosemicarbazone) reduces CD133+ highly tumorigenic cells and metastatic ability of mouse colon carcinoma. Nuclear Medicine and Biology. 2011;38(2):151–157. | ||
Miranda-Lorenzo I, Dorado J, Lonardo E, et al. Intracellular autofluorescence: a biomarker for epithelial cancer stem cells. Nature Methods. 2014;11(11):1161–1169. | ||
Li M, Zhang B, Zhang Z, et al. Stem cell-like circulating tumor cells indicate poor prognosis in gastric cancer. BioMed Research International. 2014;2014:981261. | ||
Galanzha EI, Kim JW, Zharov VP. Nanotechnology-based molecular photoacoustic and photothermal flow cytometry platform for in-vivo detection and killing of circulating cancer stem cells. Journal of Biophotonics. 2009;2(12):725–735. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.